

Abstract
Bipolar Disorder (BD) is a common mood disorder characterized by recurrent episodes of mania and depression. Both genetic and environmental factors have been implicated in BD etiology, but the biological underpinnings remain elusive. Recently, genome-wide association studies (GWAS) of neuropsychiatric disorders have identified a risk locus for BD containing the SYNE1 gene, a large gene encoding multiple proteins. The BD association signal spans, almost exclusively, the part of SYNE1 encoding CPG2, a brain-specific protein localized to excitatory postsynaptic sites, where it regulates glutamate receptor internalization. Here we show that CPG2 protein levels are significantly decreased in postmortem brain tissue from BD patients, as compared to control subjects, as well as schizophrenia and depression patients. We identify genetic variants within the postmortem brains that map to the CPG2 promoter region, and show that they negatively affect gene expression. We also identify missense single nucleotide polymorphisms (SNPs) in CPG2 coding regions that affect CPG2 expression, localization, and synaptic function. Our findings link genetic variation in the CPG2 region of SYNE1 with a mechanism for glutamatergic synapse dysfunction that could underlie susceptibility to BD in some individuals. Few GWAS hits in human genetics for neuropsychiatric disorders to date have afforded such mechanistic clues. Further, the potential for genetic distinction of susceptibility to BD from other neuropsychiatric disorders with overlapping clinical traits holds promise for improved diagnostics and treatment of this devastating illness.
INTRODUCTION
Bipolar disorder (BD) is a common, chronic mood disorder characterized by recurrent episodes of mania and depression. The lifetime prevalence is estimated at 1–3% of the population1 and high mortality rates2, mainly caused by suicide3, makes BD a major public health problem. Treatment for BD is limited, consisting mainly of pharmaceutical mood stabilizers, antidepressants, and antipsychotic drugs discovered decades ago. Their efficacy in only a subset of BD patients highlights the need for development of new drugs based on a molecular understanding of disease etiology4, 5.
The neuropsychiatric disorders BD, schizophrenia and major depression, as well as other neurodevelopmental disorders such as autism spectrum disorder (ASD) and attention deficit hyperactivity disorder (ADHD), substantially overlap in clinical traits6. For example, many BD patients suffer from cognitive deficits and psychotic symptoms qualitatively resembling those of schizophrenia patients7, and from depressed mood states resembling those of major depression patients. For this reason, precise diagnosing often requires extensive psychiatric evaluation based on clusters of symptoms6, and in some cases even erroneous pharmacological treatment attempts8. Although there is a wide consensus for differential brain structural and connectivity impairments9–11, there is little specific evidence describing neuronal substrates and mechanisms differentiating neuropsychiatric disorders at the cellular level. Consequently, in the absence of more conclusive biological markers, diagnosing is almost exclusively based on psychiatric evaluation12.
Despite robust evidence of genetic susceptibility to BD13, with heritability estimated as high as 70–80% based on twin studies13–16, only a few genetic susceptibility factors have been identified over decades of research13, 17, with little evidence for BD-specific risk genes17. Joint genome-wide association studies (GWAS) show substantial commonalities in risk loci for the major psychiatric disorders, especially between BD and schizophrenia18–20, suggesting they overlap not only in clinical symptoms but also in their contributing genetic factors20–22. A handful of common genetic variants, identified by GWASs as robustly associated with BD and replicated across independent studies, are single-nucleotide polymorphisms (SNPs) in the genes CACNA1C, ANK3, ODZ4, SYNE1, and TRANK123–28. Some of the identified loci show penetrance for two or more of the neuropsychiatric disorders, underlining a shared genetic etiology25, 29–31. The largest GWAS to date for identifying risk loci for neuropsychiatric disorders including BD, schizophrenia, and major depression, identified the region encompassing SYNE1 as the strongest BD association locus in the genome32. Meta-analyses included in the study identified SNPs in SYNE1 with genome-wide statistically significant association to BD at P=4.27×10−9 25–27, 32, 33.
Increasing evidence, mainly from genetic and pharmacological studies, has implicated abnormal glutamatergic neurotransmission and synaptic plasticity in the etiology of BD24, 25, 34–36. Notably, SNPs in GRIA2, which encodes the GluA2 subunit of AMPARs, have been associated with time to recurrence of mood episodes in BD patients37. Studies have also shown differences in glutamate levels as well as glutamate receptor expression or function between individuals with mood disorders and control subjects38–42.
SYNE1 has thus far drawn less attention in relation to neuropsychiatric disorders compared to e.g. CACNA1C, which encodes voltage-gated calcium channels well-known to play a role in synaptic functions29. Human SYNE1 is a large gene comprising 145 exons with multiple transcripts43. The most described gene products of SYNE1, the nesprins, are mainly related to muscle function44, 45, and are unlikely to explain how this locus influences susceptibility to BD. Interestingly, the BD association signal in SYNE1 maps near the transcription start site for candidate plasticity gene 2 (CPG2). CPGs are activity-regulated genes identified as potential mediators of synaptic plasticity46. CPG2 is a brain-specific transcript of the SYNE1 gene. We recently reported the existence of two human CPG2 transcripts expressed in neocortex, hippocampus, and striatum, encoding proteins that localize to the postsynaptic endocytic zone of excitatory synapses in dendritic spines43, 47. Here they facilitate glutamate receptor cycling, consistent with a role in synaptic plasticity. The identification of CPG2 as a risk locus for BD and CPG2’s known function in regulating glutamate receptor internalization, suggest that variation in CPG2 function may affect glutamate receptor cycling in a way that would influence susceptibility to BD.
Here, we combine multiple strategies to identify genetic variations within the CPG2 locus of SYNE1 that influence expression or function of the CPG2 protein. Linking BD associated variation with an underlying cellular dysfunction is an opportunity afforded by only a few GWAS hits in human genetics for neuropsychiatric disorders to date. We show that CPG2 protein levels are significantly decreased in postmortem brain tissue of BD patients as compared to schizophrenia and depression patients, as well as control subjects. By deep-sequencing the CPG2 region of SYNE1 from the same human subjects, we identified genetic variants within promoter and enhancer regions that negatively affect gene expression. We further show that certain genetic variants in the CPG2 coding region identified by exome sequencing affect CPG2 expression, subcellular localization, and synaptic function.
MATERIALS AND METHODS
Human brain tissue
Fresh frozen human brain tissue samples from BA9/10 and hippocampus were kindly provided by the Harvard Brain Tissue Resource Center, the Stanley Medical Research Institute Neuropathology Consortium collection, Mount Sinai NIH Brain and Tissue Repository (schizophrenia patients), University of Maryland Brain and Tissue Bank (controls), and Massachusetts Alzheimer’s Disease Research Center (controls). Information on age, postmortem index (PMI), gender, partial information on medication history, and relative CPG2, Arc and PSD95 protein expression levels is summarized in supplementary table ST1. Informed consent was obtained from all tissue donors or legal signatories.
