

Abstract
Genome sequencing has revealed an increasing number of genetic variations that are associated with neuropsychiatric disorders. Frequently, studies limit their focus to likely gene-disrupting mutations because they are relatively easy to interpret. Missense variants, instead, have often been undervalued. However, some missense variants can be informative for developing a more profound understanding of disease pathogenesis and ultimately targeted therapies. Here we present an example of this by studying a missense variant in a well-known autism spectrum disorder (ASD) causing gene SHANK3. We analyzed Shank3’s in vivo phosphorylation profile and identified S685 as one phosphorylation site where one ASD-linked variant has been reported. Detailed analysis of this variant revealed a novel function of Shank3 in recruiting Abelson interactor 1 (ABI1) and the WAVE complex to the post-synaptic density (PSD), which is critical for synapse and dendritic spine development. This function was found to be independent of Shank3’s other functions such as binding to GKAP and Homer. Introduction of this human ASD mutation into mice resulted in a small subset of phenotypes seen previously in constitutive Shank3 knockout mice, including increased allogrooming, increased social dominance, and reduced pup USV. Together, these findings demonstrate the modularity of Shank3 function in vivo. This modularity further indicates that there is more than one independent pathogenic pathway downstream of Shank3 and correcting a single downstream pathway is unlikely to be sufficient for clear clinical improvement. In addition, this study illustrates the value of deep biological analysis of select missense mutations in elucidating the pathogenesis of neuropsychiatric phenotypes.
INTRODUCTION
Autism spectrum disorders (ASD) are a group of neurodevelopmental disorders characterized by impaired social communication, poor language development, repetitive behaviors, and restricted interests. Genome sequencing has revealed an increasing number of genes responsible for ASD by identification of de novo likely gene-disrupting (LGD) mutations. Missense mutations, instead, have been considered far less informative because most of them are of unknown significance.
LGD mutations in SHANK3 are estimated to contribute to ~1% of all ASD cases 1–3. Moreover, epigenetic dysregulation of SHANK3 has been reported in up to 15% of individuals with ASD 4. Thus, SHANK3 is a well-established autism risk gene, and detailed mechanistic studies provide insights into ASD pathogenesis. Individuals with mutations in SHANK3 often manifest intellectual disability, autism, hypotonia, and motor delay 5,6. Shank3 serves as a scaffolding protein within the postsynaptic density (PSD) of dendritic spines, which are actin-rich protrusions from dendrites 7. The protein has a PSD95/discs large/ZO-1 (PDZ) domain that binds guanylate kinase-associated proteins (GKAPs), which further bridge Shank3 to PSD-95, another major scaffold in the PSD 7. Shank3 also has a large proline-rich domain containing ABI1, IRSp53, Homer and cortactin binding motifs 7–11. Knock down or knockout (KO) of Shank3 reduces dendritic spine density and maturation 12–16, whereas overexpression of Shank3 enhances spine development 12,17–19. Most excitatory synapses are built on dendritic spines. As expected, loss of Shank3 causes excitatory synaptic transmission deficits 13–16,20–27. Such synaptic dysfunction further leads to abnormal behaviors including altered social interaction, motor coordination deficits and repetitive behaviors in Shank3 KO mouse models 13,14,28,15,16,20,21,23,25–27.
Efforts have been made to elucidate the underlying molecular mechanisms for developing targeted treatments for individuals with ASD due to a SHANK3 mutation. Multiple pathways, including CLK2, Ih channel, mGluR5, and PAK signaling have been reported to be dysregulated upon loss of Shank3 in model systems 28–31. However, it is unknown if individual dysregulated molecular pathways are independently responsible for a subset of physiological and behavioral phenotypes. If so, this would represent a modularity to Shank3 function. This information is particularly important regarding targeted treatment of SHANKopathies because if phenotypes are indeed independently mediated by distinct modules of Shank3 function, correcting a single downstream molecular defect is unlikely to be sufficient for improving all the symptoms. A modularity of Shank3 function has not been investigated because most, if not all, studies were done using Shank3 heterozygous or homozygous null models. Although these models retain construct validity of LGD mutations, it is difficult to pinpoint the specific contribution of each pathway due to the confounding effects of other dysregulated pathways in the same model. To understand the functional modularity of Shank3, missense variants, each only affecting one residue, could be informative.
Here, by analyzing a missense variant of SHANK3 found in an individual with autism, we identify a downstream effector that interacts with Shank3 in a phosphorylation-dependent manner. We describe generation and characterization of a knock-in (KI) mouse model of this variant with this interaction specifically disrupted. Using this model, we further determine how this effector and its associated pathway mediate a subset of SHANKopathy phenotypes. We demonstrate the modularity of Shank3 function in vivo and highlight the value of studying missense mutations in autism.
MATERIALS AND METHODS
Reagents and resources
For resources such as antibodies and primers, please see Supplementary Table 8 for more information.,
A detailed description of the materials and methods is provided in Supplementary Information.
RESULTS
Phosphorylation of Shank3 at S685 modulates its interaction with ABI
Phosphorylation is a common regulatory mechanism to control key protein-effector interactions, and Shank3 phosphorylation has been reported before 32. Therefore, to identify direct downstream effectors of Shank3, we first examined the phosphorylation state of Shank3 in vivo. As no commercial Shank3 antibody consistently worked for immunoprecipitation, we generated a 2×Flag-Shank3 transgenic mouse to efficiently purify Shank3 in vivo (Supplementary Figure 1a). We characterized the 2×Flag-tagged Shank3 and found that it, like endogenous Shank3, was enriched in synaptic fractions (P2 and PSD I) (Supplementary Figure 1b). The regional and temporal expression pattern of 2×Flag-Shank3 paralleled those of endogenous Shank3 (Supplementary Figure 1c,d). Compared with wild-type (WT) mice, the transgenic mice had a 1.4-fold greater abundance of the α isoform and a 1.3-fold more in total Shank3 in the crude synaptosomal fraction (Supplementary Figure 1e).
We immunoprecipitated 2×Flag-Shank3 with an anti-Flag antibody from the cortex, hippocampus, and striatum of the transgenic mice (Supplementary Figure 1f). Immunoprecipitation followed by mass spectrometry analysis identified 41 phosphorylated residues of Shank3 in vivo (Figure 1a and Supplementary Table 1). We next evaluated whether any phosphorylated residues are in close proximity to known Shank3 protein-binding domains/motifs. We found that the phosphorylated site S685 is adjacent to a previously identified proline-rich motif (PRM) that is responsible for Shank3 interaction with the SH3 domain of ABI1, an indispensable subunit of the WAVE regulatory complex (WRC) 10,33 (Figure 1b and Supplementary Figure 2a). To further confirm that phosphorylation at S685 occurs in vivo, we generated a phospho-specific antibody to S685 (Supplementary Figure 2b). This antibody recognized purified Shank3 from mouse brain but failed to recognize Shank3 after it was treated with phosphatase (Supplementary Figure 2c). Given S685’s high degree of conservation among vertebrates (Figure 1c), we considered it highly likely to have functional importance.
Figure 1.
