

Abstract
Herein we report the self-amplified depolymerization of an aryl oligo(thiourethane) (OTU) for the release of COS/H2S. The OTU was synthesized via polyaddition of 4-isothiocyanatobenzyl alcohol and end-capped with an aryl azide. The aryl azide chain-end was reduced by tris(2-carboxyethyl)phosphine or H2S to the corresponding aniline, resulting in depolymerization (i.e., self-immolation) and the release of COS/H2S. Depolymerization was monitored by 1H NMR and UV-Vis spectroscopy, and the released COS was converted into H2S by the ubiquitous enzyme carbonic anhydrase in aqueous media.
Graphical Abstract
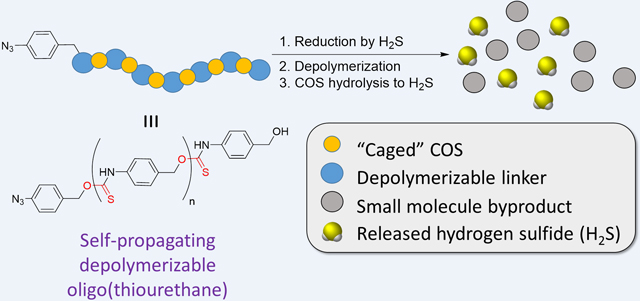
Depolymerizable or degradable polymers (i.e., self-immolative polymers) are a class of materials which depolymerize in the presence of a specific stimulus, typically resulting in the release of small molecules.1 These stimuli-responsive depolymerizable polymers are comprised of three discrete portions: a triggering moiety, a spacer, and an output.2 The triggering moiety is a functional group that responds to a specific stimulus such as light,3 redox reactions,4 or a small molecule.5, 6 Application of the stimulus results in the formation of an unstable intermediate, typically on the polymer chain-end, which causes the depolymerization of a single monomer unit and the subsequent regeneration of the unstable intermediate (Scheme 1A). This process repeats until each monomer unit in the polymer chain has depolymerized. The utility of depolymerizable polymers derives from the release of the output molecule, which occurs concurrently with depolymerization. Output molecules are often quantifiable (i.e., fluorescent small molecules), making depolymerizable polymers intriguing motifs for signal detection and amplification.7, 8 Despite this progress in depolymerizable polymers for detection of biological events, few depolymerizable polymers have been developed that release biologically active output molecules.1, 9 Here we envisioned developing an depolymerizable polymer capable of releasing hydrogen sulfide (H2S), a biological signalling gas.
Scheme 1.
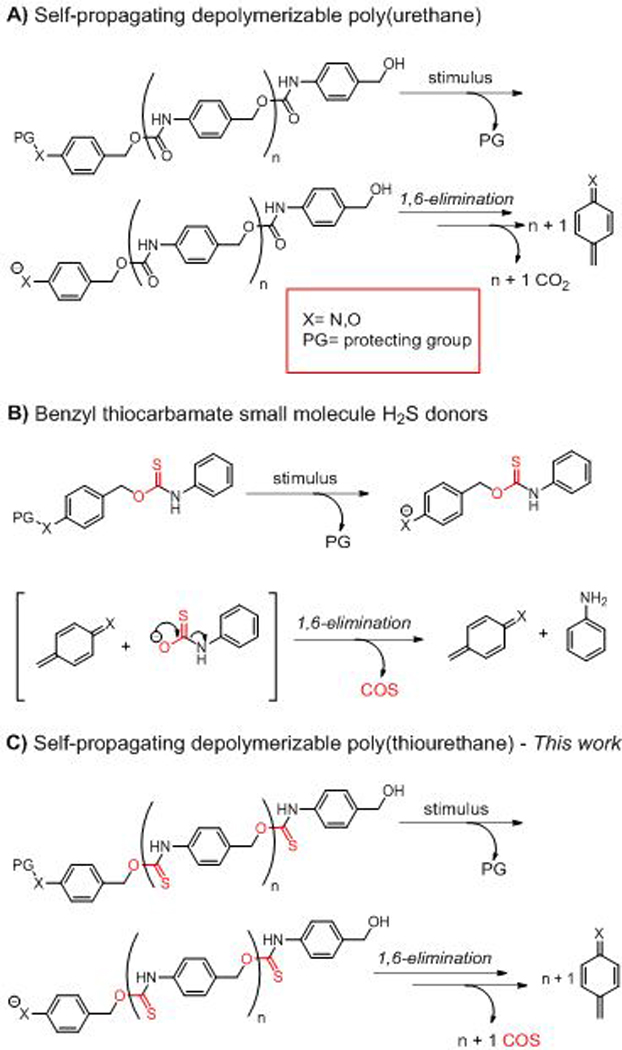
A) Depolymerizable aryl poly(urethanes). B) Small molecule benzyl thiocarbamate COS/H2S donors developed by Pluth and coworkers. C) Proposed COS/H2S releasing depolymerizable polyurethane.
