

Abstract
The membrane-bound P-glycoprotein (Pgp) transporter plays a major role in human disease and drug disposition because of its ability to efflux a chemically diverse range of drugs through ATP hydrolysis and ligand-induced conformational changes. Deciphering these structural changes is key to understanding the molecular basis of transport and to developing molecules that can modulate efflux. Here, atomic force microscopy (AFM) is used to directly image individual Pgp transporter molecules in a lipid bilayer under physiological pH and ambient temperature. Analysis of the Pgp AFM images revealed “small” and “large” protrusions from the lipid bilayer with significant differences in protrusion height and volume. The geometry of these “small” and “large” protrusions correlated to the predicted extracellular (EC) and cytosolic (C) domains of the Pgp X-ray crystal structure, respectively. To assign these protrusions, simulated AFM images were produced from the Pgp X-ray crystal structures with membrane planes defined by three computational approaches, and a simulated 80 Å AFM cantilever tip. The theoretical AFM images of the EC and C domains had similar heights and volumes to the “small” and “large” protrusions in the experimental AFM images, respectively. The assignment of the protrusions in the AFM images to the EC and C domains was confirmed by changes in protrusion volume by Pgp-specific antibodies. The Pgp domains showed a considerable degree of conformational dynamics in time resolved AFM images. With this information, a model of Pgp conformational dynamics in a lipid bilayer is proposed within the context of the known Pgp X-ray crystal structures.
Keywords: Atomic force microscopy (AFM), Multidrug resistance transporters, Membrane-bound protein modeling, Protein conformation, Drug distribution
1. Introduction
P-glycoprotein (Pgp; ABCB1) transporter is a part of the ATP Binding Cassette (ABC) transporter superfamily [1]. The transporter is concentrated in the plasma membranes and functions by effluxing a chemically and structurally diverse range of drugs from the cytosol (C) to the extracellular (EC) space [2-4]. The transporter is found in virtually all tissues, but is concentrated in the brain, kidneys, intestines and liver [5]. As a result, it plays a major role in drug absorption, distribution and excretion [6-9]. In addition, the transporter is a major driver of anti-cancer drug resistance [10], precludes penetration of drugs across the blood brain barrier (BBB) [11], and is a major contributor of adverse drug reactions (ADRs) from cardiovascular therapeutics by modulating excretion through the kidneys [12,13]. Therefore, there has been keen interest in unraveling the molecular and structural basis of drug transport by Pgp. This knowledge is critical to the development of novel transport inhibitors and drugs with desirable transport properties as well as improving predictions of drug disposition from in vitro experiments.
Our most detailed understanding of the transporter structure comes from X-ray crystal structures of the mouse [14, 15] and C. elegans [16] Pgp, and a recent cryo-electron microscopy (cryo-em) reconstruction of human Pgp [17]. These structures revealed that Pgp is comprised of a 140 KD pseudo-symmetric monomer consisting of twelve transmembrane helices and two nucleotide binding domains (NBDs). Pgp can assume a conformation with the NBDs separated with the drug binding cavity exposed to the cytosolic side of the transporter, which is often referred to as the “open” or “inward-facing” Pgp conformation [14-16]. The recent cryo-em structure revealed a conformation where the NBDs are together with the drug binding cavity exposed to the extracellular side of the transporter, which is commonly referred to the as the “closed” or “outward-facing” Pgp conformation [17]. Based on similar conformations observed in the analogous bacterial transporters, an alternating access model for ligand transport was proposed [18, 19]. A caveat of these studies is that they were performed with Pgp solubilized with detergent [14-17], which may have large effects on the transporter’s conformation [20,21]. The lipid bilayer membrane that is associated with Pgp is essential for its function [21-23]. To understand the transport process, there is a real need to determine the Pgp conformational changes in a lipid bilayer under near-native conditions and physiological pH between pH 6.0 and 8.0. Pgp investigations within this pH range are important because Pgp-mediated ATP hydrolysis is known to be severely inhibited outside of this range [24], and charged Pgp ligands are known to be sensitive to pH [25-27].
To overcome this knowledge gap, Pgp in a lipid bilayer was investigated experimentally using atomic force microscopy (AFM). AFM was originally developed within the solid-state physics community [28], but has found increasing use in biology [29]. Advances in AFM technology have allowed biological investigations to be performed under physiologically relevant conditions such as ambient temperature and pressure [30]. This has been particularly helpful for investigations of membrane proteins reconstituted into lipid bilayers [31,32]. In fact, studies of Pgp by AFM have already been performed [33-36]. Unfortunately, these studies achieved relatively low resolution [33-36]. As a result, the punctate features in the AFM images were not assigned to specific domains of the transporter [33-36]. However, AFM images of human Pgp taken with MM4.17 and MRK-16 antibodies that are specific for the EC-side of Pgp showed that it is theoretically possible to differentiate the EC and C sides of the transporter with AFM [33].
In this study, AFM was performed of Pgp reconstituted in a lipid bilayer to positively identify the EC and C domains of Pgp from AFM images. To ensure that active Pgp was used for the AFM measurements, Pgp-mediated ATP hydrolysis activity was measured. Then AFM imaging was performed of the proteoliposomes and the major features in the images were categorized. To correlate the features in the AFM images to domains of the Pgp X-ray crystal structure, AFM simulations of the EC and C sides of Pgp were performed on the “open” (PDB ID: 4M1M, [14, 15]) and “closed” (PDB ID: 6C0V, [17]) conformations of Pgp positioned in the membrane by three different methods [37-39]. These were compared to representative AFM images to determine the most likely conformation and position in a lipid bilayer. The assignment of the Pgp domains in the experimental AFM images was confirmed by Pgp specific antibodies. Pgp conformational dynamics was investigated using a high-speed AFM instrument that provided time resolved AFM images on a second time scale. The results allowed us to propose a two-state model of Pgp conformational dynamics in a lipid bilayer. This work provides a foundation to study the structure and dynamics of Pgp in a lipid bilayer.
2. Materials and methods
2.1. Reagents
Cholesterol and Tris-HCl were purchased from Amresco (Solon, OH). Escherichia (E.) coli lipid extract powder was purchased from Avanti Polar Lipids Inc. (Alabaster, AL). The C219 antibody was purchased from Novus Biologicals (Littleton, CO). The detergent n-dodecyl-β-D-maltoside (DDM), buffer 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), NaCl and MgCl2 were purchased from MilliporeSigma (Burlington, MA) (Formerly, Calbiochem Research Biochemicals and EMD Millipore). All the remaining chemicals were purchased from ThermoFisher Scientific (Waltham, MA) including the 6X His tag antibody.
2.2. Expression and purification of Pgp
The his-tagged wildtype mouse Pgp (Abcb1a, MDR3) was cloned and expressed from Pichia (P.) pastoris as previously described [40-42]. As previously noted [43], three asparagines on the N-terminus of this Pgp construct were mutated to glutamine to prevent N-linked glycosylation in this Pgp construct and still retained wild-type Pgp activity [43]. P. pastoris cells were grown and induced by methanol at the Bioexpression and Fermentation Facility (BFF) at the University of Georgia. The his-tagged protein was lysed through a series of freeze thaw cycles, extracted with excess n-dodecyl-β-D-maltoside detergent (DDM) and was purified in two steps: nickel-nitrilotriacetic acid (Ni-NTA) (Thermo Fisher Scientific, Waltham, MA) and DE52 cellulose resin (Thermo Fisher Scientific, Waltham, MA) [41]. The purity of detergent solubilized Pgp determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was > 95% pure (data not shown). Detergent solubilized Pgp was concentrated in Amicon Ultra-15 100kD cutoff spin concentrators. The concentration was measured using the extinction coefficient of 1.28 ml mg−1 cm−1 and the DC Protein Assay Kit II (Bio-Rad, Hercules, CA) [41]. Purified protein was stored at −80 °C in 10 mM Tris-HCl, 30% glycerol, pH 8.0.