Western blotting
Protein extraction and Western blotting were performed as described previously43. Blots were incubated with guinea pig polyclonal anti-CPG2 (1:1000, A002396; np913, NEO Peptide), mouse anti-Arc (1:1000; Synaptic Systems), mouse anti-PSD95 (1:20,000; UC Davis) or mouse anti-β-actin (1:2000; SIGMA) antibodies and visualized using an Odyssey infrared imaging system (LI-COR). Western blot signal intensities were quantified using a FIJI (ImageJ) gel lane plot profile tool and groups were compared using one-way ANOVA and Tukey’s post hoc test for multiple comparisons or t-tests (two groups). Sample lysates were loaded in a randomized manner and quantified blind to sample identity. For SMRI samples, subject identifying information was revealed to the experimenter only after submission of the final dataset to the SMRI consortium. One sample was below detection level and excluded from the protein level analyses. The Western blots for CPG2 showed two bands that were quantified in combination due to insufficient signal segregation and apparent co-regulation.
Targeted region deep-sequencing and variant calling
Genomic DNA was extracted from human brain tissue using a ChargeSwitch gDNA Mini Tissue Kit (Invitrogen) and purified by ethanol precipitation. High-quality gDNA samples were deep-sequenced in the extended CPG2 region of SYNE1 (NM_182961: exon 8–70 incl. introns) by CD Genomics (sequencing depth: 30–100x).
The raw sequencing data was mapped to human genome assembly GRCh37 using BWA aligner48. The BAM-files were indexed and the mapped reads within the SYNE1 gene locus (chr6:152740704–152831506) were selected using the SAMtools view module49. The generated SYNE1 BAM-files were piled up using SAMtools mpileup module for union genotype calling using BCFtools with –mv and –Oz options50. Variant annotation was performed using ANNOVAR tool51. Human brain region-specific chromatin states were collected52 and visualized using the web-based NeuVar tool http://bioinfo5pilm46.mit.edu:318/neuvar. Statistical correlation of genetic variants with CPG2 protein expression was tested using Mann-Whitney binary tests.
Molecular cloning
Putative promoter DNA regions from purified control or patient gDNA samples were amplified using LongAmp Taq polymerase and proofreading Q5 polymerase (NEB). PCR products from promoter regions were separated on agarose gels, excised, purified and cloned into the pGL3-Basic vector (Promega) in front of the Firefly Luciferase (Luc+) gene using KpnI and XhoI restriction sites.
For rescue experiments in the absence or presence of CPG2 coding variants, full-length human CPG2 (hCPG2) was cloned into a lentiviral transfer vector for rat Cpg2 KD (pFUGW-Cpg2-shRNA), in which the stop codon after GFP was removed and hCPG2 inserted using EcoRI and XhoI restriction sites yielding a GFP-hCPG2 fusion protein (pFUGW-GFP-hCPG2)43. Human CPG2 coding variants were introduced into pFUGW-GFP-hCPG2 by site-directed mutagenesis. All constructs were validated by Sanger sequencing.
Neuronal cultures
Rat cortical or hippocampal cultures were prepared as described previously43, 53.
Luciferase assay
At 8 DIV, cortical neurons were transfected by calcium phosphate precipitation for 1 hour with 1µg of various Firefly Luciferase plasmids and 1.2µg of the normalization control pRL-TK Renilla Luciferase (RLuc) vector plasmid (Promega)54. At 14 DIV, neurons were lysed and assessed using a Dual-Luciferase® Reporter Assay System (Promega) according to the manufacturer’s protocol. Plates were read on an EnSpire® Multimode Plate Reader (PerkinElmer). Where indicated, neurons were activated with KCl stimulation solution (170mM KCl, 2mM CaCl2, 1mM MgCl2, 10mM HEPES (pH 8.3)) diluted in culture media 1:6 for final concentrations of 28mM KCl and 50µM picrotoxin for 6 hours before cells were lysed and assayed. Relative Luciferase activity was statistically compared using one-way or two-way (KCl+PcTx) ANOVAs and Tukey’s post hoc tests for multiple comparisons.
Spine localization assay
At 8 DIV, neurons were infected with lentivirus for KD of rat Cpg2 (pFUGW-Cpg2-shRNA) or at 15 DIV with lentivirus for replacement of the endogenous rat CPG2 with the GFP-hCPG2 fusion reference protein (pFUGW-GFP-hCPG2) or mutated variants. At 21 DIV cells were fixed in 4% formaldehyde for 15 min and then permeabilized for 25 min with 0.2% saponin in PBS with 10% goat serum. Cells were then incubated for 3 h with primary antibodies; mouse anti-CPG2 (1:500; Nedivi lab) and rabbit anti-GFP (1:3000; Abcam), followed by 1 h incubation with secondary antibodies; goat anti-mouse IgG-Alexa Fluor 555 (1:500) and goat anti-rabbit IgG-Alexa Fluor 488 (1:500; Molecular Probes). Coverslips were mounted using Fluoromount-G (Southern Biotech) and imaged using a Nikon Eclipse E600 upright microscope with a 40×/1.40 Plan Apo oil immersion objective (Nikon).
Quantification of immunocytochemistry was conducted using ImageJ software to obtain pixel intensity values in a linear range within regions of interest (ROI). ROIs were positioned over spine heads and dendritic regions based on GFP-stained neuronal morphology and the corresponding staining intensity was measured. Spine localization of GFP-hCPG2 or mutated variants was quantified as the ratio between fluorescence intensity in spine heads and adjacent dendritic shafts. For quantification of spine size, a threshold was applied to make the fluorescence signal binary, and signal areas were quantified and calibrated to the 40X objective specific pixel size (µm2). All quantifications were done blind to sample identity. Statistical difference from controls was tested using one-way ANOVA and Dunnett’s post hoc test.
Surface receptor internalization assay
The receptor internalization assay was performed as described previously47 with the following modifications: Cortical cultures were infected at 8 DIV with lentivirus for CPG2 KD (pFUGW-Cpg2-shRNA) and at 10 DIV with lentivirus for GFP-hCPG2 (pFUGW-GFP-hCPG2) reference protein or mutated variant molecular replacement. Western blots were probed with rabbit anti-GluA2 (1:1000; Abcam), mouse anti-GluN1 (1:2000; Temicula) or mouse anti-TfR (1:1000; Invitrogen) primary antibodies and developed using the Odyssey infrared system (LI-COR). Receptor internalization was quantified as described47 and groups were compared using one-way ANOVA and Tukey’s post hoc test for multiple comparisons.
General statistics and data analysis
All statistical analyses were performed with GraphPad Prism software (version 7). The data is graphed as the mean ± standard error of the mean (SEM). Appropriate statistical tests were chosen based on the experimental conditions (see assay specific methods). Gaussian distribution of non-normalized data was assumed in all but the genetic variant analyses. Here, non-parametric tests were used as described. Brown-Forsythe and Bartlett’s tests were included in the ANOVAs assuring similar variance between statistically compared groups. Statistical significance was defined as *p ≤ 0.05. For cell culture experiments, poorly transfected cultures were excluded from data analysis. Test conditions were randomized across culture wells/plates.