In 1996 H2S was established as a critical signalling molecule in mammals.10 As such, alterations in H2S production in the body have systemic consequences and have been linked to a variety of disease states including cardiovascular disease,11 cystic fibrosis,12 and diabetes,13 among others.14 A commonality in these disease states is a decrease in endogenous H2S production.15 As a result, exogenous delivery of H2S through inorganic donor salts (NaSH, Na2S) or synthetic donor compounds may mitigate disease symptoms and improve healing.16–19 To aid in understanding H2S physiology and investigate possible benefits of H2S therapy, several types of small molecule and polymeric H2S donors have been developed over the past few years.20–24 However, many classes of synthetic donors release only one equivalent of H2S per equivalent of consumed trigger, which is often a redox-active thiol. This net neutral redox balance may ultimately limit the long-term efficacy of common of synthetic donors and complicate in vivo analysis.25, 26 Thus, H2S donors that release multiple equivalents of H2S per triggering event may be critical in furthering the therapeutic benefit of exogenous H2S delivery.
In an effort to develop a depolymerizable polymer that can release multiple equivalents of H2S in response to low concentrations of H2S itself, we were inspired by Pluth and coworkers’ 2016 report that introduced benzyl thiocarbamates as a class of small molecule, dual carbonyl sulfide (COS)/H2S donors based on the benzyl elimination reaction (Scheme 1B).27 These benzyl thiocarbamates released COS via a 1,6-elimination reaction triggered by a reducing stimulus, such as H2S itself. The elimination reaction led to release of the desired COS payload via an unstable thiocarbamic acid intermediate (Scheme 1B). The released COS was then rapidly hydrolyzed to H2S via the action of the ubiquitous enzyme carbonic anhydrase (CA). Since this seminal work on small molecule thiocarbamates as dual COS/H2S donors, Pluth and coworkers have demonstrated the ability to trigger COS/H2S release in the presence of other stimuli including hydrogen peroxide,28 cysteine,29 light,30 and others.29, 31 We envisioned that leveraging benzyl thiocarbamates as the repeat unit of a depolymerizable polymer would provide an exciting opportunity for a platform from which endogenous H2S production may be amplified, creating a self-amplified depolymerizable polymer (SADP) (Scheme 1C).
In order to synthesize a COS/H2S-donating SADP, we first set out to prepare monomer that could undergo a step-growth polyaddition to make the desired poly(thiourethane) (PTU). Typically, PTUs are prepared as S-alkyl thiocarbamates through the reaction of thiols and isocyanates mediated by the soft Lewis-acid catalyst dibutyltin dilaurate (DBTDL).32–34 Unfortunately, the S-alkyl thiocarbamate isomer is a less efficient COS donor than the O-alkyl isomer, likely stemming from an unfavorable Gibb’s free energy (ΔG) for the COS-releasing reaction.35 In contrast, the O-alkyl thiourethane isomer readily decomposes to form COS via the 1,6-benzyl elimination. Accordingly, synthesis of a depolymerizable O-alkyl thiourethane repeating unit would require a monomer containing both an aryl isothiocyanate (Ar–NCS) and a benzyl alcohol to facilitate efficient COS release.