2.3. Reconstitution of Pgp in liposomes
Detergent solubilized Pgp was integrated into liposomes as previously described [e.g. [44]]. Briefly, 80% w/v Avanti extract polar Escherichia (E.) coli lipids (Avanti Polar Lipids, Alabaster, AL) and 20% w/v cholesterol were solubilized in chloroform to a final concentration of 10 mg mL−1. This solution was evaporated in a Buchi Rotavapor Model R-114 (Buchi, Switzerland) until complete dryness was achieved and a lipid film was formed. This lipid film was rehydrated in 50 mM Tris-HCl and 0.1 mM ethylene glycol tetraacetic acid (EGTA), pH 7.4 and underwent 10 freeze thaw cycles in liquid nitrogen. The resuspended lipid film was extruded 11 times through a LIPEX extruder (Northern Lipids, Burnaby, British Columbia, Canada) with a 400 nm cutoff Millipore filter (EMD Millipore Billerica, MA). The purified Pgp product was dialyzed to remove excess detergent against HEPES buffer (20 mM HEPES, 100 mM NaCl, 5mM MgCl2, 2 mM DTT, pH 7.4). Dialyzed Pgp at 25 μM was passively integrated into 4 mg ml−1 lipososomes to a final concentration of 6.25 μM Pgp mg−1 ml liposomes. These proteoliposomes were dialyzed against HEPES buffer to remove the remaining detergent and stored at −80 °C.
2.4. ATPase activity measurements
The ATPase activity of reconstituted Pgp was measured by the Chifflet et al. method on a FlexStation 3 spectrometer (Molecular Devices, Sunnyvale, CA) as done previously [41, 45]. The production of inorganic phosphate (Pi) after ATP hydrolysis was measured by the absorbance signal at 850 nm as the Pi-molybdenum complex is formed. The Pgp-mediated ATPase activity was measured for the Pgp probe substrate verapamil in the presence of 50 nM reconstituted Pgp in Chifflet Buffer (150 mM NH4, 5 mM MgSO4, 0.02% w/v NaN3, 50 mM Tris-HCl, pH 7.4).
Non-linear regression was used to fit Pgp-coupled ATP hydrolysis kinetics with Igor Pro 6.2 (Wavemetrics, Tigard, OR) and the following equations. The modified Michaelis-Menten equation was used to fit hyperbolic ATP hydrolysis curves [41]:
| (1) |
where Vmax is the maximum ATP hydrolysis, L is the concentration of ligand, Km is the Michaelis-Menten constant and vbasal is the basal AT-Pase activity that serves as an offset that is independent of Pgp occupancy. For Pgp-coupled ATP hydrolysis curves that exhibit biphasic substrate inhibition, the following equation was used to fit the curve [41]:
| (2) |
where Ki is the inhibitory constant.
2.5. Atomic force microscope (AFM) imaging
Proteoliposome stock solutions were diluted to 100 nM Pgp in 20 mM Hepes at pH 7.4, 100 mM NaCl, 5mM MgCl2, which will be called imaging buffer. These solutions were immediately deposited on a freshly cleaved mica surface (Grade v1; Ted Pella, Redding, CA) and incubated for 45 min at ~ 30 °C. During this time, the proteoliposomes rupture and form a planar lipid bilayer supported by the mica surface [32]. The C-side and EC-side Pgp protein domains protrude out from the bilayer surface making them accessible to AFM imaging. For the antibody experiments, 2 μl of antibody (C219 at 0.07 mg/ml or 6 × His tag antibody at 1 mg/ml) was added to 198 μl of 100 nM Pgp proteoliposomes 15 min prior to incubation on mica. All samples were rinsed two times with ~ 100 μl volumes of imaging buffer prior to recording images, as described previously [31,32]. AFM images were acquired in imaging buffer, in tapping mode, using the Cypher or Cypher VRS (Asylum Research, Santa Barbara, CA) and tips (BL-AC40TS, Olympus, Tokyo Japan, or USC-F0.3-k0.3, Nanoworld, Neuchatel Switzerland). Images were acquired at ~ 32 °C with an estimated tip-sample force < 100 pN.
2.6. AFM image analysis
The protrusions in AFM images of Pgp reconstituted proteoliposomes were analyzed using scripts written in the scientific analysis package Igor Pro 7 (Wavemetrics, Portland, OR) or using the Gwyddion AFM analysis package [46]. Scripts implementing the Hessian blob algorithm within Igor Pro 7 were used to identify protrusions in the AFM images [47]. To accurately estimate the protrusion height at a given pixel of the AFM image, the Laplace interpolation was used to determine the membrane boundary underneath the protrusion [48]. The protrusion volume was estimated using a two-dimensional trapezoidal integration algorithm and the heights within the Gwyddion software [46]. The height for a given protrusion was defined as its maximum height above the membrane boundary.
2.7. Simulations of AFM image data
Simulated AFM images aided in assignment of the EC and C domains of Pgp in the experimental images and were generated using scripts written in Igor Pro 7. They were created using a 3-dimensional outline approach called morphological dilation [49,50]. The boundaries of the AFM images were defined by the van der Waals surface of the Pgp X-ray crystal structures [14,15,17] and the membrane boundaries were defined by three computational approaches: TMDET algorithm [37], Orientations of Proteins in Membranes (OPM) algorithm [38] and O’Mara and coworkers method [39]. With the TMDET and OPM algorithms an additional 4.8 Å was added to each side of the hydrophobic regions defined by these methods to account for the phosphate head groups of the membranes. To approximate the experimental AFM cantilever tip, the simulated tip had cone shape with a truncated hemispherical tip (cone angle = 17.5°, tip radius = 80 Å), which corresponds to the manufacturer’s nominal specifications. This simulated tip was run across the solvent exposed van der Waals surface and the calculated membrane boundary to generate the AFM image of Pgp in a membrane.
2.8. Analysis and visualization of the PDB structures
The PDB structures of mouse Pgp (PDB ID: 4M1M) [14, 15], of human Pgp (PDB ID: 6V0C) [17] and of mouse Pgp (PDB ID: 4M1M) embedded in a phosphatidylcholine bilayer [39] were analyzed and visualized using the PyMOL v1.8 (Schrödinger, New York, NY) molecular viewer. The dimensions of the protein structures and the phosphatidylcholine bilayer were estimated using the PyMOL script Draw_-Protein_Dimensions.py (https://pymolwiki.org/index.php/Draw_Protein_Dimensions).
3. Results
3.1. ATPase activity of liposome reconstituted Pgp
Fig. 1 shows the Pgp-coupled ATPase activity in the presence of the Pgp probe substrate verapamil. At 0 μM verapamil, the Pgp-coupled ATPase activity was 590 ± 15 nmol min−1 mg−1, which is similar to previous results [41,51]. The basal ATPase activity decreased to 130 ± 4.6 nmol min−1 mg−1 in the presence of 200 μM of the inhibitor orthovanadate (data not shown). The residual ATPase activity in the presence of saturating orthovanadate is due to non-competitive inhibition between the metal and ATP [51,52]. The Pgp-coupled ATP hydrolysis activity was measured with the probe Pgp substrate verapamil. The ATP hydrolysis kinetics was biphasic with ATPase activation and inhibition at low and high concentrations, respectively (Fig. 1, open squares). The verapamil-stimulated ATPase activity reached a maximum at 2011 ± 23 nmol min−1 mg−1 at 16 μM. Fitting the biphasic curve to Eq. (2) generated a Vmax, Km and Ki values of 2412 ± 122 nmol min−1 mg−1, 2.5 ± 0.7 μM and 819 ± 305 μM, respectively. These values are similar to Vmax, Km and Ki values we previously reported for verapamil-stimulated ATPase activity [41,51].
Fig. 1.
