

Abstract
Protein-protein interactions (PPIs) occur in complex networks. These networks are highly dependent on cellular context and can be extensively altered in disease states such as cancer and viral infection. In recent years, there has been significant progress in developing inhibitors that target individual PPIs either orthosterically (at the interface) or allosterically. These molecules can now be used as tools to dissect PPI networks. Here, we review recent examples that highlight the use of small molecules and engineered proteins to probe PPIs within the complex networks that regulate protein homeostasis. Researchers have discovered multiple mechanisms to modulate PPIs involved in host/viral interactions, deubiquitinases, the ATPase p97/VCP, and HSP70 chaperones. However, few studies have evaluated the effect of such modulators on the target’s network or have compared the biological implications of different modulation strategies. Such studies will have an important impact on next generation therapeutics.
Introduction
Proteins function in the context of protein-protein interaction (PPI) networks. PPIs serve to regulate a protein’s activity, to scaffold multi-protein complexes, and to link enzymes to their protein substrates. Analysis of human proteomic data suggests that 20% of proteins serve as network hubs, binding to at least 24 interactors [1]. Given their importance and complexity, it is not surprising that PPI networks can be altered in disease states [2–4]; hence, targeting PPIs that are aberrantly formed (or not formed) could be a powerful approach to understand and treat disease. In addition, some PPIs within a network may be linked to disease, while others are homeostatic. Perhaps PPI-modulatory drugs should target subsets of a protein’s network to safely and effectively treat the disease.
The past twenty years of research have generated a diverse set of PPI inhibitors, including proteins [5–7] and small molecules [8–12] that act through orthosteric or allosteric mechanisms. Current questions can now focus on the effect of these PPI modulators on the broader networks. Do different mechanisms of PPI modulation have different biological effects? If so, are these differential effects due to functional selectivity within the PPI’s network?
This review highlights recent, illustrative examples in which small molecules and engineered proteins have targeted PPIs within protein homeostasis networks. Protein homeostasis (proteostasis) describes the dynamic equilibrium of protein expression, folding, localization, and degradation. Proteostasis is regulated by networks of PPIs, including chaperones that modulate protein folding and macromolecular assembly [13–16], and the ubiquitin/proteasome system (UPS) that targets ubiquitin (Ub)-labeled proteins (Ub-clients) for degradation (Figure 1a) [17,18]. Additionally, ubiquitylation can have a regulatory (non-degradation) role. The complexity of these networks offers many opportunities to modulate PPI via different mechanisms and to examine the biological consequences.
Figure 1. Proteostasis systems are targeted by viruses.

(a) Overview of the UPS. Ub ligases (green) add Ub (yellow) to client proteins (blue), which are then degraded by the proteasome. DUBs (red) remove Ub from the target protein, rescuing it from proteasomal degradation. (b) Viral PPIs (magenta) hijack normal APOBEC (blue) function. The normal function of the APOBECs is to hypermutate viral mRNA or ssDNA (green arrow). Viral proteins form PPIs with Ub ligases to either promote or inhibit APOBEC degradation. (c) The structure of IMB-301, which inhibits the HIV Vif-APOBEC PPI. See references in the main text. Abbreviations: HIV, human immunodeficiency virus; HPV, human papillomavirus; VCBC, HIV viral infectivity factor PPI complex.
Misregulated proteostasis is also important in a range of disease states, from cancer to neurodegeneration. Imbalances can lead either to premature protein degradation or to delayed degradation and hence aggregation – causing a loss of normal cellular function in both cases. Cancer cells, for example, are under proteostatic stress due to dysregulated protein synthesis and expression of mutated proteins [19]; additionally, some oncogenes are dependent on chaperones for stability [20]. In neurodegeneration, age-related decline in the protein degradation machinery leads to accumulation of damaged proteins [21]. However, even if targeting a particular aspect of the proteostasis network (e.g., one chaperone/client interaction) could be therapeutic, related homeostatic processes (the chaperone’s interaction with other clients) must be maintained. Lack of selectivity could underlie the low therapeutic index for catalytic inhibitors of central proteostatic regulators like heat-shock protein (HSP)-90 [22]. Hence, being able to selectively manipulate a node in the proteostatic network is also an important challenge for drug discovery.
2. Viral proteins hijack host PPIs to alter APOBEC fate
The interaction of the human proteostasis network with viral proteins represents a classic case of PPI-network modulation in disease. In particular, the “apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like” (APOBEC) proteins are targets of viruses (Figure 1b). The APOBECs are a family of cytidine deaminases that hypermutate viral genomes, leading to stimulation of the innate immune system [23–25]. Viruses can benefit from either hyperactivating or inhibiting APOBECs: hyperactivation could lead to viral evolution and survival, while inhibition could shelter viruses from the immune system.
Several viruses hijack parts of the human proteostasis system either to upregulate or downregulate levels of human APOBECs. For instance, the cancer-causing human papillomavirus (HPV) stimulates APOBEC3A activity. The HPV E7 protein forms a complex with a human ubiquitin ligase (Ub-ligase), thereby blocking APOBEC3A binding and inhibiting its ubiquitylation and degradation [26]. Interestingly, high risk HPV genotypes (HPV16, HPV18) more efficiently stabilize APOBEC3A than low risk genotypes (HPV6, HPV11), suggesting a role for hypermutation in carcinogenesis. In contrast to HPV, human immunodeficiency virus (HIV) utilizes viral/human PPIs to inhibit APOBEC activity. The HIV viral infectivity factor (Vif) complex (VCBC) interacts with the human ubiquitin-ligase machinery to ubiquitylate and degrade several isoforms of APOBEC [27,28].
Two recent studies described small-molecule and antibody approaches to disrupt Vif-mediated APOBEC ubiquitylation/degradation. Ma et al. used virtual screening to identify IMB-301. This small molecule bound to APOBEC in vitro, disrupted the Vif/APOBEC3G PPI (Figure 1c), and restored APOBEC expression levels in HIV-infected cells [29]. Binning et al. used phage display to select fragments of antigen-binding (Fabs) that bound to VCBC and inhibited APOBEC degradation via separate pathways [30]. One Fab (1D1) bound to Vif at the Vif/Ub-ligase interface, breaking up the complex and inhibiting both ubiquitylation and virion packaging of multiple APOBEC isoforms. The other Fab (3C9) bound to Vif in the intact Vif/ligase/APOBEC complex and selectively inhibited APOBEC3F degradation, but allowed the complex to be packaged into the virion. These studies clearly demonstrated that small molecules and antibody fragments could alter APOBEC degradation by different mechanisms. More broadly, viral proteins exploit the host proteostasis networks to create new PPIs with unique properties; by manipulating only these neocomplexes, one might selectively treat viral infection.