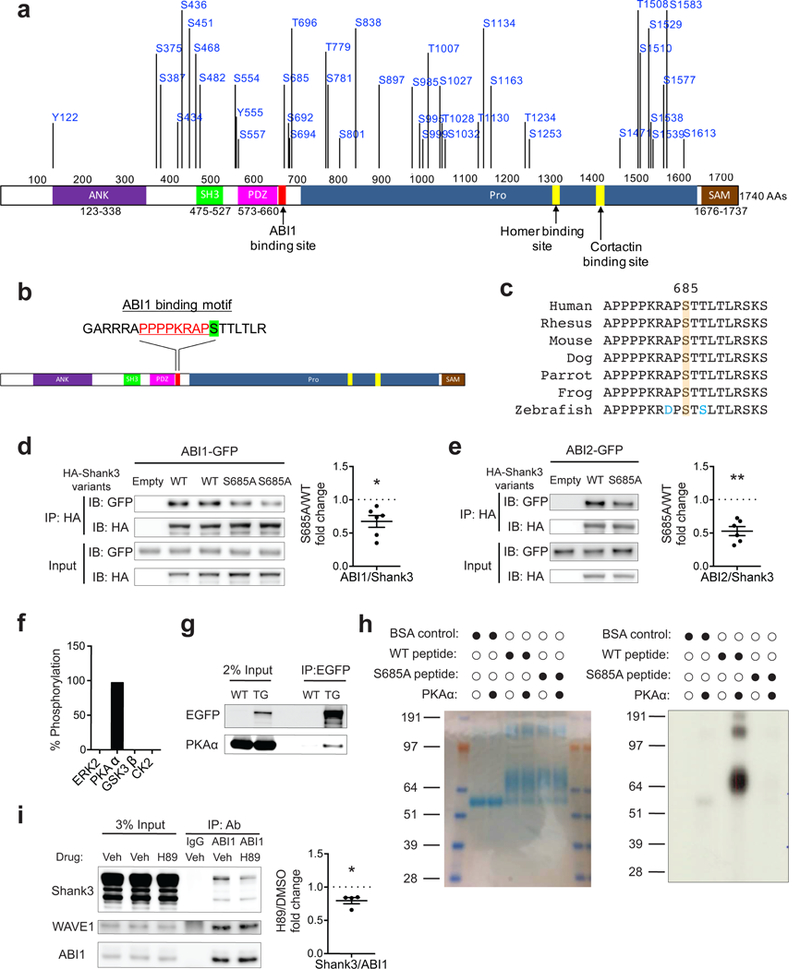
S685 phosphorylation modulates Shank3-ABI interaction. (a) Diagram of Shank3 protein (NP_067398) with 41 in vivo phosphorylated residues identified by mass spectrometry. (b) Diagram of Shank3 protein with S685 (green highlighted) adjacent to a previously identified ABI1 binding site (red text) on Shank3. (c) Protein sequence alignment of Shank3 surrounding S685 among vertebrates. (d,e) Immunoblots of ABI1-GFP (d) or ABI2-GFP (e) and HA-Shank3 following immunoprecipitation of HA-Shank3 variants in the co-immunoprecipitation assay in 293T cells (n=6, n=6 experiments). (f) Percentage of peptides with S685 phosphorylation in Shank3 mass-spectrometry-based in vitro kinase assay by ERK2, PKAα, GSK3β, and CK2. (g) Immunoblots of PKA α-catalytic subunit following immunoprecipitation of EGFP-tagged Shank3 from EGFP-Shank3 transgenic mice striatal lysate. TG, EGFP-Shank3 transgenic mice. (h) In vitro kinase assay with radiolabeled ATP using recombinant PKAα and Shank3 peptides including S685. Left, coomassie blue staining of BSA-conjugated peptides (aa 668–691). Right, autoradiography blot of kinase reaction product. (i) Immunoprecipitation of ABI1 in primary cortical neurons treated with vehicle (DMSO) or H89 (10 μM) followed by immunoblotting for Shank3, WAVE1, and ABI1 (n=4 experiments). WAVE1-ABI1 co-immunoprecipitation was used as positive control. Veh, vehicle (DMSO). All data are presented as mean ± s.e.m. *p<0.05, **p<0.01; paired two-tailed Student’s t-test.
To determine whether phosphorylation of S685 modulates the Shank3-ABI interaction, we performed a co-immunoprecipitation assay in HEK293T cells. Consistent with previous findings 10, ABI1 co-immunoprecipitated with WT Shank3 (Figure 1d). Mutation of the S685 residue of Shank3 to a non-phosphorylatable alanine (S685A) caused significantly less ABI1 to co-immunoprecipitate with Shank3 (Figure 1d). The S685A mutation also decreased the interaction between Shank3 and the ABI1 homolog ABI2, to a similar extent (Figure 1e). These results suggest that S685 phosphorylation is important for the Shank3-ABI interaction.
We next sought to identify kinases that can phosphorylate Shank3 at S685. We expanded our previously published datasets of the Shank3 interactome 17 by new immunoprecipitation-mass spectrometry experiments and public data curation, which resulted in a more comprehensive Shank3 interactome consisting of 793 proteins (see Methods and Supplementary Table 2). We identified 30 kinases among the 793 interactors. To narrow the list of candidate kinases, we searched the literature for their expression patterns and functions, which led us to four candidate kinases: Extracellular signal-regulated kinase 2 (ERK2), Protein kinase A alpha catalytic subunit (PKAα), Glycogen synthase kinase 3 beta (GSK3β), and Casein kinase 2 (CK2). Because its large size makes full-length Shank3 difficult to purify from bacteria, we purified a partial fragment of Shank3 (fragment 4, aa 668–858) that included Shank3 PRM from bacteria, and used it in an in vitro kinase assay (Supplementary Figure 2d). Mass spectrometry analysis of the kinase reaction products showed that ~97% of all peptides covering S685 were phosphorylated by PKAα (Figure 1f and Supplementary Figure 2e), and S685 is located within a consensus PKA phosphorylation motif (RXXS/T) 34. None of the other three kinases phosphorylated this site. Co-immunoprecipitation using the striatal tissue of EGFP-Shank3 transgenic mice revealed that Shank3 and PKAα interacted with each other (Figure 1g). To further validate our mass spectrometry findings, we performed a second kinase assay using synthesized Shank3 peptides (aa 668–691) conjugated with BSA and radiolabeled ATP (see Methods). We found that PKAα phosphorylated WT peptides, while the negative control (BSA alone) and S685A mutant peptides were not phosphorylated (Figure 1h). To examine whether PKA modulates Shank3-ABI interaction in neurons, we performed co-immunoprecipitation using dissociated cortical neurons while blocking PKA activity. Application of small molecule inhibitor H89 inhibited PKA in cortical neurons, as indicated by a decrease in CREB S133 phosphorylation (Supplementary Figure 2f). Inhibition of PKA reduced the amount of Shank3 co-immunoprecipitated with ABI1, while not altering ABI1’s interaction with WAVE1, the core subunit of WRC (Figure 1i). Together, these findings suggest that phosphorylation of S685 of Shank3 by PKA modulates Shank3-ABI interaction.
An autism-linked S685I mutation specifically diminishes Shank3-ABI interaction
Because SHANK3 haploinsufficiency is strongly linked to ASD, we sought mutations in or around S685 that occur in ASD individuals. An existing ASD database, the Autism Sequencing Consortium, reported several missense mutations in SHANK3 in autistic individuals 35. We found a G>T substitution at chromosome 22: 51,144,527 (hg build 19), which resulted in an S686I (S685 in mouse) missense mutation. S686I has not been observed in the gnomAD database of over 120,000 individuals without neurologic diseases, indicating it is not a common variant 36. Moreover, both Polyphen-2 and SIFT mutation analysis algorithms predicted this mutation to be damaging to Shank3 function (Supplementary Table 5) 37,38.