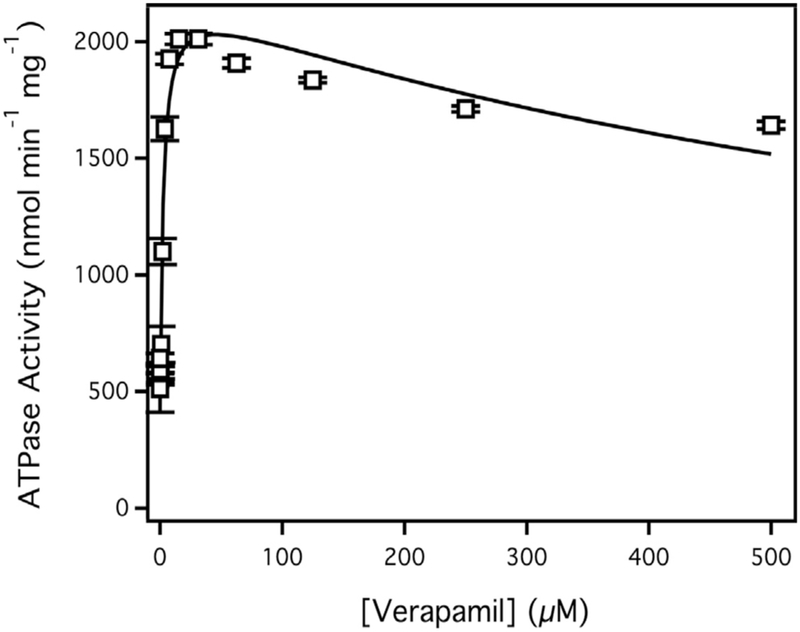
Verapamil stimulated ATPase activity of Pgp reconstituted in liposomes. The points represent an average of at least three experiments and the bars are the standard deviation. The solid line is the fit to Eq. (2).
3.2. Features of AFM images with Pgp embedded in a lipid bilayer
AFM images of Pgp reconstituted in proteoliposomes and liposomes applied to a mica surface are shown in Fig. 2. Fig. 2A shows a representative AFM image that has major features that were typically found in the AFM images. The dark area on the left side of the image reflects a void formed from the absence of a lipid bilayer and was used as an internal control to verify the presence of the bilayer. The height of the cross-section that crosses the void in Fig. 2A is shown in Fig. 2B. The average depth of ten similar voids was 37.3 ± 2.5 Å (mean ± standard deviation), which correlates with the thickness of a lipid bilayer [30]. The cross-section also revealed “small” and “large” protrusions with heights of ~ 30 Å and ~ 70 Å, respectively. The AFM image in Fig. 2C shows a number of “small” (highlighted with circles) and “large” (squares) protrusions. The “small” and “large” protrusions correlate to the asymmetric EC and C protein domains predicted from the X-ray crystal structure of Pgp (PDB IDs: 4M1M and 6C0V [14, 15, 17]), respectively. Inspection of several AFM images revealed that the “small” protrusions significantly outnumbered the “large” ones. This observation suggests that the reconstituted Pgp transporters were preferentially facing a single direction, which is consistent with previous characterization of the transporter [41,44]. In contrast, liposomes prepared in an identical manner, but without Pgp, essentially lacked any protrusions (Fig. 2D). This suggests that the punctate features in the AFM images in Fig. 2A and 2C are most likely protein domains of Pgp protruding from the lipid bilayer.
Fig. 2.

AFM images and analysis of Pgp reconstituted in liposomes. The (A) AFM image and (B) cross-section (indicated by the dashed line in panel A) shows a void as well as a “large” and a “small” protrusion. (C) A representative 1 × 1 μm AFM image showing “large” protrusions (squares), “small” protrusions (circles) and aggregates (arrow). (D) AFM image and line scan of liposomes prepared without Pgp shows no discernable features. A downward step of approximately 40 Å (bottom right) is consistent with the presence of bilayer.
3.3. Distribution of “small” and “large” protrusions in the experimental AFM images
To quantitatively characterize the protrusions, the average height and volume of 50 representative “small” and “large” protrusions were analyzed. Analysis of the static AFM images of Pgp revealed significant variations in the heights and volumes of the “small” and “large” protrusions implying conformational heterogeneity. The average height and volume of the “small” protrusions were 26.4 ± 4.9 Å and 2.0 ± 0.8 × 105 Å3, respectively. The average height and volume of the “large” protrusions were considerably bigger at 67.3 ± 7.8 Å and 1.22 ± 0.63 × 106 Å3, respectfully. To determine the distribution of “small” and “large” protrusions in the experimental AFM images, a histogram was made of the protrusion volumes as a function of probability density from 2571 individual AFM features collected from 100 AFM images in Fig. 3. In Fig. 3A, there is a prominent population of protrusions exhibiting volumes of around 2.5 × 105 Å3, which correlate to the “small” protrusions. There is also a shoulder-like subpopulation beginning at 8 × 105 Å3 and extending beyond 1.1 × 106 Å3 that correlates to the “large” protrusions. Aggregates, identified by features exhibiting volumes > 3.0 × 106 Å3 were not included in the analysis (data not shown). To determine the fraction of “small” and “large” protrusions, the fraction of a given population was plotted as a function protrusion volume in Fig. 3B. Greater than 85% of the protrusions in the AFM images were “small” and correlated to volumes less than 8 × 105 Å3. The remaining ~ 15% of the protrusions were “large”. Therefore, this suggests that 85% of NBDs are outward facing in our proteoliposome preparations. If 100% of NBDs were outward facing, the Vmax of verapamil-induced activaton of Pgp-mediated ATP hydrolysis would increase about ~ 20% to a value of 2838 nmol min−1 mg−1.
Fig. 3.

AFM protrusion volume analysis of Pgp reconsituted in liposomes. (A) Histogram showing the distribution of volumes of Pgp protrusions. N is the number of features that were measured. (B) A normalized plot of the fraction of a given feature versus the volume. In both panels, the regions corresponding to the “small” and “large” protrusions are labeled; the “large” are colored gray.
3.4. Correlating simulated AFM images of Pgp domains to experimental AFM features
Theoretical AFM images were produced from the X-ray crystal structures of Pgp in the “open” conformation (PDB ID: 4M1M [14, 15]) and the “closed” conformation (PDB ID: 6C0V [17]). The membrane position of Pgp in the AFM images was determined by three different computational methods [37-39]. The measured heights and volumes of the simulated AFM images, and the measured membrane widths are shown in Table 1.
Table 1.
AFM simulation analysis of the EC and C domains of Pgp X-ray crystal structures (PDB IDs: 4M1Ma, 6C0Vb).
| Protein Positioning method |
||||||
|---|---|---|---|---|---|---|
| PDB ID | Domain | Protrusion Measurement | TMDETd | O’Marae | OPMf | Avg. ± S.D. |
| 4M1Ma | EC | Height (Å) | 26.7 | 13.4 | 24.1 | 21.4 ± 7.0 |
| Volume-106 (Å3) | 0.117 | 0.023 | 0.095 | 0.078 ± 0.049 | ||
| Mc | Width (Å) | 39.8 | 58.3 | 40.8 ± 1.2 | 46.3 ± 10.4 | |
| C | Height (Å) | 67.5 | 72.1 | 77.3 | 72.3 ± 4.9 | |
| Volume 106 (Å3) | 1.062 | 1.147 | 1.232 | 1.147 ± 0.085 | ||
| 6C0Vb | EC | Height (Å) | 19.2 | 15.5 | 17.8 | 17.5 ± 1.9 |
| Volume 106 (Å3) | 0.080 | 0.053 | 0.075 | 0.069 ± 0.014 | ||
| Mc | Width (Å) | 43.3 | 58.3 | 42.2 ± 1.2 | 47.9 ± 9.0 | |
| C | Height (Å) | 78.4 | 73.9 | 74.8 | 75.7 ± 2.4 | |
| Volume 106 (Å3) | 1.256 | 1.159 | 1.185 | 1.200 ± 0.050 | ||
| 4M1Ma and 6C0Vb | EC | Height (Å) | 19.5 ± 5.1 | |||
| Volume 106 (Å3) | 0.074 ± 0.033 | |||||
| Mc | Width (Å) | 47.1 ± 8.7 | ||||
| C | Height (Å) | 74.0 ± 3.9 | ||||
| Volume 106 (Å3) | 1.174 ± 0.069 | |||||
The AFM image was simulated using the “open” mouse Pgp X-ray crystal structure (PDB ID: 4M1M, [14,15]).