3. DUB inhibition leads to degradation of oncogenes and stabilization of tumor suppressors
Ub-dependent degradation is rescued by deubiquitinases (DUBs), proteases that selectively remove Ub from Ub-client conjugates. Inhibition of specific DUBs could have therapeutic benefit in cancer, but the tumor context is critical. For instance, wild-type p53 is a tumor suppressor, but some mutants of p53 gain oncogenic properties and/or lose tumor-suppressor function [31]. Thus, either raising or lowering the levels of p53 may be therapeutic, depending on its mutational state. The ubiquitin-specific protease 7 (USP7) is a negative regulator of p53. USP7 removes Ub from MDM2, a Ub-ligase that ubiquitylates - and thus signals the degradation of - p53. Therefore, inhibition of USP7 should cause degradation of MDM2, which, in turn, should increase the stability of p53 and suppress cancer progression in tumors with wild-type p53 (Figure 2a). On the other hand, USP10, a p53 deubiquitinase, serves as a positive regulator of p53; inhibition of USP10 could suppress cancer by degrading oncogenic p53 mutants (Figure 2b)[32].
Figure 2. Examples of modulating the Ub-ligase/deubiquitinase network in cancer.
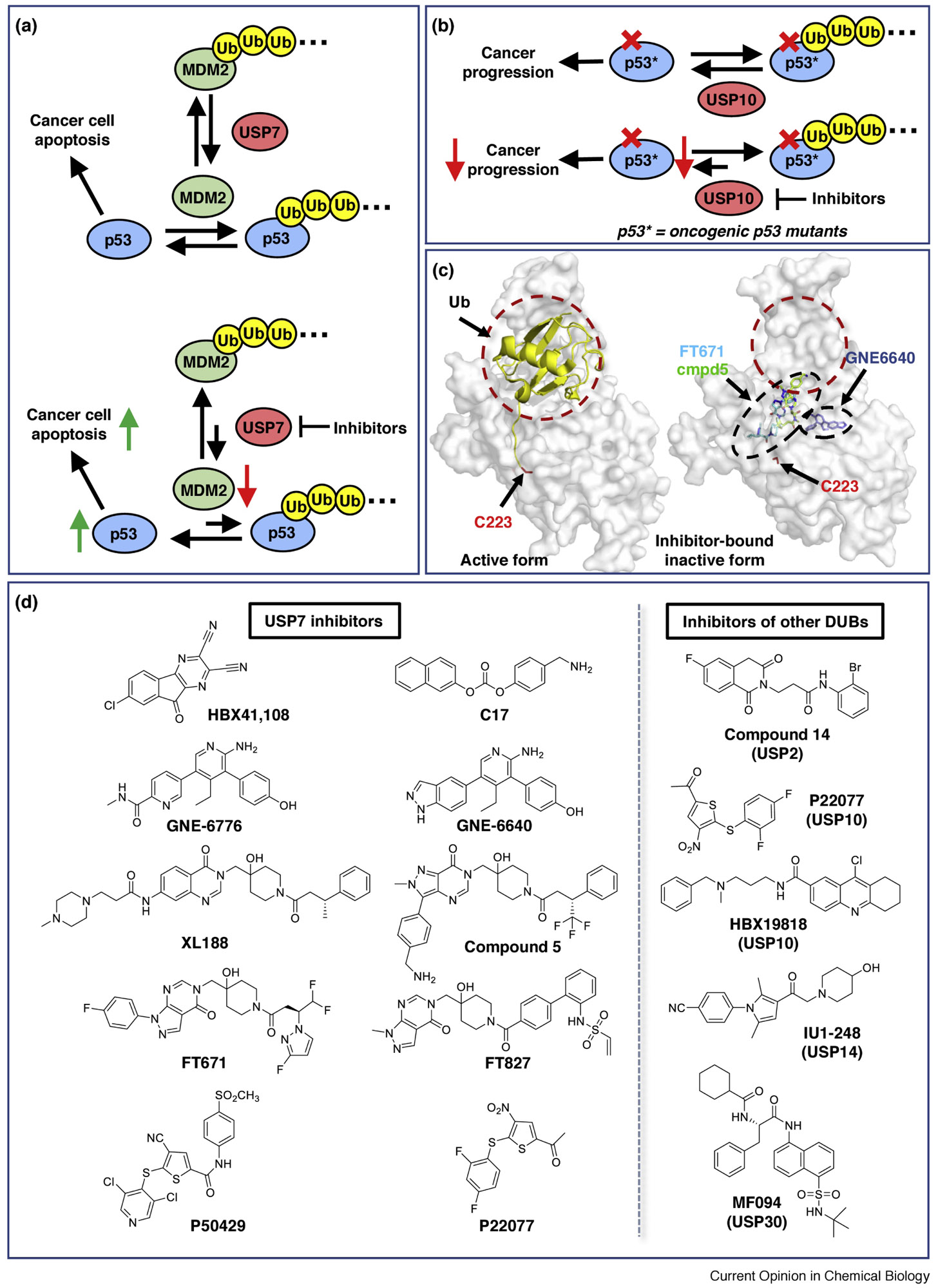
(a) USP7 functions as a positive regulator of tumor suppressor p53 by deubiquitylating MDM2 (top). Inhibition of USP7 should destabilize MDM2, causing an upregulation of p53 and hence tumor suppression (bottom). (b) USP10 deubiquitylates wild-type and oncogenic mutant p53 (top). Inhibition of USP10 increases the turnover of both wild-type and mutant p53 and prevents cancer progression in p53-mutant-driven cancer cells (bottom). (c) Illustration of allosteric small molecules binding to the USP7 catalytic domain. Ubiquitin (yellow cartoon) binds to USP7 (grey) at a broad, dynamic interface (red circle) with its C-terminus inserted into a catalytic pocket, and induces USP7 to adopt an active conformation (left, PDB 1NBF). Representative small molecules (sticks) overlaid on an inactive form of USP7 (right, PDB 5N9R). GNE6640 (blue, PDB 5UQV), FT671 (cyan, PDB 5NGE), and compound 5 (green, 5N9T) bind at distal regions from the Ub-binding interface, forcing USP7 to adopt an inactive conformation that sterically occludes ubiquitin binding. The catalytic Cys223 is highlighted. (d) Left panel: recent examples of USP7 inhibitors that are discussed in this review. Right panel: examples of other DUB inhibitors (right, more details see [17]).