RESULTS
Low CPG2 protein expression in brains of postmortem BD patients.
To investigate if the CPG2 region of SYNE1 could play a role in susceptibility for BD, we first asked whether CPG2 protein expression was compromised in BD patients. To this purpose, we isolated synaptic protein fractions from postmortem human brain tissue of control subjects, as well as BD, schizophrenia, and major depression patients (see supplementary table ST1 for source information). Since fMRI and postmortem studies have implicated the prefrontal cortex in BD55–58, we ran Western blots on tissue from Brodmann Area 9/10 (Fig. 1A), and compared CPG2 protein levels in the three patient groups with that of control subjects (Fig. 1B). We found CPG2 protein levels to be significantly lower in the BD group as compared to control subjects (One-way ANOVA: F(3,95)=2.74, *p=0.047; Tukey’s post hoc: control vs. BD *p=0.032, n=102 subjects) but not in other patient groups tested (Tukey’s post hoc: control vs. depression p=0.96, control vs. schizophrenia p=0.76) (Fig. 1C). The Western blots for CPG2 show two bands, which we speculate could represent protein products from different CPG2 transcripts, or could be a result of, as yet, unidentified posttranslational modifications. Both bands are expressed in the synaptic protein fraction, and their relative signal intensity is constant independent of patient group or other variables, suggesting that expression of the two protein products is regulated by the same mechanism either at the transcriptional, posttranscriptional, or posttranslational level.
Figure 1: CPG2 protein levels are reduced in postmortem brain tissue from BD patients.

A) Synaptic protein fractions were extracted from a total of 102 postmortem human brain tissue samples from Brodmann Area 9/10 (image adapted from commons.wikimedia.org). For SDS-PAGE, 20µg of total protein was loaded. B) Representative Western blots comparing CPG2, Arc, and PSD95 protein expression in controls, depression, schizophrenia, and BD patients. Arc is another glutamatergic synaptic protein that like CPG2 is the product of an activity-regulated gene, and the synaptic protein PSD95 serves as a positive marker of glutamatergic synapse presence. C-E) Quantification of CPG2, Arc, and PSD95 protein levels, respectively, shows that CPG2 levels are significantly lower in the BD patient population as compared to control subjects, whereas Arc is decreased in both schizophrenia and BD groups. Comparable PSD95 levels in all groups indicates that reduced CPG2 or Arc expression does not reflect synaptic loss. (*p<0.05, One-way ANOVAs, Tukey’s post hoc tests). F) Synaptic protein fractions were extracted from a total of 22 postmortem human brain tissue samples from hippocampus (image adapted from commons.wikimedia.org). G) Representative Western blots showing CPG2 protein expression in controls and BD patients. The synaptic marker protein PSD95 and β-actin serve as controls. H) Quantification of CPG2 protein expression. I) Quantification of PSD95 protein expression. J) Quantification of β-actin protein expression. K) Direct comparison of CPG2 protein expression in cortical and hippocampal tissue samples from individual subjects (black dots represent controls and red dots represent BD patients). As in neocortex, CPG2 protein expression is significantly decreased in hippocampal tissue from BD patients. (*p<0.05, Student’s t-tests).
Neuropsychiatric disorders are generally associated with lower activity in the prefrontal cortex10, 11, 59. To confirm that low levels of CPG2 were not secondary to reduced neural activity and a resulting reduction in levels of this activity-regulated transcript46, we tested for levels of another activity-regulated gene product, Arc, a protein that like CPG2 is localized to glutamatergic synapses60. We observed a positive correlation of CPG2 and Arc levels (Fig. S1A; linear reg.: F(1,95)=52.4, p<0.0001 and Fig. S1B). However, unlike CPG2, which was reduced exclusively in the BD population, Arc was significantly decreased also in other patient groups (One-way ANOVA: F(3,94)=4.13, **p=0.0085; Tukey’s post hoc: control vs. BD *p=0.020, control vs. schizophrenia *p=0.022, control vs. depression p=0.14, n=102 subjects) (Fig. 1D). Levels of the postsynaptic density protein PSD95, a marker of glutamatergic synapses, was similar among the patient groups and as compared to control subjects (One-way ANOVA: F(3,95)=0.55, p=0.65, n=102 subjects). The correlation between CPG2 and PSD95 levels was slight enough (Fig. S1C; linear reg.: F(1,96)=16.2, p=0.0001) to indicate that reduced CPG2 or Arc expression is not merely caused by synaptic loss in BD and/or Schizophrenia patients (Fig. 1E and Fig. S1D; One-way ANOVA: F(3,95)=4.51, **p=0.0083; Tukey’s post hoc: control vs. BD **p=0.0027, n=102 subjects). We did not observe significant correlation of Arc and PSD95 levels (Fig. S1E; linear reg.: F(1,95)=1.92, p=0.17 and Fig. S1F; One-way ANOVA: F(3,95)=4.83, **p=0.0036; Tukey’s post hoc: control vs. BD *p=0.012, control vs. schizophrenia **p=0.0017, n=102 subjects). Further, we found no statistically significant correlation between CPG2, Arc, or PSD95 protein expression and variation in patient age (linear reg.: F(1,97)=0.64, p=0.43; F(1,97)=0.16, p=0.69; F(1,97)=2.0, p=0.16, respectively), postmortem index (PMI) (linear reg.: F(1,97)=0.023, p=0.88; F(1,97)=1.2, p=0.28; F(1,97)=3.2, p=0.079, respectively) or gender (t-tests: t(97)=1.7, p=0.094; t(97)=1.8, p=0.073 and t(97)=0.52, p=0.61, respectively) (Fig. S2). Thus, our findings show a specific correlation between low CPG2 levels and incidence of BD that is not shared with schizophrenia or major depression patients.
To test if low CPG2 protein expression in BD patients was specific to prefrontal cortex, we also isolated synaptic protein fractions from postmortem hippocampal tissue (Fig. 1F). Western blotting showed significantly lower CPG2 protein levels in hippocampal tissue from BD patients compared to controls (t-test: t(21)=2.5, *p=0.021, n=22 subjects) (Fig. 1G-H), with no change in level of either of the two control proteins, PSD95 (t-test: t(21)=1.1, p=0.29), or the loading control β-actin (t-test: t(20)=0.15, p=0.88) (Fig. 1I-J). Cortical and hippocampal CPG2 protein levels were well correlated between tissue samples from the same individuals (Fig. 1K). This suggests that lower CPG2 protein levels is a feature common across different brain regions of BD patients.
Low CPG2 protein expression correlates with the presence of common genetic variants in CPG2 regulatory regions
Low CPG2 protein levels in the BD patient group could derive from several factors, including reduced CPG2 gene expression. We hypothesized that genetic variation associated with BD might affect regulatory elements of the gene that are important for transcription, such as promoter/enhancer regions, transcription factor binding sites, or in the CPG2 untranslated regions (UTRs). To test this hypothesis, we purified gDNA from all samples and performed targeted deep-sequencing in the SYNE1 region (NM_182961: exon 8–70 incl. introns) encompassing the CPG2 locus. Our sequencing data yielded hundreds of genetic variants within the CPG2 locus (see supplementary information, ST5), most of which were common variants also represented in population databases (1000 genomes, HapMap). Of all the identified variants, five SNPs were previously shown to be BD associated (rs452309661, rs774796062, rs937160126, rs21497233, and rs21500633; supplementary table ST2).