To meet this challenge, we designed and synthesized a bifunctional monomer containing the desired aryl isothiocyanate and benzyl alcohol functional groups (SADM1, Scheme 2). Starting from 4-nitrobenzaldehyde, a one-pot reduction of both the nitro and aldehyde was accomplished by addition of sodium borohydride (NaBH4) and palladium on carbon (Pd/C, 5 mol %) in water to give 4-aminobenzyl alcohol (4-AB). To access the aryl isothiocyanate, a method developed by Boas and coworkers36 was employed wherein the aniline of 4-AB was converted into the corresponding dithiocarbamate salt by reaction with carbon disulfide in the presence of triethylamine, followed by addition of Boc anhydride, which led to the spontaneous evolution of COS gas and tert-butyl alcohol and the formation of the desired aryl isothiocyanate (SADM1).
Scheme 2.
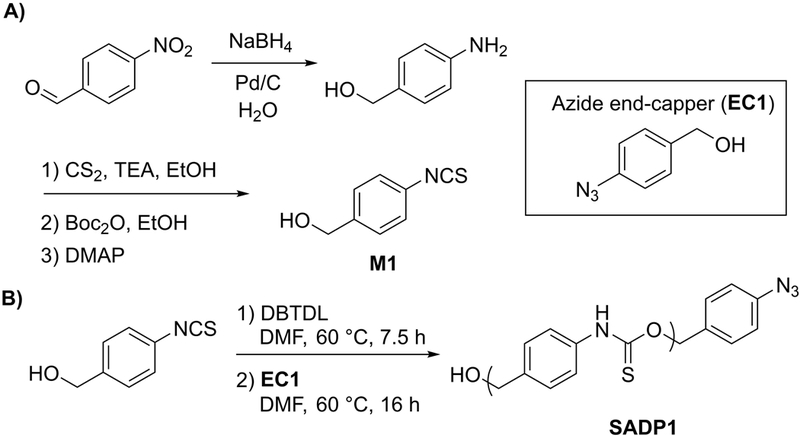
A) Synthesis of SADM1. B) Synthesis of aryl azide end-capped SADP1.
With the desired AB monomer in hand, we envisioned that it would undergo polyaddition in the presence of DBTDL, as this catalyst has been used successfully in analogous, non-sulfur-containing systems. Thus, the polymerization of SADM1 was carried out in dry DMF (1 M) at 60 °C under N2 in the presence of DBTDL (5 mol %). Under these conditions we observed a plateau in monomer conversion (p) after 7.5 h at approximately 85 % (Figure S11). At this stage in the polymerization, 1 equiv of 4-azidobenzylalcohol (EC1) was added as an end-capping reagent, and the reaction mixture was allowed to stir overnight. The azide end-capped SADP (SADP1) was then isolated as a yellow powder after precipitating from Et2O. The presence of the aryl azide on the oligomer chain end was confirmed by FTIR spectroscopy (Figure S9), and the oligomer Mn was measured to be 1.6 kg/mol by 1H NMR end-group analysis (degree of polymerization (Xn) of ~7), which is consistent with the expected Mn for 85% conversion. Size exclusion chromatography (SEC) with light scattering detection was attempted, but the low molecular weight of the oligomer coupled with its low dn/dc value in the elution solvent (THF) prevented accurate analysis.