DUBs recognize specific Ub-client conjugates, but their Ub-binding domains are well conserved [33,34]. To systematically dissect the role of DUBs, researchers have developed orthosteric inhibitors based on the Ub scaffold. Ubiquitin binds weakly to DUBs, with dissociation constants in the range of 100 μM. Using phage display and computational design, Corn and coworkers engineered a Ub variant that tightly and specifically bound to USP7 (KD ~ 60 nM), leading to stabilization of p53 in cells [35,36]. Using a similar strategy, Sidhu and coworkers developed both positive and negative regulators of p53 by selectively targeting USP7 and USP10[32]. As predicted, inhibition of USP7 caused a net upregulation of wild type p53, while inhibition of USP10 led to nuclear export and degradation of p53 in cancer cells [32]. In another study, Gorelik, et al. developed Ub variants that suppressed Ub-ligase activity by preventing the assembly of an E3 ligase complex, leading to the stabilization of the downstream substrates (e.g. cyclin E and c-Myc) [37]. In these and additional studies [5,6,38], the ubiquitin scaffold was demonstrated to be a versatile tool to inhibit Ub/enzyme complexes and test the biological functions of particular DUBs and Ub-ligases.
Translating this information into small-molecule drug discovery has been challenging, but there has been impressive progress in the past three years. Difficulty with targeting USPs was attributed to the flat Ub-binding surfaces of the USPs and the observation that many unbound enzymes were in an inactive conformation, where the active site was closed and the catalytic cysteine in an unfavorable position to bind covalent inhibitors [34]. Among the DUBs, USP7 has received the most attention due to its role in regulating turnover of p53 (vide supra). Recent successes have inhibited USP7 with good selectivity and potency by stabilizing its inactive conformation, thereby preventing Ub binding and/or inhibiting catalysis (Figures 2c)[39–41]. Cell-active USP7 inhibitors demonstrate the expected activities of lowering MDM2 levels and reactivating p53. USP10 is another emerging target. Weisberg et al. have performed a phenotypic screen and identified compounds originally discovered for USP7 that inhibit USP10 and induce degradation of the oncogenic kinase FLT3 [42]. Based on these observations, it would be valuable to compare/contrast the biological outcomes of inhibiting USP7 and USP10 with Ub-variants and small molecules, especially in terms of selectivity and generating resistance. Other USPs have also recently been inhibited by small molecules with various mechanisms. As predicted, cell-active USP inhibitors induce hyper-ubiquitylation and subsequent degradation of cognate oncogenes, including MDM2 (a USP7 substrate)[39–41,43–46][47]. Flt3 (a USP10 substrate)[42], c-Myc (a USP28 substrate) [48](Figures 2d)[17,49,50]. Excitingly, the tools and strategies are now in place to validate specific Ub-client/enzyme complexes as well as identify novel substrates in the Ub ligase/DUB networks.
4. p97 inhibitors block protein degradation and alter p97/adaptor complexes
The “ATPases Associated with diverse cellular Activity” (AAA) enzymes are molecular machines that convert the energy of ATP hydrolysis into work, such as protein unfolding and membrane remodeling. The AAA enzyme p97, for example, prepares Ub-clients for degradation by extracting them from protein complexes and membranes, initiating protein unfolding through the enzyme’s central pore [51–56], and remodeling membranes such as the autophagosome and golgi [52,57]. p97 is a therapeutic target for cancer, largely due to its role in endoplasmic-reticulum associated degradation (ERAD), which is commonly upregulated in cancer cells due to ongoing proteotoxic stress[55,58]. Point mutations in p97 also cause a disease called Multisystem Proteinopathy 1 (MSP1) [59].
To accomplish its many functions, p97 binds to a set of adaptor proteins that link the enzyme to various clients and subcellular locations. ATP hydrolysis induces changes in p97 conformation that alter the binding of these adaptor proteins [53,60–66]. Hence, p97 functions are governed by PPIs, which are in turn altered by ATPase-dependent changes in p97 conformation. This linkage provides diverse opportunities to modulate p97 function with small molecules. As with other central proteins in the proteostasis network, therapeutic inhibitors may need to spare some p97-dependent functions to maintain healthy proteostasis. In the case of MSP1, cells show proteostatic derangements suggesting both loss-of-function and gain-of-function changes, leading to the hypothesis that the disease should be treated with small-molecule ‘normalizers’ of p97 [67], rather than pure inhibitors or activators.
Several classes of p97 ATPase inhibitors have been reported [58,68–71]. Interestingly, these inhibitors vary in their enzymatic mechanisms and binding sites (Figure 3), and it is enticing to consider whether these mechanistic differences lead to changes in biological effects. Among the most potent inhibitors are the ATP-competitive inhibitors ML240 [72] and CB-5083 [58,68] and the allosteric inhibitors NMS-873 [69] and UPCDC-30245 [70,71]. These compounds have not been exhaustively compared to each other, but some mechanistic similarities and differences are emerging. NMS-873, ML240, and CB-5083 each show strong inhibition of Ub-client degradation and stimulation of the unfolded protein response in cells, consistent with inhibition of ERAD [58,69]. Other p97-dependent functions seem to be differently altered; for instance, these same compounds show different effects on cell cycle and autophagy [58,69,73]. In biochemical assays, ML240 loses inhibitory activity in the presence of the adaptor protein p47, whereas NMS-873 is insensitive to the binding of p47 to p97 [74]. Additional cellular and biochemical studies are needed to connect the compounds’ mechanism of inhibition to their modulation of cellular functions.
