

Abstract
Oncogenesis is a pathologic process driven by genomic aberrations, including changes in nucleotide sequences. The majority of these mutational events fall into two broad categories: inactivation of tumor suppressor genes (hypomorph, antimorph or amorph) or activation of oncogenes (hypermorph). The recent surge in genome sequence data and functional genomics research has ushered in the discovery of aberrations in a third category: gain-of-novel-function mutation (neomorph). These neomorphic mutations, which can be found in both tumor suppressor genes and oncogenes, produce proteins with entirely different functions from their respective wild-type proteins and the other morphs. The unanticipated phenotypic outcomes elicited by neomorphic mutations imply that tumors with the neomorphic mutations may not respond to therapies designed to target the wild-type protein. Therefore, understanding the functional activities of each genomic aberration to be targeted is crucial to devising effective treatment strategies that will benefit specific cancer patients.
Introduction
Cancer development involves a complex, yet specific series of genetic events that must be defined in order to truly advance cancer biology and develop novel therapies. The current explosion of genomic data presents an unprecedented opportunity to identify genomic aberrations as molecular targets or markers for cancer therapies. However, the translation of data into clinical practice is not straightforward. Only a small fraction of mutations in a tumor cell are functional drivers that might be paired with therapeutic options. The remainder are neutral passengers that do not confer growth advantages and/or are not targetable in the clinic. Over 2 million different coding region mutations have been identified across patients with only a small fraction of these being characterized functionally. Broadly, the majority of characterized drivers at the nucleotide level are either (1) hypomorphic, antimorphic or amorphic resulting in decreased or complete loss of protein activity or expression, or (2) hypermorphic, resulting in enhanced activity or expression. Accumulating reports now support the prevalence of a third important category of driver mutations in cancer: neomorph.
Neomorph was first described in Drosophila by the Nobel Prize winning geneticist, H.J. Muller in 1932. Neomorph represents a “change in the nature of the gene at the original locus, giving an effect not produced, or at least not produced to an appreciable extent, by the original normal gene…giving a gene that produces a new effect, foreign to the original gene, and not competing with the latter.”1 Essentially, unlike other morphs that result in either gain or loss of the activity mediated by the wild-type (WT) protein, neomorphs carry new and unanticipated functions through inducing unexpected alterations of cellular signaling pathways and networks. The neomorphic effects can alter the response to therapeutic targeting, even potentially converting a drug that inhibits the functional consequences of the WT protein into a drug that increases the outcomes driven by the neomorphic mutation. It is important the note that hypermorphic mutations by increasing the duration and magnitude of signaling or by being insensitive to feedback regulatory events can rewire intracellular networks in unexpected manners. This pseudo-neomorphic activity may contribute to unexpected lack of response to drugs targeted pathways normally activated by the parent WT molecule.
This review explores the increasingly appreciated role of neomorphic mutations in cancer promotion. These neomorphic mutations can be recurrent or infrequent mutations in cancer genes and are primarily discovered by experimental characterization. However, suggestions of neomorphic activity can frequently be gleaned by determining associations of specific mutations with unexpected transcriptional profiles or proteomic readouts in patient samples or cell lines. We also discuss the mechanisms engendered by an example set of neomorphic activities and the potential strategies to target tumors bearing these neomorphic mutations.
IDH1 and IDH2
Isocitrate dehydrogenases (IDHs) are key enzymes of the Krebs cycle, catalyzing the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) with concomitant reduction of NAD+ or NADP+ to NADH or NADPH respectively.2–4 There are two NADP(+)-dependent IDH isoforms in eukaryotic cells. IDH1 localizes to the cytoplasm while IDH2 is in the mitochondria. α-KG represents a key substrate in the Krebs cycle and also provides building blocks for the synthesis of fatty acids and amino acids. The products of IDH isoforms regulate dioxygenase-mediated hydroxylation reactions. The NADPH generated during the reaction by IDH1 or IDH2 fuels lipid biosynthesis and protects cells against oxidative damage.4, 5 Under hypoxic conditions, the reaction catalyzed by IDH1/2 is reversed and α-KG is converted to isocitrate which can in turn be converted to acetyl-CoA to support lipid metabolism.6, 7
To date, IDH1 mutations have been identified in up to 82% of grade II-III gliomas and secondary glioblastomas in multiple cohorts.8–11 IDH1 mutations have also been detected in other cancers including acute myeloid leukemia (AML; 10%) and prostate cancer (1–3%).9, 10, 12, 13 The frequency of IDH2 mutations is much lower than that of IDH1 in glioma (0–7%9–11) but is slightly higher in AML (15%10, 14, 15). The most common IDH1 mutation occurs at R132, which is located in the enzyme active site and forms hydrogen bonds with the α- and β-carboxylic groups of isocitrate.16 Initial studies suggested that IDH1 R132H was an antimorph (dominant negative mutant) by inhibiting the activity of WT IDH1 because R132H led to reduction in the affinity for isocitrate and α-KG production.17 Interestingly, however, a study by Dang et al. suggested that R132H is a neomorph that alters binding affinity for substrates.18 The authors first analyzed metabolites from R132H-expressing glioblastoma cells using liquid chromatography-electrospray ionization-mass spectrometry. The cells displayed elevated level of R(−)-2-hydroxyglutarate (2-HG), a molecule structurally similar to α-KG that thereby functions as a competitive inhibitor of α-KG-dependent enzymes.18 Analysis of the crystal structure of IDH1 suggested that R132H caused a shift in the active site resulting in decreased binding affinity of the enzyme for isocitrate and increased affinity for NADPH.18 Together, this promoted NADPH-dependent reduction of α-KG to 2-HG (Figure 1).
Figure 1. IDH1 R132H and IDH2 R172K/R140Q reduce α-KG to the oncometabolite 2-HG.

WT IDH2 and IDH2 reduce NADP+ and convert isocitrate into α-KG, which is a co-substrate of α-KG-dependent enzymes including PHD, TET2, and JHDM to regulate HIF-1α stability and epigenetic modifications. IDH1 R132H and IDH2 R172K/R140Q do not catalyze the conversion of isocitrate into α-KG but instead reduce α-KG to 2-HG. Competition of 2-HG with α-KG for binding to the α-KG-dependent enzymes results in DNA/histone methylation, HIF-1α stabilization and subsequent gene transcription (e.g. VEGF) to promote stem cell maintenance and angiogenesis.
The neomorphic function of IDH1 R132H was further extended to the mitochondrial homolog IDH2 by Ward and colleagues, who observed elevated 2-HG in a number of AML patients who did not have IDH1 mutations.14 Some of the tumors that lacked IDH1 mutation carried IDH2 R172K or R140Q mutation.9, 11, 14 IDH2 R172K is an analogue of IDH1 R132H and is the most frequent IDH2 hotspot mutation. These studies suggested that the production of the oncometabolite 2-HG may be a common consequence of the IDH1 and IDH2 hotspot mutations seen in cancers. Due to the functional redundancy of IDH1 and IDH2, mutations of the two gene were mutually exclusive.10 All reported IDH1 or IDH2 mutations are heterozygous, therefore the conversion of isocitrate into α-KG can still be fulfilled by the retained WT IDH1 or IDH2 allele. Intriguingly, these IDH1 or IDH2 neomorphic mutations tended to co-occur with TP53 mutation but demonstrate mutual exclusivity with aberrations of PTEN, EGFR, CDKN2A, or CDKN2B.9 The interactions of the IDH1 or IDH2 neomorph with the function of these genes remain to be investigated.
Several mechanisms underlying the oncogenic activities of the IDH1 and IDH2 neomorphs have been proposed. Specifically, 2-HG competes with α-KG for binding to the active sites of dioxygenases that use α-KG as a co-substrate for catalysis. Recent studies have identified at least 3 classes of α-KG–dependent dioxygenases that are targeted by 2-HG (Figure 1).19, 20 The first class includes prolyl hydroxylases (PHDs) which promote the degradation of HIF-1α, an essential transcription factor in response to hypoxia.21 IDH1 R132H upregulates HIF-1α and increases HIF-1α-inducible genes including VEGF, presumably by competing with α-KG and preventing PHD-mediated degradation of HIF-1α.17, 22 The second class involves jumonji C domain-containing histone demethylases (JHDMs) which target methylated lysine residues on histones thereby regulating gene expression. 2-HG competitively inhibits JHDMs including JMJD2A.23–25 The IDH1/2 neomorphs therefore inhibit histone demethylation, which in turn blocks the differentiation of non-transformed lineage-specific progenitor cells.24 Thirdly, IDH1/2 neomorphs inhibit TET2, a DNA demethylase enzyme through catalyzing 5-methylcytosine hydroxylation, thereby leading to global hypermethylation and epigenetic alterations.26, 27 The latter two mechanisms have broad implications for gene regulation.