Active promoter and enhancer elements within the CPG2 gene region were mapped based on histone methylation states seen in ChIP-seq data from human cultured neurons and from human prefrontal cortex43, 52 (Fig. 2A). Interestingly, three of the BD associated SNPs (rs4523096, rs7747960 and rs9371601) map in or near the proposed CPG2 promoter region. The two other SNPs (rs214972 and rs215006) map near the 3’UTR of a short CPG2 isoform and near the 3’UTR end of a long CPG2 isoform, respectively43.
Figure 2. Human CPG2 variants identified by deep-sequencing.

Brain tissue gDNA was extracted from all patient and control subjects, and deep-sequenced in the CPG2 locus to identify genetic variants. Common BD associated variants identified from GWASs26, 33, 61, 62 were statistically tested for correlation with CPG2 protein expression levels. A) The genomic position (GRCh37 assembly) of five SNPs identified as BD associated by GWAS mapped onto the CPG2 region of SYNE1 (dark blue vertical bars represent exons) shown in the context of previously published ChIP-sequencing data from human neurons identifying active promoter (green) and enhancer (purple) regions43. The five SNPs are rs4523096 (green*), rs7747960 (red*), rs9371601 (blue*), rs214972 (yellow*) and rs215006 (grey*). B) The allele frequencies of the five BD associated SNPs were quantified for high and low CPG2 expression subjects. C) Six LD proxies (rs9478332, rs12055686, rs4343926, rs4318888, rs7771568, and rs6908747) for the five BD SNPs map to the CPG2 TSS flanking region (color-matched to origin SNPs). D) The LD allele frequencies of the six SNP proxies were quantified for high and low CPG2 expression subjects. Four alleles (rs4523096[T], rs7747960[T], rs9478332[T] and rs4343926[C]) were trending towards higher allele frequency in low expressing subjects. E) The frequency of having at least one of the four non-reference alleles was compared between high and low CPG2 expression subjects, and F) between BD patients with low CPG2 and control subjects (Mann-Whitney binary tests). Note: rs4523096[T] and rs4343926[C] have high allele frequencies (>0.4) and were quantified for homozygous subjects in E, F and G. G) CPG2 protein expression levels (from figure 1) are displayed on a continuum from low (red) to high (blue) expression, where each colored bar represents one subject and each of the four identified variants enriched in the low CPG2 population shown as dark grey bars. The threshold between high and low CPG2 expression (dashed red line) was defined at the mean CPG2 protein expression level of the BD group as displayed in Figure 1C.
To quantify whether the identified BD associated SNPs correlate with low CPG2 expression, we defined mean CPG2 expression in the BD group as the threshold between high and low expression subjects. Within our limited sample set, we did not find statistically significant overrepresentation of any single SNP allele in the low CPG2 expression subjects when compared to the high CPG2 expression subjects (Fig. 2B). However, the BD associated alleles of the two SNPs (rs4523096[T] and rs7747960[T]) closest to the CPG2 transcription start site (TSS) trended towards higher frequency in the low, as compared to high, CPG2 expression subjects. This prompted us to focus on the locus encompassing the CPG2 TSS region. From our sequence data, we identified ~25 genetic variants within this region (supplementary table S3). Based on allele frequencies from the 1000 Genomes database, we found that six SNPs (rs9478332, rs12055686, rs4343926, rs4318888, rs7771568, and rs6908747) within the CPG2 TSS flanking region were in linkage disequilibrium (LD) with BD associated SNPs (D’=~1, r2=>0.8) (Fig. 2C, and supplementary table ST2). One of the six SNPs, rs9478332 was also found in the UCSC database of annotated transcription factor binding sites based on ENCODE ChIP-seq data52, and mapped within a HFH-1 consensus motif upstream of the annotated CPG2 TSS.
We then tested whether any of the six CPG2 TSS flanking region SNPs in LD with the BD associated SNPs had alleles overrepresented in our low CPG2 expression subjects (Fig. 2D). Two of the non-reference alleles, rs9478332[T] and rs4343926[C]), trended towards higher frequency within our limited pool of low CPG2 expression subjects, when compared to high CPG2 expression subjects. When considering all four non-reference allele SNPs with higher frequency in low versus high CPG2 expression subjects, (rs4523096[T], rs7747960[T], rs9478332[T] and rs4343926[C]), we found a trend for low CPG2 expression subjects to have at least one, or more, of the four alleles (Mann-Whitney test: U=942, p=0.058, n=60 (any of the four alleles) of 102 subjects) (Fig. 2E). Furthermore, ~75% of the low CPG2 expression BD subjects had at least one of the identified alleles, as compared to ~50% of subjects in the control group (Mann-Whitney test: U=195, p=0.068, n=36 (any of the four alleles) of 50 subjects)) (Fig. 2F).
The representation of the non-reference alleles with higher frequency in low CPG2 expression subjects is illustrated in Figure 2G for all patient groups. Here, CPG2 protein expression for individual subjects in the different patient groups is displayed on a continuum ranging from low to high expression, where each colored bar represents CPG2 levels in a single subject. Grey bars illustrate the presence of each of the four selected alleles in individual subjects. The dashed line demarcates the threshold between low and high CPG2 expression (as defined above, mean CPG2 expression level for the BD group). Our data suggests that no single allele is associated with the low CPG2 protein expression found in the BD patient subjects. Nonetheless, the presence of a handful of common alleles in or near the proposed CPG2 promoter region might correlate with low CPG2 protein expression in a larger sample set and could potentially mark BD susceptibility loci within the gene.
Non-reference SNP alleles within the CPG2 promoter region and their effects on gene expression.
To examine whether genetic variants in CPG2 regulatory loci can influence transcription, we first functionally mapped CPG2 regulatory domains. We cloned putative promoter and enhancer regions from human brain gDNA samples based on genomic mapping of the CPG2 locus in SYNE1 (Fig. 3A), and tested their ability to drive expression of a Luciferase reporter gene in cultured cortical neurons. When plasmids, containing potential CPG2 promoter regions cloned in front of the Luciferase gene, were transfected into primary neurons, we found a ~2kb region encompassing the CPG2 TSS with robust promoter activity (Figs. 3B and S3). Subsequent deletion analysis on this fragment resolved two regions with promoter activity: One within the CPG2 5’UTR, and one within the SYNE1 intron before exon 16 (i16), both downstream of the predicted TSS (One-way ANOVA: F(12,188)=167, ****p<0.0001, Tukey’s post hoc tests, ****p<0.0001, n=6–15 transfections, 3–6 cultures) (Figs. 3B and S3).
Figure 3. Non-reference SNP alleles in the CPG2 promoter region and their effect on gene expression.