The AFM image was simulated using the “closed” human Pgp X-ray crystal structure (PDB ID: 6C0V, [17]).
The membrane width. With the TMDET and OPM approaches, the membrane width was calculated by taking the sum of the hydrophobic thickness and 9.6 Å to account for the two phosphate head groups. With the O’Mara et al. protocol, the membrane dimensions were estimated using a PyMOL script as described in the Materials and Methods with the membrane-bound Pgp models in [35].
Pgp was positioned in the membrane using the TMDET method [33]
Pgp was positioned in the membrane according to O’Mara et al. [35].
Pgp was positioned in the membrane according to the OPM method [34].
The first column of the table shows the PDB ID that was used to simulate the AFM image. The next two columns show the Pgp domain that was used to produce the AFM image and the type of measurement. This is followed by the computational approach that was used to position Pgp in the membrane and define the membrane boundaries. The last column shows the average and standard deviation of these measurements.
The first row of the table shows the analysis of the theoretical AFM images produced from the 4M1M Pgp X-ray crystal structure. Analysis revealed that protrusions of the C-side AFM simulations were about 3-fold higher than protrusions from the EC-side AFM images. The degree of protrusion depended on how the algorithms positioned Pgp in the membrane and how they defined the membrane. The protrusion produced from the EC-side of the 4M1M Pgp X-ray crystal structure varied more than 10 Å with an average height of 21.4 ± 7.0 Å. These differences are primarily the result of differences in the simulated membrane width. The simulated membrane width was 50% larger using the O’Mara et al. approach than the TMDET and OPM approaches. This membrane width is also considerably larger than the experimentally determined membrane width from the void depths in the AFM images. If we examine the C-domain on the opposite side of the membrane, there was less variation with an average height of 72.3 ± 4.9 Å, regardless of the computational approach.
The next row shows the analysis of AFM images produces from the 6C0V Pgp X-ray crystal structure. Like the AFM simulations of the 4M1M X-ray crystal structure, the measured heights and volumes were several-fold higher for the AFM images produced from the C-side domain than the EC-side domain of Pgp. In contrast to the other simulation, the protein positioning method did not have as pronounced effect on the simulated AFM heights, despite differences in the membrane width. The average heights from the simulated AFM images of the EC- and C-side of Pgp were 17.5 ± 1.9 Å and 75.7 ± 2.4 Å, respectively.
To tentatively assign the “small” and “large” features of the experimental AFM images to a Pgp domain, an average of averages of the simulated AFM images was calculated (Table 1, last row). The simulated AFM images of the EC domain had an average height and volume of 19.5 ± 5.1 Å and 0.74 ± 0.33 × 105 Å3, respectively. This height is about 25% smaller than the average height of the representative “small” features in the experimental AFM images, where the average height and volumes were 26.4 ± 4.9 Å and 2.0 ± 0.8 × 105 Å3, respectively. However, this is within the range of the average heights and volumes in AFM simulations of the EC domain from the “open” 4M1M Pgp X-ray crystal structure (Table 1). The AFM images simulated using the TMDET [37] and OPM [38] algorithms correlate best to the “small” features in the experimental AFM images. Therefore, the “small” features in the experimental AFM images were assigned to the EC-side of Pgp. The average membrane width was 47.1 ± 8.7 Å, which is a little bit larger than the experimentally determined membrane width. If we exclude the high membrane width predicted using the O’Mara et al. approach, the average membrane width decreases to 41.5 ± 1.5 Å, which is in range of experimentally determined values. The “large” features in the AFM images were within the error of the average height and volume of the theoretical AFM images produced from the C domain of Pgp. The average height and volume of these simulated AFM images were 74.0 ± 3.9 Å and 1.174 ± 0.069 × 106 Å3, respectively. Thus, the “large” features in the experimental AFM images were assigned to the C-side of Pgp.
The simulated AFM images were compared to the “large” and “small” features in the experimental images in Fig. 4. Fig. 4A shows the topographical surface produced from the C-side of the 4M1M Pgp X-ray crystal structure and membrane boundaries defined by the OPM algorithm. The surface extends out over 70 Å from the membrane surface with two broad lobes formed from the NBDs. A contour plot of the topographical surface is shown (Fig. 4B) where 0 Å represents the position of the membrane and lighter shades reflect a greater protrusion height. For comparison, a representative “large” AFM feature is shown in Fig. 4C. To compare the simulated to the experimental AFM image, a cross-section was taken of each of these contour plots and is shown in Fig. 4D. The simulated and experimental curves both show two relatively broad lobes. The Pearson correlation (R) between the simulated and the experimental AFM images was 0.96. This is consistent with the “large” feature in the experimental AFM images being the C-side of Pgp. Topography of the EC-side formed by the 4M1M Pgp X-ray crystal structure and the membrane defined by the OPM algorithm is shown in Fig. 4E. The surface protrudes about a third as high as the simulated AFM image produced from the C-side of Pgp. A contour plot of this surface is shown in Fig. 4F and a representative “small” feature in the AFM images is shown next to it in Fig. 4G. Cross-sections of these plots are shown in Fig. 4H. The simulated image of the EC side had a slightly smaller footprint than the selected experimental AFM image. This is likely due to the fact that the Pgp conformation used to produce the theoretical AFM image is not identical to the Pgp conformation that produced the experimental AFM image. However, the R correlation was still 0.86 between the simulated and experimental AFM images. This is in line with the “small” AFM feature correlating to the EC domain of PgP.
Fig. 4.

Simulated versus experimental AFM images of Pgp. The 4M1M Pgp X-ray crystal structure (light gray van der Waals surface) and the simulated topographic surface (black) produced of the (A) C and (E) EC domains of Pgp using membrane positioning by OPM algorithm. Simulated AFM gray scale images of the (B) C and (F) EC domains are colored according to the protrusion height of the topographical surfaces shown in panels A and E, respectively. A representative (C) “large” and (G) “small” protrusion feature in the experimental AFM images. The white horizontal scale bar indicates 100 Å. Cross-sections of the (D) C-side simulated AFM image versus the “large” experimental AFM feature and (H) EC-side simulated AFM image versus the “small” AFM feature. The simulated and experimental cross-sections are shown as dashed black lines and gray lines, respectively. The lines in these panels correspond to the black dashed lines shown in the simulated and experimental contour plots of panels B, C, F and G.
3.5. Confirming the assignment of Pgp-related features with Pgp-specific antibodies
In the previous section, the “large” and “small” Pgp features in the experimental AFM images were assigned to the C and EC domains of Pgp, respectively. To confirm the assignment, the effect of Pgp-specific antibodies on the protrusions in the experimental AFM images of Pgp were examined. Although there are EC-side specific antibodies for human Pgp [53], no EC-side specific antibodies currently exist for mouse Pgp. However, there are at least two Pgp-specific antibodies that can target the C-side of Pgp. The 6X His tag antibody [54] that targets the hexahistidine sequence on the C-terminal end of Pgp and the C219 antibody that targets a specific sequence in the C-side protein domains of Pgp [55,56].
