

Abstract
Background
This study aimed at identifying genomic regions that underlie genetic variation of worm egg count, as an indicator trait for parasite resistance in a large population of Australian sheep, which was genotyped with the high-density 600 K Ovine single nucleotide polymorphism array. This study included 7539 sheep from different locations across Australia that underwent a field challenge with mixed gastrointestinal parasite species. Faecal samples were collected and worm egg counts for three strongyle species, i.e. Teladorsagia circumcincta, Haemonchus contortus and Trichostrongylus colubriformis were determined. Data were analysed using genome-wide association studies (GWAS) and regional heritability mapping (RHM).
Results
Both RHM and GWAS detected a region on Ovis aries (OAR) chromosome 2 that was highly significantly associated with parasite resistance at a genome-wise false discovery rate of 5%. RHM revealed additional significant regions on OAR6, 18, and 24. Pathway analysis revealed 13 genes within these significant regions (SH3RF1, HERC2, MAP3K, CYFIP1, PTPN1, BIN1, HERC3, HERC5, HERC6, IBSP, SPP1, ISG20, and DET1), which have various roles in innate and acquired immune response mechanisms, as well as cytokine signalling. Other genes involved in haemostasis regulation and mucosal defence were also detected, which are important for protection of sheep against invading parasites.
Conclusions
This study identified significant genomic regions on OAR2, 6, 18, and 24 that are associated with parasite resistance in sheep. RHM was more powerful in detecting regions that affect parasite resistance than GWAS. Our results support the hypothesis that parasite resistance is a complex trait and is determined by a large number of genes with small effects, rather than by a few major genes with large effects.
Electronic supplementary material
The online version of this article (10.1186/s12711-019-0479-1) contains supplementary material, which is available to authorized users.
Background
Gastrointestinal nematode infections (GNI) are one of the most important health problems that affect sheep and other grazing ruminants in Australia and worldwide. The annual cost associated with nematode infections in the Australian sheep industry has been estimated at $436 million for lost production and treatment costs [1]. The effects of parasitism on the health and productivity of grazing ruminants are well documented and include loss of weight, diarrhoea, anorexia, scours, anaemia, and death [2]. During the past few decades, the sheep industry has become increasingly dependent on anthelmintic treatments as a method of parasite control. However, anthelmintic treatments are expensive and often not very effective. The frequent use of these treatments has also resulted in a rapid increase in anthelmintic resistance in sheep worldwide [3, 4]. Breeding sheep for enhanced resistance has been suggested as a viable method of parasite control. The majority of breeding programs for parasite control are based on indicator traits, in particular worm egg count (WEC) in faeces. However, recording WEC is time-consuming, costly, and unattractive. Therefore, it would be useful to select directly for parasite resistance without the need for nematode challenge.
The identification of genes or genomic regions that are responsible for parasite resistance could greatly improve the accuracy of genomic prediction and therefore result in genetic improvement for this trait [5, 6]. Genomic improvement for parasite resistance would benefit from greater knowledge about how sheep are able to mount effective immune responses against parasite infection and the genetic architecture behind the trait. Initial quantitative trait loci (QTL) mapping studies for parasite resistance in sheep were performed using microsatellite markers, e.g. [7–10]. In the last decade, genome-wide association studies (GWAS) using dense single nucleotide polymorphism (SNP) arrays have been used to identify QTL for most of the economically important traits in livestock species. To date, several GWAS have been reported for parasite resistance in different sheep breeds, e.g. [11–14]. Minimal consistency has emerged from these studies, probably due to the physiological complexity of the trait, and the fact that these studies are very diverse in terms of methodologies, statistical approaches, sheep breeds and parasite species.
Genome-wide significant SNPs identified from GWAS for complex traits in sheep, and other species such as humans, have generally failed to explain most of the genetic variation, e.g. [11, 15]. Such studies typically test the association with a phenotype of each SNP individually. In GWAS, the association between each SNP and the trait depends on the existence of linkage disequilibrium (LD) between the observed SNP and the causal loci that underlie the trait. Because of the large number of statistical tests performed in GWAS, very stringent thresholds are applied to avoid spurious associations. These stringent thresholds minimize false positive associations but also lead to many false negatives since variants with small effects or incomplete LD with the SNPs will fail to pass the stringent statistical threshold and remain undetected. Attempts to increase the power of GWAS have focused on increasing the number of observations for each experiment and the density of SNP arrays. Optimizing power in GWAS is both crucial and challenging. Without increasing the number of observations, power could be gained by testing a cumulative effect of multiple variants in a given region of a genome rather than testing each variant individually. Regional heritability mapping (RHM) has been suggested as a better approach to capture more of the genetic variation [16]. RHM facilitates the capture of genetic variation for a given region in the genome by integrating the effects of common and rare variants. Thus, the RHM has the ability to capture some of the genetic variation that is not detected by conventional GWAS methods. The aim of this study was to identify genomic regions with effects on parasite resistance in a large population of sheep naturally challenged with mixed strongyle nematode species (Teladorsagia circumcincta, Haemonchus contortus and Trichostrongylus colubriformis) using both GWAS and RHM approaches.
Methods
Phenotypes and population structure
Parasite resistance, as measured by worm egg counts (WEC), was investigated in lambs from a large multi-breed sheep population from the information nucleus (IN) flock of the Australian Sheep Cooperative Research Centre (CRC). Details on the IN flock, design and trait measurements are described in Van der Werf et al. [17]. Lambs were not drenched with anthelmintic until after sampling. When, after weaning, a random faecal sample within a management group exceeded a threshold of 1000 eggs per gram (epg) in sites predominated by H. contortus, or 500 epg in sites predominated by other species, faecal samples were collected from all individual animals. Worm eggs were then counted using a modified McMaster counting technique [18]. Worm eggs for three strongyle species were identified, i.e. T. circumcincta, H. contortus and T. colubriformis. The analysis included 7539 animals with both phenotype and genotype data. The distribution of the frequency of WEC records across different ages (days) is shown in Fig. 1.
Fig. 1.

Number of records across ages (per day)
Various breeds were represented in the population but with a significant proportion of Merino genes (70.0%), and only this breed had a substantial proportion of purebred animals (45.2%). The remaining breeds were represented mainly by crossbred offspring of their sires in crosses with Merino or Border Leicester × Merino ewes. Breed group size ranged from 3493 sheep for purebred Merino to 97 for a Poll Dorset/Suffolk/White Suffolk/White Dorper/Border Leicester/Merino cross. The breed content of the population is in Table 1.
Table 1.
Proportions of different breeds’ ancestry in the population
| Breed | BL | COR | SUF | WS | BRL | WD | PD | TEX | PS | MER |
|---|---|---|---|---|---|---|---|---|---|---|
| Proportion (%) | 10.9 | 0.8 | 2.5 | 1.7 | 0.7 | 0.4 | 10.0 | 1.8 | 1.2 | 70.0 |
BL Border Leicester, COR Corriedale, WD White Dorper, PD Poll Dorset, TEX Texel, PS Prime Samm, MER Merino, SUF Suffolk, WS White Suffolk, BRL Booroola
Genotypes and quality controls
50 k genotypes
Animals were genotyped with the Illumina 50 K SNP panel (Illumina Inc., San Diego, CA, USA). Several quality measures were applied to the 50 k SNP data. SNPs were removed if they had a minor allele frequency (MAF) lower than 0.01, a call rate lower than 90%, an Illumina Gentrain score (GC) lower than 0.6, a p value testing Hardy–Weinberg equilibrium less than 10−15, if the heterozygosity rate deviated by more than 3 standard deviations from the population mean and if they were located on the X and Y chromosomes. Furthermore, an individual was removed if the correlation of the genotypes with another sample (animal) was equal or higher than 0.99. After applying the quality control measures, 48,599 SNPs were retained for the analyses.
High-density (HD) genotypes
All animals with WEC phenotypes were then imputed from 50 K genotypes to the 600kOvine Infinium® HD SNP BeadChip panel (International Sheep Genomic Consortium and FarmIQ Project NZ). The high-density (HD) genotypes were imputed using a reference set of 1881 animals with real HD genotypes. This reference set of HD genotyped animals represented four main breeds (Merino, Poll Dorset, Border Leicester, and White Suffolk): 1042 represented various crosses of these breeds, while purebreds included 677 Merino, 47 White Suffolk, 44 Poll Dorset, 32 Border Leicester, and 39 from 10 other breeds. After applying the same quality measures as above, 510,065 SNPs were retained, and these 1881 HD animals were then used as a reference set to impute the 50 K genotypes to HD using Minimac3 [19]. Prior to imputation, phasing was performed on both the 50 K-genotyped and HD-genotyped animals separately using Eagle2 [20]. The accuracy of imputation to HD, which was tested within subsets of animals with observed HD genotypes, was on average high (0.98) across the whole genome.