The identification of 2-HG as a point of convergence for IDH1 and IDH2 neomorphic effects allows for the potential use of 2-HG as a clinical biomarker of IDH1/2 mutations. In fact, Ward et al. used 2-HG to screen AML patients for IDH1/2 mutations leading to the discovery of the function of IDH2 R140Q and R172K,14 demonstrating the potential of 2-HG measurement for prediction of IDH1/2 mutation status. Moreover, in vivo detection of 2-HG in body fluids could potentially be used as a surrogate for more invasive and less frequently obtained histopathologic assessments. Using proton magnetic resonance spectroscopy, 2-HG levels were detected in glioma and AML patients and correlated with the presence of IDH1/2 mutations.15, 28–31 IDH1/2 mutations also remain exciting therapeutic targets because the altered active sites are potentially selectively targetable by small molecule inhibitors, the mutations are stable even as the cancer progresses and IDH1/2 neomorphic mutations appear relatively early in cancer development.9, 32 The overall objectives would be to either inhibit the mutant enzymes to prevent 2-HG production, restore normal IDH1/2 function, deplete 2-HG and/or replace α-KG. Multiple preclinical studies have shown that IDH1/2 mutant-specific inhibitors could reverse the activity of the mutants and 2-HG accumulation.33–38 Remarkably, the growth inhibitory effect of the inhibitor was specific to neomorph-bearing cells but not to cells with WT IDH1/2.36 Indeed, some of these inhibitors have entered clinical trials and are showing encouraging results in patients with AML.39 Inhibitors that target dioxygenases have also been developed.40 Targeting the downstream targets of 2-HG presents may provide another opportunity. For example, blocking VEGF function in glioma patients with IDH1 neomorphic mutations resulted in improved progression free survival and overall survival.41
PIK3CA
Phosphatidylinositide-3-kinases (PI3Ks) are lipid kinases that catalyze the phosphorylation of phosphoinositides at the 3’-hydroxyl group generating second messengers that activate AKT and other pleckstrin homology domain-containing proteins to regulate diverse cellular activities.42–45 Among the three classes of PI3Ks, class 1, particularly the 1A subclass, is most widely studied for its role in oncogenesis. Class 1A PI3Ks are obligate heterodimers composed of a catalytic subunit (p110α, p110β, or p110δ) and a regulatory subunit p85 (p85α, p85β, p85γ and 4 additional splice variants). p85 stabilizes and inhibits p110 activity under quiescent conditions but the inhibition is relieved upon activation of growth factor signaling.46–49
PIK3CA, which encodes the p110α catalytic subunit, is frequently aberrant in human cancers with more than 5000 reported mutations.10 PIK3CA is within the 4th most amplified region across 11 cancer types50 and is the 2nd most frequently mutated gene and most commonly mutationally activated gene across 12 cancer types in the Cancer Genome Atlas (TCGA).51 PIK3CA was shown to be amplified in 1999,52 which was followed by demonstration of mutations in glioblastoma, and colon gastric, breast and lung cancers in 2004.53 PIK3CA mutations have subsequently been identified in other neoplasms including cancers of the endometrium, ovary, liver, stomach, and brain.54, 55 Although somatic mutations are scattered throughout PIK3CA, the majority of mutations cluster in three hotspots: E542 and E545 in the helical domain encoded by exon 9 and H1047 on exon 20 which encodes the kinase domain. E542 and E545 are commonly substituted with lysine while H1047 is often changed to arginine. These mutants are thought to be hypermorphs due to altered interaction with p85 or conformational changes of the activation loop, leading to constitutive low level activity of the E542 and E545 variants and increased sensitivity of the H1047 mutant to ligand activation and thus increased tumorigenicity. 56–59
Intriguingly, a recent study by Hao et al. demonstrated a neomorphic activity of the E545K mutant.60 Using mass spectrometry to identify interacting partners of the pulled-down wild-type p110α and E545K mutant, E545K was found to associate with insulin receptor substrate 1 (IRS1) independent of its canonical partner p85 (Figure 2). Indeed, p85 might compete with IRS1 for binding to E545K mutant p110α. Instead of solely relying on bound p85 for stabilization and membrane localization, the interaction with IRS1 stabilized E545K mutant p110α and enhanced the membrane association of E545K p110α, resulting in activation of this p110α neomorph and the PI3K pathway. The experiments were performed in serum-depleted conditions, suggesting a growth factor independent activation of neomorph signaling. Interestingly, other hot-spot PIK3CA mutations in the helical domain, including E542K, E545A, E545G, and Q546K, also gained interaction with IRS1 suggesting a generalizable activity. In stark contrast, this neomorphic effect was absent with the hot-spot mutations in the kinase domain (H1047R, H1047L, and G1049R).60 Two other independent studies showed that cell lines with E542K or E545K had lower phosphorylated AKT levels compared to those with kinase domain mutation or PTEN protein loss.61, 62 Instead, E542K or E545K induced robust activation of PDK1, which could be a consequence of enhanced membrane association of PDK1 driven by E542K or E545K.61 PDK1 promoted anchorage independent growth of breast cancer cells through the activity of downstream substrate SGK3.61 Whether this induction of PDK1 signaling is linked to the neomorphic p110α-IRS1 interaction warrants investigation. Further, some relatively rare PIK3CA mutations (so-called PIK3CA tail mutations) increased the phosphorylation of MEK1/2 (upstream kinase of ERK1/2),63 suggesting potential neomorphic activities of these mutations although this remains to be verified with future mechanistic studies.
Figure 2. p110α helical domain neomorphs bind to IRS1 for stabilization and activation independent of its canonical partner p85.
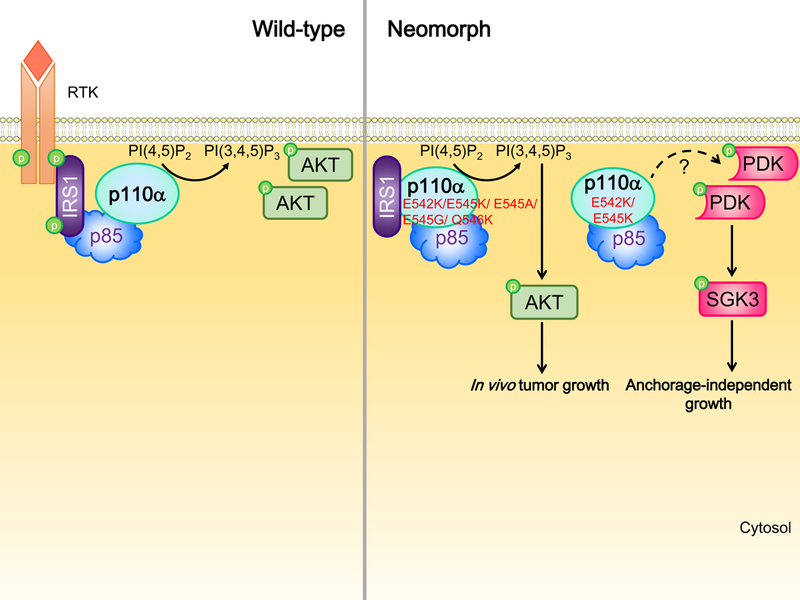
Upon RTK activation, p485 binds to phosphorylated IRS1 that thereby activates p110α. p110α neomorphs in the helical domain directly associate with IRS1 independent of p85 and growth factor stimulation. This association stabilizes and activates p110α, thereby promoting PI(3,4,5)P3 production and in vivo tumor growth through AKT signaling. Another independent study showed that these mutants increase membrane localization and activation of PDK1, which in turn promotes anchorage-independent growth through SGK3 activation. The detailed mechanism underlying the activation of PDK1 and whether PDK1 activation is a consequence of neomorphic activity remain to be revealed. RTK, receptor tyrosine kinase.
As these recurrent PIK3CA mutations are present in a large number of cancer patients, understanding the neomorphic functions of the helical domain mutants is required to capitalize on additional and perhaps more facile and effective targets for cancer therapy that are not associated with the H1047R mutation. Hao et al. alluded to these possibilities with a hydrocarbon-stapled mutant peptide that disrupted the neomorphic interaction between IRS-1 and the E545K mutant.60 Strikingly, the peptide inhibited AKT phosphorylation and xenograftic tumor growth of cancer cells with the neomorph.60 This exciting result points to a potential therapeutic approach allowing cancer patients carrying the neomorph to be treated with a small molecule drug that inhibits this interaction and that may have a high therapeutic index as they do not inhibit WT PIK3CA. Small molecule inhibitors (e.g. Akt/PDK dual inhibitors, PI3K/mTOR dual inhibitors, and MAPK inhibitors) targeting the downstream signaling pathways activated by the PIK3CA neomorphs could also represent potential therapeutic approaches.