We tested whether the S685I mutation could alter Shank3-ABI interaction. In a co-immunoprecipitation assay, the S685I mutation diminished Shank3 interaction with ABI1 by at least 75% (Figure 2a). The S685I substitution had a similar effect on Shank3-ABI2 interaction (Figure 2b). It is worth noting that S685I did not alter Shank3 interaction with several other well-known binding partners, including cortactin, GKAP, Homer, and IRSp53 (Supplementary Figure 3a-d).
Figure 2.

The autism-linked Shank3 S685I mutation specifically diminishes Shank3-ABI interaction and causes selective social phenotypes. (a,b) Immunoblots of ABI1-GFP (a) or ABI2-GFP (b) and HA-Shank3 following immunoprecipitation of HA-Shank3 variants in the co-immunoprecipitation assay in 293T cells (n=4, n=4 experiments). (c) Schematic diagram for S685I KI mice generation by CRISPR/Cas9. Top: single strand oligodeoxynucleotides used as a repair template; red: codon encoding isoleucine; blue: synonymous mutation for genotyping. Middle: genomic sequence spanning Shank3 S685; green: codon encoding S685; scissors: Cas9 cutting site. Bottom: structure of wild-type mouse Shank3 gene. (d) Sequencing chromatograms of the S685I allele. (e,f) Immunoprecipitation of ABI1 in cortical (e) or striatal (f) lysate of KI-HET or KI-HOM mice followed by immunoblotting for Shank3, WAVE1, and ABI1 (n=3, n=3 experiments). (g) Time spent sniffing or allogrooming the intruder C57 mice in the resident-intruder assay (n=10, 13 mice). (h) Number of mounting events on the intruder C57 mice in the resident-intruder assay (n=10, 13 mice). (i) Time spent attacking or biting the intruder C57 mice in the resident-intruder assay (n=10, 13 mice). (j) Time spent allogrooming the intruder A/J mice in the resident-intruder assay (n=20, 8, 14 mice). (k) Percentage of wins in test pairs between indicated genotypes in the tube test (n=36, 32, 30, 24 matches). (l) Number of ultrasonic calls from pups separated from their dams (n=53, 26, 27 pups). All data are presented as mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, NS, not significant; (a,b) paired two-tailed Student’s t-test; (e,f) paired and unpaired two-tailed Student’s t-test; (g,h) unpaired two-tailed Student’s t-test; (i) Mann-Whitney U test; (j) Kruskal-Wallis test with post hoc Dunn’s test; (k) two-tailed binomial test; (l) two-way ANOVA with post hoc Tukey’s tests.
To better understand the effect of the S685I mutation, we performed kinetic analysis using surface plasmon resonance (Supplementary Figure 3e). Analysis of the binding curves of Shank3 fragment 4 with full-length recombinant ABI1 indicated that the S685I mutation significantly decreased the on-rate constant and caused a ~4-fold increase in the dissociation constant (Supplementary Figure 3f). Thus, the autism-linked Shank3 S685I variant appears to specifically disrupt Shank3-ABI interaction by changing a critical amino acid in the interacting motif.
Loss of Shank3-ABI interaction causes altered social behaviors
To validate our findings and understand the functional importance of Shank3-ABI interaction in vivo, we generated KI mice with the S685I substitution (Figure 2c,d). Both heterozygous KI (KI-HET) and homozygous KI (KI-HOM) mice are viable and fertile. They also have a similar body weight compared with WT mice (Supplementary Figure 4a). In addition, both protein abundance and isoform-specific expression pattern of S685I Shank3 were not changed compared with WT control (Supplementary Figure 4b).
Next, to confirm that S685I mutation disrupts Shank3-ABI interaction in vivo, we performed a co-immunoprecipitation assay in KI mice. As expected, significantly less Shank3 was co-immuoprecipitated with ABI1 in the cortex of KI-HET and KI-HOM mice compared with WT mice, with KI-HOM showing more prominent effect (Figure 2e). A similar reduction in Shank3-ABI1 co-immunoprecipitation was observed in the striatum (Figure 2f). In addition, Shank3-GKAP and Shank3-Homer interactions were not affected (Supplementary Figure 4c,d). These results indicate that S685I mutation specifically disrupts Shank3’s interaction with ABI1 in vivo.
Because S685I mutation is linked to autism, we next asked whether there are any social deficits in KI mice. We first tested sociability by the three-chamber test 17. Both WT and KI mice showed a significant preference for a stranger mouse to a novel object (Supplementary Figure 4e), indicating normal sociability.
To determine if there are any specific abnormal social behaviors other than sociability in KI mice, we performed a resident-intruder assay using stranger C57BL/6J mice as intruders 39. We quantified three categories of social behaviors: active social behavior (sniffing/allogrooming), threatening behavior (mounting), and aggressive behavior (attacking/biting). While there was no difference in threatening and aggressive behaviors, we found an increase in sniffing/allogrooming behavior in KI-HOM mice (Figure 2g-i). The continuous moving and escaping of the C57 intruders made it difficult to differentiate allogrooming from sniffing. Thus, we repeated the assay using submissive A/J mice as intruders 40. We found that KI-HOM mice displayed a robust increase in allogrooming behavior on A/J intruders (Figure 2j and Supplementary Video 1 and 2).
Excessive allogrooming behavior has been associated with increased social dominance 41. To evaluate social dominance, we performed the tube test. In this test, the dominant mouse will push its opponent out of the tube and “win” the match 21,42. We found that both male and female KI-HOM mice won most of the matches when tested against their WT opponents (Figure 2k), indicating increased social dominance. Only male KI-HET mice displayed the same phenotype (Figure 2k).
Social communication deficit is another major feature of ASD, especially those caused by mutations in SHANK3. To evaluate social communication phenotype in KI mice, we performed isolation-induced pup ultrasonic vocalization (USV) recording. As previously reported 43, the USVs of WT pups peaked around postnatal day 6 (P6) to P8 and stopped around P14 (Figure 2l). KI-HOM pups, however, emitted significantly fewer USVs on P2, P4, and P6. Interestingly, on P14, while most WT pups stopped emitting USVs, KI-HOM pups continued to do so. This is not due to eye-opening defects as both WT and KI-HOM pups opened their eyes around P13-P14. These data suggest that USV development is altered in KI-HOM mice.
We also performed open field assay and rotarod to evaluate motor function; elevated plus maze to evaluate anxiety; self-grooming and marble burying to evaluate repetitive behaviors; acoustic startle and prepulse inhibition to evaluate sensory-gating; adult USV to evaluate social communication in the adult. All have previously been shown to be abnormal in Shank3 null mice 15,20,21. We did not observe any difference between WT and KI mice in these tests (Supplementary Figure 5a-j and Supplementary Table 6). Together, these results show that loss of Shank3-ABI interaction causes abnormal social behavior/social communication that mimic a subset of phenotypes found in mice heterozygous or homozygous for Shank3 null alleles 15,21.