Genome-wide association studies (GWAS)
In order to reduce computation time, a two-step association analysis was performed. First, phenotypes were pre-adjusted for fixed effects using the following model:
| 1 |
where is a vector of cube-root transformed WEC records; is the overall mean; is a design matrix of fixed effects; is a vector of fixed effects and is a vector of residuals assumed to be distributed as , where is the identity matrix and is the residual variance. The fixed effects included in the models to determine the corrected phenotypes were age of animals at WEC recording, age of dam, gender, rearing type birth type, contemporary groups (combination of flock, birth year and management group effects) and breed composition, which were fitted as covariates, one for each contributing breed. Second, residuals obtained from Model 1 were treated as corrected phenotypes for a single-SNP regression where each SNP was fitted separately, and a pedigree relationship matrix was fitted to account for population and pedigree structure. A linear mixed model was performed using the GEMMA program [21] as follows:
| 2 |
where is a vector of adjusted phenotypes (residuals) obtained from Eq. 1, is the overall mean, is a vector of genotypes for (coded as 0, 1, or 2 for the genotypes 00, 01/10, or 11, respectively), is the effect size of the th SNP (allele substitution effect), is a design matrix of random additive genetic effects, is a vector of random additive genetic effects assumed to be distributed as , where is the numerator relationship matrix calculated from available pedigree using the pedigree package in R [22], and is the vector of residuals. The false discovery rate (FDR) was applied to adjust for multiple SNP testing. Significant SNPs were determined by using the genome-wise FDR of 5%.
Regional heritability mapping (RHM)
RHM analyses were carried out on the whole genome using the MTG2 software [23]. Each chromosome was divided into regions that contained a predefined number of SNPs, and the additive genetic variance attributable to the joint SNP effects within each window was estimated. Window sizes of 1000 SNPs (~ 5 Mbp), 500 SNPs (~ 2.5 Mbp) and 200 SNPs (~ 1 Mbp) were used to build the genomic relationship matrices (GRM) for the specific regions and the windows were then shifted along the genome in steps of 500, 250 and 100 SNPs, respectively. To test the significance of each window, a likelihood ratio test (LRT) was applied to compare the full model, which includes the regional effect (Eq. 3) with the reduced model with no regional variance in that window (Eq. 4):
| 3 |
| 4 |
where the terms are as defined in Eqs. 1 and 2, except , which is the additive genetic effect of the window genotype estimated from SNPs within region and assumed to be distributed as , where is the regional genomic relationship matrix constructed from SNPs within region , and is the genomic variance explained by the SNPs in region . Phenotypic variance was given by and therefore the regional genomic heritability was estimated as .
For the RHM approach, LRT was assumed to follow a mixture of and distributions [16]. In total, 980, 2005 and 5025 windows were tested across the genome using RHM with window sizes of 1000, 500 and 200 SNPs, respectively. Due to the large number of windows tested across the genome, FDR was applied to correct for multiple testing. Significant windows were selected by using the genome-wise FDR of 5%.
Conditional GWAS and RHM analyses
Conditional GWAS and RHM analyses were carried out to determine if significant SNPs and regions were independent. First, GWAS analyses were performed on the regions of Ovis aries (OAR) chromosomes 2 and 6 by including the most significant SNP as a fixed covariate in the model and testing all SNPs in the region that were not in strong LD with the conditional SNP (). Second, RHM analyses were performed by adding the most significant regions to the model in a stepwise manner. To obtain a more conservative and probably better heritability estimate for a given region, both from the significant regions, and its complementary GRM (), which is based on all the remaining SNPs in the 600 K SNP panel, were fitted jointly in one model. Significant regions were added to the model sequentially where, each time, a new was built from all regions combined and a new was also built from all the SNPs on the 600 K panel, excluding the unique fitted SNPs in . Third, RHM analyses were performed conditionally on the top SNP from each region, which were then added to the model sequentially as fixed covariates, and the proportion of variance explained by all regions combined was estimated.
Haplotype construction and analysis
Haplotypes were constructed for SNPs located within the statistically significant windows using the Fimpute algorithm [24]. Once the haplotypes were constructed, LD between SNPs was calculated as the D′ statistics using the Haploview software [25], with haplotype blocks defined based on the criteria of Gabriel et al. [26]. Haplotype analysis was then carried out using the following model:
| 5 |
where is the adjusted phenotypic (residual) value for the th animal; is the overall mean; is the effect of the th haplotype; is the haplotype score (0, 1, or 2) of the haplotype for the animal, is the number of haplotypes segregating in the population for that haplotype block; is the vector of random additive genetic effects of individual and is the vector of random residual effects.
Principal component analysis
Principal component analysis (PCA) was performed on the genomic relationship matrix using MTG2. The first two principal components (PC) of the genotyped animals were plotted and samples were coloured according to their breed compositions, as known from pedigree information.
Gene annotation and functional information
Candidate genes in the significant regions were obtained from Ensembl (http://www.ensembl.org/biomart) and UCSC Genome Bioinformatics (http://genome.ucsc.edu). Because the sheep genome is not completely annotated compared to the human genome, human orthologous genes were used to explore their molecular functions. Biological pathways associated with these identified genes were obtained using the BioSystem Tools (https://www.ncbi.nlm.nih.gov/biosystems), which contains pathways from the main databases including: KEGG (Kyoto Encyclopedia of Genes and Genomes), Pathway Interaction Database (PID), WikiPathways and Reactome.
Results
Association analyses
A Manhattan plot of GWAS results for parasite resistance in sheep is in Fig. 2. Three SNPs (rs421630816, rs424521894, and rs413835864) were statistically significant at a genome-wise FDR of 5% (Table 2). The quantile–quantile (Q–Q) plot shows that, for these three significant SNPs, the deviation from their expected values is larger, which indicates a strong association between these SNPs and parasite resistance (Fig. 3). SNP rs421630816 is located within the PALLD gene at 110.8 Mbp on OAR2, and the rs424521894 and rs413835864 SNPs are located within the GALNTL6 gene at 107.3 and 107.4 Mbp on OAR2, respectively.
Fig. 2.

Manhattan plot of GWAS results for parasite resistance in Australian sheep. The y-axis shows the (p-values) of single-SNP association and the x-axis shows the position of the SNPs across the 26 chromosomes. Genome-wide significant SNPs (FDR of 5%) are highlighted by red dots
Table 2.
List of significant SNPs identified by GWAS for parasite resistance in Australian sheep
| OAR | SNP | Variant type | Position (chr:bp) | Nearest gene | p-value | FDR | |
|---|---|---|---|---|---|---|---|
| Name | Distance | ||||||
| 2 | rs421630816 | Intron | 2:110875234 | PALLD | Within | 6.7 × 10−8 | 0.02 |
| 2 | rs424521894 | Downstream gene | 2:107301187 | GALNTL6 | Within | 8.6 × 10−8 | 0.02 |
| 2 | rs413835864 | Intron | 2:107458856 | GALNTL6 | Within | 1.1 × 10−7 | 0.03 |
Significant SNPs were selected at a FDR of 5%
Fig. 3.