Fig. 5 shows the effect of C-side specific antibodies on AFM images of Pgp. In Fig. 5A and B, the effect of the C219 and 6x His tag antibodies on the distribution of features in the AFM images of Pgp was examined. This analysis was used to confirm the existence of C-side features in the AFM images as well as the potency of antibodies to bind Pgp under the conditions used for AFM. If there were no significant changes in feature distribution in the AFM images, that would indicate a lack of C-side Pgp domain features or a lack of antibody potency. Instead, there was a significant rightward shift in the feature distribution in the presence of C219 (Fig. 5A) and 6 × His tag (Fig. 5B) antibodies that starts around 40 Å and extending out beyond 80 Å.
Fig. 5.

The effect of C-side Pgp domain specific antibodies on the AFM images. Height histograms of Pgp reconstituted in liposomes in the presence (gray dashed lines) and absence (black) of (A) the C219 and (B) the 6 × His antibodies. N is the number of features that were measured. Representative AFM images in grayscale of the “large” Pgp feature without (C, G) and with (D) C219 and (H) 6X His antibodies, and the “small” Pgp feature without (E, I) and with (F) C219 and (J) 6X His antibodies. The white horizontal scale bar reflects 200 Å.
The lower part of Fig. 5 shows representative AFM images of the “large” and “small” features in the experimental AFM images with and without antibodies. Fig. 5C-F illustrate the effects of the C219 antibody on the AFM images. A “large” feature in AFM image that was assigned to the C-side is shown in Fig. 5C. When the C219 antibody is added in Fig. 5D, the “large” feature increases more than 30 Å in height, which is consistent with antibody binding. In contrast, the height of the “small” feature in the AFM image that was assigned to the EC domain of Pgp in Fig. 5E is unaffected by the C219 antibody in Fig. 5F. Fig. 5G-J show the effect of the 6X His tag antibody on representatives of Pgp. Like the C219 antibody, the 6X His tag antibody increases the height of a representative “large” feature by about 30 Å (cf. Fig. 5G and 5H) and has no effect on a representative “small” AFM feature (cf. Fig. 5I and J). These results confirm that the “large” AFM feature is the C-side domain of Pgp and, by process of elimination, show that the “small” AFM feaure is most likely the EC-side domain of Pgp.
3.6. Conformational dynamics of Pgp in a lipid bilayer
In the previous sections, the “large” and the “small” features in the AFM images were shown to likely be the C and EC domains of Pgp, respectively. Therefore, the “large” and “small” features in the AFM images will henceforth be referred to as C-side and EC-side AFM features, respectively. The distribution of heights and volumes of these features in Fig. 3 demonstrated that Pgp in a lipid bilayer has a significant amount of conformational heterogeneity. A number of investigations with Pgp have suggested that conformational changes play a key role in transport [57-61], so we hypothesized that the conformational heterogeneity of Pgp observed in the lipid bilayer reflects Pgp dynamics.
To test the hypothesis, a representative C-side and EC-side AFM feature were monitored over time using a high speed AFM (i.e. Cypher VRS) with 1 s time resolution in Fig. 6. The change of height and volume of a single representative C-side feature as a function of time is shown in Fig. 6A and B, respectively. The C-side feature shows a considerably large amount of conformational dynamics over 25 s. During this time, the height of the feature varies ~ 10 Å from ~ 70 Å to ~ 80 Å, while the volume varied between 1.0 × 106 Å3 and 1.3 × 106 Å3. The average heights and volumes during this time period were 77 ± 4 Å and 1.151 ± 0.013 × 106 Å3, respectively. From these AFM images, six between 19 and 24 s were selected that showed a significant amount of conformational dynamics (Fig. 6C). The C-side feature shows bilobal structure suggestive of NBDs. Between 19 s and 20 s, the C-side feature height changes about ~ 10 Å from 70 Å to 80 Å. During this time, there was also a small ~0.05 × 106 Å3 increase in protrusion volume. The largest changes in protrusion volume occurred between 21 s and 22 s with a ~ 0.2 × 106 Å3 volume increase. The change of height and volume of a single representative EC-side feature as a function of time is shown in Fig. 6D and E, respectively. Fig. 6D shows that the EC-side feature vacillates around 10 Å with an average height of 31 ± 4 Å. These changes in EC-side feature height roughly correlate to changes in protrusion volume in Fig. 6E, which oscillates around an average of 0.22 ± 0.05 × 106 Å3. Individual AFM images between 2 and 7 s are displayed in Fig. 6F. These images demonstrate notable changes in height and volume. The largest differences with these AFM images were observed between 2 and 3 s. During this time, the height decreases more than 10 A and the volume decreases almost 3-fold.
Fig. 6.

Conformational dynamics of Pgp reconsituted in liposomes investigated by high speed AFM imaging. The (A) height and (B) volume of a representative C-side feature in the AFM images as a function of time. (C) Oblique 3D renderings of the C-side feature in the AFM image between 19 and 24 s. The (D) height and (E) volume of a representative EC-side feature in the AFM images as a function of time. (F) Oblique 3D renderings of the EC-side feature in the AFM image between 2 and 7 s.
4. Discussion
Structural investigations of Pgp in detergent micelles and in soluble nanoscale phosphoplipid bilayers called Nanodiscs have provided details of the conformational dynamics of the transporter [e.g. [14,57,62]]. However, a question still remains whether Pgp in a detergent micelle and Nanodiscs accurately mimics the behavior of Pgp in an unrestricted bilayer. Previous AFM studies of Pgp in lipid bilayers have identified Pgp-specific, albeit unassigned, protrusions [29-32]. In this study, experimental AFM images of Pgp embedded in a lipid bilayer revealed distinct “small” and “large” Pgp-specific protrusions. AFM simulations of Pgp X-ray crystal structures and changes to the experimental AFM images in the presence of Pgp-specific antibodies suggested that these “large” and “small” features were the C and EC domains of Pgp, respectively. On a seconds time scale, high speed AFM images (Fig. 6) showed that the features undergo significant changes in height and volume.
From the results, a simplified model of Pgp conformational dynamics involving the “open” and “closed” Pgp conformations in a lipid bilayer is provided in Fig. 7. This model was developed using the heights and volumes from the simulated (Table 1) and time resolved AFM images (Fig. 6). The model focused on simulated AFM images produced with the TMDET and OPM algorithms because the simulated membrane widths and the EC domain protrusion heights correlated best to the membrane width deduced from the experimental AFM images. A cartoon of the changes in protrusion height of Pgp C-domain and the corresponding AFM envelope is shown in Fig. 7A. In the “open” conformation (Fig. 7A, left), the C domain of the 4M1M Pgp X-ray crystal structure using the TMDET and OPM algorithms extends approximately 70 A above the membrane plane. This is close to the minimum protrusion height of the “large” feature measured in the individual and time resolved experimental AFM images. In the lipid bilayer, a shift to the “closed” conformation increases the C domain height by ~10 Å (Fig. 7A, right). This is based on the C domain height difference with simulations performed with the “open” 4M1M and “closed” 6C0V Pgp X-ray crystal structures (Table 1). This also correlates to the 10 Å fluctuation in the “large” C-side feature observed in the time resolved AFM images (Fig. 6A-C). This fluctuation is exemplified by the ~10 Å height increase of AFM images between 19 and 20 s (Fig. 6C). Interestingly, the volume of the C-side AFM feature remains relatively constant during this transition. That is because the increase in height between 19 and 20 s has a corresponding decrease in width of the same feature. In kind, the volume of the theoretical AFM images of the C-domain using the TMDET and OPM algorithms with the 4M1M and 6C0V Pgp X-ray crystal structures was also relatively constant within the error (Table 1). The average volumes produced from the C-domain of the 4M1M and the 6C0V X-ray crystal structures was 1.147 ± 0.120 × 106 Å3 and 1.221 ± 0.050 × 106 Å3, respectively, using the TMDET and OPM algorithms. A similar degree of conformational dynamics was observed for the EC-domain and is shown diagrammatically in Fig. 7B. In the “open” conformation (Fig. 7B, left), the EC-domain extends out about 30 Å with the TMDET and OPM algorithms. Like the C-domain, the time resolved AFM images revealed that the “small” feature height fluctuates by 10 Å (Fig. 6D). Logically, if the C-domain increases by ~ 10 Å in the “closed” conformation (Fig. 7A, right), then the height of the EC-domain should decrease by ~ 10 Å as shown in the right panel of Fig. 7B. Indeed, the theoretical AFM images of the EC domain using the TMDET and OPM algorithms indicate a ~ 7 Å decrease in height from 25.4 ± 1.8 Å to 18.5 ± 1.0 Å. This correlates with the greater than 10 Å decrease in the time resolved AFM images between 2 and 3 s (Fig. 6F). At least with these two time points, there is also a corresponding several fold decrease in the protrusion volume with height. The theoretical AFM images from the 4M1M and the 6C0V Pgp X-ray crystal structures show a less pronounced ~ 30% reduction in protrusion volume from 0.106 ± 0.016 × 106 Å3 to 0.078 ± 0.004 × 106 Å3. For the other time points in the time resolved AFM images (Fig. 6D-F), the changes in the “small” EC-side feature height do not necessarily correspond well to changes to volume. For example, for AFM images at 14 and 15 s (Fig. 6F), there were virtually no changes to the height (Fig. 6D), but large changes to the volume (Fig. 6E). Discrepancies between the theoretical and experimental AFM images likely reflect the fact that Pgp can assume many different conformations in a lipid bilayer. Thus, this model only reflects two discrete conformational states in an ensemble of Pgp conformations in a lipid bilayer revealed in the AFM images.