Shank3-ABI interaction is critical for dendritic spine development and synaptic transmission
To understand how disruption of Shank3-ABI interaction impairs neuronal function at a cellular level, we first investigated the functional relationship between Shank3 and ABIs in dendritic spine development. Loss of either ABI1 or ABI2 leads to reduced spine maturation and spine density 10,44, similar to the effect of Shank3 deficiency 13,14,24. We created distinct lines of mouse dissociated hippocampal neurons, either overexpressing Shank3, depleting Abi1/Abi2 using siRNAs, or combining the two (Figure 3a and Supplementary Figure 6a). Analysis of the morphology of dendritic spines revealed that knock down of Abi1 and Abi2 decreased stubby spine, mushroom-like spine (mature form), and total spine densities (Figure 3a,b and Supplementary Figure 6b). Overexpression of Shank3 increased stubby spine, mushroom-like spine, and total spine densities, but concurrent Shank3 overexpression and Abi1/Abi2 depletion resulted in a spine morphology that was indistinguishable from Abi1/Abi2 depletion alone. These results demonstrate that ABIs are downstream of Shank3 in spine development.
Figure 3.
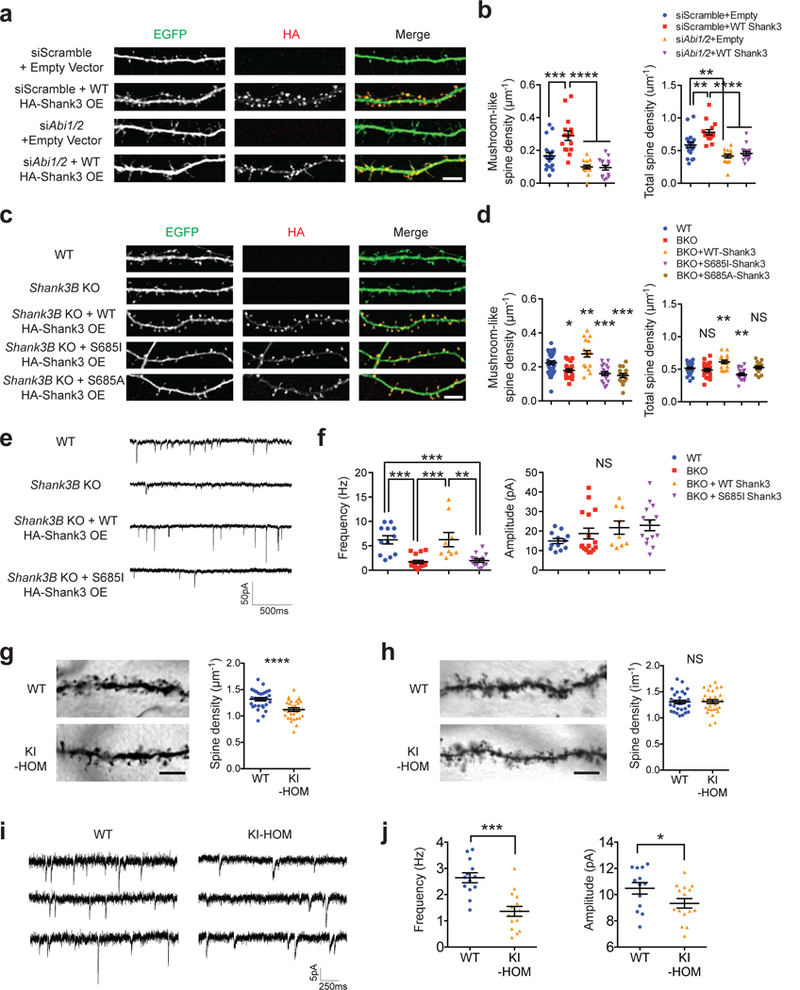
Shank3-ABI interaction is critical for dendritic spine development and synaptic transmission. (a) Fluorescent images of dendrites from days in vitro (DIV) 14 dissociated mouse hippocampal neurons co-transfected with siRNAs (scramble or against mouse Abi1 and Abi2), pEGFP-C1 plasmids and vectors expressing HA-Shank3 (empty or WT) on DIV 7. Scale bar, 5μm; OE, overexpression. (b) Quantification of dendritic spine densities in a (n=19, 15, 15, 15 neurons). (c) Fluorescent images of dendrites from DIV 14 dissociated mouse hippocampal neurons (WT or Shank3B KO) transfected with pEGFP-C1 plasmids and vectors expressing HA-Shank3 (empty, WT, S685I, or S685A) on DIV 7. Scale bar, 5μm. OE, overexpression. (d) Quantification of dendritic spine densities in c (n=32, 22, 18, 20, 15 neurons). (e) Typical mEPSC recordings of DIV 12–16 primary hippocampal neurons from WT or Shank3B KO mice transfected with pEGFP-C1 plasmids and vectors expressing HA-Shank3 (empty, WT or S685I). (f) Quantification of mEPSC frequency and amplitude in e (n=12, 16, 9, 14 neurons). (g) Representative images and quantification of spine densities of dorsal striatal medium spiny neurons of mice with indicated genotypes (n=30, 30 neurons). Scale bar, 5μm. (h) Representative images and quantification of spine densities of hippocampal CA1 pyramidal neurons of mice with indicated genotypes (n=30, 30 neurons). Scale bar, 5μm. (i) Typical mEPSC recordings of dorsal striatal medium spiny neurons from WT or KI-HOM mice. (j) Quantification of mEPSC frequency and amplitude in h (n=13, 15 neurons). All data are presented as mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, NS, not significant; (b,f) one-way ANOVA with post hoc Tukey’s tests; (d) one-way ANOVA with post hoc Dunnett’s tests (multiple comparisons were carried against WT group as control); (g,h) unpaired two-tailed Student’s t-test; (j) Mann-Whitney U test.
Next, the necessity of the Shank3-ABI protein interaction for Shank3’s function in spine development was assayed using a molecular replacement method. Neurons from Shank3B KO mice 13 had a greater number of immature thin spines and fewer mushroom-like spines (Figure 3c,d and Supplementary Figure 6c). Overexpression of WT Shank3 in Shank3B KO neurons rescued all the altered spine characteristics and further increased the number of stubby spines, mushroom-like spines, and total spines compared with WT neurons. In contrast, both S685I and S685A Shank3 failed to rescue the decreased mushroom-like spines. These data indicate that Shank3 interaction with ABI requires phosphorylation at S685, which, in turn, is necessary for Shank3 to promote mushroom-like, more mature spine formation.
Conversely, the role of the Shank3-ABI interaction for ABI’s function in spine formation was assayed. To test this, the residues critical for ABI to bind Shank3 were identified. Based on the structure of the ABI2 SH3 domain (PDB: 2ED0) and sequence homology search with HHpred 45 (see Methods), N446 and L461 of ABI2 were predicted to bind S685 of Shank3. We confirmed the importance of these residues for Shank3-ABI interaction by co-immunoprecipitation (Supplementary Figure 6d). Overexpression of WT ABI1 significantly increased stubby spine, mushroom-like spine, and total spine densities (Supplementary Figure 6e). However, overexpression of the Shank3 binding-deficient ABI1 (N447A L462A) failed to increase spine densities of any category, which further confirmed the importance of Shank3-ABI interaction in spine development.