To explain the limited conversion of monomer under these conditions, the reaction Gibbs energy for a model small molecule reaction between benzyl alcohol and phenyl isothiocyanate was calculated using density functional theory. The 60 oC reaction Gibbs energy using the M06–2X functional and aug-cc-pVDZ basis with implicit DMF solvation is -2.28 kcal/mol. Combining Carother’s equation for step-growth polymerizations with the reaction Gibbs energy relationship to the equilibrium constant (K) (equation 1) we calculated p to be 0.85 for this system under ideal conditions. These calculations indicate a maximum degree of polymerization (Xn) of 6.7. The polymerization of SADM1 routinely yielded oligomers with Xn ~7, in good agreement with the predictions. Therefore, the limited conversion observed experimentally appears to be due to the small polymerization exoergicity under the experimental conditions.
| (Equation 1) |
We next investigated the depolymerization of azide-terminated SADP1 by 1H NMR spectroscopy. For these experiments, tris(2-carboxyethyl)phosphine (TCEP, 1.7 equiv with respect to azide chain ends) was employed as an organo-soluble reducing agent to facilitate the reduction of the chain-end aryl azide, leading to depolymerization and ultimately COS release. In order to follow the reaction, changes in the peaks attributed to SADP1 were monitored as well as the generation of 4-AB, an expected major byproduct of SADP1 depolymerization. Due to the low water solubility of SADP1 at concentrations required for NMR spectroscopy, 1H NMR analysis was performed in DMSO-d6, which dramatically decreases reaction rates for 1,6-elimination reactions relative to water.37 However, despite the slow reaction rate, 1H NMR analysis revealed a decrease in the broad heteroatomic peak attributed to the thiocarbamate repeating unit proton (Ar–NHC(S)O) as well as the appearance of well resolved aromatic doublets consistent with 4-AB. Under the same conditions, depolymerization of a benzyl alcohol end-capped control SADP (Ctrl-SADP) occurred more slowly in the presence of TCEP (Figures S12 and S13), indicating that reduction of the chain-end azide is critical for initiating depolymerization.
Monitoring depolymerization by UV-Vis spectroscopy allowed for use of aqueous media because lower concentrations of SADP1 could be employed than in the 1H NMR spectroscopy experiments. For these experiments, SADP1 was dissolved in PBS buffer (pH 7.4) containing DMSO (2 % v/v) and cetrimonium bromide (CTAB) to aid in solubility. Prior to adding a reducing agent, a broad absorbance for SADP1 was observed at 284 nm (Figure 1A). Upon addition of a reducing agent (TCEP or Na2S), the absorbance maximum began to gradually shift to lower wavelength over the course of 2 h. We attribute the shift in λmax for the oligomer to depolymerization and the generation of 4-AB. Isosbestic points at 240 and 271 nm were observed, although they shifted slightly during the course of the analysis, which we attribute to low MW SADP species and oxidized byproducts of 4-AB.
Figure 1.
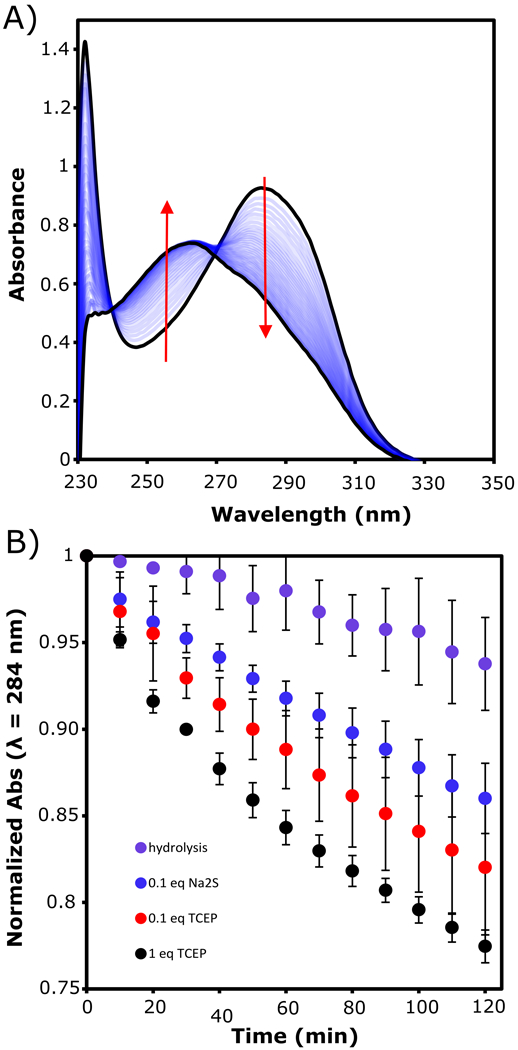
A) Representative UV-Vis spectra of SADP1 (10 μM) prior to the addition of reducing agent (λmax = 284 nm) and 2 h after the addition of reducing agent (λmax = 254 nm). B) Change in absorbance of SADP1 at 284 nm over time in the presence and absence of reducing agents. Data are normalized to the absorbance at t0, prior to the addition of reducing agent. All depolymerization experiments were run in PBS buffer (pH 7.4) with 2% DMSO, 1 mM CTAB, and 300 nM CA.