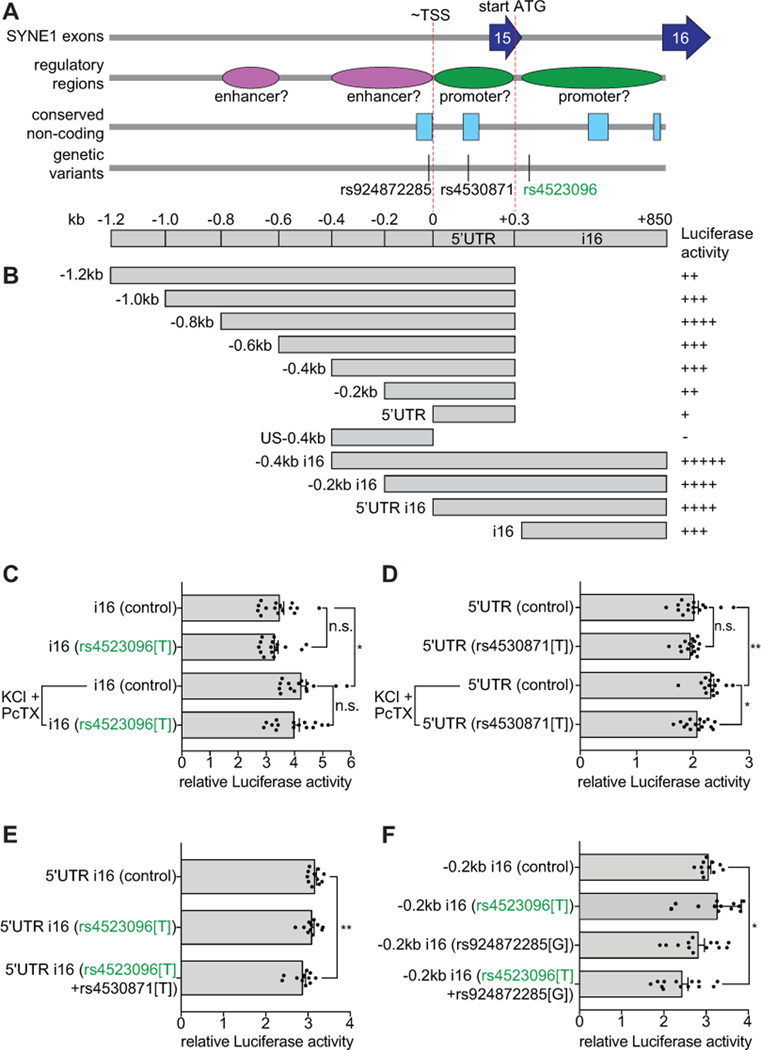
CPG2 gene regions with promoter activity were identified using a Luciferase gene expression assay in cultured cortical neurons. A) Schematic depiction of the CPG2 promoter region in SYNE1 (−1.2kb to +850kb from proposed CPG2 transcription start site (~TSS)43). Dark blue arrows indicate SYNE1 exons and red dashed lines mark the approximate transcription start site (~TSS) and the translation start site (start ATG). Green and purple ovals mark identified regulatory regions with promoter or enhancer activity, respectively. Light blue boxes indicate mammalian conserved intronic regions and black vertical lines mark identified variants within the region. B) Overview of constructs with CPG2 promoter region fragments and their relative activity in the Luciferase expression assay. C) Effects of SNP allele rs4523096[T] on activity of the i16 promoter region with or without KCl+PcTX-induced neuronal stimulation. D) Effects of SNP allele rs4530871[T] on activity of the 5’UTR region with or without KCl+PcTX-induced neuronal stimulation. The 5’UTR SNP allele rs4530871[T] significantly attenuated activity-induced gene expression. E) Effect of SNP alleles rs4523096[T] and rs4530871[T] combined, on relative Luciferase activity of the 5’UTR i16 region. The combination of the two non-reference SNP alleles significantly affects gene expression at basal neuronal activity levels as compared to control. F) Effect of SNP alleles rs4523096[T] and rs924872285[G] combined, on relative Luciferase activity of the −0.2kb i16 region. This combination of non-reference alleles also significantly affects gene expression as compared to control. (*p<0.05, **p<0.01, One-way or Two-way (KCl+PcTX) ANOVAs, Tukey’s post hoc tests).
Of the SNP alleles with higher prevalence in the low CPG2 population, SNP allele rs4523096[T] mapped within the i16 promoter region. To test whether its presence could influence promoter function, we introduced rs4523096[T] into the i16 promoter luciferase construct, and performed the luciferase reporter assay on transfected primary neurons (Fig. 3C). Because CPG2 is an activity-regulated gene46, we used a protocol with mild depolarizing KCl in the presence of the GABA receptor inhibitor picrotoxin (KCl+PcTX) to stimulate activity in the cultures. This protocol increased the relative Luciferase activity promoted by the i16 construct (Two-way ANOVA: F(1,56)=19, ****p<0.0001, Tukey’s post hoc: *p=0.011, n=15 transfections, 5 cultures), but we found no effect of the rs4523096[T] i16 SNP allele, either on basal or activity-induced gene expression in our cell culture assay (Two-way ANOVA: F(1,56)=1.6, p=0.22, Tukey’s post hoc: control vs. rs4523096[T] p=0.89 (basal), p=0.73 (KCl+PcTx), n=15 transfections, 5 cultures).
Recent studies suggest that risk genes containing common variants with low effect often also contain rarer variants with larger effect63, 64. In our limited sample set, we could not use statistical methods to identify rare variants. Instead, from our deep sequencing data of patient gDNA we identified several genetic variants within the putative CPG2 promoter regions, and tested all observed non-reference alleles in our Luciferase reporter assay. One SNP allele, rs4530871[T], located within a mammalian conserved sequence of the 5’UTR CPG2 promoter region significantly attenuated activity-induced gene expression (Two-way ANOVA: F(1,56)=7.0, *p=0.011, Tukey’s post hoc: control vs. rs4530871[T] p=0.86 (basal), *p=0.022 (KCl+PcTx), n=15 transfections, 5 cultures) (Fig. 3D).
Since the rs4530871[T] allele does not exist in our dataset without rs4523096[T], we tested the combined effect of the two alleles on relative Luciferase activity of the 5’UTR i16 promoter construct, which encompasses both SNP locations. We found that the combination of the two alleles significantly affects gene expression, even at basal activity levels of the cultured neurons (One-way ANOVA: F(2,30)=6.26, **p=0.0053, Tukey’s post hoc: control vs. rs4523096[T]+rs4530871[T] **p=0.0054, n=11 transfections, 3 cultures) (Fig. 3E).
We also tested the effect of another rare non-reference SNP allele rs924872285[G] on promoter activity of the −0.2kb i16 construct. This allele was identified in a single BD patient with low CPG2 expression in our dataset, and is situated in a conserved enhancer region immediately upstream of the TSS. This allele too, is only present together with rs4523096[T]. When we tested the combination of both alleles in the context of the −0.2kb i16 promoter construct, we saw a significant reduction in gene expression at basal activity levels as compared to controls (One-way ANOVA: F(3,45)=6.83, ***p=0.0007, Tukey’s post hoc: control vs. rs4523096[T]+ rs924872285[G] *p=0.015, n=13 transfections, 3 cultures) (Fig. 3F).