PIK3R1
PIK3R1 is the 11th most commonly mutated gene across cancer lineages in TCGA.10 Somatic mutations in PIK3R1 are particularly prevalent in endometrial cancer (20–34%),55, 64 metastatic prostate cancer (11.5%),10 glioblastoma (11%),65 and colon cancer (4.5–10%).10, 66 PIK3R1 encodes p85α, which binds and regulates the p110 catalytic subunit as described above. To be more specific, the nSH2 and iSH2 domains of p85α bind to the helical and C2 domain of p110 respectively, to prevent p110 from thermal degradation and to inhibit its catalytic activity.67, 68 Approximately two-thirds of PIK3R1 mutations are located in the iSH2 domain of p85α. Driver mutations within this domain, including E439del, KS459delN, DKRMNS560del, D560Y, N564D, Q572*, R574fs, T576del, and QYL579L, are hypomorphs that have lost the ability to inhibit the catalytic activity of p110, thereby allowing constitutive, ligand-independent activation of the PI3K pathway.69 Other mutations at the N-terminal domains, such as the BH domain, which does not interact with p110, activate PI3K signaling through mechanisms independent of p110. p110α-unbound WT p85α forms homodimers via intermolecular interactions mediated by the SH3 and BH domains.55, 70, 71 The p85α homodimer binds to and stabilizes PTEN, resulting in inhibition of the PI3K pathway. Cancer patient-derived PIK3R1 mutations in this region (e.g. I133N, E160*, and I177N) are hypomorphs or antimorphs that disrupt the formation of p85α homodimers leading to PTEN destabilization and subsequent PI3K signaling activation.71
The hypomorphic or antimorphic mutations described above exclusively target PI3K pathway activation albeit through altering functions of different targets: p110 and PTEN. We have recently discovered a subset of PIK3R1 truncation mutations that activate MAPK pathways independent of p110.72 This group of neomorphic mutations, which clusters around the nSH2 domain and lacks intact SH2 domains, is found in cancers of the breast, colon, uterus, and ovary. Among these mutations, R348* is one of the most frequent recurrent PIK3R1 mutations (about 10% of all PIK3R1 mutations)10 and was first described as a driver mutation because of its ability to transform the interleukin 3 (IL3)-dependent murine myeloid BaF3 cells into IL3-independence.55 Subsequently, screening of the R348*-expressing cells against a library of “informer” therapeutic compounds targeting several major signaling pathways revealed increased sensitivity to AKT inhibition and unexpectedly to multiple inhibitors of MEK and JNK.72 Pathway activation profiling by reverse-phase protein arrays revealed that R348* and the neighboring mutation, L370fs, increased phosphorylation of members of the ERK and JNK pathways in addition to members of the PI3K pathway.72 In contrast, WT p85α or other mutants (E160*, DKRMNS560del, R574fs, and T576del) had no effect on MAPK pathway activation or sensitivity to MAPK inhibitors. The activation of MAPK signaling was independent of the canonical role of p85α in regulating PI3K or of its ability to regulate PTEN.72 This neomorphic role correlates with the production of truncated proteins that lack the p110-interacting SH2 domains. The activation of ERK occurs in the cytosol and involves upstream kinases including MEK1, MEK2 and BRaf, whereas the activation of JNK is a nuclear event mediated by the kinases MKK7 and MLK3. The intact BH domain and the absence of SH2 domains in the p85α neomorphs appear to provide the key to nuclear localization. The BH domain of the truncation protein interacts with Rho GTPases family members Cdc42 and Rac1, which facilitates translocation of the mutant from the cytosol to the nucleus (Figure 3).72 In the nucleus, the truncated p85 molecule acts as a scaffold for the tethering of signaling molecules along the JNK pathway, thereby promoting JNK activation. The activities of Cdc42 and Rac1 are also required for the activation of the ERK and JNK pathways.
Figure 3. PIK3R1 neomorphs activate PI3K and MAPK signaling.
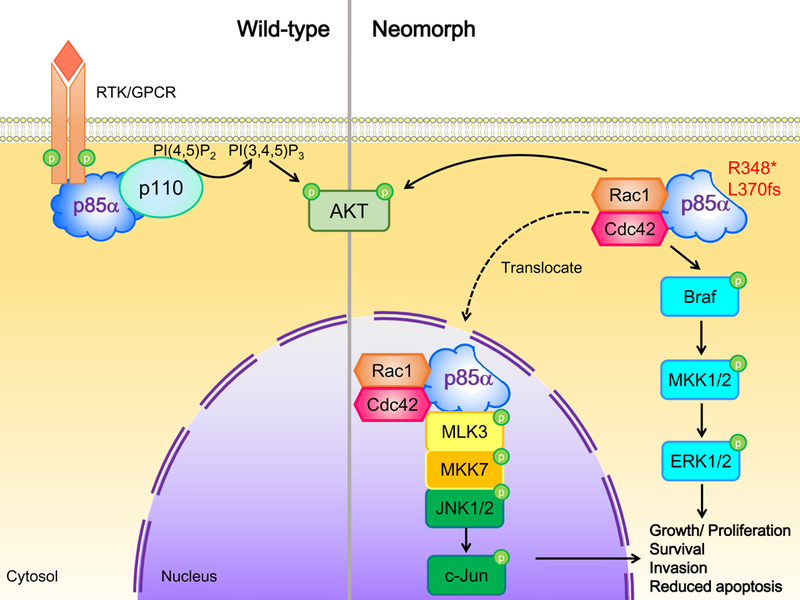
p6α neomorphs activate ERK and JNK signaling pathways to promote cell growth, survival, invasion and inhibit apoptosis. Binding of neomorphs to Cdc42 and Rac1 leads to phosphorylation of Braf, MKK1/2 and ERK1/2 in the cytoplasm. Cdc42 and Rac1 also facilitate the translocation of p85α neomorphs from the cytoplasm to the nucleus, where the neomorphs act as scaffolds for a MLK3/MKK7/JNK1/JNK2 complex that activates nuclear JNK signaling. The neomorphs also activate AKT but the mechanism is unknown. RTK, receptor tyrosine kinase. GPCR, G protein-coupled receptor.
Co-activation of both the MAPK and PI3K pathways can act as a compensatory survival mechanism to bypass the inhibition of either the MAPK or PI3K pathway.73, 74 Thus combinatorial treatment against both the MAPK and PI3K pathways may be required for optimal patient benefit in the context of the p85 neomorph.75 Indeed, dual blockade of the MAPK and PI3K pathways demonstrated enhanced anti-tumor effects in cancer cell lines and preclinical mouse models with KRAS mutation or co-mutations in both MAPK and PI3K pathways.76, 77 This approach is currently being explored in ongoing clinical trials.75, 78, 79 Whether PIK3R1 neomorphic mutations could be potential biomarkers predictive of clinical outcomes and response to pathway inhibitors awaits investigation.
PTEN
PTEN, a lipid and protein phosphatase, is one of the most frequently aberrant bona-fide tumor suppressors across a wide spectrum of tumor types. PTEN is the 9th most significantly deleted gene 50 and 3rd most frequently mutated gene across cancer lineages 51 in TCGA. WT PTEN is primarily a phosphoinositide 3-phosphatase with the key target being the PI3K pathway wherein PTEN opposes the action of PI3K by dephosphorylating the signaling lipids PIP(3,4,5)P3 to PI(4,5)P2 and PI(3,4)P2 to PI(4)P (Figure 4).80 In addition, PTEN possesses protein phosphatase activity that inhibits cancer cell invasion, however, whether this is a critical function of PTEN suppressor activity remains controversial.81–83 The tumor-suppressing activities of PTEN also extend to the nucleus, where PTEN, independent of its catalytic activity, induces p53-independent apoptosis and maintains genomic stability through preserving heterochromatin structure.84, 85 Hypomorphic mutants that lead to the inactivation of these diverse functions of PTEN are frequent in cancers.82, 84–86 Mutations leading to reduced PTEN expression have been reported. The C-terminus of PTEN regulates PTEN stability and cancer patient-derived truncated mutants that lack the C-terminus were unstable resulting in loss of PTEN expression.87, 88 WT PTEN activities are also suppressed by antimorphic PTEN mutants that bind to and inhibit the WT PTEN protein.87
Figure 4. PTEN A126G has altered enzymatic activity.
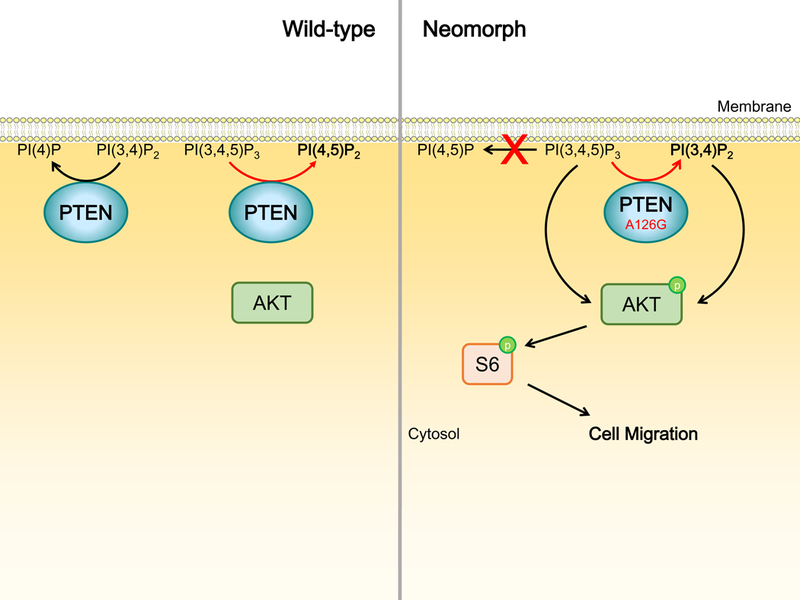
WT PTEN inhibits the activation of PI53K/AKT signaling by dephosphorylating PI(3,4,5)P3 and PI(3,4)P2 to PI(4,5)P2 and PI(4)P respectively. PTEN A126G, when compared to WT PTEN, dephosphorylates PI(3,4,5)P3 at reduced rate. Moreover, PTEN A126G displays a shift in enzymatic specificity toward 5’ phosphoinositides, resulting in the production of PI(3,4)P2 rather than PI(4,5)P2 which is produced by WT PTEN. The accumulation of PI(3,4,5)P2 and PI(3,4)P2 together leads to AKT and S6 phosphorylation and ultimately cell motility.