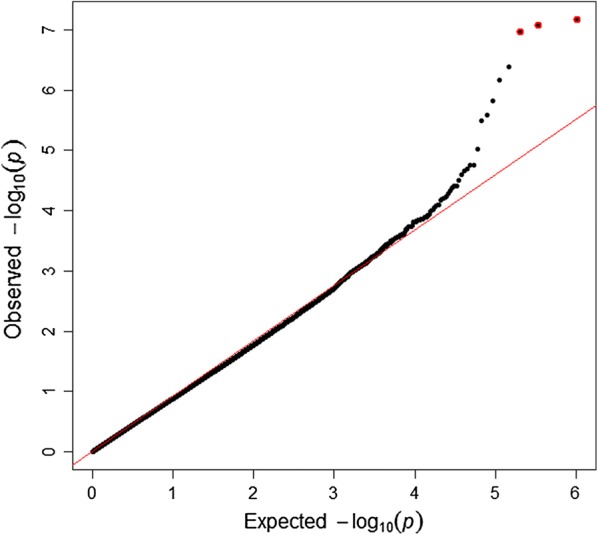
Quantile-quantile (Q–Q) plot of the observed p-values from the expected p-values of GWAS results (λ = 0.95). The observed (p-values) are shown by black dots, and the expected values under null distribution are shown by a red line. Significant SNPs (FDR of 5%) are highlighted by red dots
The results from the RHM analyses using window sizes of 1000, 500 and 200 SNPs are in Table 3 and Fig. 4. Q–Q plots of observed versus expected p-values of RHM analyses are in Fig. 5. RHM analysis using 1000-SNP windows identified three overlapping windows between 106.4 and 118.7 Mbp on OAR2, and another three overlapping windows between 32.7 and 42.3 Mbp on OAR6 that were significantly associated with parasite resistance at the genome-wide level. RHM analysis with 200-SNP windows identified three regions on OAR2: two overlapping windows between 106.9 and 108.4 Mbp, three overlapping windows between 110.1 and 113.3 Mbp, and one window between 117.01 and 118.13 Mbp. The two overlapping windows between 106.9 and 108.4 Mbp on OAR2 contains the second and third most significant SNPs (rs413835864 and rs424521894) identified by the GWAS, whereas the window between 110.59 and 112.38 Mbp contains the top significant SNP (rs421630816). Fine-mapping analysis also identified a significant region between 34.7 and 39.2 Mbp on OAR6, which included the sixth top ranked SNP (rs416517011; p = 7.87 × 10−8) based on GWAS. In addition, RHM using 200-SNP windows identified a region that contains two overlapping windows between 17.6 and 18.9 Mbp on OAR18, and a region on OAR24 between 40.4 and 41.9 Mbp.
Table 3.
Summary of significant windows identified by RHM using window sizes of 1000, 500 and 200 SNPs
| Window # | OAR | Position (bp) | (SE) | LRT | −logP | FDR | |
|---|---|---|---|---|---|---|---|
| Start | End | ||||||
| 1000-SNP window analysis | |||||||
| 159 | 2 | 106479658 | 112388825 | 0.007 | 19.2 | 5.23 | 0.001 |
| 160 | 2 | 109055156 | 115988999 | 0.007 | 17.2 | 4.76 | 0.003 |
| 161 | 2 | 112409311 | 118712363 | 0.010 | 22.6 | 5.99 | 0.001 |
| 412 | 6 | 32700889 | 37820042 | 0.009 | 20.0 | 5.40 | 0.001 |
| 413 | 6 | 35275766 | 40133729 | 0.010 | 24.8 | 6.49 | 0.000 |
| 414 | 6 | 37828214 | 42360019 | 0.007 | 13.6 | 3.93 | 0.019 |
| 500-SNP window analysis | |||||||
| 318 | 2 | 106479658 | 109049182 | 0.006 | 16.7 | 4.66 | 0.006 |
| 319 | 2 | 107706511 | 110358002 | 0.007 | 13.6 | 3.93 | 0.021 |
| 320 | 2 | 109055156 | 112388825 | 0.007 | 15.9 | 4.46 | 0.009 |
| 321 | 2 | 110360839 | 114468943 | 0.006 | 17.0 | 4.71 | 0.006 |
| 322 | 2 | 112409311 | 115988999 | 0.007 | 14.7 | 4.19 | 0.013 |
| 323 | 2 | 114470485 | 117276436 | 0.012 | 18.3 | 5.02 | 0.004 |
| 324 | 2 | 115995299 | 118712363 | 0.008 | 18.9 | 5.17 | 0.003 |
| 325 | 2 | 117294302 | 119911849 | 0.006 | 15.0 | 4.26 | 0.012 |
| 830 | 6 | 34069188 | 36548480 | 0.007 | 13.1 | 3.82 | 0.025 |
| 831 | 6 | 35275766 | 37820042 | 0.009 | 21.0 | 5.64 | 0.002 |
| 832 | 6 | 36552018 | 38994028 | 0.007 | 23.2 | 6.14 | 0.001 |
| 833 | 6 | 37828214 | 40133729 | 0.008 | 19.0 | 5.18 | 0.003 |
| 200-SNP window analysis | |||||||
| 798 | 2 | 106987063 | 107924749 | 0.005 | 13.13 | 3.83 | 0.051 |
| 799 | 2 | 107447474 | 108441185 | 0.007 | 18.8 | 5.14 | 0.007 |
| 804 | 2 | 110107622 | 111043903 | 0.007 | 13.6 | 3.95 | 0.048 |
| 805 | 2 | 110590464 | 112388825 | 0.006 | 20.2 | 5.46 | 0.005 |
| 806 | 2 | 111047822 | 113380103 | 0.006 | 13.1 | 3.82 | 0.055 |
| 814 | 2 | 117019488 | 118134501 | 0.007 | 17.5 | 4.83 | 0.009 |
| 2088 | 6 | 35275766 | 36251932 | 0.005 | 13.6 | 3.95 | 0.047 |
| 2089 | 6 | 35743335 | 36802158 | 0.005 | 13.5 | 3.95 | 0.047 |
| 2090 | 6 | 36252650 | 37275709 | 0.005 | 15.0 | 4.27 | 0.030 |
| 2091 | 6 | 36805051 | 37820042 | 0.010 | 22.4 | 5.94 | 0.003 |
| 2092 | 6 | 37286366 | 38279284 | 0.008 | 24.6 | 6.45 | 0.002 |
| 2093 | 6 | 37828214 | 38766020 | 0.006 | 19.8 | 5.36 | 0.005 |
| 4122 | 18 | 17645871 | 18425906 | 0.007 | 17.8 | 4.91 | 0.009 |
| 4123 | 18 | 18057085 | 18957804 | 0.005 | 13.2 | 3.84 | 0.051 |
| 4879 | 24 | 40476812 | 41995998 | 0.008 | 18.3 | 5.02 | 0.008 |
: regional genomic heritability; SE: standard error; -logP:
Significant windows were selected at a FDR of 5%
Fig. 4.

Regional heritability mapping (RHM) across the genome. The y-axis shows the associated with each window and the x-axis shows the window number across the genome. Windows were analyzed using 1000-SNP (top plot), 500-SNP (middle plot) and 200-SNP windows (lower plot). Genome-wide significant windows (FDR of 5%) are highlighted by red dots
Fig. 5.

Quantile-quantile (Q–Q) plot of the observed p-values from the expected p-values of RHM using (1) 1000-SNP window size (λ = 1.06), (2) 500-SNP window size (λ = 1.08), and 200-SNP window size (λ = 1.09). The observed (p-values) are shown by black dots, and the expected values under null distribution are shown by a red line. Significant windows (FDR of 5%) are highlighted by red dots
Figure 6 shows a comparison of the RHM results among the three window sizes used (1000-SNP, 500-SNP and 200-SNP) for the target regions on OAR2 and OAR6. The 200-SNP window RHM analysis improved the mapping resolution of the identified regions (regions became narrower). However, the overall power was not higher compared to the larger window sizes since the LRT values did not increase when window sizes were shifted from 1000 to 200 SNPs.
Fig. 6.
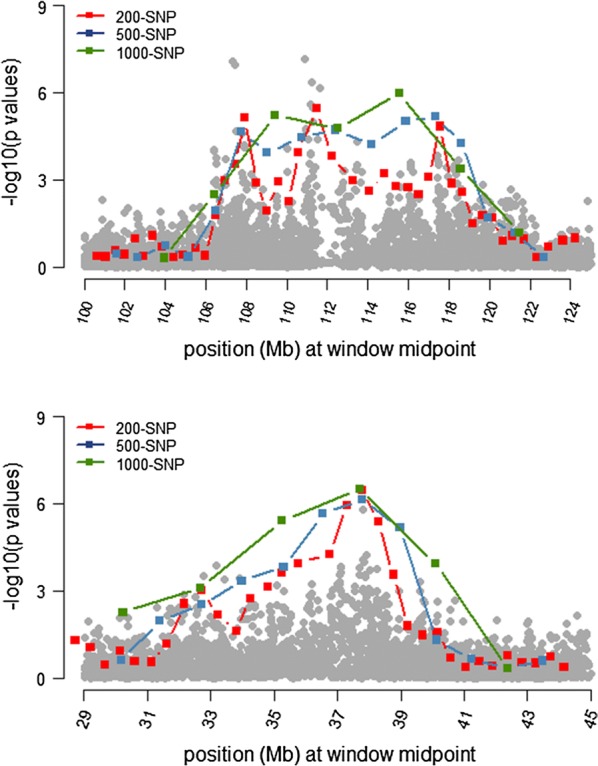
RHM and GWAS results of the identified region on OAR2 between 100 and 124 Mb (top plot) and on OAR6 between 28 and 44 Mb (bottom plot). The solid lines show the RHM results using three window sizes (1000 SNPs = green; 500 SNPs = blue; 200 SNPs = red), where each window is positioned at the window midpoint. Grey dots show the GWAS results within each region
The significant regions identified by RHM were re-analysed using 200-SNP windows for Merino sheep only to validate whether the same target regions persisted within the Merino sheep population. The results show that the significance of the peaks for the target regions on OAR2, 18, and 24 decreased, whereas the peak in the OAR6 region was maintained (Fig. 7). These results are likely due to the reduced sample size, with half the animals being Merino sheep, which resulted in the statistical power being inadequate to confirm the association in these target regions.