The protein expression data, together with the identification of genetic variants in CPG2 regulatory regions from BD patients that negatively affect gene expression, implicate low CPG2 expression in susceptibility to BD.
Non-reference SNP alleles in the CPG2 coding region affect spine localization of the CPG2 protein.
While low CPG2 levels could lead to dysregulation of synaptic glutamate receptor internalization43, 47, synapse dysregulation could also derive from CPG2 protein variants with reduced function.
From public databases (1000 Genomes, gnomAD), we identified 12 common missense SNPs within the CPG2 coding region (Table 1). The reference alleles for ten of the SNPs in the dataset are conserved among mammalian species.
Table 1.
Genetic variants in CPG2 exons
| Genome Position (GRCh37) chr6: |
SNP | Variant allele |
MAF (1000G) |
SYNE1 exon # |
AA residue Substitution in human CPG2 |
effect on spine localization |
effect on GluA2/GluN1 internalization |
|---|---|---|---|---|---|---|---|
| 152784621 | rs9397509 | A/G | G=0.004 | 19 | Q171R | no | no |
| 152779933 | rs34610829 | C/T | T=0.011 | 22 | R359C* | n/a | n/a |
| 152777095 | rs17082709 | T/G | G=0.043 | 23 | L401V* | n/a | n/a |
| 152776571 | rs76646638 | G/A | A=0.002 | 24 | R477Q | no | yes |
| 152776571 | rs201146062 | C/T | T=0.001 | 24 | R477W | n/a | n/a |
| 152774753 | rs148346599 | G/A | A=0.002 | 25 | E515K | n/a** | n/a** |
| 152772264 | rs214976 | T/C | T=0.39 | 26 | V551A | yes | no |
| 152771967 | rs141464488 | T/C | C=0.001 | 27 | V579A | n/a | n/a |
| 152768738 | rs116939102 | C/T | T=0.0002 | 29 | T691I | no | yes |
| 152768726 | rs117461489 | A/C | C=0.0008 | 29 | E695A | no | no |
| 152762307 | rs149109801 | T/A | A=0.0002 | 32 | F885L | n/a | n/a |
| 152757224 | rs34028822 | C/T | 0.0029 | 33 | R904W | yes | yes |
Not conserved between rat and human.
Low expression in cellular assays
To gauge the extent to which individual coding SNPs could influence CPG2 cellular localization and/or function in the general population, we tested seven representative mutations (corresponding to the identified missense SNP non-reference alleles). We used a previously described knockdown (KD)47 and replacement strategy43, where endogenous CPG2 was knocked down in cultured cortical rat neurons and replaced with human reference or mutated CPG2. We have previously used this molecular replacement strategy to show that human CPG2 can replace endogenous rat CPG2, both in terms of its cellular localization to dendritic spines and its ability to regulate internalization of synaptic glutamate receptors43. The seven representative mutations were individually introduced into the full length human CPG2 coding sequence and cloned as GFP fusion constructs into a lentiviral vector also expressing a previously validated rat Cpg2-specific small hairpin RNA (shRNA)47. These replacement viruses were used to infect cultured neurons, and mutated CPG2 localization was compared to human reference CPG2. As previously shown, uninfected neurons showed enriched CPG2 localization in dendritic spines (Fig. 4A), adjacent to the postsynaptic density protein PSD9543, 47. Consistent with previous findings43, neurons infected with the shRNA virus showed robust KD of CPG2 protein (Fig. 4B), and molecular replacement with plasmids co-expressing the Cpg2-specific shRNA and a shRNA-resistant GFP-hCPG2 fusion construct showed a punctate anti-GFP staining pattern, resembling the spine localization of the endogenous protein (Fig. 4C).
Figure 4. Effect of human coding variants on CPG2 spine localization.

Schematic depiction of the CPG2 coding region in SYNE1. Dark blue arrows indicate SYNE1 exons (note: the i34 intron is not spliced out in CPG2). Purple indicates the CPG2 coding region and orange marks the 5’ and 3’UTRs in the CPG2 mRNA. Light blue depicts the predicted CPG2 protein structure with dark blue regions indicating coiled-coil domains and red circles marking two PKA phosphorylation sites known to affect CPG2 protein function65. Black vertical lines label the positions of identified missense SNPs within the CPG2 coding region with their amino acid residue exchange indicated. Cultured hippocampal neurons were lentivirus infected with shRNA for KD of endogenous CPG2 and replaced with GFP-fused human CPG2 reference protein or mutated variants. Representative images showing dendritic segments of neurons expressing endogenous rCPG2 (A) and shRNA rCPG2 KD (B) stained with anti-CPG2 monoclonal antibodies. C-I) Representative images showing dendritic segments of neurons expressing GFP-hCPG2 control or mutated variants stained with anti-GFP antibodies. Scale bar: 2µm. J) Spine localization was quantified as the ratio between fluorescence intensity in spine regions and in 10µm of adjacent dendrite. K) Quantification of spine size defined as spine head area (µm2). Spine localization of the V551A and R904W variants is significantly decreased while spine size is comparable to control. (*p<0.05, **p<0.01, One-way ANOVAs, Dunnett’s post hoc tests).
Six of the seven tested GFP-hCPG2 protein variants were expressed at levels comparable to the WT GFP-hCPG2 protein (Fig. 4D-I). Interestingly, expression of the E515K variant was very low and did not allow proper evaluation of spine localization (data not shown), suggesting this coding mutation could also impact CPG2 levels or protein stability. When compared to the WT GFP-hCPG2 protein, immunostaining with anti-GFP antibodies showed significantly decreased spine localization for the V551A and R904W GFP-CPG2 protein variants (One-way ANOVA: F(6,193)=3.6, **p=0.002, Dunnett’s post hoc: WT vs. V551A **p=0.0089, WT vs. R904W *p=0.044, n=4 independent experiments) (Fig. 4J). Significant differences in spine size was not observed for the tested variants (One-way ANOVA: F(6,213)=2.1, *p=0.049, Dunnett’s post hoc: p>0.05, n=4 independent experiments) (Fig. 4K).
Non-reference SNP alleles within the CPG2 coding region affect glutamate receptor internalization.