Intriguingly, a recent study has demonstrated neomorphic activity of a PTEN mutation detected in a prostate cancer patient.89 This neomorphic mutation, A126G, is not frequently observed and was only identified in one breast cancer case across all TCGA samples.10 The amino acid residue A126 is in close proximity to the catalytic C124 and R130 residues. These residues together constitute the phosphatase signature motif (referred to as the catalytic “P-loop”) at the bottom of the active site pocket. Interestingly, A126G results in an amino acid sequence identical to the family of voltage-sensing phosphatases, which are typical phosphoinositide 5-phosphatases. In silico modeling of the substrate-protein complex using available crystal structures of PTEN predicted a change in enzymatic specificity of A126G and this was confirmed by in vivo activity assays.89 In contrast to WT PTEN which dephosphorylates 3’ phosphoinositides, the mutant demonstrated a shift in specificity toward 5’ phosphoinositides, resulting in the production and accumulation of PI(3,4)P2 from PI(3,4,5)P3 (Figure 4). Consistent with a previous independent study 86, the mutant dephosphorylated PI(3,4,5)P3 at a reduced rate relative to WT PTEN. Moreover, the neomorphic activity is unique to A126G as mutation of A126 to several other amino acid residues (A126P, A126V and A126S) revealed no neomorphic effect. PTEN-null prostate cancer cells PC-3 transfected with A126G displayed increased AKT and S6 phosphorylation as well as cell migration compared with those expressing WT PTEN.89 These consequences could be due to a decrease in catalysis of PI(3,4,5)P3 compared with WT PTEN. Alternatively, the neomorphic product, PI(3,4)P2, can also bind to and activate a subset of PH domains, including that of AKT.90, 91
Accumulating evidence has shown that tumors with aberrations of PTEN including PTEN protein loss or PTEN mutation are more susceptible to treatment with PI3K pathway inhibitors.29, 92, 93 Further, PTEN aberrations can dictate the dependency on individual Class IA PI3K isoforms.29, 94 It would therefore be instructive to examine whether A126G-bearing cells are sensitive to PI3K pathway inhibition and particularly to any isoform-specific inhibitors of PI3K. Along this line, A126G-induced migration of PC-3 cells was abrogated by a PI3Kα inhibitor (BYL-719) and a PI3Kβ inhibitor (AZD6482).89 A broader array of PI3K pathway inhibitors and cell models will bring us closer to an answer to this question and to potential clinical applications.
TP53
TP53 encodes p53, which has been extensively studied for its role in tumorigenesis. TP53 is the most frequently mutated gene in human cancers (with the three most frequent hotspot mutations at R175, R248 and R273).50, 95 p53 was initially identified in complex with simian virus 40 large T-antigen and was increased in tumors relative to normal cells.96–99 Subsequently, many studies described the tumorigenic properties of p53,100, 101 only later to realize that these experiments had actually used mutated p53 isolated from tumor cells.102–104 It was demonstrated in 1989 that greater than half of colorectal tumors had loss of heterozygosity at the TP53 locus.105 Additional experiments proved that WT p53 overexpression was sufficient to suppress tumorigenesis and thereafter TP53 was considered as a tumor suppressor gene.104, 106–108
Cellular stress activates and stabilizes p53, which is present at very low levels in non-stressed normal cells. Activated p53 functions as a sequence specific tetrameric transcription factor that induces cell cycle arrest, apoptosis, senescence, DNA repair, and alters metabolism.109–112 As with most tumor suppressors, loss of p53 expression can result from frameshift or nonsense mutations leading to truncated or deleted proteins.113 However, most TP53 mutations in cancers are missense mutations resulting in single-base pair changes that still allow for the translation of full-length protein. These missense mutations, including the vast majority of hotspot mutations, generally cluster within the highly conserved DNA binding domain.10, 114, 115 Some of these missense mutations are antimorphs impairing the DNA-binding activity of the WT p53 by binding to WT protein.,116, 117 The idea that some TP53 mutations are not simply an antimorphs was suggested by Dittmer et al. in 1993 when p53 mutants (including R175H and R273H) were expressed in p53-null cells, resulting in a more aggressive tumorigenic phenotype compared with parental cells.118 Since then, evidence supporting TP53 mutations as neomorphs continues to accumulate108, 119.
p53 neomorphs can interact with a number of transcription factors and modulate their transcriptional output. These transcription factors include the p53-related proteins p63 and p73. Despite a remarkable structural similarity among the three family members (about 60% identity in the DNA-binding domain), whether p63 or p73 act as tumor suppressor genes or as oncogenes remains controversial.120 Remarkably, while p73 and p63 can regulate p53 target genes, p63 and p73 also have a number of unique target genes that are distinct from p53.121, 122 p63 and p73 form heterotetramers through the oligomerization domain but WT p53 does not interact with p63 or p73.123, 124 However, neomorphic mutations of p53 cause conformational changes in the p53 protein that allow interaction of mutant p53 with p63 and p73.125, 126 This physical interaction restrains p63 and p73 from binding to DNA and thereby blocks the activation of p63/p73 target genes leading to chemoresistance, migration, invasion, and metastasis (Figure 5a).125, 127–131 Neomorphic p53 mutants may also selectively recruit other cofactors into the transcriptional machinery resulting in aberrant gene regulation. WT p53 and p53 neomorphs (R175H, R273H and D281G) bind the transcription factor NF-Y but different cofactors are recruited (Figure 5b).132 WT p53 recruits cofactor histone deacetylase HDAC1, whereas p53 neomorphs recruit histone acetyltransferase p300. The p53 neomorph/NF-Y/p300 complexes lead to aberrant transcription of NF-Y target genes including the cell cycle control genes CCNA, CCNB, CDK1, and CDC25C.132 In contrast, the recruitment of HDAC1 by WT p53 represses transcription of these cell cycle genes.133 Thus, analogous to IDH1/2, WT and neomorphic mutant p53 can exert diametrically opposite effects. Further, p53 neomorphs can interact with a number of proteins that are not transcription factors. For example, the nuclease Mre11 interacts with two hotspot p53 neomorphs (R248W and R273H) but not WT p53 (Figure 5c).134 Mre11 is a component within the Mre11-Rad50-NBS1 (MRN) complex that acts as a sensor of DNA double-strand breaks (DSB) and is responsible for the activation of ataxia-telangiectasia mutated (ATM) kinase involved in DSB repair and other cellular functions.135, 136 The physical interaction between the p53 neomorphs and Mre11 impairs the recruitment of MRN to DNA damages sites and decreases ATM activation, thereby contributing to genetic instability.134
Figure 5. p53 neomorphic mutants gain novel interacting partners leading to pro-oncogenic activities.
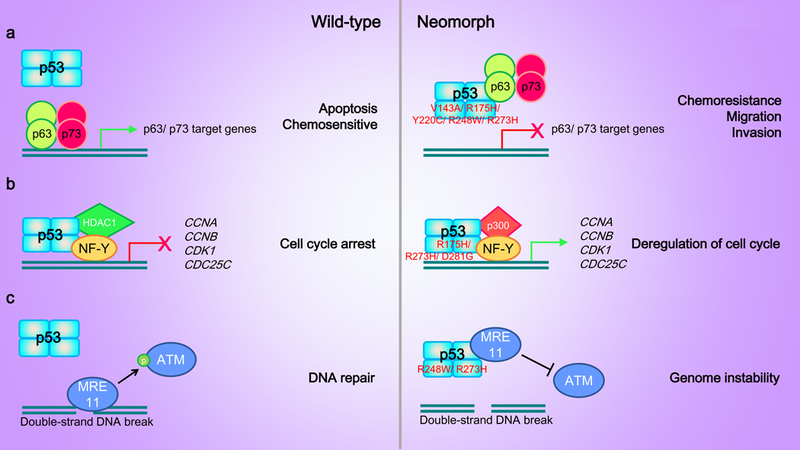
(a) p6 and p73 form homotetramers or heterotetramers that activate transcription of genes promoting cell apoptosis and sensitivity to chemotherapeutic drugs. p63 or p73 do not interact with WT p53. Neomorphic p53 mutants bind to p63 and p73 homotetramers or heterotetramers and inhibit p63-/p73-mediated gene transcription leading to increased chemoresistance, cell migration and invasion. (b) WT p53 binds to the transcription factor NF-Y and recruits the histone deacetylase HDAC1 as a co-factor to inhibit the transcription of NF-Y target genes involved in cell cycle progression such as CCNA, CCNB, CDK1, and CDC25C. Neomorphic p53 mutants recruit histone acetyltransferase p300, an opposing histone modifying enzyme, to promote the transcription of NF-Y target genes. (c) Neomorphic p53 mutants, but not WT p53, bind to Mre11. The interaction impairs the recruitment of Mre11 to double-strand DNA breaks and inhibits the activation of ATM thereby preventing DNA repair.