Fig. 7.

Comparison of RHM results using 200-SNP windows between the Merino population and the entire sheep population for the target regions on (1) OAR2, (2) OAR18, (3) OAR6, and (4) OAR24. The red lines represent the RHM results for the entire sheep population and blue lines represent the RHM results for Merino sheep population
Conditional association analyses
Results of conditional GWAS analyses for the significant regions on OAR2 and 6 are shown in Figs. 8 and 9, respectively. GWAS analysis conditioned on the first top significant SNP (rs421630816) removed the peaks in the region between 110 and 112 Mbp on OAR2. The p-values of the fourth and fifth top ranked SNPs (rs403231265 SNP; p = 4.2 × 10−7 and rs405353352 SNP; p = 6.7 × 10−7) at 111 Mbp on OAR2 became 0.38, whereas the p-values for the second and third significant SNPs (rs424521894 and rs413835864) at 107 Mbp remained lower than 10−4. When GWAS was conditioned on rs424521894 SNP, the p-value for rs413835864 became 0.9, whereas p-values for rs421630816 remained lower than 2 × 10−5. GWAS analysis conditioned on rs425769499 SNP, which is the top ranked SNP in the region between 117 and 118 Mbp, was also performed and the p-values for all significant SNPs on OAR2 remained lower than 10−4. LD between rs424521894, rs421630816, and rs425769499, i.e. the top SNP in each of the three regions between 107 and 108, 110 and 112, and 117 and 118 Mbp, was zero for all pairwise comparisons. However, LD between rs424521894 and rs413835864 SNPs of the same region was moderate (), whereas LD between rs421630816 SNP and either of the rs403231265 and rs405353352 SNPs of the same region was moderate () and strong (), respectively.
Fig. 8.

Conditional GWAS plots for the region between 100 and 120 Mbp on OAR2. The first plot shows GWAS results with no conditioning. Subsequent plots show GWAS results conditioned on the first and second significant SNPs (rs421630816 and rs424521894) as well as SNP rs425769499, the top ranked SNP in a region between 117 and 118 Mbp
Fig. 9.

Conditional GWAS plots for the region between 29 and 44 Mbp on OAR6. The left plot shows GWAS results with no conditioning. Right plot shows GWAS results conditioned on rs416517011, the sixth top ranked SNP based on GWAS
The proportion of phenotypic variance explained by the significant regions gradually increased with each additional region fitted in the model (Table 4), ranging from 0.004 (s.e. = 0.002) by fitting only the region between 107 and 108 Mbp on OAR2 to 0.030 (s.e. = 0.008) by fitting all five significant regions combined in one GRM. The model fit, as measured by LRT, gradually improved with each additional region fitted in the model.
Table 4.
RHM analysis conditioned on adding significant regions to the model sequentially
| Scenario | (SE) | (SE) | Logl | Logl null | LRT |
|---|---|---|---|---|---|
| R1 | 0.004 (0.002) | 0.19 (0.02) | − 10667 | − 10671 | 8 |
| R1 + R2 | 0.006 (0.003) | 0.18 (0.02) | − 10666 | − 10672 | 12 |
| R1 + R2 + R3 | 0.008 (0.003) | 0.18 (0.02) | − 10664 | − 10672 | 16 |
| R1 + R2 + R3 + R4 | 0.015 (0.005) | 0.17 (0.02) | − 10656 | − 10674 | 36 |
| R1 + R2 + R3 + R4 + R5 | 0.024 (0.007) | 0.16 (0.02) | − 10649 | − 10674 | 50 |
| R1 + R2 + R3 + R4 + R5 + R6 | 0.030 (0.008) | 0.16 (0.02) | − 10644 | − 10675 | 62 |
R1: between 107 and 108 Mbp on OAR2; R2: between 110 and 113 Mbp on OAR2; R3: between 117 and 118 Mbp on OAR2; R4: between 34 and 39 Mbp on OAR6, R5: between 17 and 18 Mbp on OAR18; and R6: between 40 and 41 Mbp on OAR24
: variance due to regions defined in each scenario and estimated with a GRM constructed from SNPs in these regions
: is the complementary GRM containing all SNPs from the 600 k excluding the SNPs fitted in
Logl: log likelihood for the tested model which includes both and
Logl null: log likelihood for the null model which includes only
SE: standard error
The proportion of phenotypic variance explained by all combined regions gradually decreased from 0.030 to 0.028 when RHM was conditioned only on the rs421630816 SNP, and from 0.030 to 0.019 when RHM was conditioned on the top six SNPs (Table 5). The model fit also improved with each additional SNP fitted as a fixed covariate except when the rs425769499 SNP was included.
Table 5.
RHM analysis conditioned on adding the top SNPs from GWAS to the model sequentially
| Scenario | (SE) | (SE) | Logl | LRT |
|---|---|---|---|---|
| No condition | 0.030 (0.008) | 0.16 (0.02) | − 10644 | – |
| Cond. on S1 | 0.028 (0.008) | 0.16 (0.02) | − 10643 | 2 |
| Cond. on S1 + S2 | 0.026 (0.007) | 0.16 (0.02) | − 10642 | 4 |
| Cond. on S1 + S2 + S3 | 0.026 (0.007) | 0.16 (0.02) | − 10644 | 0 |
| Cond. on S1 + S2 + S3 + S4 | 0.023 (0.007) | 0.16 (0.02) | − 10641 | 6 |
| Cond. on S1 + S2 + S3 + S4 + S5 | 0.020 (0.006) | 0.16 (0.02) | − 10637 | 14 |
| Cond. on S1 + S2 + S3 + S4 + S5 + S6 | 0.019 (0.006) | 0.16 (0.02) | − 10636 | 16 |
GRMi and GRMc as in Table 4
S1: rs421630816; S2: rs424521894; S3: rs425769499; S4: rs416517011; S5: rs404837788; S6: rs413573644
Logl: log likelihood
SE: standard error
Haplotype analysis
Haplotype analysis was performed for the region between 106.4 and 118.7 Mbp on OAR2 and the region between 32 and 36 Mbp on OAR6. Using the criteria described by Gabriel et al. [26], SNPs that showed high LD within each region were grouped together in haplotype blocks. Twenty-six and 10 haplotype blocks were identified in the regions on OAR2 and 6, respectively. Haplotype block sizes ranged from 2 to 141 kb. Using Eq. 5, only block 6 (Fig. 10) located between 107.33 and 107.38 Mbp on OAR2 had a significant effect on parasite resistance (p-value = 0.003). Six distinct haplotypes were identified in this haplotype block (Table 6). Haplotypes TTTG and CTTA had positive significant effects on parasite resistance, while haplotype CTTG had a marginally negative effect.
Fig. 10.
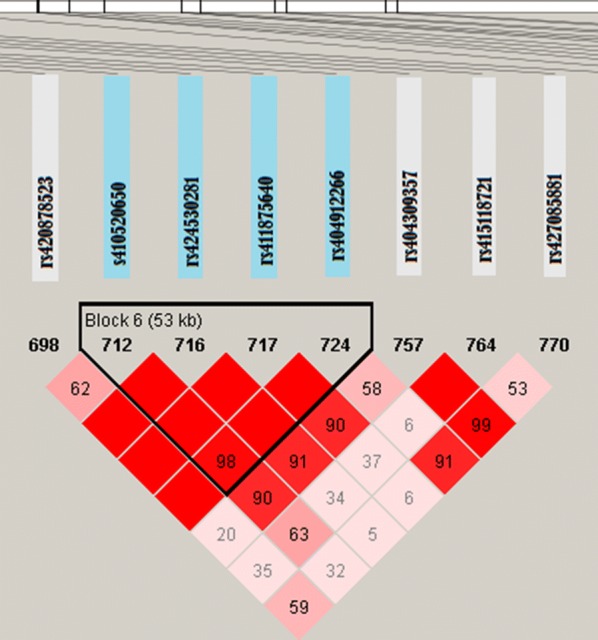
Linkage disequilibrium (LD) map for the region between 107.33 and 107.38 Mbp on OAR2. The haplotype block (block 6) containing the four SNPs (rs410520650, rs424530281, rs411875640 and rs404912266) is highlithed in blue boxes
Table 6.