Figure 3. Examples of p97 small molecule inhibitors.

The center cartoon shows the domain structure of p97, with arrows pointing to the binding sites for allosteric and ATP-competitive inhibitors. Each monomer in the p97 hexamer has three folded domains: the N-terminal PPI domain (purple), the D1 ATPase domain (orange), and the D2 ATPase domain (blue). Competitive inhibitors ML240 and CB5083 (left panel) bind at the ATP-binding pocket of p97 D2 domain (light blue). Inhibitors UPCDC30245 and NMS-873 (right panel) bind at allosteric sites in the D2 ATPase domain, near the D1-D2 linker region (red). The binding site for allosteric inhibitors has been defined by cross-linking, resistant mutants, and cryo-EM. See references in the main text.
To learn how catalytic inhibitors alter the p97 PPI network, two proteomics studies have measured the binding of adaptor proteins to p97 in the presence and absence of NMS-873 [75,76]. By immunoprecipitation and mass spectrometry, Her, et al. [75] and Xue, et al. [76] showed that NMS-873 reduces binding of some adaptors (e.g. SAKS1 and Erasin), while others are unaffected (e.g. p37 and p47) and several others are increased (e.g., NPL4/UFD1 and FAF1). The dimeric adaptor NPL4/UFD1 (NU) is particularly interesting because it is central to p97’s unfoldase activity.
Increased binding in the presence of NMS-873 could be due to allosteric stabilization of the p97/NU complex or to the inhibition of unfoldase activity, which could result in the stabilization of trapped p97/NU/Ub-client complexes. Formation of trapped NU complexes and reduction in p97/Erasin complexes are both consistent with NMS-873-mediated inhibition of ERAD. These proteomic data provide initial insight into allosteric PPI modulation and pose a number of interesting questions. For instance, do all ATPase inhibitors alter the PPI network in the same way, or are there mechanistic differences? Can these alterations in PPI be modeled using biochemically defined systems? Development of function-specific modulators will require an iterative evaluation of p97 function and PPI-complex formation in vitro and in cells. As a first step, Chou, et al. have screened a set of ML240/CB-5083 analogs in the presence of p47 and NU, seeking to select p97 inhibitors that act in the context of particular protein complexes [74]. While this compound set did not yield adaptor-selective inhibitors, extending these approaches may provide novel, PPI-dependent inhibitors that could be used to dissect the p97/PPI functional network in the context of cancer and degenerative disease.
5. The Heat Shock Protein 70 (HSP70) network provides many avenues for small-molecule modulation
HSP70 chaperones help maintain proteostasis by stabilizing folding-competent clients or shuttling misfolded proteins to be degraded (Figure 4a). The HSP70 family associates with co-chaperones such as J-proteins (HSP40s), tetratricopeptide repeat (TPR) proteins (including HSP70-HSP90 organizing protein, or HOP), HSP90, and nucleotide exchange factors (NEFs). HSP70 undergoes dramatic conformational changes during its ATPase cycle (see reviews [15,77]), leading to binding and release of client proteins, J-proteins and NEFs. Modulating any of these PPIs would affect ATPase cycle turnover, and therefore could act as checkpoints in altering HSP functions.
Figure 4. Multiple approaches to targeting the chaperone HSP70.

(a) The HSP70 catalytic cycle offers several opportunities for inhibition. HSP70 is shown schematically in light blue, with potential sites of inhibition in light gray. J-proteins (orange) help deliver client proteins (red) to the HSP70 substrate-binding domain (SBD) and accelerate ATP hydrolysis. The HSP70 LID domain adopts an “open” conformation in its ATP-bound state, and a “closed” position in its ADP-bound state (gray dots). The nucleotide exchange factors (NEFs; light yellow) are involved with ADP exchange, thereby helping release client proteins from the network. HSP70 also transfers protein substrates for further processing and refolding to HSP90 (yellow) via an interaction with HOP (black). (b) Examples of HSP70 PPI inhibitors targeting various checkpoints. See references in the main text. Abbreviations: HOP, HSP70-HSP90 organizing protein; LID, LID domain; NBD, nucleotide binding domain; NEF, nuclear exchange factor; SBD, substrate binding domain.
Members of the HSP family are frequently overexpressed under stress conditions and may serve as biomarkers of disease states [78]; furthermore, HSP70 can be beneficial or disease-associated. In neurodegenerative diseases, HSP70 plays a neuroprotective role by interacting with intrinsically unstructured proteins that are prone to aggregation (see reviews [79,80]). In cancer, HSP70 binding can stabilize and thereby activate oncogenes (see reviews [81,82]). There is also evidence for chaperone-PPI rewiring in cancers: Rodina et al., define an “epichaperome” containing large complexes including HSP70, HSP90, and their co-chaperones [83]. The presence of this epichaperome correlates with sensitivity to HSP90 inhibitors, independent of the abundance of HSP90. Together, these data suggest that modulating particular complexes within HSP90 and HSP70 networks is a potentially powerful therapeutic strategy [11].
To develop a chemical toolbox for HSP70, Gestwicki and coworkers have reported allosteric PPI modulators that target multiple HSP70 sites (Figure 4b), offering an alternative approach to existing ATP-competitive inhibitors such as VER-155008 [84]. In particular, a series of optimized benzothiazole rhodacyanine compounds (JG-231, JG-294, and JG-237) that bind near the nucleotide binding site were reported to allosterically disrupt HSP70-NEF PPIs by “rigidly stopping” concerted motion within HSP70 [85,86]. Additionally, a de novo high throughput screen yielded two scaffolds that inhibited either the HSP70/DnaJA2 or HSP70/NEF PPIs. The HSP70/NEF inhibitor (IT2-144) blocked refolding of a model client using multiple NEF variants in combination with HSP70 in a nucleotide-independent manner similar to JG-231 [87]. Peptide-based inhibitors that modulated HSP70-HOP PPIs via two separate mechanisms were also reported [88]. One peptide (C1) stabilized HSP70-HOP binding, trapping HSP70 in a complex that inhibited protein folding. A second series of peptides (S7, S8) inhibited HSP70-HOP PPIs, thereby preventing formation of the protein-folding complex. Taken together, these molecules suggest that different modes of binding to HSP70 may have different biochemical and biological effects.