WT p53 is maintained at low cellular levels through a negative feedback loop involving the E3 ubiquitin ligase Mdm2, however, p53 mutants are stabilized and are expressed at high levels in tumor cells.137 R175H binds to heat shock protein 70 (HSP70) or HSP90, which inhibits the ability of Mdm2 to promote ubiquitination of the p53 mutant.138, 139 Therefore, one therapeutic strategy has been to destabilize the p53 neomorphs by either directly targeting HSP proteins or indirectly inhibiting upstream regulators of HSPs such as histone deacetylases (HDACs), which positively regulate HSP.140 SAHA, a FDA-approved HDAC inhibitor, selectively downregulates p53 neomorphs (R175H and R273H) but not WT p53.141 Inhibition of HSP90 through inhibiting HDAC6 releases HSP90 from p53 neomorphs, which then become susceptible to Mdm2-mediated degradation.141 HDAC inhibitors may also suppress expression of p53 neomorphs by decreasing the expression of the TP53 transcriptional activator HoxA5.141, 142 Other approaches that lead to the destabilization of p53 neomorphs may also provide effective antitumor strategies. For example, p53 neomorphs including R175H are positively regulated by acetylation.143 Deacetylation of the mutants through activating deacetylase sirtuins (SIRTs) reduces p53 neomorph levels and inhibits proliferation of breast cancer cell lines carrying TP53 neomorphs.144 Another elegant therapeutic strategy utilizes the mutant p53 structure to develop peptides or small molecules that restore WT p53 protein folding and activities.145 Alternatively, given the broad range of effects resulting from interaction of p53 neomorphs with p63 and p73, efforts to disrupt this interaction include a screen that identified RETRA (reactivation of transcriptional reporter activity) as being able to block interactions. RETRA disrupted the p53 neomorph:p73 complex reconstituting p73-mediated transcription, however it is unclear whether eliminating the neomorphic effect of the p53:p73 complex will be equivalent to restoring WT p53 function.146 A number of small molecule drugs including ZMC1, PRIMA1 or APREA-246 and COTI2 have been proposed to allow folding of unfolded p53 mutant proteins thus allowing reconstitution of p53 function including reconstitution of DNA binding and transcriptional activity.147 Understanding the interaction between the p53 neomorphs with their binding proteins will shed light on more approaches that target the activity of the mutants.
MYOD1
Myogenic Differentiation 1 (MYOD1) and c-Myc are both members of the large basic helix-loop-helix (bHLH) family of transcription factors, in which a 12–15 amino acid segment containing basic residues comprises a DNA-binding domain adjacent to a HLH domain that mediates dimerization to enhance DNA binding.148–150 Nearly 25 years ago, Van Antwerp et al. mutated amino acids in the basic domain of MYOD1 to analogous amino acids in c-Myc to identify residues mediating DNA-binding specificity.151 Electrophoretic mobility-shift assays revealed that three of six mutants with a single amino acid mutated were neutral, having no impact on DNA binding activity.151 One mutant was hypomorphic with reduced capacity for DNA binding and most interestingly, one mutant (L122R) gained an unexpected ability to bind to the c-Myc recognition site (5’-CACGTG-3’) but was still able to bind to the consensus MYOD1 site (5’-CACCTG-3’),151 suggesting that L122 of MYOD1 is critical to maintaining DNA specificity. Indeed, L122 is conserved in all myogenic bHLH transcription factors and is analogous to arginine in the MYC family, providing an explanation for the neomorphic activity of L122R. Remarkably, the mutant also competed with WT MYOD1 for the DNA binding site without leading to target gene transactivation.151 L122R may therefore serve as both an antimorph and a neomorph.
Because MYOD1 is a key player in muscle differentiation,152 it was postulated that L122R would lead to inhibition of differentiation and promotion of proliferation, especially in skeletal muscles. This postulate was confirmed by Kohsaka et al in 2014 wherein whole-exome sequencing and Sequenom analysis revealed an overall 10% (10/104) prevalence of MYOD1 L122R in 104 embryonal rhabdomyosarcoma (ERMS) tumors.153 ERMS tumors with MYOD1 L122R demonstrated decreased survival with histology revealing increased cellularity and mitotic activity.153 In vitro, mouse myoblast C2C12 cells with L122R were less differentiated and displayed increased anchorage-independent growth compared to those with WT MYOD1.153 Strikingly, chromatin immunoprecipitation sequencing (CHIP-Seq) and gene expression microarrays also demonstrated a genome-wide shift towards a Myc transcriptional program.153 No MYOD1 mutation was found in patients with alveolar rhabdomyosarcoma, the other major subtype of rhabdomyosarcoma which is driven by fusion genes comprised of the anchor gene forkhead box protein O1 (FOXO1) and fusion partners including paired box 3 (PAX3), paired box 7 (PAX7) or fibroblast growth factor receptor 1 (FGFR1).154, 155 MYOD1 mutations could be detected in other cancer types but L122R was found exclusively in ERMS. Interestingly MYOD1 L122M was found in one lung cancer case.10 Whether L122M exhibits the same neomorphic activity as L122R remains unknown.
Aberrations along the PI3K pathway including hotspot mutations in PIK3CA or PTEN gene deletion occurred exclusively with MYOD1 L122R in ERMS.153, 156 Co-expression of p110α H1047R and MYOD1 L122R had an additive effect on in vitro and in vivo tumorigenicity,153 implicating cooperativity of the two aberrations and suggesting that inhibiting their activities may be a potential anti-tumor strategy for this molecularly distinct and high risk cohort of patients. Further mechanistic characterization of MYOD1 L122R is imperative to identify actionable targets associated with this neomorph.
YY1
Yin Yang 1 (YY1) is a ubiquitous zinc-finger transcription factor involved in cell proliferation, apoptosis, cell cycle control, differentiation and hematopoiesis.157, 158 Depending on interacting partners, YY1 can either activate or repress gene expression and hence the name “Yin Yang”. YY1 regulates gene transcription through direct binding to gene promoters, by acting as a transcription cofactor, by interfering with other transcription factors or by altering chromatin structure.158,159
Our understanding of the role of YY1 in tumorigenesis is evolving. It is overexpressed in diverse cancer types.160–166 YY1 modulates the expression or transcriptional activity of a number of key players in the pathogenesis of malignancies, for example, MYC and ERBB2.167–169 Also, YY1 binds to the silencer region of the FAS promotor, thereby repressing FAS gene expression and conferring resistance to Fas-mediated apoptosis.170 Moreover, YY1 is a negative regulator of p53, disrupting the interaction between p53 and its coactivator p300.171 YY1 interacts with p53 and inhibits transcription of a subset of p53 target genes regulating cell cycle arrest and apoptosis.172, 173 Overexpression of YY1 stimulates p53 ubiquitination and degradation though assembly of a p53-Mdm2 complex.173, 174 Intriguingly, YY1 overexpression does not mediate the same clinical effects in every tissue. In ovarian cancer, YY1 overexpression correlates with improved overall survival, whereas decreased survival was observed in breast cancers where YY1 protein expression was associated with ERBB2 expression.161, 162, 166
Insulinoma is a type of pancreatic neuroendocrine tumor that inappropriately secretes insulin independent of serum glucose, thereby resulting in hypoglycemia. Whole exome sequencing of 113 sporadic insulinomas revealed a recurrent YY1 mutation at T372R in 30% of tumors.175 This recurrent mutation was also detected in an independent tumor cohort consisting of benign and malignant insulinomas, suggesting a role of the mutation in early carcinogenesis.176 The crystal structure of DNA-bound YY1 revealed that T372 locates within a highly conserved zinc finger domain that makes direct contact with DNA. Intriguingly, using CHIP-Seq, Cromer et al. demonstrated that T372R alters the DNA motif bound by YY1 and therefore transcription factor specificity.176 In contrast to the consensus DNA sequence 5’-GCCATNTT-3’ bound by WT YY1, T372R recognized 5’-CCATC-3’. Gene expression microarray analysis using cells with WT YY1 or T372R identified 149 genes that were differentially expressed by at least 10 fold. Two genes, ADCY1 (adenylyl cyclase 1), which catalyzes cAMP synthesis, and CACNA2D2, which encodes the α−2-δ−2 auxiliary (pore-forming) subunit of a high-voltage gated calcium channel, are not targets of WT YY1 but were highly expressed in cells with T372R. The mechanism underlying this neomorphic effect is unknown. ADCY1 and CACNA2D2 are involved in cAMP and calcium signaling; both processes are important for insulin secretion. Concomitant expression of these genes in pancreatic β-cell lines increased insulin release. Further, YY1 T372R was not found in pancreatic adenocarcinomas or malignant primitive neuroectodermal tumors that did not secrete insulin.177, 178 Consistently, although YY1 mutations were detected across tumor types in the TCGA including cancers of endometrium, head and neck, stomach, prostate breast and melanomas, none of these mutations was T372R.10 These data together suggest that the neomorphic mutation is specific to insulinomas. The molecular pathology underlying insulinomas remains unclear, and therefore whether targeting the YY1 neomorph and its associated signaling effects will provide an effective treatment is unknown. However, understanding the pathogenesis of insulinoma development allows for the possibility of eventually inducing insulin secretion in the β-cells of patients with diabetes.