Haplotype association analysis of block 6 (107.33–107.38 Mbp on OAR2) with parasite resistance
| Haplotype | Effectsa () | -value |
|---|---|---|
| TTTG | 0.093388 | 0.00601 |
| TTTA | 0.002921 | 0.89287 |
| CTTG | − 0.083304 | 0.02507 |
| CATG | − 0.045274 | 0.83761 |
| CACG | − 0.028541 | 0.39638 |
| CTTA | 0.816840 | 0.00592 |
The overall association between haplotypes and the trait was significant ( < 0.003). Significance level at 0.05.
aEstimates of regression coefficients () in phenotypic standard deviation (STD) units of the trait
2The for testing null hypothesis of
Principal components
The first two principal components (PC1 and PC2) are plotted and annotated by breed composition of the animals (Fig. 11). Only Purebred Merino and other breeds that make up 50% and more of the animals breed composition were annotated.
Fig. 11.
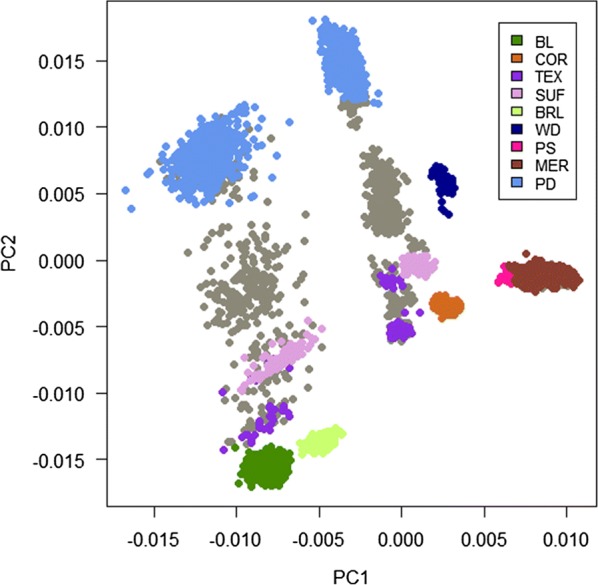
Clustering of animals based on the plot of principal components (PC1 and PC2). Animals were coloured based on their breed composition of purebred Merino and composition of other breeds, with the breed that makes up more than 50% of the breed proportion. BL Border Leicester; COR Corriedale; TEX Texel; SUF Suffolk; BRL Booroola; WD White Dorper; PS Prime Samm; PD Poll Dorset; and MER pure Merino. The grey colour represents animals of different crosses but with less than 50% of a particular breed composition
Pathway analysis was performed to link genes within the significant regions on OAR2, 6, 18 and 24 to their biological pathways using the BioSystem Tools from NCBI. The analysis linked 84 genes (Table 7) to 271 unique pathways (see Additional file 1: Table S1). The number of genes in the pathways ranged from 1 to 10 genes, with a median size of 1. Pathways with the largest sizes were ‘Metabolism’ (10 genes), ‘Immune system’ (8 genes), ‘Signal transduction’ (8 genes), ‘Antigen processing: Ubiquitination and proteasome degradation’ (6 genes), ‘Class I MHC mediated antigen processing and presentation’ (6 genes), ‘Adaptive immune system’ (6 genes), ‘Transmembrane transport of small molecules’ (5 genes) and ‘Gene expression’ (5 genes). Genes involved in major immune pathways were extracted and are listed in Table 8, which shows that 13 genes are linked to 16 pathways with major roles in the innate and acquired immunity as well as cytokine signalling in the immune system.
Table 7.
List of genes in the significant regions
| OAR | Position (Mbp) | Genes |
|---|---|---|
| 2 | 106.9–108.4 | GALNTL6 |
| 2 | 110.1–113.3 | C2H4orf27, CLCN3, NEK1, SH3RF1, CBR4, PALLD, DDX60, ANXA10, MFSD14B, LGSN, OCA2, HERC2, NIPA1, NIPA2, CYFIP1, TUBGCP5, PTPN18, AMER3, ARHGEF4 |
| 2 | 117.0–118.1 | MAP3K2, ERCC3, LOC101118856, BIN1, LOC105608784, NAB1, NEMP2, MFSD6, HIBCH, INPP1, C2H2orf88, MSTN |
| 6 | 34.7–39.2 | GPRIN3, TIGD2, FAM13A, NAP1L5, HERC3, PYURF, PIGY, HERC5, HERC6, PPM1 K, ABCG2, PKD2, SPP1, MEPE, IBSP, FAM184B, MED28, NCAPG, DCAF16, LCORL |
| 18 | 17.6–18.9 | NTRK3, DET1, MRPS11, MRPL46, ISG20, LOC101123585 |
| 24 | 40.4–41.9 | IQCE, TTYH3, LFNG, GRIFIN, CHST12, LOC105604828, LOC101112623, EIF3B, SNX8, NUDT1, MRM2, MAD1L1, LOC105604879, PSMG3, TMEM184A, MAFK, INTS1, LOC105604880, GPR146, C24H7orf50, LOC105604882, LOC101120042, GET4, SUN1, LOC105604834, LOC105604883, LOC106991921, DNAAF5, LOC106991898, FAM20C |
Table 8.
List of genes involved in immune responses and the immune pathways they belong to.
| Pathways | OAR2 | OAR6 | OAR18 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SH3RF1 | HERC2 | MAP3K2 | CYFIP1 | PTPN18 | BIN1 | HERC3 | HERC5 | HERC6 | IBSP | SPP1 | ISG20 | DET1 | |
| Immune system | x | x | x | x | x | x | x | x | |||||
| Adaptive immune system | x | x | x | x | x | x | |||||||
| Class I MHC mediated antigen processing & presentation | x | x | x | x | x | x | |||||||
| Antigen processing: ubiquitination and proteasome degradation | x | x | x | x | x | x | |||||||
| Innate immune system | x | x | |||||||||||
| B cell receptor signaling pathway | x | ||||||||||||
| Toll-like receptor signaling pathway | x | ||||||||||||
| Regulation of toll-like receptor signaling pathway | x | ||||||||||||
| Fc gamma R-mediated phagocytosis | x | ||||||||||||
| Fcgamma receptor (FCGR) dependent phagocytosis | x | ||||||||||||
| Interferon signaling | x | x | |||||||||||
| Cytokine signaling in immune system | x | x | |||||||||||
| Interferon alpha/beta signaling | x | ||||||||||||
| Antiviral mechanism by IFN-stimulated genes | x | ||||||||||||
| Interleukin-11 signaling pathway | x | ||||||||||||
| IL-1 signaling pathway | x |
Ovine chromosomes OAR (Ovis aries)
Gene symbols in italics
Discussion
Our study aimed at detecting genomic regions with effects on parasite resistance in a large population of sheep naturally challenged in the field with mixed parasite species. Both RHM and GWAS identified the region on OAR2 as being significant (FDR of 5%). However, RHM identified additional significant regions at the FDR of 5%, i.e. the regions on OAR6, 18 and 24, which were not detected by GWAS. Both methods use genomic information in different ways, and their power to detect genomic regions depends on the genetic architecture behind the trait. According to Nagamine et al. [16], when trait variation is due to a few causal variants, and those variants are in complete LD with the SNP, then GWAS should be the most powerful approach. However, most complex traits are polygenic with trait variation being explained by variants in many loci, each with a small effect. For such polygenic traits, RHM may be more efficient than the conventional GWAS approach. In principle, RHM facilitates the capture of genetic variance for each region in the genome by integrating the effects of both rare and common variants in a joint analysis. Thus, the RHM approach is potentially capable of identifying loci that cannot be detected by a conventional GWAS analysis. Furthermore, RHM captures the effect of the region, which may also include cis-interaction effects between causal genes in that given region. Our results show that some candidate genes within a given region share similar mechanisms related to the immune system, which suggests that some possible interaction effects take place between those genes for protecting the host against parasite infections.