Recent efforts have focused on the importance of HSP70 in stabilizing androgen receptors (ARs) in prostate cancer. Increased levels of full-length AR and AR-V7, a constitutively active splice variant that lacks the C-terminal, androgen-binding domain, were correlated with poor prognosis in castration-resistant prostate cancer (CRPC) patients. AR-V7 is particularly problematic because approved prostate cancer therapies, like enzalutamide (Figure 4b), block production or binding of the androgen hormone [89]. Hence, cancers expressing AR-V7 are resistant to current therapies. Proteomic approaches indicate that HSP70 binds similarly to both AR and AR-V7 in multiple CPRC cell lines [90,91]. Treatment with either VER-155008 or quercetin (Figure 4b), an inhibitor of HSP70 expression, reduced both AR-FL and AR-V7 levels in LNCaP95 cells [90]. Disruption of the HSP70/J-protein activity using compounds that bind to either HSP40 (C86) or HSP70 (JG-98) also led to destabilization and reduced transcriptional activity of AR-FL and AR-V7 in 22Rv1 cells [92]. A detailed dissection of the HSP70/AR PPI pinpointed the interaction to be centered at the N-terminal domain of AR, explaining why inhibition of HSP70 reduces transactivation of both AR variants [91]. Thus, multiple modes of HSP70 inhibition were effective at reducing AR in CRPC. Despite the demonstrated importance of HSP70 in cancer, catalytic HSP70 inhibitors have so far not been effective in human clinical trials. Further studies could determine whether inhibitors that target selective HSP70 PPI, e.g., by blocking interactions with J-proteins or with clients directly, show improved therapeutic index compared to ATPase inhibitors.
6. Conclusions and Outlook
Modulating PPIs with drug-like molecules had been akin to the holy grail in drug discovery; however, the field has matured significantly in the past several years. In the small-molecule realm, there are now numerous examples of orthosteric and allosteric inhibitors of PPIs. For protein therapeutics, there are emerging technologies for transporting recombinant proteins/antibodies into cells, which would open up this therapeutic modality for intracellular PPIs [5]. Taken together, we conclude that existing and emerging technologies are enabling drug discovery for PPIs.
The field is now progressing into a compelling new phase, where the tools exist to address complex questions concerning mechanisms of inhibition and proteome-wide effects of PPI modulation. As highlighted here, there are now multiple small-molecule and protein-based inhibitors targeting hub proteins such as APOBEC, deubiquitinases, p97, and HSP70. These inhibitors have different modes of binding and could therefore show distinct mechanisms of action in cells and animals. Future work will systematically compare the effects of inhibitory mechanisms on functional specificity, toxicity, and mechanisms of resistance, so that the best therapeutic modality can be selected for a given clinical indication. Additionally, proteomic approaches allow researchers to evaluate the effect of an inhibitor on the broader PPI network. Finally, while this review has focused on inhibitors, the development of PPI stabilizers - using bivalent antibodies and small-molecule ‘molecular glues’ - is gaining traction. Together, these developments will yield next generation therapeutics that selectively and effectively modulate PPI networks.
Acknowledgements
The authors wish to acknowledge Dr. John Kenneth Morrow for helpful discussions of the manuscript. Funding was provided by the National Institutes of Health (GM130145-01) and The Ono Pharma Foundation Breakthrough Award.
Footnotes
Conflict of Interest
Nothing declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Recommended Reading
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Hu G, Wu Z, Uversky VN, Kurgan L: Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int J Mol Sci 2017, 18:2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, et al. : A subcellular map of the human proteome. Science 2017, 356:806–807. [DOI] [PubMed] [Google Scholar]
- 3.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. : Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z, Ivanov AA, Su R, Gonzalez-Pecchi V, Qi Q, Liu S, Webber P, McMillan E, Rusnak L, Pham C, et al. : The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat Commun 2017, 8:14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miersch S, Sidhu SS: Intracellular targeting with engineered proteins. F1000Research 2016, 5:1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin HL, Bedford R, Heseltine SJ, Tang AA, Haza KZ, Rao A, McPherson MJ, Tomlinson DC: Non-immunoglobulin scaffold proteins: Precision tools for studying protein-protein interactions in cancer. New Biotechnology 2018, 45:28–35. [DOI] [PubMed] [Google Scholar]
- 7.Wuo MG, Arora PS: Engineered protein scaffolds as leads for synthetic inhibitors of protein–protein interactions. Current Opinion in Chemical Biology 2018, 44:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arkin MR, Tang Y, Wells JA: Small-molecule inhibitors of protein-protein interactions: progressing towards the reality. Chem Biol 2014, 21:1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosch J: PPI inhibitor and stabilizer development in human diseases. Drug Discov Today Technol 2017, 24:3–9. [DOI] [PubMed] [Google Scholar]
- 10.Andrei SA, Sijbesma E, Hann M, Davis J, O’Mahony G, Perry MWD, Karawajczyk A, Eickhoff J, Brunsveld L, Doveston RG, et al. : Stabilization of protein-protein interactions in drug discovery. Expert Opin Drug Discov 2017, 12:925–940. [DOI] [PubMed] [Google Scholar]
- **11.Gestwicki JE, Shao H: Inhibitors and chemical probes for molecular chaperone networks. J Biol Chem 2018, doi: 10.1074/jbc.TM118.002813.A recent review of small molecule compounds which serve as probes for multiple heat shock protein sub-networks.
- 12.Breen ME, Mapp AK: Modulating the masters: chemical tools to dissect CBP and p300 function. Curr Opin Chem Biol 2018, 45:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernández-Fernández MR, Gragera M, Ochoa-Ibarrola L, Quintana-Gallardo L, Valpuesta JM: Hsp70 - a master regulator in protein degradation. FEBS Lett 2017, 591:2648–2660. [DOI] [PubMed] [Google Scholar]
- 14.Fernández-Fernández MR, Valpuesta JM: Hsp70 chaperone: a master player in protein homeostasis. F1000Res 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freilich R, Arhar T, Abrams JL, Gestwicki JE: Protein-Protein Interactions in the Molecular Chaperone Network. Acc Chem Res 2018, 51:940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Genest O, Wickner S, Doyle SM: Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J Biol Chem 2018, doi: 10.1074/jbc.REV118.002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **17.Wertz IE, Wang X: From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem Biol 2018, doi: 10.1016/j.chembiol.2018.10.022.A comprehensive review of recent progress of small molecule inhibitor development of UPSs, with an emphasis on novel ways to target the ubiquitin system for therapies.