Conclusion
To date, most mutations are commonly reported as hypomorphs or hypermorphs; however, for some genes, like TP53, neomorphic mutations appear to be more frequent. Herein, we have presented a series of examples of neomorphic mutations, some of which engender therapeutic responses distinct from WT and other types of morphs. Whether subsets of hypermorphic mutations, through high level activity or activity that is insensitive to feedback loops rewire cell signaling sufficiently to activate pathways not normally activated by WT proteins (pseudo-neomorphs) remains a distinct possibility. This underscores the significance of functional characterization of mutations to distinguish whether they are hypomorphic, hypermorphic, or neomorphic. It is likely that the examples we described represent only a subset of the neomorphic mutations that exist. While computational prediction and experimental transformation assays help differentiate between functional drivers and passengers, several experimental approaches can be appreciated for their robustness in the functional characterization of neomorphs we have discussed. The first involves the ability to create and express large numbers of mutant open reading frames to allow evaluation of their activities. CRISPR approaches allow the creation of specific mutations in the endogenous gene and provide an opportunity to characterize the effect of mutations in their normal expression context. Subsequent profiling of gene expression levels, protein levels, and metabolites as well as sensitivity to informer drug libraries or genomic interaction screens can facilitate the identification of signaling outcomes of the aberrations and provides insight regarding potential therapeutic approaches. While similar profile approaches in human tumors can give hints of neomorphic activities, many of the neomorphic mutations are sufficiently rare as to challenge this approach and thus require careful experimental validations. Additional strategies to determine the functional consequences of neomorphic mutations include analyses of available crystal structures or prediction modeling from conserved protein features. Characterization of protein binding partners as well as DNA binding sites can also provide evidence for neomorphic activities. We expect that more tools geared towards unraveling the functional impact of aberrations will become available. For example, a kinome data-driven systematic approach facilitated the identification of the amino acid residues that govern peptide specificity towards substrates or interacting proteins.179, 180 Based on this analysis, PKCγ mutants were shown to gain the capacity to phosphorylate de novo substrates.179 Further, mutations leading to neomorphic phosphorylation sites were also discovered.179 In these modeling studies, the description of neomorphs was extended from the concept presented in this review of aberrant downstream consequences of a mutational event to include altering the spectrum of upstream signals or environmental cues that active specific kinases. Under this definition, the number of neomorphic mutations are likely much higher than presented herein.
All oncogenic neomorphic mutations identified to date are substitution or truncation mutations. Insertion or deletion mutations leading to alteration of protein domains and gene fusion resulting in hybrid genes represent other interesting opportunities for future investigation. Indeed, whether the BCR-ABL fusion that alters the location of Abl activation is a neomorph should be considered. Our enhanced understanding of these deviant pathways and players provides us with new targets for rational drug design, novel biomarkers with which to assess therapeutic effects, and the ability to make molecularly-based prognostic classifications. Most importantly, the potential for neomorph-bearing tumors to be insensitive or to potentially bypass therapeutic agents active against the WT molecule or other types of morph raises an urgent need to determine which of the over 2 million point mutations that have been identified in cancer patients function as neomorphs. As genomic data continues to be generated and analyzed, we believe that the neomorphic mutations described in this review will represent just a few of the many more waiting to be identified.
Acknowledgement
We apologize to colleagues whose work was not cited owing to space constraints or our oversight. This work was supported by U54HG008100, U01CA168394, P50 CA098258, P50CA083639 and the Adelson Medical Research Foundation.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Muller HJ. Further studies on the nature and causes of gene mutations. Proceedings of the 6th International Congress of Genetics, vol. 1. Brooklyn Botanic Gardens Menasha, Wisconsin, 1932, pp 213–255. [Google Scholar]
- 2.Dalziel K Isocitrate dehydrogenase and related oxidative decarboxylases. FEBS Lett 1980; 117 Suppl: K45–55. [DOI] [PubMed] [Google Scholar]
- 3.Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem 2001; 276: 16168–16176. [DOI] [PubMed] [Google Scholar]
- 4.Lee SM, Koh HJ, Park DC, Song BJ, Huh TL, Park JW. Cytosolic NADP(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic Biol Med 2002; 32: 1185–1196. [DOI] [PubMed] [Google Scholar]
- 5.Kim SY, Lee SM, Tak JK, Choi KS, Kwon TK, Park JW. Regulation of singlet oxygen-induced apoptosis by cytosolic NADP+-dependent isocitrate dehydrogenase. Mol Cell Biochem 2007; 302: 27–34. [DOI] [PubMed] [Google Scholar]
- 6.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012; 481: 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res 2012; 25: 375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360: 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 2009; 118: 469–474. [DOI] [PubMed] [Google Scholar]
- 12.Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer 2009; 125: 353–355. [DOI] [PubMed] [Google Scholar]
- 13.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361: 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010; 17: 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 2010; 207: 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E et al. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem 2004; 279: 33946–33957. [DOI] [PubMed] [Google Scholar]
- 17.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009; 324: 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462: 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hausinger RP. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol 2004; 39: 21–68. [DOI] [PubMed] [Google Scholar]
- 20.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boulahbel H, Duran RV, Gottlieb E. Prolyl hydroxylases as regulators of cell metabolism. Biochem Soc Trans 2009; 37: 291–294. [DOI] [PubMed] [Google Scholar]
- 22.Williams SC, Karajannis MA, Chiriboga L, Golfinos JG, von Deimling A, Zagzag D. R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1alpha upregulation in adult glioma. Acta Neuropathol 2011; 121: 279–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 2011; 12: 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012; 483: 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18: 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011; 145: 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pope WB, Prins RM, Albert Thomas M, Nagarajan R, Yen KE, Bittinger MA et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J Neurooncol 2012; 107: 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med 2012; 18: 624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalinina J, Carroll A, Wang L, Yu Q, Mancheno DE, Wu S et al. Detection of “oncometabolite” 2-hydroxyglutarate by magnetic resonance analysis as a biomarker of IDH1/2 mutations in glioma. J Mol Med (Berl) 2012; 90: 1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andronesi OC, Kim GS, Gerstner E, Batchelor T, Tzika AA, Fantin VR et al. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med 2012; 4: 116ra114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 2010; 115: 2749–2754. [DOI] [PubMed] [Google Scholar]
- 33.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013; 340: 622–626. [DOI] [PubMed] [Google Scholar]
- 34.Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F et al. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med Chem Lett 2012; 3: 850–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013; 339: 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013; 340: 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol 2015; 11: 878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crunkhorn S Cancer: Novel IDH1 mutant inhibitors identified. Nat Rev Drug Discov 2015; 14: 820. [DOI] [PubMed] [Google Scholar]
- 39.Clark O, Yen K, Mellinghoff IK. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin Cancer Res 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Marholz LJ, Wang X. Novel Scaffolds of Cell-Active Histone Demethylase Inhibitors Identified from High-Throughput Screening. J Biomol Screen 2015; 20: 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lv S, Teugels E, Sadones J, Quartier E, Huylebrouck M, S DUF et al. Correlation between IDH1 gene mutation status and survival of patients treated for recurrent glioma. Anticancer Res 2011; 31: 4457–4463. [PubMed] [Google Scholar]
- 42.Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002; 296: 1655–1657. [DOI] [PubMed] [Google Scholar]
- 43.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006; 7: 606–619. [DOI] [PubMed] [Google Scholar]
- 44.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2: 489–501. [DOI] [PubMed] [Google Scholar]
- 45.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005; 4: 988–1004. [DOI] [PubMed] [Google Scholar]
- 46.Cuevas BD, Lu Y, Mao M, Zhang J, LaPushin R, Siminovitch K et al. Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3-kinase. J Biol Chem 2001; 276: 27455–27461. [DOI] [PubMed] [Google Scholar]