Genomic regions that were identified by RHM as significant explained only a small proportion of the trait variation, for each region ranged from 0.003 (1.5% of the trait heritability) to 0.01 (5% of the trait heritability), and for all regions combined was equal to 0.030 (15% of the trait heritability). The small proportion of phenotypic variance explained by the significant regions suggests that parasite resistance is a polygenic trait with a large number of variants involved in the mechanism of resistance. This result is in agreement with Kemper et al. [11], Riggio et al. [12], Riggio et al. [27], and Lee et al. [28] who reported that parasite resistance is a complex trait influenced by a large number of genes each with a relatively small effect. The phenotype as measured in this study was subject to a strict measuring protocol, however, worm egg counts were recorded at different ages and from different parasite species. This likely affects the power and accuracy of the detection of causal variants. However, previous studies have shown that there are high genetic correlations between parasite challenges from different parasite species [29], as well as between WEC measurements at different ages [30].
Regions with a higher impact on parasite resistance were found on OAR2 and OAR6. RHM using 1000-SNP windows identified two regions between 106.4 and 121.1 Mbp on OAR2 and between 30.1 and 42.3 Mbp on OAR6 that were significantly associated with parasite resistance at the genome-wide significance level. Fine-mapping of the region on OAR6 with 200-SNP windows revealed a large region between 35.27 and 38.76 Mbp that passed the genome-wide significance level (FDR ≤ 0.05). Fine-mapping of the region on OAR2 with 200-SNP windows revealed three adjacent regions between 106.9 and 108.4 Mbp, 110.1 and 113.3 Mbp, and 117.0 and 118.1 Mbp that were significant at the genome-wide significance level. Stepwise conditional analyses showed that the proportion of phenotypic variance explained by significant regions gradually increased from 0.4% when regional heritability was based on SNPs from the region between 106.9 and 108.4 Mbp to 0.8% when regional heritability was based on SNPs from all three significant regions on OAR2. These results suggest that several causal mutations are likely responsible for the genetic variation in the OAR2 region.
A comparison with previous studies showed that the region on OAR2 fell within the QTL region (61.7–137.9 Mbp) reported by Crawford et al. [31] for resistance to T. colubriformis, which was identified in an outcross of Romney and Coopworth sheep, and which partially overlapped with the QTL region (117.9–133.9 Mbp) reported by Davies et al. [32] for resistance to Nematodirus spp. in Scottish blackface sheep. Furthermore, the region on OAR6 has been reported by Riggio et al. [12] for resistance to Strongyles in Scottish Blackface sheep. The authors identified a genome-wide significant SNP for Strongyles fecal egg count (FEC) in this region on OAR6 using GWAS, and confirmed a QTL region between 33 and 39 Mbp by RHM analysis. The identified region on OAR6 also corresponds to the QTL (25.1-62.6 Mbp) region reported by Silva et al. [10] for resistance to H. contortus and Trichostrongylus spp in a backcross population of Red Maasai and Dorper sheep. Interestingly, both regions on OAR2 and OAR6 have also been reported as being under selection in a large number of sheep breeds from the Sheep HapMap dataset [33, 34]. RHM analysis also identified a significant region on OAR18 (17.64-18.95 Mbp) and two novel regions between 36.96 and 37.84 Mbp and between 40.47 and 41.99 on OAR24. The identified region on OAR18 partially overlapped with a QTL region identified by Marshal, et al. [35].
GALNTL6 (polypeptide N-acetylgalactosaminyltransferase-like 6) was the only gene annotated in the region between 106.9 and 108.4 Mbp on OAR2. GALNTL6 harbours the second and third most significant SNPs detected by GWAS analysis and contains haplotype block 6 (107.33–107.38 Mbp), the only haplotype block identified as having a significant effect on parasite resistance. GALNTL6 is a member of a highly conserved family of proteins that are responsible for the synthesis of mucin-type O-glycans. Several genes from this family, such as GALNT1, GALNT4 and GALNT8, have also been reported as being of importance for sheep resistance to gastrointestinal parasite infections [10, 14, 35]. Abomasum mucus, with mucin as its main component, is considered to be the first line of host defence against invading gastrointestinal parasites [36, 37]. Mucus production during parasite infections is under the immune control of type-2 cytokines [38], with interleukin-4 (IL-4), IL-13, and IL-22 altogether playing the major role in host protection [39–41]. Furthermore, Newlands et al. [42] found that sheep immunized by daily oral challenge with H. contortus had unchanged gastric mucin profiles two days after infection, which demonstrates that animals are able to control mucin levels based on their immunological status. In vitro studies showed that parasite feeding and motility are restricted when larvae were co-cultured with sheep intestinal mucus [43].
The region between 110.1 and 113.3 Mbp on OAR2 harbours four genes that are directly involved in immune pathways: SH3 domain containing ring finger 1 (SH3RF1); E3 ubiquitin-protein ligase HERC2 (HERC2); presentation, and cytoplasmic FMR1 interacting protein 1 (CYFIP1); and protein tyrosine phosphatase, non-receptor type 18 (PTPN18). SH3RF1 and HERC2 are involved in the MHC class I mediated antigen processing and presentation pathway. This pathway activates type-1 T lymphocytes (Th1), which is characterized by the production of the cytokine interferon (IFN)-gamma among other cytokines, providing effective cellular response and protection against chronic parasite infection [44]. CYFIP1 encodes a protein involved in the Fcgamma receptor (FCGR) dependent phagocytosis pathway, a crucial event in the immune system that permits effector cells such as macrophages to uptake and eliminate infectious pathogens. This event is mediated by immunoglobulin (IgG) binding to Fc gamma receptors (Fc gamma R) on the effector cells [45]. PTPN18 is involved in the B cell receptor signalling pathway, which is essential for the expression of other genes involved with B cell differentiation, proliferation and immunoglobulin (Ig) production.
Other potential genes of interest in this region include: paladin (PALLD), and probable ATP-dependent RNA helicase DDX60 (DDX60). PALLD contains the top significant SNPs identified by GWAS analysis. The role of PALLD is poorly understood, although it has been found to play an essential role in organizing the skeletal muscle [46]. DDX60 is important for the production of inflammatory cytokines such as type 1 interferon [47]. In humans, an increased expression of DDX60 has been detected following viral infections [48, 49], which suggests that DDX60 is essential to initiate the innate antiviral mechanism.
The region between 117.0 and 118.1 Mbp on OAR2 overlaps with two important genes involved in immune pathways: bridging integrator 1 (BIN1), which encodes a protein being involved in Fc gamma R-mediated phagocytosis pathway, and mitogen-activated protein kinase kinase kinase 2 (MAP3K2), which is involved in the IL-1 signalling pathway. MAP3K2 plays an important role in the IκB kinase (IKK) activation, which is essential for NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) signalling [50]. NF-κB regulates the expression of many genes associated with immune responses in the gastrointestinal tract. For instance, NF-κB plays an essential role in the transcriptional regulation of many cytokine genes, including interferon (IFN)-gamma, IL-1, IL-2, IL-6, and IL-12, in epithelial cells, lymphocytes and monocytes [51]. The Κ light-chains of NF-κB are also critical components of immunoglobulins, making NF-κB a key regulator of humoral immune responses [52].
The region between 35.27 and 38.76 Mbp on OAR6 overlaps with three genes from the HERC family of ubiquitin ligases (HERC3, HERC5 and HERC6) that are associated to four biological pathways (Table 8) including: ‘Immune system’; ‘Antigen processing: Ubiquitination and proteasome degradation’; ‘Class I MHC mediated antigen processing and presentation’; and ‘Adaptive immune system’ pathways.
The OAR6 region also contains two immune genes: secreted phosphoprotein 1 (SPP1) and integrin binding sialoprotein (IBSP). SPP1 encodes a protein involved in toll-like receptors (TLR) signalling. TLR are innate immune receptors that detect pathogen invasion in the intestinal mucosa and are essential for mounting a type-2 immune response [53, 54]. This gene also plays an important role in wound healing [54, 55]. IBSP is involved in the interleukin-11 (IL-11) signalling pathway. IL-11 accelerates platelet recovery [56], which is important to maintain adequate blood volume levels following parasite infection. Gastrointestinal parasites, especially H. contortus, can cause severe blood loss, leading to haemorrhagic anaemia. Maintaining haemostasis is important for sheep recovery following infection as a way to minimise anaemia. Furthermore, haemostasis serves as a defence mechanism to expel parasites from the host body since clotting at the infection site significantly reduces blood supply to adult worms, inhibiting feeding and survival at the infection sites [54].
Other candidate genes in the OAR6 region include: ligand dependent nuclear receptor corepressor like (LCORL) and ATP binding cassette subfamily G member 2 (ABCG2). LCORL contains the sixth most significant SNP detected by GWAS analysis. In most mammalian species, LCORL contains trinucleotide repeats in the coding region, resulting in an expanded polyalanine tract in the amino-terminal region of its encoded protein [57]. The extreme expansions of trinucleotide repeats can alter protein function and cause genetic diseases such as the fragile X syndrome and Huntington’s disease [58, 59]. At present, the function and the propensity of repeat expansions in the coding region of the ovine LOCRL are not known.