- 18.Hanna J, Guerra-Moreno A, Ang J, Micoogullari Y: Protein Degradation and the Pathologic Basis of Disease. Am J Pathol 2019, 189:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai C, Sampson SB: HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol 2016, 26:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR: Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci 2006, 31:164–172. [DOI] [PubMed] [Google Scholar]
- 21.Kaushik S, Cuervo AM: Proteostasis and aging. Nat Med 2015, 21:1406–1415. [DOI] [PubMed] [Google Scholar]
- 22.Johnson ML, Yu HA, Hart EM, Weitner BB, Rademaker AW, Patel JD, Kris MG, Riely GJ: Phase I/II Study of HSP90 Inhibitor AUY922 and Erlotinib for EGFR-Mutant Lung Cancer With Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. J Clin Oncol 2015, 33:1666–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salter JD, Bennett RP, Smith HC: The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem Sci 2016, 41:578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang B, Li X, Lei L, Chen J: APOBEC: From mutator to editor. Journal of Genetics and Genomics 2017, 44:423–437. [DOI] [PubMed] [Google Scholar]
- 25.Zou J, Wang C, Ma X, Wang E, Peng G: APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci 2017, 7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westrich JA, Warren CJ, Klausner MJ, Guo K, Liu C-W, Santiago ML, Pyeon D: Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J Virol 2018, 92:e01318–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **27.Olson ME, Harris RS, Harki DA: APOBEC Enzymes as Targets for Virus and Cancer Therapy. Cell Chem Biol 2018, 25:36–49.An extensive review of APOBECs role in HIV and cancer, as well as descriptions of known and potential therapeutic routes.
- 28.Bennett RP, Salter JD, Smith HC: A New Class of Antiretroviral Enabling Innate Immunity by Protecting APOBEC3 from HIV Vif-Dependent Degradation. Trends in Molecular Medicine 2018, 24:507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma L, Zhang Z, Liu Z, Pan Q, Wang J, Li X, Guo F, Liang C, Hu L, Zhou J, et al. : Identification of small molecule compounds targeting the interaction of HIV-1 Vif and human APOBEC3G by virtual screening and biological evaluation. Sci Rep 2018, 8:8067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binning JM, Smith AM, Hultquist JF, Craik CS, Caretta Cartozo N, Campbell MG, Burton L, La Greca F, McGregor MJ, Ta HM, et al. : Fab-based inhibitors reveal ubiquitin independent functions for HIV Vif neutralization of APOBEC3 restriction factors. PLoS Pathog 2018, 14:e1006830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ: Gain of function mutations in p53. Nature Genetics 1993, 4:42–46. [DOI] [PubMed] [Google Scholar]
- *32.Zhang W, Sartori MA, Makhnevych T, Federowicz KE, Dong X, Liu L, Nim S, Dong A, Yang J, Li Y, et al. : Generation and Validation of Intracellular Ubiquitin Variant Inhibitors for USP7 and USP10. J Mol Biol 2017, 429:3546–3560.Several Ub-based positive and negative regulators of p53 that bound USP7 or USP10 and inhibited deubiquitination activity are reported. Over-expression of the discovered clones in cancer cells induces cell death, indicating the therapeutic value of this approach.
- 33.Reyes-Turcu FE, Wilkinson KD: Polyubiquitin binding and disassembly by deubiquitinating enzymes. Chem Rev 2009, 109:1495–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ronau JA, Beckmann JF, Hochstrasser M: Substrate specificity of the ubiquitin and Ubl proteases. Cell Res 2016, 26:441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips AH, Zhang Y, Cunningham CN, Zhou L, Forrest WF, Liu PS, Steffek M, Lee J, Tam C, Helgason E, et al. : Conformational dynamics control ubiquitin-deubiquitinase interactions and influence in vivo signaling. Proceedings of the National Academy of Sciences 2013, 110:11379–11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **36.Zhang Y, Zhou L, Rouge L, Phillips AH, Lam C, Liu P, Sandoval W, Helgason E, Murray JM, Wertz IE, et al. : Conformational stabilization of ubiquitin yields potent and selective inhibitors of USP7. Nature Chemical Biology 2013, 9:51–58.A series of Ub-based proteins with nanomolar binding affinity and high selectivity of USP7 is reported. The developed variants can selectively inhibit the deubiquitylation activity of USP7 in vitro and in cells, and consequently stabilize p53 levels in cancer cells.
- 37.Gorelik M, Orlicky S, Sartori MA, Tang X, Marcon E, Kurinov I, Greenblatt JF, Tyers M, Moffat J, Sicheri F, et al. : Inhibition of SCF ubiquitin ligases by engineered ubiquitin variants that target the Cul1 binding site on the Skp1–F-box interface. PNAS 2016, 113:3527–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang W, Ben-David M, Sidhu SS: Engineering cell signaling modulators from native protein–protein interactions. Current Opinion in Structural Biology 2017, 45:25–35. [DOI] [PubMed] [Google Scholar]
- *39.Gavory G, O’Dowd CR, Helm MD, Flasz J, Arkoudis E, Dossang A, Hughes C, Cassidy E, McClelland K, Odrzywol E, et al. : Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nature Chemical Biology 2018, 14:118–125.Discovery of non-covalent inhibitors that bind at a novel allosteric pocket of inactive form of USP7 with high cellular specificity and MDM2 antagonistic activities.
- **40.Kategaya L, Di Lello P, Rougé L, Pastor R, Clark KR, Drummond J, Kleinheinz T, Lin E, Upton J-P, Prakash S, et al. : USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 2017, 550:534–538.A series of compounds that can selectively inhibit USP7 and induce tumor cell death is described. Structural study revealed that those compounds bind at an allosteric site on USP7 and constrain it in an inactive form, therefore block the ubiquitin binding. This sheds lights on new means to achieve selectivity over other DUBs.