- 47.Backer JM, Myers MG Jr., Shoelson SE, Chin DJ, Sun XJ, Miralpeix M et al. Phosphatidylinositol 3’-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J 1992; 11: 3469–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol 1998; 18: 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo J, Cantley LC. The negative regulation of phosphoinositide 3-kinase signaling by p85 and it’s implication in cancer. Cell Cycle 2005; 4: 1309–1312. [DOI] [PubMed] [Google Scholar]
- 50.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet 2013; 45: 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet 1999; 21: 99–102. [DOI] [PubMed] [Google Scholar]
- 53.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554. [DOI] [PubMed] [Google Scholar]
- 54.Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F et al. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle 2009; 8: 1352–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov 2011; 1: 170–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A 2006; 103: 1475–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 2007; 318: 1744–1748. [DOI] [PubMed] [Google Scholar]
- 58.Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res 2005; 65: 10992–11000. [DOI] [PubMed] [Google Scholar]
- 59.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A 2005; 102: 18443–18448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hao Y, Wang C, Cao B, Hirsch BM, Song J, Markowitz SD et al. Gain of interaction with IRS1 by p110alpha-helical domain mutants is crucial for their oncogenic functions. Cancer Cell 2013; 23: 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009; 16: 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 2008; 68: 6084–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dogruluk T, Tsang YH, Espitia M, Chen F, Chen T, Chong Z et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Res 2015; 75: 5341–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al. The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 2007; 317: 239–242. [DOI] [PubMed] [Google Scholar]
- 68.Wu H, Shekar SC, Flinn RJ, El-Sibai M, Jaiswal BS, Sen KI et al. Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110alpha and are disrupted in oncogenic p85 mutants. Proc Natl Acad Sci U S A 2009; 106: 20258–20263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quayle SN, Lee JY, Cheung LW, Ding L, Wiedemeyer R, Dewan RW et al. Somatic mutations of PIK3R1 promote gliomagenesis. PLoS One 2012; 7: e49466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harpur AG, Layton MJ, Das P, Bottomley MJ, Panayotou G, Driscoll PC et al. Intermolecular interactions of the p85alpha regulatory subunit of phosphatidylinositol 3-kinase. J Biol Chem 1999; 274: 12323–12332. [DOI] [PubMed] [Google Scholar]
- 71.Cheung LW, Walkiewicz KW, Besong TM, Guo H, Hawke DH, Arold ST et al. Regulation of the PI3K pathway through a p85alpha monomer-homodimer equilibrium. Elife 2015; 4: e06866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cheung LW, Yu S, Zhang D, Li J, Ng PK, Panupinthu N et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell 2014; 26: 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ihle NT, Lemos R Jr., Wipf P, Yacoub A, Mitchell C, Siwak D et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res 2009; 69: 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res 2009; 69: 4286–4293. [DOI] [PubMed] [Google Scholar]
- 75.Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res 2012; 18: 2316–2325. [DOI] [PubMed] [Google Scholar]
- 76.Pitts TM, Newton TP, Bradshaw-Pierce EL, Addison R, Arcaroli JJ, Klauck PJ et al. Dual pharmacological targeting of the MAP kinase and PI3K/mTOR pathway in preclinical models of colorectal cancer. PLoS One 2014; 9: e113037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res 2015; 21: 396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol 2013; 71: 1395–1409. [DOI] [PubMed] [Google Scholar]
- 79.Temraz S, Mukherji D, Shamseddine A. Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers. Int J Mol Sci 2015; 16: 22976–22988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res 2001; 264: 29–41. [DOI] [PubMed] [Google Scholar]
- 81.Cai XM, Tao BB, Wang LY, Liang YL, Jin JW, Yang Y et al. Protein phosphatase activity of PTEN inhibited the invasion of glioma cells with epidermal growth factor receptor mutation type III expression. Int J Cancer 2005; 117: 905–912. [DOI] [PubMed] [Google Scholar]
- 82.Tibarewal P, Zilidis G, Spinelli L, Schurch N, Maccario H, Gray A et al. PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci Signal 2012; 5: ra18. [DOI] [PubMed] [Google Scholar]
- 83.Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 2003; 3: 117–130. [DOI] [PubMed] [Google Scholar]
- 84.Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007; 128: 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gong L, Govan JM, Evans EB, Dai H, Wang E, Lee SW et al. Nuclear PTEN tumor-suppressor functions through maintaining heterochromatin structure. Cell Cycle 2015; 14: 2323–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rodriguez-Escudero I, Oliver MD, Andres-Pons A, Molina M, Cid VJ, Pulido R. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet 2011; 20: 4132–4142. [DOI] [PubMed] [Google Scholar]
- 87.Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014; 157: 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 2000; 20: 5010–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Costa HA, Leitner MG, Sos ML, Mavrantoni A, Rychkova A, Johnson JR et al. Discovery and functional characterization of a neomorphic PTEN mutation. Proc Natl Acad Sci U S A 2015; 112: 13976–13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol 1997; 17: 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma K, Cheung SM, Marshall AJ, Duronio V. PI(3,4,5)P3 and PI(3,4)P2 levels correlate with PKB/akt phosphorylation at Thr308 and Ser473, respectively; PI(3,4)P2 levels determine PKB activity. Cell Signal 2008; 20: 684–694. [DOI] [PubMed] [Google Scholar]
- 92.DeGraffenried LA, Fulcher L, Friedrichs WE, Grunwald V, Ray RB, Hidalgo M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann Oncol 2004; 15: 1510–1516. [DOI] [PubMed] [Google Scholar]
- 93.Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep 2014; 6: 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A 2008; 105: 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408: 307–310. [DOI] [PubMed] [Google Scholar]
- 96.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979; 278: 261–263. [DOI] [PubMed] [Google Scholar]
- 97.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979; 17: 43–52. [DOI] [PubMed] [Google Scholar]
- 98.DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci U S A 1979; 76: 2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rotter V p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells. Proc Natl Acad Sci U S A 1983; 80: 2613–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Parada LF, Land H, Weinberg RA, Wolf D, Rotter V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 1984; 312: 649–651. [DOI] [PubMed] [Google Scholar]
- 101.Eliyahu D, Raz A, Gruss P, Givol D, Oren M. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 1984; 312: 646–649. [DOI] [PubMed] [Google Scholar]
- 102.Hinds P, Finlay C, Levine AJ. Mutation is required to activate the p53 gene for cooperation with the ras oncogene and transformation. J Virol 1989; 63: 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hinds PW, Finlay CA, Quartin RS, Baker SJ, Fearon ER, Vogelstein B et al. Mutant p53 DNA clones from human colon carcinomas cooperate with ras in transforming primary rat cells: a comparison of the “hot spot” mutant phenotypes. Cell Growth Differ 1990; 1: 571–580. [PubMed] [Google Scholar]
- 104.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9: 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989; 244: 217–221. [DOI] [PubMed] [Google Scholar]
- 106.Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci U S A 1989; 86: 8763–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989; 57: 1083–1093. [DOI] [PubMed] [Google Scholar]
- 108.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev 2012; 26: 1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer 2002; 2: 594–604. [DOI] [PubMed] [Google Scholar]
- 110.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137: 413–431. [DOI] [PubMed] [Google Scholar]
- 111.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9: 402–412. [DOI] [PubMed] [Google Scholar]
- 112.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 2009; 9: 691–700. [DOI] [PubMed] [Google Scholar]
- 113.Eiriksdottir G, Barkardottir RB, Agnarsson BA, Johannesdottir G, Olafsdottir K, Egilsson V et al. High incidence of loss of heterozygosity at chromosome 17p13 in breast tumours from BRCA2 mutation carriers. Oncogene 1998; 16: 21–26. [DOI] [PubMed] [Google Scholar]
- 114.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994; 265: 346–355. [DOI] [PubMed] [Google Scholar]
- 115.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 2007; 26: 2157–2165. [DOI] [PubMed] [Google Scholar]
- 116.Chan WM, Siu WY, Lau A, Poon RY. How many mutant p53 molecules are needed to inactivate a tetramer? Mol Cell Biol 2004; 24: 3536–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991; 65: 765–774. [DOI] [PubMed] [Google Scholar]
- 118.Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M et al. Gain of function mutations in p53. Nat Genet 1993; 4: 42–46. [DOI] [PubMed] [Google Scholar]
- 119.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9: 701–713. [DOI] [PubMed] [Google Scholar]
- 120.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004; 2: 371–386. [PubMed] [Google Scholar]
- 121.Lokshin M, Li Y, Gaiddon C, Prives C. p53 and p73 display common and distinct requirements for sequence specific binding to DNA. Nucleic Acids Res 2007; 35: 340–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Perez CA, Ott J, Mays DJ, Pietenpol JA. p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene 2007; 26: 7363–7370. [DOI] [PubMed] [Google Scholar]