Rat CPG2 is known to localize to the endocytic zone of dendritic spines, where it regulates endocytosis of synaptic glutamate receptors47, 65. We have recently shown that human CPG2 is functionally equivalent to rat CPG2 in facilitating glutamate receptor internalization in cultured neurons, suggesting a conserved function for CPG2 in rat and human brain43. To test whether any of the six human CPG2 coding variants could affect the ability of human CPG2 to functionally replace endogenous rat CPG2, we used an internalization assay for biotinylated surface receptors. Consistent with previous data43, 47, 65, we found that 6.0 ± 0.9% of GluA2 containing AMPARs were constitutively internalized after 30 min in uninfected neurons, and KD of CPG2 decreased GluA2 internalization by approximately half (3.5 ± 0.8%, *p< 0.05) (Fig. 5A-F). Viral replacement with the GFP-hCPG2 reference protein rescued GluA2 internalization to levels comparable to endogenous CPG2 (6.0 ± 1.0%). When testing the six mutated CPG2 variants, we found significantly decreased GluA2 internalization rates for the R477Q (One-way ANOVA: F(3,37)=5.7, **p=0.0027, Tukey’s post hoc: WT vs. R477Q *p=0.014, n=8), T691I (One-way ANOVA: F(3,35)=5.6, **p=0.003, Tukey’s post hoc: WT vs. T691I *p=0.039, n=7) and R904W (One-way ANOVA: F(3,34)=7.2, ***p=0.0008, Tukey’s post hoc: WT vs. R904W *p=0.012, n=12) variants (Fig. 5B, D and F).
Figure 5. Effect of human CPG2 coding variants on glutamate receptor internalization.

CPG2 coding variants were tested in an internalization assay for biotinylated surface receptors. Schematic depiction of predicted CPG2 protein structure with dark blue regions indicating coiled-coil domains and red circles marking two PKA phosphorylation sites. Black vertical lines label the positions of identified missense SNPs with their amino acid residue exchange indicated. A-F) Cultured cortical neurons were either uninfected (control), infected with shRNA for KD of endogenous CPG2 (KD) or infected with shRNA for CPG2 KD together with GFP-fused human CPG2 reference protein (hCPG2) or mutated variants (as indicated). Representative Western blots showing the internalized fraction of biotinylated surface receptors probed with GluA2 antibodies. Quantification of internalized GluA2 is presented as percent internalization calibrated to levels of surface receptors. Human CPG2 replacement shows receptor internalization rates comparable to control. CPG2 variants R477Q, T691I, and R904W show significantly decreased GluA2 internalization. (*p<0.05, **p<0.01, One-way ANOVAs, Tukey’s post hoc tests).
Similar to its effect on GluA2 internalization, CPG2 KD significantly decreased constitutive internalization of GluN1 containing NMDARs (*p<0.05), while viral replacement with human CPG2 also rescued internalization of GluN1 to control levels (Fig. S4A-F). GluN1 receptor internalization rates were significantly decreased for the same CPG2 variants that affected GluA2 internalization (One-way ANOVAs: F(3,29)=6.2, **p=0.0022 (R477Q); F(3,37)=5.7, **p=0.0026 (T691I); F(3,36)=6.7, **p=0.001 (R904W), Tukey’s post hoc tests: WT vs. R477Q *p=0.035, n=8; WT vs. T691I *p=0.036, n=7; WT vs. R904W *p=0.025, n=12, respectively) (Fig. S4 B, D and F). Neither CPG2 KD, nor replacement with WT or mutated human CPG2 significantly affected the constitutive internalization of transferrin receptor (TfR) (p>0.05) (Fig. S3G-L). Our data suggest that low frequency but relatively common CPG2 coding variants (MAF>0.001 present in the population) can have significant effects on CPG2 synaptic function.
DISCUSSION
Recently, large consortium GWAS studies have shed new light on the genetics of neuropsychiatric disorders. However, genetic variants identified so far account for only a fraction of disease liability, a phenomenon generally characteristic of complex genetic traits. Purely genomic approaches such as GWAS and linkage studies are only the first step in elucidating the complex neurobiology of BD. They need to be followed and complemented with molecular and cellular studies of defective neuronal function to obtain a more complete understanding of disease etiology.
Identification of risk loci for BD has firmly implicated dysregulated excitatory neurotransmission as a key component of BD etiology24, 25, 34–36. Given the role of CPG2, a SYNE1 gene product, in facilitating glutamate receptor internalization and regulating excitatory synaptic strength43, 47, 65, this gene might be an important player in the neurobiological underpinnings of BD. Here, we show that low levels of the CPG2 protein are more prevalent in BD patients as compared to controls and other patient groups, and use multiple strategies to identify genetic variants within CPG2 regulatory or protein-coding regions that negatively affect gene expression or disrupt protein function, respectively.
Our finding that CPG2 protein levels are significantly lower in postmortem prefrontal cortex from BD patients as compared to control subjects, a phenomenon not shared with schizophrenia or major depression patients, supports specificity of CPG2 perturbation in BD. This is consistent with GWAS findings that genetic variation in SYNE1 is more closely associated with risk for BD23, 26. Low CPG2 levels across brain regions of BD patients suggests it is likely to derive from genetic causes. The clustering of common genetic variants in CPG2 regulatory regions identified in the low CPG2 expression BD subjects further supports this notion, although we cannot exclude additional causes such as brain region-specific reduced activity in BD disease state.
It is often noted that common risk variants identified by GWAS overwhelmingly reside in large introns and in sequences immediately upstream of the implicated genes66. This suggests that disease-associated variation may derive not mainly from disrupted protein function, but from dysregulated gene expression. For example, the largest number of disease associations found by GWAS in schizophrenia are in regulatory regions, such as promoter or enhancer sequences67, 68. Studies of expression quantitative trait loci (eQTLs) in human tissues69, 70 also show that disease implicated genetic variants tend to associate with quantitative differences in expression levels of the same genes, especially when gene expression is measured in the tissue relevant to the disease68, 69. Unfortunately, we were not able to find eQTL data for the CPG2 gene region in public databases (GTEx, ExSNP). Progress in the genome-scale analysis of chromatin states now reveals hundreds of thousands of sites across the genome that contain dynamic chromatin marks suggestive of tissue-specific promoter or enhancer activity, with the ability to regulate the expression of nearby genes in specific tissues52, 71. The regulatory elements mainly found in introns are often less conserved between humans and rodents, as well as across all of evolution. Interestingly, the CPG2 promoter region is highly conserved among mammalian species as compared to other intronic regions within the gene (see supplementary table ST4 and sequence alignment for quantification), suggesting that this region has a conserved activity-dependent regulatory function in mammalian brains.
Our data suggest that no single genetic variation is associated with the low CPG2 protein expression found in BD patient subjects. This is in accordance with the pleiotropic and polygenic nature of BD, and other neuropsychiatric disorders, where many genetic risk factors each contributing a small effect cumulatively add up to a larger disease susceptibility. Common regulatory variants can also result in milder phenotypes that reflect tissue- or cell type specific gene expression. For example, a common variant in a CACNA1C regulatory region associated with approximately 15% increased risk of schizophrenia and BD, has no apparent association to cardiac or immune phenotypes66. By analogy, common regulatory variants influencing expression of brain-specific CPG2 transcripts from the SYNE1 gene would confer increased risk for developing BD, without significant impact on muscular phenotypes associated with other SYNE1 transcripts.