Fig. 7.
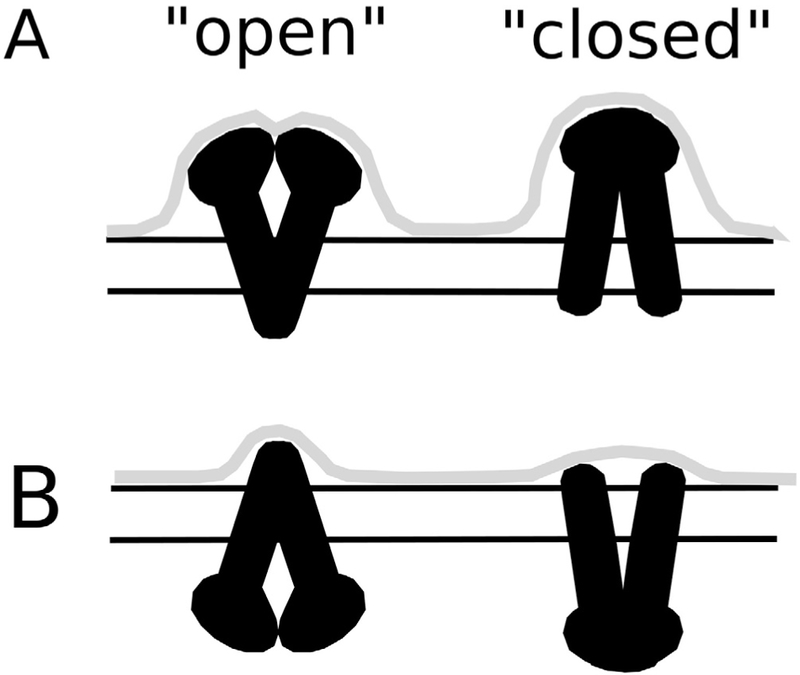
Model of Pgp conformational dynamics in a lipid bilayer. The protrusion envelope (gray line) produced from the (A) C and (B) EC domains of Pgp (black) with cartoons of the “open” (left) and “closed” (right) Pgp structure. The two horizontal lines roughly reflect the membrane position deduced using the OPM or TMDET algorithms.
The static and time resolved AFM images of Pgp in this study revealed that the transporter undergoes a range of conformations within the bilayer. This conformational range is compatible with the “open” Pgp ligand-free X-ray crystal structure [14,15] and the “closed” cryo-electron microscopy (cryo-em) Pgp structure in the presence of ATP [17]. This also suggests that ATP is not required to drive Pgp into the “closed” conformation. In addition, because substrates are believed to access the transporter from the lipid bilayer [10], the vertical Pgp movement revealed from the time resolved AFM images and the computer simulations using the X-ray crystal structures provides insight into how substrates ingress and egress into the transmembrane region of the transporter. The Pgp conformation(s) identified in the AFM images combined with known Pgp X-ray crystal structures or structural information from NMR can be used to accurately dock Pgp ligands to the transporter. This information is essential because drugs are known to induce significant conformational changes of the transporter [e.g. [64-66]], which can potentially alter their drug binding surfaces.
AFM is a powerful tool to monitor Pgp conformational dynamics in a lipid bilayer, but has certain limitations. In comparison to the cryo-em technique [67], AFM has angstrom resolution along the vertical dimension for Pgp, but is currently limited to ~ 1 nm along the lateral dimensions because of cantilever tip sharpness [68]. Furthermore, crevasses and indentations of the Pgp surface remain inaccessible because of instrumentation limitations [68]. The time resolved AFM images of Pgp produced in this study in Fig. 6 represents a major advance in AFM technology [69]. However, the AFM images are acquired on a seconds time scale, when important Pgp conformational changes may occur on a submicrosecond time scale as suggested by molecular dynamics (MD) simulations of the transporter [39,70,71]. Therefore, the structural information acquired from this approach is best exploited, when combining it with other techniques. For example, the EC and C domain volumes determined from the AFM images can be used as potential restraints for MD simulations of the Pgp X-ray crystal or cryo-em structures in a lipid bilayer. This will require development of computational approaches to combine and manipulate AFM images like is currently done with the cryo-em technique [67]. Ultimately, this work has provided insight into the structures and the conformational dynamics of Pgp in a lipid bilayer. It also serves as a foundation for future structural studies investigating the effect of ligands and nucleotides on Pgp in its natural membrane environment and under physiological conditions.
Acknowledgements
We would like to give thanks Dr. Ina L. Urbatsch of Texas Tech University Health Sciences Center for her generous gift of Pichia (P.) pastoris with the wild-type mouse Pgp transporter gene. Without their contribution and generosity, this research would not be possible. This work was supported by the National Institutes of Health (1R01CA204846-01A1, A.G.R.), the National Science Foundation (1054832, G.M.K.) and the University of Missouri Research Board. We thank Asylum Research Inc. for the loan of the video rate scanning (VRS) Cypher instrument.