- 41.Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL, Tonkin LM, Townsend EC, Buker SM, Lancia DR, et al. : Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550:481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weisberg EL, Schauer NJ, Yang J, Lamberto I, Doherty L, Bhatt S, Nonami A, Meng C, Letai A, Wright R, et al. : Inhibition of USP10 induces degradation of oncogenic FLT3. Nature Chemical Biology 2017, 13:1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN, Camonis J, et al. : Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther 2009, 8:2286–2295. [DOI] [PubMed] [Google Scholar]
- 44.Lamberto I, Liu X, Seo H-S, Schauer NJ, Iacob RE, Hu W, Das D, Mikhailova T, Weisberg EL, Engen JR, et al. : Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7. Cell Chem Biol 2017, 24:1490–1500.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen C, Song J, Wang J, Xu C, Chen C, Gu W, Sun H, Wen X: Synthesis and biological evaluation of thiazole derivatives as novel USP7 inhibitors. Bioorg Med Chem Lett 2017, 27:845–849. [DOI] [PubMed] [Google Scholar]
- 46.Long MJC, Lawson AP, Baggio R, Qian Y, Rozhansky L, Fasci D, El Oualid F, Weerapana E, Hedstrom L: Diarylcarbonates are a new class of deubiquitinating enzyme inhibitor. Bioorg Med Chem Lett 2019, 29:204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *47.Pozhidaeva A, Valles G, Wang F, Wu J, Sterner DE, Nguyen P, Weinstock J, Kumar KGS, Kanyo J, Wright D, et al. : USP7-Specific Inhibitors Target and Modify the Enzyme’s Active Site via Distinct Chemical Mechanisms. Cell Chemical Biology 2017, 24:1501–1512.e5.Discovery of USP7-specific small molecule inhibitors with covalent mechanism. Compounds label the catalytic Cys223 on USP7 with high selectivity in cells and destabilize USP7-specific substrates.
- 48.Wang X, Liu Z, Zhang L, Yang Z, Chen X, Luo J, Zhou Z, Mei X, Yu X, Shao Z, et al. : Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis 2018, 9:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tavana O, Gu W: Modulation of the p53/MDM2 interplay by HAUSP inhibitors. J Mol Cell Biol 2017, 9:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan T, Yan F, Ying M, Cao J, He Q, Zhu H, Yang B: Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front Pharmacol 2018, 9:1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kloppsteck P, Ewens CA, Förster A, Zhang X, Freemont PS: Regulation of p97 in the ubiquitin–proteasome system by the UBX protein-family. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2012, 1823:125–129. [DOI] [PubMed] [Google Scholar]
- 52.Meyer H, Bug M, Bremer S: Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nature Cell Biology 2012, 14:117–123. [DOI] [PubMed] [Google Scholar]
- 53.Blythe EE, Olson KC, Chau V, Deshaies RJ: Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proceedings of the National Academy of Sciences 2017, 114:E4380–E4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bodnar NO, Rapoport TA: Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 2017, 169:722–735.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proceedings of the National Academy of Sciences 2005, 102:14132–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stein A, Ruggiano A, Carvalho P, Rapoport TA: Key Steps in ERAD of Luminal ER Proteins Reconstituted with Purified Components. Cell 2014, 158:1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uchiyama K, Kondo H: p97/p47-Mediated biogenesis of Golgi and ER. J Biochem 2005, 137:115–119. [DOI] [PubMed] [Google Scholar]
- **58.Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, et al. : Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 2015, 28:653–665.Description of first clinical-stage p97 inhbitor.
- 59.Al-Obeidi E, Al-Tahan S, Surampalli A, Goyal N, Wang AK, Hermann A, Omizo M, Smith C, Mozaffar T, Kimonis V: Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet 2018, 93:119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bulfer SL, Chou T-F, Arkin MR: p97 Disease Mutations Modulate Nucleotide-Induced Conformation to Alter Protein-Protein Interactions. ACS Chem Biol 2016, 11:2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuetz AK, Kay LE: A Dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife 2016, 5:e20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schütz AK, Rennella E, Kay LE: Exploiting conformational plasticity in the AAA+ protein VCP/p97 to modify function. Proceedings of the National Academy of Sciences 2017, 114:E6822–E6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang T, Xu W, Qin M, Yang Y, Bao P, Shen F, Zhang Z, Xu J: Pathogenic Mutations in the Valosin-containing Protein/p97(VCP) N-domain Inhibit the SUMOylation of VCP and Lead to Impaired Stress Response. Journal of Biological Chemistry 2016, 291:14373–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang X, Gui L, Zhang X, Bulfer SL, Sanghez V, Wong DE, Lee Y, Lehmann L, Lee JS, Shih P-Y, et al. : Altered cofactor regulation with disease-associated p97/VCP mutations. Proc Natl Acad Sci USA 2015, 112:E1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rijal R, Arhzaouy K, Strucksberg K-H, Cross M, Hofmann A, Schröder R, Clemen CS, Eichinger L: Mutant p97 exhibits species-specific changes of its ATPase activity and compromises the UBXD9-mediated monomerisation of p97 hexamers. Eur J Cell Biol 2016, 95:195–207. [DOI] [PubMed] [Google Scholar]
- 66.Rao MV, Williams DR, Cocklin S, Loll PJ: Interaction between the AAA+ ATPase p97 and its cofactor ataxin3 in health and disease: Nucleotide-induced conformational changes regulate cofactor binding. J Biol Chem 2017, 292:18392–18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang T, Mishra P, Hay BA, Chan D, Guo M: Valosin-containing protein (VCP/p97) inhibitors relieve Mitofusin-dependent mitochondrial defects due to VCP disease mutants. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang WK, Odzorig T, Jin W, Xia D: Structural Basis of p97 Inhibition by the Site-Selective Anti-Cancer Compound CB-5083. Mol Pharmacol 2018, doi: 10.1124/mol.118.114256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, et al. : Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nature Chemical Biology 2013, 9:548–556. [DOI] [PubMed] [Google Scholar]
- 70.Alverez C, Bulfer SL, Chakrasali R, Chimenti MS, Deshaies RJ, Green N, Kelly M, LaPorte MG, Lewis TS, Liang M, et al. : Allosteric Indole Amide Inhibitors of p97: Identification of a Novel Probe of the Ubiquitin Pathway. ACS Medicinal Chemistry Letters 2016, 7:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. *.Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. : 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 2016, 351:871–875.First published high-resolution structure of p97/VCP with a small-molecule inhibitor bound. Structures of inhibitor-bound, ATP, ADP, and mixed ATP/ADP show previously known and newly identified changes in protein conformation, and suggest the mechanism of allosteric inhibition. One of the first small-molecule/protein structures to be solved at high-resolution using Cryo-EM.