To test whether these morphological changes translate into functional consequences, we recorded miniature excitatory postsynaptic currents (mEPSCs) in dissociated pyramidal-like hippocampal neurons from WT and Shank3B KO mice. Compared with WT neurons, Shank3B KO neurons had reduced mEPSC frequency, without significant changes in mEPSC amplitude (Figure 3e,f). This reduction in mEPSC frequency can be rescued by transfecting these neurons with WT Shank3, but not with S685I Shank3, suggesting that Shank3-ABI interaction contributes to the formation of functional synaptic sites.
To validate these findings in vivo, we assessed spine development and synaptic transmission in KI-HOM mice. Because Shank3 is highly expressed in the striatum, and previous reports found both spine development and synaptic transmission deficits in the dorsal striatum of Shank3 KO mice 13,15, we quantified dendritic spine densities of medium spiny neurons (MSNs) in the dorsal striatum. KI-HOM mice displayed a significant reduction in spine density (Figure 3g). A reduction in spine density was not observed in hippocampal CA1 pyramidal neurons (Figure 3h), probably due to compensation from Shank2 which is highly expressed in the hippocampus and interacts with ABIs. Recording mEPSCs of MSNs in the dorsal striatum determined that both mEPSC frequency and amplitude are reduced in KI-HOM mice (Figure 3i,j), similar to what has been reported in Shank3 KO mice 13,21. Together, these results indicate that interaction with ABI is critical for Shank3 function in both spine maturation and excitatory synaptic transmission.
Shank3-ABI interaction is critical for actin nucleation
KI-HOM mice have reduced dendritic spines, impaired synaptic transmission, altered social behaviors, but intact motor function and lack repetitive self-grooming, indicating that only a subset of Shank3 function (a module) is mediated by Shank3-ABI interaction. To determine the module Shank3-ABI interaction belongs to, we compared the interactomes of Shank3 and ABI1 to find their shared functional partners. First, we applied a method similar to that which we used to build Shank3 interactome to build an ABI1 interactome consisting of 151 proteins (Supplementary Table 3). Next, we compared the two interactomes and found significant overlap (Figure 4a; Fisher’s exact test, p = 2.11 × 10−13, odds ratio = 6.21). Gene ontology analysis of the 31 common interactors suggested that regulation of the Arp2/3 complex and actin nucleation may be one of their principal functions (Figure 4b and Supplementary Table 7).
Figure 4.
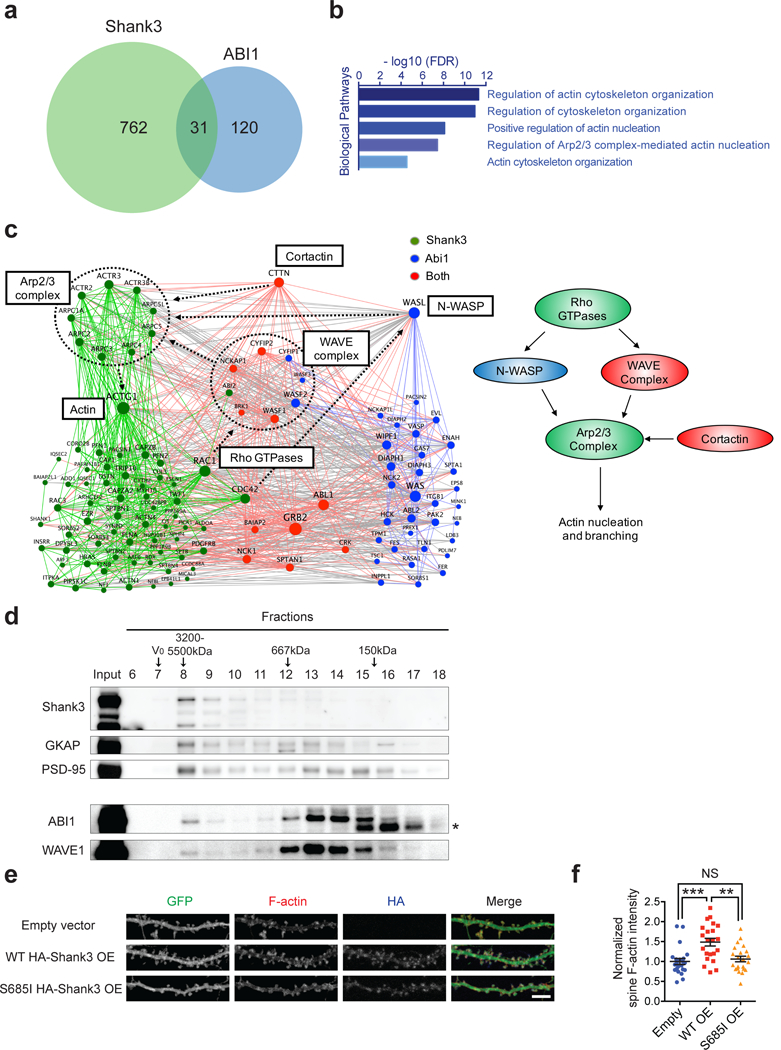
Shank3 and ABI1 functions converge on actin nucleation. (a) Venn diagrams showing the overlap between Shank3 interactome and ABI1 interactome. (b) Gene ontology analysis of the intersection of Shank3 and ABI1 interactomes. (c) Actin cytoskeleton-related sub-network of the Shank3-ABI1 interactome. Green nodes indicate Shank3 specific interactors, blue nodes indicate ABI1 specific interactors, and red nodes indicate the shared interactors between Shank3 and ABI1. Green edges indicate interactions within the Shank3 specific interactors, blue edges indicate interactions within the ABI1 specific interactors, red edges indicate interactions associated with the shared interactors, and gray edges indicate interactions between the Shank3 specific interactors and the ABI1 specific interactors. (d) Immunoblots following size-exclusion chromatography of mouse striatal lysate. V0, void volume; *, non-specific band. (e) Fluorescent images of dendrites from DIV 21 dissociated rat hippocampal neurons transfected with pEGFP-C1 plasmids and vectors expressing HA-Shank3 (empty, WT, or S685I) on DIV 6. Scale bar, 5μm. OE, overexpression. (f) Quantification of normalized integrated spine F-actin intensity in e (n=22, 22, 22 neurons). OE, overexpression. All data are presented as mean ± s.e.m. **p<0.01, ***p<0.001, NS, not significant; (f) one-way ANOVA with post hoc Tukey’s tests
To pinpoint the specific roles of Shank3 and ABI1 in the actin nucleation pathway, we integrated the two interactomes to generate a sub-network of actin-related proteins (Figure 4c and Supplementary Table 2, 3, and 4). Most components of the Arp2/3-mediated actin nucleation pathway were found in the network. Rho GTPases such as RAC1 and CDC42 activate nucleation-promoting factors (NPF) such as WRC and N-WASP, which then activate the Arp2/3 complex to initiate actin nucleation and branching 46. Cortactin is another type of NPF that facilitates Arp2/3 activation 46. As shown in the network, Shank3 binds the Arp2/3 complex, its upstream regulators (Rho GTPases, WRC, cortactin), and its downstream effector actin, potentially serving as a hub for actin nucleation. In contrast, ABI1, as a subunit of WRC, interacts with other WRC components (WAVE1/2/3, CYFIP1/2, NCKAP1, BRK1) and cortactin but not with the Arp2/3 complex itself. Thus, ABI1 might participate in actin nucleation in a supercomplex centered on Shank3. Consistent with this, size-exclusion chromatography showed that some ABI1 and WAVE1 were present in the same high-molecular-weight fraction (fraction 8) as the Shank3-GKAP-PSD-95 complex (Figure 4d).