In order to better evaluate the depolymerization kinetics of SADP1, a series of UV-Vis experiments were conducted using varying amounts of TCEP, Na2S, and no trigger (hydrolysis) in the presence of CA (which converts COS into H2S). By plotting the change in absorbance at 284 nm (λmax of SADP1) over time, a clear trend in reaction kinetics was observed. Addition of 1 equiv TCEP relative to aryl azide SADP1 chain-end gave the greatest decrease in λmax for SADP1 over the course of 2 h. When the amount of TCEP was reduced to 0.1 equiv, a concomitant decrease in rate was observed. Addition of Na2S (0.1 equiv) as an alternative means of reducing the SADP1 chain-end gave data similar to that for the addition of 0.1 equiv TCEP, indicating that TCEP and Na2S are roughly equal in reduction capacity under these conditions and that both generate multiple equivalents of H2S per equivalent of added trigger. Additionally, the same shift in λmax was observed without the addition of any reducing agent, albeit at a much slower rate, indicating that hydrolysis, likely of the thiourethane units, contributes to the depolymerization of SADP1.
In light of the data indicating that hydrolysis is a viable mechanism for depolymerization of SADP1, UV-Vis experiments of Ctrl-SADP were conducted to investigate whether the reducing agent (Na2S) or the H2S released in the depolymerization reaction might also contribute to degradation of the benzyl thiocarbamate moiety. The spectral profile of Ctrl-SADP is very similar to SADP1, with a broad absorbance centred at 284 nm (Figure S14). However, there was no significant change the λmax of Ctrl-SADP in the presence or absence of Na2S, indicating that sulfide does not degrade the benzyl thiocarbamate moiety. Therefore, we conclude that the observed enhanced rate of SADP1 depolymerization in the presence of Na2S compared with water is due to the reduction of the chain-end azide and subsequent depolymerization.
Lastly, analysis of the H2S release profile for SADP1 was performed using an H2S-selective electrochemical probe. H2S release experiments were performed in PBS buffer (pH 7.4) at 100 μM SADP1 with the addition of CTAB, similar to the UV-Vis experiments. Addition of Na2S (0.1 equiv) as the reducing agent to a solution of SADP1 containing CA generated an initial spike in H2S due to the presence of Na2S, followed by a rapid decrease in H2S concentration, followed by steady generation of H2S, ultimately reaching a peak concentration after 220 min (Figure 2, purple curve). In contrast, addition of Na2S (0.1 equiv) to a solution of SADP1 in the absence of CA generated a spike in H2S concentration followed by a rapid return to baseline, similar to probe response when only Na2S is added (orange curve). This result indicates that the released COS from SADP1 was not converted into H2S, as expected when CA is not present. The apparently low peak H2S concentration (0.9 μM) is due to the long peaking time of SADP1, where low peak concentration is a result of slow H2S generation combined with COS/H2S volatilization and H2S oxidation. Taken together, results from these H2S release experiments demonstrate that SADP1 successfully generates H2S in the presence of submolar quantities of a Na2S trigger, acting in an autoinductive self-propagating amplification reaction38 where H2S derived from hydrolysis of COS generates increasing amounts of H2S.
Figure 2.