- 72.Chou T-F, Bulfer SL, Weihl CC, Li K, Lis LG, Walters MA, Schoenen FJ, Lin HJ, Deshaies RJ, Arkin MR: Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. J Mol Biol 2014, 426:2886–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chou T-F, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ: Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem 2013, 8:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gui L, Zhang X, Li K, Frankowski KJ, Li S, Wong DE, Moen DR, Porubsky PR, Lin HJ, Schoenen FJ, et al. : Evaluating p97 Inhibitor Analogues for Potency against p97–p37 and p97–Npl4–Ufd1 Complexes. ChemMedChem 2016, 11:953–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **75.Her N-G, Toth JI, Ma C-T, Wei Y, Motamedchaboki K, Sergienko E, Petroski MD: p97 Composition Changes Caused by Allosteric Inhibition Are Suppressed by an On-Target Mechanism that Increases the Enzyme’s ATPase Activity. Cell Chem Biol 2016, 23:517–528.Proteomic study demonstrating the effect of small-molecule inhibitor on p97's PPI network. Mechanisms of resistance to NMS-873 are also described.
- **76.Xue L, Blythe EE, Freiberger EC, Mamrosh JL, Hebert AS, Reitsma JM, Hess S, Coon JJ, Deshaies RJ: Valosin-containing protein (VCP)–Adaptor Interactions are Exceptionally Dynamic and Subject to Differential Modulation by a VCP Inhibitor. Molecular & Cellular Proteomics 2016, 15:2970–2986.Proteomic study demonstrating the effect of small-molecule inhibitor on p97's PPI network. Multiple PPI-capturing strategies are compared, giving a more comprehensive picture of p97 PPI and highlighting the challenges with mapping dynamically changing complexes.
- 77.Zuiderweg ERP, Hightower LE, Gestwicki JE: The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 2017, 22:173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qu B, Jia Y, Liu Y, Wang H, Ren G, Wang H: The detection and role of heat shock protein 70 in various nondisease conditions and disease conditions: a literature review. Cell Stress Chaperones 2015, 20:885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brehme M, Voisine C: Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity. Dis Model Mech 2016, 9:823–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **80.Penke B, Bogár F, Crul T, Sántha M, Tóth ME, Vígh L: Heat Shock Proteins and Autophagy Pathways in Neuroprotection: from Molecular Bases to Pharmacological Interventions. Int J Mol Sci 2018, 19:325.An extensive review describing the relationships of heat shock proteins and the ubiquitin-proteasome system in neurodegenerative diseases. Also includes drug candidates used in modulating chaperone, UPS, and autophagy networks.
- 81.Chatterjee S, Burns TF: Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int J Mol Sci 2017, 18:1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S: Heat Shock Proteins and Cancer. Trends Pharmacol Sci 2017, 38:226–256. [DOI] [PubMed] [Google Scholar]
- **83.Rodina A, Wang T, Yan P, Gomes ED, Dunphy MPS, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H, et al. : The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538:397–401.Description of interrelated chaperone networks in tumor cells, with HSC70 and HSP90 serving as "nucleating sites". Chaperone networks are rewired by Myc to form stable high-molecular weight complexes, increasing survivability of tumor cells.
- 84.Williamson DS, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Foloppe N, Francis GL, Graham CJ, Howes R, et al. : Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J Med Chem 2009, 52:1510–1513. [DOI] [PubMed] [Google Scholar]
- 85.Shao H, Li X, Moses MA, Gilbert LA, Kalyanaraman C, Young ZT, Chernova M, Journey SN, Weissman JS, Hann B, et al. : Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70). J Med Chem 2018, 61:6163–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rinaldi S, Assimon VA, Young ZT, Morra G, Shao H, Taylor IR, Gestwicki JE, Colombo G: A Local Allosteric Network in Heat Shock Protein 70 (Hsp70) Links Inhibitor Binding to Enzyme Activity and Distal Protein-Protein Interactions. ACS Chem Biol 2018, 13:3142–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taylor IR, Dunyak BM, Komiyama T, Shao H, Ran X, Assimon VA, Kalyanaraman C, Rauch JN, Jacobson MP, Zuiderweg ERP, et al. : High-throughput screen for inhibitors of protein-protein interactions in a reconstituted heat shock protein 70 (Hsp70) complex. J Biol Chem 2018, 293:4014–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zaiter S, Huo Y, Tiew FY, Gestwicki JE, McAlpine SR: Designing de novo small molecules that control Heat shock protein 70 (Hsp70) and Heat shock organizing protein (HOP) within the chaperone protein-folding machinery. J Med Chem 2018, doi: 10.1021/acs.jmedchem.8b01436. [DOI] [PubMed] [Google Scholar]
- 89.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al. : Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324:787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kita K, Shiota M, Tanaka M, Otsuka A, Matsumoto M, Kato M, Tamada S, Iwao H, Miura K, Nakatani T, et al. : Heat shock protein 70 inhibitors suppress androgen receptor expression in LNCaP95 prostate cancer cells. Cancer Sci 2017, 108:1820–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dong J, Wu Z, Wang D, Pascal LE, Nelson JB, Wipf P, Wang Z: Hsp70 binds to the androgen receptor N-terminal domain and modulates the receptor function in prostate cancer cells. Mol Cancer Ther 2019, 18:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moses MA, Kim YS, Rivera-Marquez GM, Oshima N, Watson MJ, Beebe KE, Wells C, Lee S, Zuehlke AD, Shao H, et al. : Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res 2018, 78:4022–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]