Footnotes
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- [1].George AM (Ed.), ABC Transporters – 40 Years on, first ed., Springer, New York, NY, 20152016 edition. [Google Scholar]
- [2].Kim RB, Drugs as P-glycoprotein substrates, inhibitors, and inducers, Drug Metab. Rev. 34 (2002) 47–54, 10.1081/DMR-120001389. [DOI] [PubMed] [Google Scholar]
- [3].Fu D, Where is it and how does it get there – intracellular localization and traffic of P-glycoprotein, Front. Oncol. 3 (2013) 1–5, 10.3389/fonc.2013.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM, P-glycoprotein: from genomics to mechanism, Oncogene 22 (2003) 7468–7485, 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- [5].Staud F, Ceckova M, Micuda S, Pavek P, Expression and function of P-glycoprotein in normal tissues: effect on pharmacokinetics, Methods Mol. Biol. Clifton NJ: 596 (2010) 199–222, 10.1007/978-1-60761-416-6_10. [DOI] [PubMed] [Google Scholar]
- [6].Choi YH, Yu A-M, ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development, Curr. Pharm. Des. 20 (2014) 793–807, 10.2174/138161282005140214165212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tanigawara Y, Role of P-glycoprotein in drug disposition, Ther. Drug Monit. 22 (2000) 137–140, 10.1097/00007691-200002000-00029. [DOI] [PubMed] [Google Scholar]
- [8].Zhou S-F, Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition, Xenobiotica 38 (2008) 802–832, 10.1080/00498250701867889. [DOI] [PubMed] [Google Scholar]
- [9].Pang KS, Modeling of intestinal drug absorption: roles of transporters and metabolic enzymes (for the Gillette Review series), Drug Metab. Dispos. 31 (2003) 1507–1519, 10.1124/dmd.31.12.1507. [DOI] [PubMed] [Google Scholar]
- [10].Waghray D, Zhang Q, Inhibit or evade multidrug resistance P-glycoprotein in cancer treatment, J. Med. Chem. 61 (2017) 5108–5121, 10.1021/acs.jmedchem.7b01457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Miller DS, Bauer B, Hartz AMS, Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy, Pharmacol. Rev. 60 (2008) 196–209, 10.1124/pr.107.07109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ledwitch KV, Roberts AG, Cardiovascular ion channel inhibitor drug-drug interactions with P-glycoprotein, AAPS J. 19 (2017) 409–420, 10.1208/s12248-016-0023-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wessler JD, Grip LT, Mendell J, Giugliano RP, The P-glycoprotein transport system and cardiovascular drugs, J. Am. Coll. Cardiol. 61 (2013) 2495–2502, 10.1016/j.jacc.2013.02.058. [DOI] [PubMed] [Google Scholar]
- [14].Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G, Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding, Science 323 (2009) 1718–1722, 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li J, Jaimes KF, Aller SG, Refined structures of mouse P-glycoprotein, Protein Sci. 23 (2014) 34–46, 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jin MS, Oldham ML, Zhang Q, Chen J, Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans, Nature 490 (2012) 566–569, 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim Y, Chen J, Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation, Science 359 (2018) 915–919, 10.1126/science.aar7389. [DOI] [PubMed] [Google Scholar]
- [18].Ward A, Reyes CL, Yu J, Roth CB, Chang G, Flexibility in the ABC transporter MsbA: alternating access with a twist, Proc. Natl. Acad. Sci. 104 (2007) 19005–19010, 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dawson RJP, Hollenstein K, Locher KP, Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism, Mol. Microbiol. 65 (2007) 250–257, 10.1111/j.1365-2958.2007.05792.X. [DOI] [PubMed] [Google Scholar]
- [20].Shukla S, Abel B, Chufan EE, Ambudkar SV, Effects of a detergent micelle environment on P-glycoprotein (ABCBl)-ligand interactions, J. Biol. Chem. 292 (2017) 7066–7076, 10.1074/jbc.M116.771634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Loo TW, Clarke DM, P-glycoprotein ATPase activity requires lipids to activate a switch at the first transmission interface, Biochem. Biophys. Res. Commun. 472 (2016) 379–383, 10.1016/j.bbrc.2016.02.124. [DOI] [PubMed] [Google Scholar]
- [22].Clay AT, Sharom FJ, Lipid bilayer properties control membrane partitioning, binding, and transport of P-Glycoprotein substrates, Biochemistry 52 (2013) 343–354, 10.1021/bi301532c. [DOI] [PubMed] [Google Scholar]
- [23].Doige CA, Yu X, Sharom FJ, The effects of lipids and detergents on ATPase-active P-glycoprotein, BBA-Biomembr. 1146 (1993) 65–72, 10.1016/0005-2736(93)90339-2. [DOI] [PubMed] [Google Scholar]
- [24].Al-Shawi MK, Omote H, The remarkable transport mechanism of P-glycoprotein; a multidrug transporter, J. Bioenerg. Biomembr. 37 (2005) 489–496, 10.1007/s10863-005-9497-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Aanismaa P, Seelig A, P-Glycoprotein kinetics measured in plasma membrane vesicles and living cells, Biochemistry 46 (2007) 3394–3404, 10.1021/bi0619526. [DOI] [PubMed] [Google Scholar]
- [26].Varma MVS, Panchagnula R, pH-dependent functional activity of P-glycoprotein in limiting intestinal absorption of protic drugs: kinetic analysis of quinidine efflux in situ, J. Pharm. Sci. 94 (2005) 2632–2643, 10.1002/jps.20489. [DOI] [PubMed] [Google Scholar]
- [27].Young AM, Audus KL, Proudfoot J, Yazdanian M, Tetrazole compounds: The effect of structure and pH on Caco-2 cell permeability, J. Pharm. Sci. 95 (2006) 717–725, 10.1002/jps.20526. [DOI] [PubMed] [Google Scholar]
- [28].Binnig G, Quate CF, Gerber C, Atomic force microscope, Phys. Rev. Lett. 56 (1986) 930–933, 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- [29].Müller DJ, Dufrêne YF, Atomic force microscopy: a nanoscopic window on the cell surface, Trends Cell Biol. 21 (2011) 461–469, 10.1016/j.tcb.2011.04.008. [DOI] [PubMed] [Google Scholar]
- [30].Muller DJ, Engel A, Atomic force microscopy and spectroscopy of native membrane proteins, Nat. Protocol 2 (2007) 2191–2197, 10.1038/nprot.2007.309. [DOI] [PubMed] [Google Scholar]
- [31].Whited AM, Park PS-H, Atomic force microscopy: a multifaceted tool to study membrane proteins and their interactions with ligands, Biochim. Biophys. Acta 2014 (1838) 56–68, 10.1016/j.bbamem.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sanganna Gari RR, Frey NC, Mao C, Randall LL, King GM, Dynamic structure of the translocon SecYEG in membrane: direct single molecule observations, J. Biol. Chem. 288 (2013) 16848–16854, 10.1074/jbc.M113.471870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ruspantini I, Diociaiuti M, Ippoliti R, Lendaro E, Gaudiano MC, Cianfriglia M, Chistolini P, Arancia G, Molinari A, Immunogold localisation of P-glycoprotein in supported lipid bilayers by transmission electron microscopy and atomic force microscopy, Histochem. J. 33 (2001) 305–309, 10.1023/A:1017933225226. [DOI] [PubMed] [Google Scholar]
- [34].Diociaiuti M, Molinari A, Ruspantini I, Gaudiano MC, Ippoliti R, Lendaro E, Bordi F, Chistolini P, Arancia G, P-glycoprotein inserted in planar lipid bilayers formed by liposomes opened on amorphous carbon and Langmuir-Blodgett monolayer, Biochim. Biophys. Acta 1559 (2002) 21–31, 10.1016/S0005-2736(01)00425-4. [DOI] [PubMed] [Google Scholar]
- [35].Inci F, Celik U, Turken B, Ozer HO, Kok FN, Construction of P-glycoprotein incorporated tethered lipid bilayer membranes, Biochem. Biophys. Rep. 2 (2015) 115, 10.1016/j.bbrep.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Barakat S, Gayet L, Dayan G, Labialle S, Lazar A, Oleinikov V, Coleman AW, Baggetto LG, Multidrug-resistant cancer cells contain two populations of P-glycoprotein with differently stimulated P-gp ATPase activities: evidence from atomic force microscopy and biochemical analysis, Biochem. J. 388 (2005) 563–571, 10.1042/BJ20041999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tusnády GE, Dosztányi Z, Simon I, TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates, Bioinformatics 21 (2005) 1276–1277, 10.1093/bioinformatics/btil21. [DOI] [PubMed] [Google Scholar]