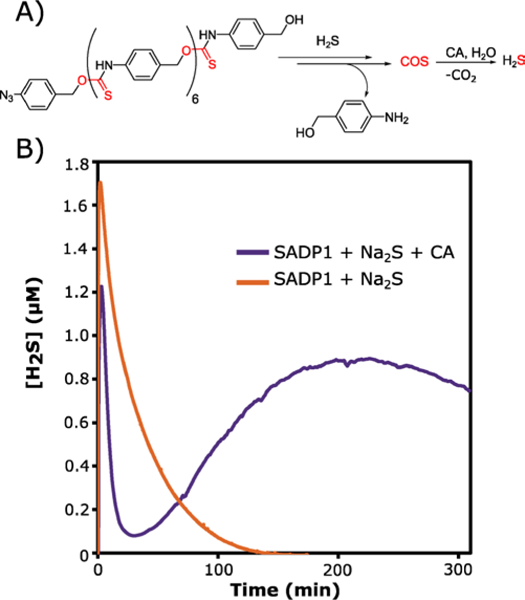
A) Scheme depicting H2S release from SADP1. B) H2S release data from SADP1 (100 μM) in the presence of Na2S (0.1 equiv) with 300 nM CA (purple curve) and without CA (orange curve).
Conclusions
The first COS/H2S-releasing SADP is reported. The depolymerizable oligo(thiourethane) was synthesized in a polyaddition reaction from a bifunctional monomer containing an aryl isothiocyanate and a benzyl alcohol. The oligomer structure was confirmed by 1H NMR and FTIR spectroscopy. Aryl azide-terminated SADP1 underwent depolymerization in the presence of reducing agents, with a greater concentration of reducing agent resulting in an enhanced reaction rate. Additionally, upon addition of submolar concentrations of reducing agents, including Na2S, SADP1 demonstrated COS release, which was converted to H2S in the presence of CA, generating multiple equivalents of H2S per triggering event in a manner consistent with signal amplification.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (DMR-1454754), the National Institutes of Health (R01GM123508), and the Dreyfus foundation. The authors acknowledge Ryan Carrazzone for help with polymer analysis.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Peterson GI, Larsen MB and Boydston AJ, Macromolecules, 2012, 45, 7317–7328. [Google Scholar]
- 2.Carl PL, Chakravarty PK and Katzenellenbogen JA, J. Med. Chem., 1981, 24, 479–480. [DOI] [PubMed] [Google Scholar]
- 3.de Groot FMH, Albrecht C, Koekkoek R, Beusker PH, Scheeren HW, Amir RJ, Pessah N, Shamis M and Shabat D, Angew. Chem. Int. Ed., 2003, 42, 4411–4411. [Google Scholar]
- 4.Weinstain R, Baran PS and Shabat D, Bioconjugate Chem., 2009, 20, 1783–1791. [DOI] [PubMed] [Google Scholar]
- 5.Seo W and Phillips ST, J. Am. Chem. Soc., 2010, 132, 9234–9235. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Yeung K, Robbins JS, Pavlick RA, Wu M, Liu R, Sen A and Phillips ST, Angew. Chem. Int. Ed., 2012, 51, 2400–2404. [DOI] [PubMed] [Google Scholar]
- 7.Sagi A, Weinstain R, Karton N and Shabat D, J. Am. Chem. Soc., 2008, 130, 5434–5435. [DOI] [PubMed] [Google Scholar]
- 8.Redy O, Kisin-Finfer E, Sella E and Shabat D, Org. Biomol. Chem., 2012, 10, 710–715. [DOI] [PubMed] [Google Scholar]
- 9.Shabat D, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1569–1578. [Google Scholar]
- 10.Abe K and Kimura H, J. Neurosci., 1996, 16, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefer DJ, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang L, Li H, Tang C, Geng B, Qi Y and Liu X, Can. J. Physiol. Pharmacol., 2009, 87, 531–538. [DOI] [PubMed] [Google Scholar]
- 13.Szabo C, Antioxidants & redox signaling, 2012, 17, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powell CR, Dillon KM and Matson JB, Biochem. Pharmacol., 2018, 149, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang R, FASEB J., 2002, 16, 1792–1798. [DOI] [PubMed] [Google Scholar]