ABCG2 is highly expressed in the canalicular membrane of the liver, kidney, colon, and in the epithelia of the small intestine [60, 61]. ABCG2 plays a major role in multidrug resistance [61], and has been identified as a candidate gene for facial eczema in sheep [62]. The expression of ABCG2 at the apical surface of the intestinal epithelium, a layer of cells that forms a physical barrier between mucosa and the gut luminal content, suggests a potential role for this gene in protecting the host from parasites that try to destroy the lining of the lumen to access the bloodstream.
The region between 17.64 and 18.92 on OAR18 overlaps with the interferon stimulated exonuclease 20 (ISG20) gene, which regulates different cytokine signalling pathways in the immune system including ‘Interferon signalling’ and ‘Interferon alpha/beta signalling’ pathways, and de-etiolated homolog 1 (DET1) gene which encodes a protein involved in the ‘class I MHC mediated antigen processing and presentation’ pathway.
Fine mapping RHM analysis using smaller windows identified a novel region on OAR24 between 40.4 and 41.9 Mbp. This region overlaps with the LFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase (LFNG) gene, which plays an important role in T lymphocyte differentiation and development through regulating notch signalling [63]. The MAF bZIP transcription factor K (MAFK) gene is another candidate gene in this region that is involved in the haemostasis pathway (see Additional file 1: Table S1), an integral response mechanism against H. contortus infection.
Conclusions
This study identified significant genomic regions on ovine chromosomes 2, 6, 18, and 24 that are associated with parasite resistance in sheep. These results show that RHM is more powerful in detecting regions for parasite resistance and capturing variance than single SNP GWAS. The identified regions overlap with candidate genes that are involved in innate and acquired immune mechanisms, as well as cytokine signalling. Genes involved with haemostasis and mucus production are also relevant for host protection against parasite infections. Our results support the hypothesis that parasite resistance is a complex trait, and is determined by a large number of genes with various roles, rather than by a few genes with a major role in resistance.
Additional file
Additional file 1: Table S1. List of candidate genes and the biological pathways they belong to.
Acknowledgements
The authors acknowledge and thank the Cooperative Research Centre for Sheep Industry Innovation for providing data and funding the project. The authors also thank Hawlader Al-Mamun for help in performing the haplotype analysis and Klint Gore for retrieving and preparing the phenotypes and genotypes.
Authors’ contributions
MA performed the Statistical analyses and wrote the manuscript. JHJV coordinated the study and supervised the statistical analysis and writing of the manuscript. All authors contributed to discussions and writing the final manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All activities and procedures involving the animals were approved by the Animal Ethics Committee for the flock “Sheep Genomics Australia” and each site of the “Information Nucleus” breeding program of the Cooperative Research Centre for Sheep Industry Innovation. All animals in the project were managed according to the Australian Code for the Care and Use of Animals for Scientific Purposes.
Consent for publication
All authors have read and accepted the paper.
Competing interests
The authors declare they do not have competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mohammad Al Kalaldeh, Email: malkala2@une.edu.au.
John Gibson, Email: jgibson5@une.edu.au.
Sang Hong Lee, Email: Hong.Lee@unisa.edu.au.
Cedric Gondro, Email: gondroce@msu.edu.
Julius H. J. van der Werf, Email: jvanderw@une.edu.au
References
- 1.Lane J, Jubb T, Shepherd R, Webb-Ware J, Fordyce G. Priority list of endemic diseases for the red meat industries. Final Report. North Sydney: Meat and Livestock Australia Limited; 2015
- 2.Padula A. Gastrointestinal nematodes of sheep and cattle: biology and control. Aust Vet J. 2011;89:54. doi: 10.1111/j.1751-0813.2010.00674.x. [DOI] [Google Scholar]
- 3.Gilleard JS. Understanding anthelmintic resistance: the need for genomics and genetics. Int J Parasitol. 2006;36:1227–1239. doi: 10.1016/j.ijpara.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan RM. Drug resistance in nematodes of veterinary importance: a status report. Trends Parasitol. 2004;20:477–481. doi: 10.1016/j.pt.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 5.McRae KM, McEwan JC, Dodds KG, Gemmell NJ. Signatures of selection in sheep bred for resistance or susceptibility to gastrointestinal nematodes. BMC Genomics. 2014;15:637. doi: 10.1186/1471-2164-15-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roeber F, Jex AR, Gasser RB. Impact of gastrointestinal parasitic nematodes of sheep, and the role of advanced molecular tools for exploring epidemiology and drug resistance-an Australian perspective. Parasit Vectors. 2013;6:153. doi: 10.1186/1756-3305-6-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beh KJ, Hulme DJ, Callaghan MJ, Leish Z, Lenane I, Windon RG, et al. A genome scan for quantitative trait loci affecting resistance to Trichostrongylus colubriformis in sheep. Anim Genet. 2002;33:97–106. doi: 10.1046/j.1365-2052.2002.00829.x. [DOI] [PubMed] [Google Scholar]
- 8.Dominik S, Hunt PW, McNally J, Murrell A, Hall A, Purvis IW. Detection of quantitative trait loci for internal parasite resistance in sheep. I. Linkage analysis in a Romney × Merino sheep backcross population. Parasitology. 2010;137:1275–1282. doi: 10.1017/S003118201000020X. [DOI] [PubMed] [Google Scholar]
- 9.Marshall K, Maddox JF, Lee SH, Zhang Y, Kahn L, Graser HU, et al. Genetic mapping of quantitative trait loci for resistance to Haemonchus contortus in sheep. Anim Genet. 2009;40:262–272. doi: 10.1111/j.1365-2052.2008.01836.x. [DOI] [PubMed] [Google Scholar]
- 10.Silva MV, Sonstegard TS, Hanotte O, Mugambi JM, Garcia JF, Nagda S, et al. Identification of quantitative trait loci affecting resistance to gastrointestinal parasites in a double backcross population of Red Maasai and Dorper sheep. Anim Genet. 2012;43:63–71. doi: 10.1111/j.1365-2052.2011.02202.x. [DOI] [PubMed] [Google Scholar]
- 11.Kemper KE, Emery DL, Bishop SC, Oddy H, Hayes BJ, Dominik S, et al. The distribution of SNP marker effects for faecal worm egg count in sheep, and the feasibility of using these markers to predict genetic merit for resistance to worm infections. Genet Res (Camb). 2011;93:203–219. doi: 10.1017/S0016672311000097. [DOI] [PubMed] [Google Scholar]
- 12.Riggio V, Matika O, Pong-Wong R, Stear M, Bishop SC. Genome-wide association and regional heritability mapping to identify loci underlying variation in nematode resistance and body weight in Scottish Blackface lambs. Heredity (Edinb). 2013;110:420–429. doi: 10.1038/hdy.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riggio V, Pong-Wong R, Sallé G, Usai MG, Casu S, Moreno CR, et al. A joint analysis to identify loci underlying variation in nematode resistance in three European sheep populations. J Anim Breed Genet. 2014;131:426–436. doi: 10.1111/jbg.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benavides MV, Sonstegard TS, Kemp S, Mugambi JM, Gibson JP, Baker RL, et al. Identification of novel loci associated with gastrointestinal parasite resistance in a Red Maasai x Dorper backcross population. PLoS One. 2015;10:e0122797. doi: 10.1371/journal.pone.0122797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagamine Y, Pong-Wong R, Navarro P, Vitart V, Hayward C, Rudan I, et al. Localising loci underlying complex trait variation using regional genomic relationship mapping. PLoS One. 2012;7:e46501. doi: 10.1371/journal.pone.0046501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van der Werf J, Kinghorn B, Banks R. Design and role of an information nucleus in sheep breeding programs. Anim Prod Sci. 2010;50:998–1003. doi: 10.1071/AN10151. [DOI] [Google Scholar]
- 18.Whitlock HV. Some modifications of the McMaster helminth egg-counting technique and apparatus. J Council Sci Ind Res. 1948;21:177–180. [Google Scholar]
- 19.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loh PR, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coster A. Package pedigree package reference manual. 2013. https://cran.r-project.org/web/packages/pedigree/pedigree.pdf.