- [38].Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI, Positioning of proteins in membranes: a computational approach, Protein Sci. 15 (2006) 1318–1333, 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].O’Mara ML, Mark AE, Structural characterization of two metastable ATP-bound states of P-glycoprotein, PLoS One 9 (2014) e91916, 10.1371/journal.pone.0091916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bai J, Swartz DJ, Protasevich II, Brouillette CG, Harrell PM, Hildebrandt E, Gasser B, Mattanovich D, Ward A, Chang G, Urbatsch IL, A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris, PLoS One. 6 (2011) e22577,, 10.1371/journal.pone.0022577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ledwitch KV, Barnes RW, Roberts AG, Unravelling the complex drug-drug interactions of the cardiovascular drugs, verapamil and digoxin, with P-glycoprotein, Biosci. Rep. 36 (2016) 1–14, 10.1042/BSR20150317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lerner-Marmarosh N, Gimi K, Urbatsch IL, Gros P, Senior AE, Large scale purification of detergent-soluble p-glycoprotein from Pichia pastoris cells and characterization of nucleotide binding properties of wild-type, walker a, and walker b mutant proteins, J. Biol. Chem. 274 (1999) 34711–34718, 10.1074/jbc.274.49.34711. [DOI] [PubMed] [Google Scholar]
- [43].Urbatsch IL, Wilke-Mounts S, Gimi K, Senior AE, Purification and characterization of N-glycosylation mutant mouse and human P-glycoproteins expressed in Pichia pastoris cells, Arch. Biochem. Biophys. 388 (2001) 171–177. [DOI] [PubMed] [Google Scholar]
- [44].Wilt LA, Nguyen D, Roberts AG, Insights into the molecular mechanism of triptan transport by P-glycoprotein, J. Pharm. Sci. 106 (2017) 1670–1679, 10.1016/j.xphs.2017.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chifflet S, Torriglia A, Chiesa R, Tolosa S, A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases, Anal. Biochem. 168 (1988) 1–4, 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- [46].Nečas D, Klapetek P, Gwyddion: an open-source software for SPM data analysis, Cent. Eur. J. Phys. 10 (2012) 181–188, 10.2478/s11534-011-0096-2. [DOI] [Google Scholar]
- [47].Marsh BP, Chada N, Gari RRS, Sigdel KP, King GM, The hessian blob algorithm: Precise particle detection in atomic force microscopy imagery, Sci. Rep. 8 (2018) 978, 10.1038/s41598-018-19379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kitanidis PK, Generalized covariance functions associated with the Laplace equation and their use in interpolation and inverse problems, Water Resour. Res. 35 (n.d.) 1361–1367. doi: 10.1029/1999WR900026. [DOI] [Google Scholar]
- [49].Udpa L, Ayres VM, Fan Y, Chen Q, Kumar SA, Deconvolution of atomic force microscopy data for cellular and molecular imaging, IEEE Signal Process. Mag. 23 (2006) 73–83, 10.1109/MSP.2006.1628880. [DOI] [Google Scholar]
- [50].Haralick RM, Sternberg SR, Zhuang X, Image analysis using mathematical morphology, IEEE Trans. Pattern Anal. Mach. Intell. PAMI-9 (1987) 532–550, 10.1109/TPAMI.1987.4767941. [DOI] [PubMed] [Google Scholar]
- [51].Ledwitch KV, Gibbs ME, Barnes RW, Roberts AG, Cooperativity between verapamil and ATP bound to the efflux transporter P-glycoprotein, Biochem. Pharmacol. 118 (2016) 96–108, 10.1016/j.bcp.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Litman T, Zeuthen T, Skovsgaard T, Stein WD, Competitive, non-competitive and cooperative interactions between substrates of P-glycoprotein as measured by its ATPase activity, BBA-Mol. Basis Dis. 1361 (1997) 169–176, 10.1016/S0925-4439(97)00027-6. [DOI] [PubMed] [Google Scholar]
- [53].Mechetner EB, Roninson IB, Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody, Proc. Natl. Acad. Sci. 89 (1992) 5824–5828, 10.1073/pnas.89.13.5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zentgraf H, Frey M, Schwinn S, Tessmer C, Willemann B, Samstag Y, Velhagen I, Detection of histidine-tagged fusion proteins by using a high-specific mouse monoclonal anti-histidine tag antibody, Nucl. Acids Res. 23 (1995) 3347–3348, 10.1093/nar/23.16.3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].van Den Elsen JM, Kuntz DA, Hoedemaeker FJ, Rose DR, Antibody C219 recognizes an alpha-helical epitope on P-glycoprotein, Proc. Natl. Acad. Sci. 96 (1999) 13679–13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liu B, Sun D, Xia W, Hung MC, Yu D, Cross-reactivity of C219 anti-p170(mdr-1) antibody with p185(c-erbB2) in breast cancer cells: cautions on evaluating p170(mdr-1), J. Natl. Cancer Inst. 89 (1997) 1524–1529, 10.1093/jnci/89.20.1524. [DOI] [PubMed] [Google Scholar]
- [57].Li MJ, Guttman M, Atkins WM, Conformational dynamics of P-glycoprotein in lipid nanodiscs and detergent micelles reveal complex motions on a wide time scale, J. Biol. Chem. (2018), 10.1074/jbc.RA118.002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ritchie TK, Kwon H, Atkins WM, Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid Nanodiscs and inhibition by the antibodies MRK16 and UIC2, J. Biol. Chem. 286 (2011) 39489–39496, 10.1074/jbc.M111.284554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang G, Pincheira R, Zhang M, Zhang JT, Conformational changes of P-glycoprotein by nucleotide binding, Biochem. J. 328 (1997) 897–904, 10.1042/bj3280897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Loo TW, Bartlett MC, Clarke DM, Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein: direct evidence for the substrate-induced fit mechanism for drug binding, J. Biol. Chem. 278 (2003) 13603–13606, 10.1074/jbc.C300073200. [DOI] [PubMed] [Google Scholar]
- [61].Bhatnagar J, Sim H-M, Georgieva E, Kapoor K, Chufan E, Ohnuma S, Borbat PP, Freed JH, Sauna ZE, Ambudkar SV, Mapping conformational changes associated with the catalytic cycle of human P-glycoprotein (ABCB1), Biophys. J. 102 (2012) 606a–607a, 10.1016/j.bpj.2011.11.3305.22325284 [DOI] [Google Scholar]
- [62].Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, Mobin A, Esser L, Earl LA, Gottesman MM, Xia D, Ambudkar SV, Subramaniam S, Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle, Mol. Pharmacol. 90 (2016) 35–41, 10.1124/mol.116.104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gibbs ME, Wilt LA, Ledwitch KV, Roberts AG, A conformationally-gated model of methadone and loperamide transport by P-glycoprotein, J. Pharm. Sci. (2018), 10.1016/j.xphs.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Loo TW, Bartlett MC, Clarke DM, Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains, J. Biol. Chem. 278 (2003) 1575–1578, 10.1074/jbc.M211307200. [DOI] [PubMed] [Google Scholar]
- [66].Sonveaux N, Vigano C, Shapiro AB, Ling V, Ruysschaert JM, Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. a tryptophan fluorescence quenching analysis, J. Biol. Chem. 274 (1999) 17649–17654. [DOI] [PubMed] [Google Scholar]
- [67].Frank J, Three-Dimensional Electron Microscopy of Macromolecular Assemblies: Visualization of Biological Molecules in Their Native State, second ed., Oxford University Press, Oxford; New York, 2006. [Google Scholar]
- [68].Eaton P, West P, Atomic Force Microscopy, Reprint ed., Oxford University Press, 2018. [Google Scholar]
- [69].Sarioglu AF, Solgaard O, Time-resolved tapping-mode atomic force microscopy, Scanning Probe Microsc Nanosci. Nanotechnol. 2, Springer, Berlin, Heidelberg, 2011, pp. 3–37 doi: 10.1007/978-3-642-10497-8_1. [DOI] [Google Scholar]
- [70].Condic-Jurkic K, Subramanian N, Mark AE, O’Mara ML, The reliability of molecular dynamics simulations of the multidrug transporter P-glycoprotein in a membrane environment, PLoS One 13 (2018) e0191882, 10.1371/journal.pone.0191882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Domicevica L, Koldsq H, Biggin PC, Multiscale molecular dynamics simulations of lipid interactions with P-glycoprotein in a complex membrane, J. Mol. Graph. Model. 80 (2018) 147–156, 10.1016/j.jmgm.2017.12.022. [DOI] [PubMed] [Google Scholar]