- 16.Zhao W, Zhang J, Lu Y and Wang R, The EMBO Journal, 2001, 20, 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whiteman M, Cheung NS, Zhu YZ, Chu SH, Siau JL, Wong BS, Armstrong JS and Moore PK, Biochem. Biophys. Res. Commun., 2005, 326, 794–798. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Yang C, Organ C, Li Z, Bhushan S, Otsuka H, Pacheco A, Kang J, Aguilar HC, Lefer DJ and Xian M, J. Med. Chem., 2015, 58, 7501–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z, Organ CL, Zheng Y, Wang B and Lefer DJ, Circulation, 2016, 134, A17903–A17903. [Google Scholar]
- 20.Foster JC, Powell CR, Radzinski SC and Matson JB, Org. Lett., 2014, 16, 1558–1561. [DOI] [PubMed] [Google Scholar]
- 21.Powell CR, Foster JC, Okyere B, Theus MH and Matson JB, J. Am. Chem. Soc., 2016, 138, 13477–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao Z, Bonnard T, Shakouri-Motlagh A, Wylie RAL, Collins J, White J, Heath DE, Hagemeyer CE and Connal LA, Chem. Eur. J., 2017, 23, 11294–11300. [DOI] [PubMed] [Google Scholar]
- 23.Ercole F, Mansfeld FM, Kavallaris M, Whittaker MR, Quinn JF, Halls ML and Davis TP, Biomacromolecules, 2016, 17, 371–383. [DOI] [PubMed] [Google Scholar]
- 24.Zheng Y, Yu B, Ji K, Pan Z, Chittavong V and Wang B, Angew. Chem. Int. Ed. Engl., 2016, 55, 4514–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Wang H and Xian M, J. Am. Chem. Soc., 2011, 133, 15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martelli A, Testai L, Citi V, Marino A, Pugliesi I, Barresi E, Nesi G, Rapposelli S, Taliani S, Da Settimo F, Breschi MC and Calderone V, ACS Med. Chem. Lett., 2013, 4, 904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steiger AK, Pardue S, Kevil CG and Pluth MD, J. Am. Chem. Soc., 2016, 138, 7256–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y and Pluth MD, Angew. Chem. Int. Ed., 2016, 55, 14638–14642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Steiger AK and Pluth MD, Chem. Commun., 2018, 54, 4951–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Bolton SG and Pluth MD, Org. Lett., 2017, 19, 2278–2281. [DOI] [PubMed] [Google Scholar]
- 31.Steiger AK, Yang Y, Royzen M and Pluth MD, Chem. Commun., 2017, 53, 1378–1380. [DOI] [PubMed] [Google Scholar]
- 32.Kultys A, Rogulska M and Pikus S, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1770–1782. [Google Scholar]
- 33.Eglin D, Griffon S and Alini M, Journal of Biomaterials Science -- Polymer Edition, 2010, 21, 477–491. [DOI] [PubMed] [Google Scholar]
- 34.Campiñez MD, Ferris C, de Paz MV, Aguilar-de-Leyva A, Galbis J and Caraballo I, Int. J. Pharm., 2015, 480, 63–72. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Y, Henthorn HA and Pluth MD, J. Am. Chem. Soc., 2017, 139, 16365–16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munch H, Hansen JS, Pittelkow M, Christensen JB and Boas U, Tetrahedron Lett., 2008, 49, 3117–3119. [Google Scholar]
- 37.Schmid KM, Jensen L and Phillips ST, J. Org. Chem., 2012, 77, 4363–4374. [DOI] [PubMed] [Google Scholar]
- 38.Sun X, Shabat D, Phillips ST and Anslyn EV, J. Phys. Org. Chem., 2018, 31, e3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.