To test if the Shank3-ABI1 interaction is critical for actin nucleation, we stained F-actin in rat primary hippocampal neurons overexpressing either WT or S685I Shank3. Overexpression of WT Shank3 greatly increased F-actin intensity in dendritic spines, but overexpression of the ABI1 binding-deficient S685I Shank3 had a minimum if any effect on F-actin intensity (Figure 4e,f). Together, these results define ABI1-dependent actin nucleation as a module of Shank3 function.
Shank3 recruits WRC to the PSD via ABI1 to promote spine maturation
Finally, we investigated the molecular mechanism by which Shank3-ABI1 interaction promotes actin nucleation in dendritic spines. Because ABI1 is the only subunit of the WRC that has previously been shown to directly bind Shank3 10, we hypothesized that it mediates the interaction between Shank3 and WAVE/WRC. To test this possibility, we performed co-immunoprecipitation assays between Shank3 and WAVE1 in HEK293T cells. Consistent with our hypothesis, S685I Shank3, which disables interaction with ABI1, also abolished Shank3 binding to WAVE1 (Figure 5a). In a complementary experiment, a WAVE1 mutant, whose binding to ABI1 was abolished by two amino acid changes 33, failed to interact with Shank3 (Figure 5b). We also tested the interaction between Shank3 and WAVE3, another WAVE family protein that is, like WAVE1, enriched in brain and localized at dendritic spines 47. Disruption of Shank3-ABI1 interaction also abolished Shank3 binding to WAVE3 (Figure 5c).
Figure 5.

Shank3 recruits WRC to the PSD via ABI1 to promote spine development. (a) Immunoblots of GFP-WAVE1 and HA-Shank3 following immunoprecipitation of HA-Shank3 variants in the co-immunoprecipitation assay in 293T cells. (b) Immunoblots of GFP-WAVE1 variants and HA-Shank3 following immunoprecipitation of HA-Shank3 in the co-immunoprecipitation assay in 293T cells. (c) Immunoblots of GFP-WAVE3 and HA-Shank3 following immunoprecipitation of HA-Shank3 variants in the co-immunoprecipitation assay in 293T cells. (d) Immunoblots following PSD fractionation using WT or Shank3B KO mice striatal lysate. PSD-95 and Shank3 were used as positive controls for each fraction. S1, supernatant 1; P2’, crude synaptosomal fraction; P3, membrane fraction; SPM, synaptic plasma membrane; PSD I, SPM after one-time Triton X-100 washout. (e) Quantifications of immunoblots in d (n=3 experiments). (f) Immunoblots following PSD fractionation using WT or KI-HOM mice striatal lysate. (g) Quantifications of immunoblots in f (n=3 experiments). (h) Fluorescent images of dendrites from DIV 14 dissociated mouse hippocampal neurons co-transfected with siRNAs (scramble or against mouse Wave1 and Wave3), pEGFP-C1 plasmids and vectors expressing HA-Shank3 (empty or WT) on DIV 7. Scale bar, 5μm; OE, overexpression. (i) Quantification of dendritic spine densities in h (n=16, 23, 24, 30 neurons). All data are presented as mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, NS, not significant; (e,g) paired two-tailed Student’s t-test; (i) one-way ANOVA with post hoc Tukey’s tests.
Given that ABI1 mediates Shank3-WRC interaction, it is conceivable that Shank3 promotes F-actin formation in dendritic spines by recruiting ABI1 and its associated WRC into spines to activate Arp2/3. To test this possibility, we overexpressed Shank3 in rat hippocampal neurons and stained endogenous ABI1 to quantify its localization. Contrary to our hypothesis, Shank3 overexpression did not change the enrichment of ABI1 in dendritic spines (Supplementary Figure 7a,b). The spine localization of ABI1 was also preserved in Shank3B KO neurons (Supplementary Figure 7c,d). These results indicate that ABI1 localization to spines might be independent of Shank3.
Since both Shank3 and ABI1 are expressed at the tip of the spine in the post-synaptic density or PSD, which occupies ~10% of the surface area of spine 48, Shank3 could interact with, recruit, and stabilize ABI1 and WRC to the PSD from other parts of the dendritic spine. Indeed, in the striatal PSD fraction from Shank3B KO mice, but not in the total homogenate, the abundance of ABI1 was dramatically lower than in WT (Figure 5d,e). WAVE1 was also lower in the PSD of Shank3B KO mice. We noticed that PSD-95 abundance was also slightly reduced, which implies a change in the PSD architecture upon loss of Shank3. Thus, it is possible that reduced WRC in the PSD is caused by some secondary effects other than loss of Shank3-ABI1 interaction. To rule out this possibility, we performed PSD fractionation on striatal tissues from KI-HOM mice. Again, we found that ABI1 and WAVE1 abundances were reduced in the PSD, but not in the total homogenate of KI-HOM mice (Figure 5f,g). Note that both Shank3 and PSD-95 abundances in the PSD fraction of KI-HOM mice remain unchanged, indicating their proper localization and intact PSD organization. These results demonstrate that recruitment of WRC to the PSD requires Shank3-ABI1 interaction.
To determine if WRC-dependent actin nucleation mediates the effect of Shank3 on spine maturation, we either overexpressed Shank3, depleted Wave1/Wave3 using siRNAs (Supplementary Figure 7e,f), or combined the two in dissociated hippocampal neurons and then analyzed the morphology of spines. Similar to Abi1/Abi2 depletion, Wave1/Wave3 depletion completely abolished the effect of Shank3 overexpression (Figure 5h,i and Supplementary Figure 7g), demonstrating that WAVE proteins are also critical downstream effectors for Shank3.
DISCUSSION
This study was initiated to identify critical protein interactions and downstream effectors of Shank3 for its function at the synapse. By combining protein interaction domain data, post-translational modification data, and ASD exome sequencing data, we discovered a residue of Shank3 critical for its interaction with ABI1 that is mutated in autism. We further validated ABI1/WRC as a bona fide, direct downstream effector of Shank3 in vivo. Shank3 recruits WRC to the PSD through phosphorylation-dependent interaction with ABI1 to facilitate actin nucleation and spine development, disruption of which impairs synaptic transmission and multiple social behaviors (Supplementary Figure 7h). The most critical finding is that the S685I mutation, which specifically disables Shank3 to promote actin nucleation, only causes a subset of phenotypes found in constitutive Shank3 KO condition. This sort of functional and phenotypic modularity was unexpected.
Phosphorylation-dependent regulation of Shank3
We explored Shank3 phosphorylation pattern in vivo and found that it is extensively phosphorylated at dozens of residues. As phosphorylation can rapidly modulate a protein’s function by altering its stability, subcellular localization, or protein interactions, our work provides a valuable resource for future studies on the post-translational regulation of Shank3 in spine remodeling and synaptic plasticity. It is worth mentioning that the phosphorylation sites were identified on the full-length isoform of Shank3. Whether other Shank3 isoforms are phosphorylated on the same residues requires further investigation.