- 23.Lee SH, Van Der Werf J. MTG2: an efficient algorithm for multivariate linear mixed model analysis based on genomic information. Bioinformatics. 2016;32:1420–1422. doi: 10.1093/bioinformatics/btw012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15:478. doi: 10.1186/1471-2164-15-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 26.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 27.Riggio V, Abdel-Aziz M, Matika O, Moreno CR, Carta A, Bishop S. Accuracy of genomic prediction within and across populations for nematode resistance and body weight traits in sheep. Animal. 2014;8:520–528. doi: 10.1017/S1751731114000081. [DOI] [PubMed] [Google Scholar]
- 28.Lee CY, Munyard KA, Gregg K, Wetherall JD, Stear MJ, Groth DM. The influence of MHC and immunoglobulins A and E on host resistance to gastrointestinal nematodes in sheep. J Parasitol Res. 2011;2011:101848. doi: 10.1155/2011/101848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woolaston RR. Selection of Merino sheep for increased and decreased resistance to Haemonchus contortus: peri-parturient effects on faecal egg counts. Int J Parasitol. 1992;22:947–953. doi: 10.1016/0020-7519(92)90052-M. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Brown DJ, Swan AA, van der Werf JHJ. Genetic parameters for faecal worm egg count at different ages in Australian sheep under natural challenge. Anim Prod Sci. 2018;59:1201–1208. doi: 10.1071/AN17833. [DOI] [Google Scholar]
- 31.Crawford AM, Paterson KA, Dodds KG, Tascon CD, Williamson PA, Thomson MR, et al. Discovery of quantitative trait loci for resistance to parasitic nematode infection in sheep: I. Analysis of outcross pedigrees. BMC Genomics. 2006;7:178. doi: 10.1186/1471-2164-7-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies G, Stear MJ, Benothman M, Abuagob O, Kerr A, Mitchell S, et al. Quantitative trait loci associated with parasitic infection in Scottish blackface sheep. Heredity (Edinb). 2006;96:252–258. doi: 10.1038/sj.hdy.6800788. [DOI] [PubMed] [Google Scholar]
- 33.Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10:e1001258. doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fariello MI, Servin B, Tosser-Klopp G, Rupp R, Moreno C, San Cristobal M, et al. Consortium ISG. Selection signatures in worldwide sheep populations. PLoS One. 2014;9:e103813. doi: 10.1371/journal.pone.0103813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marshall K, Mugambi JM, Nagda S, Sonstegard TS, Tassell CP, Baker RL, et al. Quantitative trait loci for resistance to Haemonchus contortus artificial challenge in Red Maasai and Dorper sheep of East Africa. Anim Genet. 2013;44:285–295. doi: 10.1111/j.1365-2052.2012.02401.x. [DOI] [PubMed] [Google Scholar]
- 36.Hasnain SZ, Gallagher AL, Grencis RK, Thornton DJ. A new role for mucins in immunity: insights from gastrointestinal nematode infection. Int J Biochem Cell Biol. 2013;45:364–374. doi: 10.1016/j.biocel.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 37.Moncada DM, Kammanadiminti SJ, Chadee K. Mucin and Toll-like receptors in host defense against intestinal parasites. Trends Parasitol. 2003;19:305–311. doi: 10.1016/S1471-4922(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 38.Grencis RK. Immunity to helminths: resistance, regulation, and susceptibility to gastrointestinal nematodes. Annual Rev Immunol. 2015;33:201–225. doi: 10.1146/annurev-immunol-032713-120218. [DOI] [PubMed] [Google Scholar]
- 39.McKenzie GJ, Bancroft A, Grencis RK, McKenzie AN. A distinct role for interleukin-13 in Th2-cell-mediated immune responses. Curr Biol. 1998;8:339–342. doi: 10.1016/S0960-9822(98)70134-4. [DOI] [PubMed] [Google Scholar]
- 40.Hasnain SZ, Wang H, Ghia JE, Haq N, Deng Y, Velcich A, et al. Mucin gene deficiency in mice impairs host resistance to an enteric parasitic infection. Gastroenterology. 2010;138:1763–1771. doi: 10.1053/j.gastro.2010.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner JE, Stockinger B, Helmby H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS Pathog. 2013;9:e1003698. doi: 10.1371/journal.ppat.1003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newlands GF, Miller HR, Jackson F. Immune exclusion of Haemonchus contortus larvae in the sheep: effects on gastric mucin of immunization, larval challenge and treatment with dexamethasone. J Comp Pathol. 1990;102:433–442. doi: 10.1016/S0021-9975(08)80164-8. [DOI] [PubMed] [Google Scholar]
- 43.Douch PG, Harrison GB, Buchanan LL, Greer KS. In vitro bioassay of sheep gastrointestinal mucus for nematode paralysing activity mediated by substances with some properties characteristic of SRS-A. Int J Parasitol. 1983;13:207–212. doi: 10.1016/0020-7519(83)90014-0. [DOI] [PubMed] [Google Scholar]
- 44.Else KJ, Finkelman FD, Maliszewski CR, Grencis RK. Cytokine-mediated regulation of chronic intestinal helminth infection. J Exp Med. 1994;179:347–351. doi: 10.1084/jem.179.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Garcia E, Rosales C. Signal transduction during Fc receptor-mediated phagocytosis. J Leukoc Biol. 2002;72:1092–1108. [PubMed] [Google Scholar]
- 46.Otey CA, Rachlin A, Moza M, Arneman D, Carpen O. The palladin/myotilin/myopalladin family of actin-associated scaffolds. Int Rev Cytol. 2005;246:31–58. doi: 10.1016/S0074-7696(05)46002-7. [DOI] [PubMed] [Google Scholar]
- 47.Wong MT, Chen SS. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell Mol Immunol. 2016;13:11–35. doi: 10.1038/cmi.2014.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyashita M, Oshiumi H, Matsumoto M, Seya T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol Cell Biol. 2011;31:3802–3819. doi: 10.1128/MCB.01368-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Q, Lee FS. Mitogen-activated protein kinase/ERK kinase kinases 2 and 3 activate nuclear factor-κB through IκB kinase-α and IκB kinase-β. J Biol Chem. 1999;274:8355–8358. doi: 10.1074/jbc.274.13.8355. [DOI] [PubMed] [Google Scholar]
- 51.Neurath MF, Becker C, Barbulescu K. Role of NF-κB in immune and inflammatory responses in the gut. Gut. 1998;43:856–860. doi: 10.1136/gut.43.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caamano J, Hunter CA. NF-κB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev. 2002;15:414–429. doi: 10.1128/CMR.15.3.414-429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun S, Wang X, Wu X, Zhao Y, Wang F, Liu X, et al. Toll-like receptor activation by helminths or helminth products to alleviate inflammatory bowel disease. Parasit Vectors. 2011;4:186. doi: 10.1186/1756-3305-4-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benavides MV, Sonstegard TS, Van Tassell C. Genomic regions associated with sheep resistance to gastrointestinal nematodes. Trends Parasitol. 2016;32:470–480. doi: 10.1016/j.pt.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 55.Liaw L, Birk DE, Ballas CB, Whitsitt JS, Davidson JM, Hogan B. Altered wound healing in mice lacking a functional osteopontin gene (spp1) J Clin Invest. 1998;101:1468–1478. doi: 10.1172/JCI2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du X, Williams DA. Interleukin-11: review of molecular, cell biology, and clinical use. Blood. 1997;89:3897–3908. [PubMed] [Google Scholar]
- 57.Whan V, Hobbs M, McWilliam S, Lynn DJ, Lutzow YS, Khatkar M, et al. Bovine proteins containing poly-glutamine repeats are often polymorphic and enriched for components of transcriptional regulatory complexes. BMC Genomics. 2010;11:654. doi: 10.1186/1471-2164-11-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pearson CE, Edamura KN, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 59.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sarkadi B, Homolya L, Szakács G, Váradi A. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol Rev. 2006;86:1179–1236. doi: 10.1152/physrev.00037.2005. [DOI] [PubMed] [Google Scholar]
- 61.Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chin J Cancer. 2012;31:73–99. doi: 10.5732/cjc.011.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duncan EJ, Dodds KG, Henry HM, Thompson MP, Phua SH. Cloning, mapping and association studies of the ovine ABCG2 gene with facial eczema disease in sheep. Anim Genet. 2007;38:126–131. doi: 10.1111/j.1365-2052.2006.01557.x. [DOI] [PubMed] [Google Scholar]
- 63.Tsukumo S, Hirose K, Maekawa Y, Kishihara K, Yasutomo K. Lunatic fringe controls T cell differentiation through modulating notch signaling. J Immunol. 2006;177:8365–8371. doi: 10.4049/jimmunol.177.12.8365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. List of candidate genes and the biological pathways they belong to.