- 123.Joerger AC, Rajagopalan S, Natan E, Veprintsev DB, Robinson CV, Fersht AR. Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci U S A 2009; 106: 17705–17710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Davison TS, Vagner C, Kaghad M, Ayed A, Caput D, Arrowsmith CH. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem 1999; 274: 18709–18714. [DOI] [PubMed] [Google Scholar]
- 125.Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 2001; 21: 1874–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y, Castagnoli L et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem 2000; 275: 29503–29512. [DOI] [PubMed] [Google Scholar]
- 127.Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol 1999; 19: 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A et al. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem 2001; 276: 15164–15173. [DOI] [PubMed] [Google Scholar]
- 129.Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG Jr. Chemosensitivity linked to p73 function. Cancer Cell 2003; 3: 403–410. [DOI] [PubMed] [Google Scholar]
- 130.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009; 139: 1327–1341. [DOI] [PubMed] [Google Scholar]
- 131.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009; 137: 87–98. [DOI] [PubMed] [Google Scholar]
- 132.Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006; 10: 191–202. [DOI] [PubMed] [Google Scholar]
- 133.Imbriano C, Gurtner A, Cocchiarella F, Di Agostino S, Basile V, Gostissa M et al. Direct p53 transcriptional repression: in vivo analysis of CCAAT-containing G2/M promoters. Mol Cell Biol 2005; 25: 3737–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol 2007; 9: 573–580. [DOI] [PubMed] [Google Scholar]
- 135.Maser RS, Monsen KJ, Nelms BE, Petrini JH. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol Cell Biol 1997; 17: 6087–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005; 308: 551–554. [DOI] [PubMed] [Google Scholar]
- 137.Frum RA, Grossman SR. Mechanisms of mutant p53 stabilization in cancer. Subcell Biochem 2014; 85: 187–197. [DOI] [PubMed] [Google Scholar]
- 138.Peng Y, Chen L, Li C, Lu W, Chen J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J Biol Chem 2001; 276: 40583–40590. [DOI] [PubMed] [Google Scholar]
- 139.Wiech M, Olszewski MB, Tracz-Gaszewska Z, Wawrzynow B, Zylicz M, Zylicz A. Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS One 2012; 7: e51426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kramer OH, Mahboobi S, Sellmer A. Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol Sci 2014; 35: 501–509. [DOI] [PubMed] [Google Scholar]
- 141.Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ 2011; 18: 1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yan W, Liu S, Xu E, Zhang J, Zhang Y, Chen X et al. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013; 32: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Perez RE, Knights CD, Sahu G, Catania J, Kolukula VK, Stoler D et al. Restoration of DNA-binding and growth-suppressive activity of mutant forms of p53 via a PCAF-mediated acetylation pathway. J Cell Physiol 2010; 225: 394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yi YW, Kang HJ, Kim HJ, Kong Y, Brown ML, Bae I. Targeting mutant p53 by a SIRT1 activator YK-3–237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget 2013; 4: 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Selivanova G, Wiman KG. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene 2007; 26: 2243–2254. [DOI] [PubMed] [Google Scholar]
- 146.Kravchenko JE, Ilyinskaya GV, Komarov PG, Agapova LS, Kochetkov DV, Strom E et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci U S A 2008; 105: 6302–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell 2012; 21: 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell 1989; 56: 777–783. [DOI] [PubMed] [Google Scholar]
- 149.Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L et al. Muscle-specific transcriptional activation by MyoD. Genes Dev 1991; 5: 1377–1386. [DOI] [PubMed] [Google Scholar]
- 150.Tapscott SJ, Weintraub H. MyoD and the regulation of myogenesis by helix-loop-helix proteins. J Clin Invest 1991; 87: 1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Van Antwerp ME, Chen DG, Chang C, Prochownik EV. A point mutation in the MyoD basic domain imparts c-Myc-like properties. Proc Natl Acad Sci U S A 1992; 89: 9010–9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tapscott SJ, Davis RL, Thayer MJ, Cheng PF, Weintraub H, Lassar AB. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science 1988; 242: 405–411. [DOI] [PubMed] [Google Scholar]
- 153.Kohsaka S, Shukla N, Ameur N, Ito T, Ng CK, Wang L et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet 2014; 46: 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu J, Guzman MA, Pezanowski D, Patel D, Hauptman J, Keisling M et al. FOXO1-FGFR1 fusion and amplification in a solid variant of alveolar rhabdomyosarcoma. Mod Pathol 2011; 24: 1327–1335. [DOI] [PubMed] [Google Scholar]
- 155.Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol 2002; 20: 2672–2679. [DOI] [PubMed] [Google Scholar]
- 156.Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res 2012; 18: 748–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Castellano G, Torrisi E, Ligresti G, Malaponte G, Militello L, Russo AE et al. The involvement of the transcription factor Yin Yang 1 in cancer development and progression. Cell Cycle 2009; 8: 1367–1372. [DOI] [PubMed] [Google Scholar]
- 158.Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene 2006; 25: 1125–1142. [DOI] [PubMed] [Google Scholar]
- 159.Deng Z, Wan M, Cao P, Rao A, Cramer SD, Sui G. Yin Yang 1 regulates the transcriptional activity of androgen receptor. Oncogene 2009; 28: 3746–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Seligson D, Horvath S, Huerta-Yepez S, Hanna S, Garban H, Roberts A et al. Expression of transcription factor Yin Yang 1 in prostate cancer. Int J Oncol 2005; 27: 131–141. [PubMed] [Google Scholar]
- 161.Matsumura N, Huang Z, Baba T, Lee PS, Barnett JC, Mori S et al. Yin yang 1 modulates taxane response in epithelial ovarian cancer. Mol Cancer Res 2009; 7: 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Allouche A, Nolens G, Tancredi A, Delacroix L, Mardaga J, Fridman V et al. The combined immunodetection of AP-2alpha and YY1 transcription factors is associated with ERBB2 gene overexpression in primary breast tumors. Breast Cancer Res 2008; 10: R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Baritaki S, Sifakis S, Huerta-Yepez S, Neonakis IK, Soufla G, Bonavida B et al. Overexpression of VEGF and TGF-beta1 mRNA in Pap smears correlates with progression of cervical intraepithelial neoplasia to cancer: implication of YY1 in cervical tumorigenesis and HPV infection. Int J Oncol 2007; 31: 69–79. [PubMed] [Google Scholar]
- 164.Grubach L, Juhl-Christensen C, Rethmeier A, Olesen LH, Aggerholm A, Hokland P et al. Gene expression profiling of Polycomb, Hox and Meis genes in patients with acute myeloid leukaemia. Eur J Haematol 2008; 81: 112–122. [DOI] [PubMed] [Google Scholar]
- 165.de Nigris F, Botti C, de Chiara A, Rossiello R, Apice G, Fazioli F et al. Expression of transcription factor Yin Yang 1 in human osteosarcomas. Eur J Cancer 2006; 42: 2420–2424. [DOI] [PubMed] [Google Scholar]
- 166.Castellano G, Torrisi E, Ligresti G, Nicoletti F, Malaponte G, Traval S et al. Yin Yang 1 overexpression in diffuse large B-cell lymphoma is associated with B-cell transformation and tumor progression. Cell Cycle 2010; 9: 557–563. [DOI] [PubMed] [Google Scholar]
- 167.Riggs KJ, Saleque S, Wong KK, Merrell KT, Lee JS, Shi Y et al. Yin-yang 1 activates the c-myc promoter. Mol Cell Biol 1993; 13: 7487–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shrivastava A, Yu J, Artandi S, Calame K. YY1 and c-Myc associate in vivo in a manner that depends on c-Myc levels. Proc Natl Acad Sci U S A 1996; 93: 10638–10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Begon DY, Delacroix L, Vernimmen D, Jackers P, Winkler R. Yin Yang 1 cooperates with activator protein 2 to stimulate ERBB2 gene expression in mammary cancer cells. J Biol Chem 2005; 280: 24428–24434. [DOI] [PubMed] [Google Scholar]
- 170.Garban HJ, Bonavida B. Nitric oxide inhibits the transcription repressor Yin-Yang 1 binding activity at the silencer region of the Fas promoter: a pivotal role for nitric oxide in the up-regulation of Fas gene expression in human tumor cells. J Immunol 2001; 167: 75–81. [DOI] [PubMed] [Google Scholar]
- 171.Sui G, Affar el B, Shi Y, Brignone C, Wall NR, Yin P et al. Yin Yang 1 is a negative regulator of p53. Cell 2004; 117: 859–872. [DOI] [PubMed] [Google Scholar]
- 172.Yakovleva T, Kolesnikova L, Vukojevic V, Gileva I, Tan-No K, Austen M et al. YY1 binding to a subset of p53 DNA-target sites regulates p53-dependent transcription. Biochem Biophys Res Commun 2004; 318: 615–624. [DOI] [PubMed] [Google Scholar]
- 173.Sui G, Affar el B, Shi Y, Brignone C, Wall NR, Yin P et al. Yin Yang 1 is a negative regulator of p53. Cell 2004; 117: 859–872. [DOI] [PubMed] [Google Scholar]
- 174.Gronroos E, Terentiev AA, Punga T, Ericsson J. YY1 inhibits the activation of the p53 tumor suppressor in response to genotoxic stress. Proc Natl Acad Sci U S A 2004; 101: 12165–12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cao Y, Gao Z, Li L, Jiang X, Shan A, Cai J et al. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat Commun 2013; 4: 2810. [DOI] [PubMed] [Google Scholar]
- 176.Cromer MK, Choi M, Nelson-Williams C, Fonseca AL, Kunstman JW, Korah RM et al. Neomorphic effects of recurrent somatic mutations in Yin Yang 1 in insulin-producing adenomas. Proc Natl Acad Sci U S A 2015; 112: 4062–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011; 331: 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321: 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Creixell P, Schoof EM, Simpson CD, Longden J, Miller CJ, Lou HJ et al. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell 2015; 163: 202–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Creixell P, Palmeri A, Miller CJ, Lou HJ, Santini CC, Nielsen M et al. Unmasking determinants of specificity in the human kinome. Cell 2015; 163: 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]