In this study, we focused on one specific in vivo phosphorylation site S685, because of its importance for the Shank3-ABI interaction. Because this interaction is essential for Shank3 function and S685A Shank3 failed to promote mature spine formation, S685 phosphorylation might serve as a mechanism to finely tune Shank3 function in actin nucleation at the PSD during spine maturation. We showed that PKA phosphorylates S685 and that inhibition of PKA decreased Shank3-ABI interaction in neurons. The involvement of PKA in Shank3 regulation is interesting, as short-term activation of PKA increases both spine size and spine density in hippocampal neurons 49,50. Our data suggest that Shank3 phosphorylation at S685 facilitate WRC recruitment and rapid actin cytoskeleton reorganization in dendritic spines upon activation of PKA signaling.
Shank3 as a hub for actin nucleation necessary for spine development
The role of Shank3 in spine development is well-known. Although Shank3 has long been proposed to regulate actin 51, whether Shank3 promotes spine development through regulating actin cytoskeleton has not been established, especially in vivo. We show that ABI and WAVE, both known to primarily function as actin nucleation regulators, are downstream effectors of Shank3 in spine development. Moreover, the S685I mutation, which specifically disables Shank3 for actin nucleation while does not affect its other interactions, abundance or PSD localization, still causes spine defects in vivo. Together, these results demonstrate that Shank3 function in spine development largely relies on its actin nucleation module. Based on our integrated interactome analysis, Shank3 might serve as a hub in the deeper layer of the PSD, bringing together NPFs and Arp2/3 to cooperatively facilitate actin nucleation.
The involvement of Shank3 in actin nucleation and the association of the S685I variant with autism indicate a critical role of actin nucleation in the pathogenesis of SHANK3 deficiency. Beyond that, dysregulation of actin nucleation could be a general mechanism underlying autism spectrum disorders. Supporting this idea, de novo mutations in NCKAP1 and ABI2, which encode WRC subunits, have been linked to autism 52. Moreover, copy number variation of CYFIP1, which encodes another WRC subunit, has been linked to both autism spectrum disorder and schizophrenia 53–55. Further human genetic studies to explore genes critical for actin dynamics might prove fruitful in identifying new etiologies for autism or other neurodevelopmental disorders. Detailed studies of how variation in these genes contribute to pathology will undoubtedly provide insights into not only mechanisms of the disease, but also the regulation of actin dynamics and its role in synaptogenesis.
The S685I missense mutation demonstrates the modularity of Shank3 function
While displaying increased social dominance, increased allogrooming, and delayed USV development, our Shank3 KI mice do not express all the phenotypes reported in Shank3 KO mice. For example, motor defects, increased anxiety, abnormal self-grooming behaviors, and altered sensory gating that were repeatedly reported in Shank3 KO lines 15,20,21, are intact in KI mice, suggesting that they are mediated by other protein interactions/pathways. These results are consistent with previous findings that targeting Shank3 at different domains leads to different sets of physiological and behavioral abnormalities 23,25–27. This modularity further indicates that there is more than one independent pathogenic pathway downstream of Shank3 and correcting a single downstream pathway is unlikely to be sufficient for clear clinical improvement. An example similar to this is Rett syndrome where targeting the downstream dysregulated BDNF through IGF-1 only partially rescued Rett phenotypes 56. Thus, rather than attempting to correct downstream deficits, it might be more fruitful to identify upstream regulators of dosage-dependent genes in neuropsychiatric disorders for therapeutic targeting.
Our study also highlights the value of missense variants in understanding the pathogenesis of neuropsychiatric disorders. Most mutations found in genome sequencing studies are missense mutations of unknown significance. While they are often less informative for identifying new neuropsychiatric disease-causing genes, missense mutations could lead to a greater understanding of protein functions that are linked to the disease. The difficulty lies in distinguishing informative missense mutations from non-pathogenic ones. Our strategy was to map missense variants onto post-translational modification sites and protein interaction domains to identify potentially critical residues. This approach could be used for other neuropsychiatric disease-causing genes to gain additional insight into mechanisms of the disease.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Gabriele Schuster for injection of Shank3 BAC; Yousuf O. Ali and Hui-Chen Lu for technical input on primary neuron culture; Antrix Jain and Sung Y. Jung for their help in mass spectrometry; Dinghui Yu for his help in imaging data analysis; Vicky Brandt, Aya Ito-Ishida, Callison E. Alcott, and Vincenzo A. Gennarino for editorial input. This work was supported by the Howard Hughes Medical Institute (to H.Y.Z.), a gift from Mr. Charif Souki (to the Jan and Dan Duncan Neurological Research Institute), a grant (1R01GM097207) from the National Institute of Health (to P.B.), and a supercomputer allocation (MCB120101) from XSEDE (to P.B.). The work was also supported in part by IDDRC grant number 1U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. IDDRC Neurobehavior and Neurovisualization cores were used for this project. L.W. was supported by a predoctoral fellowship from Autism Speaks (#9120). J.L.H. was supported by an NINDS K08 Award (NS091381), the Robbins Foundation, Baylor Pediatrics Pilot Award, as well as Chao Physician-Scientist Award.
Footnotes
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
REFERENCE
- 1.Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A Gradient of Severity in Cognitive Impairments. PLoS Genet 2014; 10: e1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet 2007; 81: 1289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007; 39: 25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu L, Wang X, Li X-L, Towers A, Cao X, Wang P et al. Epigenetic Dysregulation of SHANK3 in Brain Tissues from Individuals with Autism Spectrum Disorders. Hum Mol Genet 2013; : 1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cochoy DM, Kolevzon A, Kajiwara Y, Schoen M, Pascual-Lucas M, Lurie S et al. Phenotypic and functional analysis of SHANK3 stop mutations identified in individuals with ASD and/or ID. Mol Autism 2015; 6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol Syndromol 2012; 2: 186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naisbitt S, Eunjoon K, Tu JC, Xiao B, Sala C, Valtschanoff J et al. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 1999; 23: 569–582. [DOI] [PubMed] [Google Scholar]
- 8.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 1999; 23: 583–92. [DOI] [PubMed] [Google Scholar]
- 9.Bockmann J, Kreutz MR, Gundelfinger ED, Böckers TM. ProSAP/Shank postsynaptic density proteins interact with insulin receptor tyrosine kinase substrate IRSp53. J Neurochem 2002; 83: 1013–7. [DOI] [PubMed] [Google Scholar]
- 10.Proepper C, Johannsen S, Liebau S, Dahl J, Vaida B, Bockmann J et al. Abelson interacting protein 1 (Abi-1) is essential for dendrite morphogenesis and synapse formation. EMBO J 2007; 26: 1397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N. The complexity of PDZ domain-mediated interactions at glutamatergic synapses: A case study on neuroligin. Neuropharmacology 2004; 47: 724–733. [DOI] [PubMed] [Google Scholar]
- 12.Grabrucker AM, Knight MJ, Proepper C, Bockmann J, Joubert M, Rowan M et al. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J 2011; 30: 569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011; 472: 437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, McCoy P a, Rodriguiz RM, Pan Y, Je HS, Roberts AC et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet 2011; 20: 3093–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK et al. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun 2016; 7: 11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism 2010; 1: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han K, Holder JL Jr, Schaaf CP, Lu H-C, Chen H, Kang H et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 2013; 503: 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF et al. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci 2005; 25: 3560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry 2012; 17: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mei Y, Monteiro P, Zhou Y, Kim J-A, Gao X, Fu Z et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 2016; 530: 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 2016; 89: 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE et al. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J Neurosci 2012; 32: 14966–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kouser M, Speed HE, Dewey CM, Reimers JM, Widman AJ, Gupta N et al. Loss of predominant shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J Neurosci 2013; 33: 18448–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verpelli C, Dvoretskova E, Vicidomini C, Rossi F, Chiappalone M, Schoen M et al. Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J Biol Chem 2011; 286: 34839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Speed HE, Kouser M, Xuan Z, Reimers JM, Ochoa CF, Gupta N et al. Autism-Associated Insertion Mutation ( InsG ) of Shank3 Exon 21 Causes Impaired Synaptic Transmission and Behavioral Deficits. 2015; 35: 9648–9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Escamilla CO, Weaver TP et al. Novel Shank3 mutant exhibits behaviors with face validity for autism and altered striatal and hippocampal function. Autism Res 2017; 10: 42–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Liu S, Powell CM. Altered Striatal Synaptic Function and Abnormal Behaviour in Shank3 Exon4–9 Deletion Mouse Model of Autism. Autism Res 2016; 9: 350–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vicidomini C, Ponzoni L, Lim D, Schmeisser MJ, Reim D, Morello N et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol Psychiatry 2017; 22: 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bidinosti M, Botta P, Kruttner S, Proenca CC, Stoehr N, Bernhard M et al. CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science 2016; 351: 1199–203. [DOI] [PubMed] [Google Scholar]
- 30.Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 2016; 352: aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep 2015; : 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas GM, Rumbaugh GR, Harrar DB, Huganir RL. Ribosomal S6 kinase 2 interacts with and phosphorylates PDZ domain-containing proteins and regulates AMPA receptor transmission. Proc Natl Acad Sci U S A 2005; 102: 15006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM et al. Structure and control of the actin regulatory WAVE complex. Nature 2010; 468: 533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem 1991; 266: 15555–15558. [PubMed] [Google Scholar]
- 35.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lek M, Karczewski KJ, Minikel EV., Samocha KE, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 39.Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P et al. Deletion of Mecp2 in Sim1-Expressing Neurons Reveals a Critical Role for MeCP2 in Feeding Behavior, Aggression, and the Response to Stress. Neuron 2008; 59: 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brodkin ES, Goforth SA, Keene AH, Fossella JA, Silver LM. Identification of quantitative trait Loci that affect aggressive behavior in mice. J Neurosci 2002; 22: 1165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang F, Kessels HW, Hu H. The mouse that roared: Neural mechanisms of social hierarchy. Trends Neurosci 2014; 37: 674–682. [DOI] [PubMed] [Google Scholar]
- 42.Wang F, Zhu J, Zhu H, Zhang Q, Lin Z, Hu H. Bidirectional control of social hierarchy by synaptic efficacy in medial prefrontal cortex. Science 2011; 334: 693–7. [DOI] [PubMed] [Google Scholar]
- 43.Hamilton SM, Spencer CM, Harrison WR, Yuva-Paylor LA, Graham DF, Daza RAM et al. Multiple autism-like behaviors in a novel transgenic mouse model. Behav Brain Res 2011; 218: 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grove M, Demyanenko G, Echarri A, Zipfel PA, Quiroz ME, Rodriguiz RM et al. Abi2-Deficient Mice Exhibit Defective Cell Migration, Aberrant Dendritic Spine Morphogenesis, and Deficits in Learning and Memory. Mol Cell Biol 2004; 24: 10905–10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Söding J Protein homology detection by HMM-HMM comparison. Bioinformatics 2005; 21: 951–960. [DOI] [PubMed] [Google Scholar]
- 46.Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol 2006; 7: 713–26. [DOI] [PubMed] [Google Scholar]
- 47.Pilpel Y, Segal M. Rapid WAVE dynamics in dendritic spines of cultured hippocampal neurons is mediated by actin polymerization. J Neurochem 2005; 95: 1401–1410. [DOI] [PubMed] [Google Scholar]
- 48.Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2001; 2: 880–888. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Pöschel B, Faul C, Upreti C, Stanton PK, Mundel P. Essential role for synaptopodin in dendritic spine plasticity of the developing hippocampus. J Neurosci 2013; 33: 12510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu Y, Zha XM, Kim EY, Schachtele S, Dailey ME, Hall DD et al. A Kinase Anchor Protein 150 (AKAP150)-associated protein kinase A limits dendritic spine density. J Biol Chem 2011; 286: 26496–26506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sarowar T, Grabrucker AM. Actin-Dependent Alterations of Dendritic Spine Morphology in Shankopathies. Neural Plast 2016; 2016: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014; 515: 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stefansson H, Rujescu D, Cichon S, Pietiläinen OPH, Ingason A, Steinberg S et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008; 455: 232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet 2012; 8: e1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tam GWC, van de Lagemaat LN, Redon R, Strathdee KE, Croning MDR, Malloy MP et al. Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochem Soc Trans 2010; 38: 445–51. [DOI] [PubMed] [Google Scholar]
- 56.Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci 2009; 106: 2029–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 2005; 33: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci 1996; 16: 5425–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sakai Y, Shaw C a, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE et al. Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med 2011; 3: 86ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han K, Kim MH, Seeburg D, Seo J, Verpelli C, Han S et al. Regulated RalBP1 binding to RalA and PSD-95 controls AMPA receptor endocytosis and LTD. PLoS Biol 2009; 7: e100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B et al. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010; 26: 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R. Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes, Brain Behav 2005; 4: 420–430. [DOI] [PubMed] [Google Scholar]
- 63.Janz JM, Sakmar TP, Min KC. A novel interaction between atrophin-interacting protein 4 and β-p21-activated kinase-interactive exchange factor is mediated by an SH3 domain. J Biol Chem 2007; 282: 28893–28903. [DOI] [PubMed] [Google Scholar]
- 64.Ghose R, Shekhtman A, Goger MJ, Ji H, Cowburn D. A novel, specific interaction involving the Csk SH3 domain and its natural ligand. Nat Struct Biol 2001; 8: 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Corominas R, Yang X, Lin GN, Kang S, Shen Y, Ghamsari L et al. Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism. Nat Commun 2014; 5: 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Razick S, Magklaras G, Donaldson IM. iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics 2008; 9: 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bayés A, van de Lagemaat LN, Collins MO, Croning MDR, Whittle IR, Choudhary JS et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci 2011; 14: 19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bayés À, Collins MO, Croning MDR, van de Lagemaat LN, Choudhary JS, Grant SGN. Comparative Study of Human and Mouse Postsynaptic Proteomes Finds High Compositional Conservation and Abundance Differences for Key Synaptic Proteins. PLoS One 2012; 7: e46683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2005; 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate : A Practical and Powerful Approach to Multiple Testing. J R Stat Soc 1995; 57: 289–300. [Google Scholar]
- 71.Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD et al. ATAXIN-1 Interacts with the Repressor Capicua in Its Native Complex to Cause SCA1 Neuropathology. Cell 2006; 127: 1335–1347. [DOI] [PubMed] [Google Scholar]
- 72.Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008; 452: 713–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Um K, Niu S, Duman JG, Cheng JX, Tu YK, Schwechter B et al. Dynamic Control of Excitatory Synapse Development by a Rac1 GEF/GAP Regulatory Complex. Dev Cell 2014; 29: 701–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.