GWAS and genetic linkage studies, with the intrinsic requirement for very large sample sizes, are best suited to identify relatively common genetic risk variants. We found that when tested alone, the common BD associated i16 SNP allele (rs4523096[T])61 had no apparent effect on gene expression. A suggested explanation for some of the ‘missing heritability’, not yet accounted for in the genetics of complex diseases, is that common disease-associated SNPs act as sentinels for other genetic risk factors adjacent within the same region with more penetrant effects63. From our deep sequencing data of patient gDNA, we identified several genetic variants within the putative CPG2 promoter and enhancer regions. One SNP allele (rs4530871[T]), situated within a mammalian conserved sequence of the 5’UTR CPG2 promoter region significantly attenuated activity-induced gene expression when tested alone. Interestingly, (rs4523096[T] and rs4530871[T]) in combination, reduced basal gene expression. Likewise, another SNP allele, rs924872285[G] identified in a putative CPG2 enhancer, also reduced basal gene expression when combined with rs4523096[T]. This exemplifies the common notion for complex genetic diseases that cumulative effects of low penetrant variants can combine to a greater effect size. Further, the differential effect of specific SNP alleles on basal versus activity-dependent gene expression suggests that within the general population, there are low frequency but relatively common genetic variants in the CPG2 promoter region that influence neuronal activity-dependent gene expression without apparent effect on basal level gene expression. The activity-dependent requirement for gene expression could potentially provide one explanation for the influence of environmental ‘stressors’ on development of BD associated psychiatric symptoms.
While low CPG2 levels have already been shown to disrupt synaptic glutamate receptor internalization43, 47, synapse dysregulation could also derive from CPG2 protein variants with reduced function. Thus, some patients with apparently normal CPG2 levels might suffer from risk for BD due to coding region mutations. In a recent report, six nonsynonymous SNPs were identified in the CPG2 region of SYNE1 using high-resolution melt analysis72, but only predicted effects were described. The four most common of the missense SNPs overlap with our findings, namely rs34610829[T] (R359C), rs17082709[G] (L401V), rs148346599[A] (E515K), and rs214976[C] (V551A), of which the former two are not conserved across evolution and were therefore not tested here. The two latter were predicted by Polyphen and SIFT analyses to be tolerated in other SYNE1 products and likely damaging to CPG2, and benign to both gene products, respectively72. This agrees with our findings that E515K had an apparent effect on CPG2 expression or stability, and V551A had a small effect on CPG2 spine localization with no apparent effect on glutamate receptor internalization. We found three additional SNPs with significant negative effects on glutamate receptor internalization, rs76646638[A] (R477Q), rs116939102[T] (T691I), and rs34028822[T] (R904W), of which the latter also affected spine localization. The R904W variant is situated at the C-terminal of CPG2 in between two reported protein kinase A (PKA) phosphorylation sites important for CPG2 spine localization and protein function65. We speculate that the bulky tryptophan in the minor allele variant might interfere with PKA binding and CPG2 phosphorylation, and thereby disrupt CPG2 protein function. The two other functionally disruptive variants R477Q and T691I do not have apparent effects on spine localization. They are situated far from the CPG2 C-terminal f-actin binding site important for spine localization73 but might affect functional protein domains in ways that allows normal spine localization but disrupt binding to the endocytic machinery, which facilitates receptor internalization47.
In this study, we present protein functional data from a statistically significant and replicated BD associated risk locus. We show that levels of the brain-specific SYNE1 product CPG2 are lower specifically in BD patients as compared to controls as well as schizophrenia and depression patients. We identify genetic variants in the CPG2 promoter region with negative effects on gene expression, as well as low frequency coding variants in CPG2 that significantly affect CPG2 protein function. Taken together, our data fit a genetic architecture of BD, likely involving clusters of both regulatory and protein-coding variants, whose combined contribution to phenotype is an important piece of a puzzle containing other risk and protective factors influencing BD susceptibility. The ultimate goal is to allow a more scientifically informed, evidence-based approach to measure, differentiate and treat neuropsychiatric disorders, preferably with the aid of genetic and other non-invasive biomarkers.
Supplementary Material
ACKNOWLEDGEMENTS
Human postmortem brain tissue samples were generously provided by the Stanley Medical Research Institute neuropathology collection, Massachusetts Alzheimer’s Disease Research Center (funding source: P50 AG005134) and NIH Neurobiobank with contributions from Harvard Brain Tissue Resource Center, Mount Sinai NIH Brain & Tissue Repository and University of Maryland Brain & Tissue Bank. We acknowledge Picower Institute for Learning and Memory Staff Bioinformatician, Fan Gao, for invaluable help with bioinformatics and statistical analyses. Furthermore, we acknowledge Drs. Jeffrey Cottrell and Dennis Lal at the Stanley Center for Psychiatric Research, and our colleagues in the Nedivi Laboratory for valuable input and editing of the manuscript. The work was funded by the Jeffry M. and Barbara Picower Foundation (EN), The Gail Steel Fund (EN), the Carlsberg Foundation (MR), Lundbeck Foundation (MR), and the Danish Council for Independent Research (MR).
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Merikangas KR, Akiskal HS, Angst J, Greenberg PE, Hirschfeld RM, Petukhova M et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch Gen Psychiatry 2007; 64(5): 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crump C, Sundquist K, Winkleby MA, Sundquist J. Comorbidities and mortality in bipolar disorder: a Swedish national cohort study. JAMA Psychiatry 2013; 70(9): 931–939. [DOI] [PubMed] [Google Scholar]
- 3.Jamison KR. Suicide and bipolar disorder. J Clin Psychiatry 2000; 61 Suppl 9: 47–51. [PubMed] [Google Scholar]
- 4.Bavamian S, Mellios N, Lalonde J, Fass DM, Wang J, Sheridan SD et al. Noncoding RNAs connect genetic risk factors to the neurodevelopmental basis of bipolar disorder. Mol Psychiatry 2015; 20(5): 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bavamian S, Mellios N, Lalonde J, Fass DM, Wang J, Sheridan SD et al. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol Psychiatry 2015; 20(5): 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jablensky A Psychiatric classifications: validity and utility. World Psychiatry 2016; 15(1): 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schretlen DJ, Cascella NG, Meyer SM, Kingery LR, Testa SM, Munro CA et al. Neuropsychological functioning in bipolar disorder and schizophrenia. Biol Psychiatry 2007; 62(2): 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott J, Leboyer M. Consequences of delayed diagnosis of bipolar disorders. Encephale 2011; 37 Suppl 3: S173–175. [DOI] [PubMed] [Google Scholar]
- 9.Strakowski SM, Adler CM, Almeida J, Altshuler LL, Blumberg HP, Chang KD et al. The functional neuroanatomy of bipolar disorder: a consensus model. Bipolar Disord 2012; 14(4): 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delvecchio G, Fossati P, Boyer P, Brambilla P, Falkai P, Gruber O et al. Common and distinct neural correlates of emotional processing in Bipolar Disorder and Major Depressive Disorder: a voxel-based meta-analysis of functional magnetic resonance imaging studies. Eur Neuropsychopharmacol 2012; 22(2): 100–113. [DOI] [PubMed] [Google Scholar]
- 11.Delvecchio G, Sugranyes G, Frangou S. Evidence of diagnostic specificity in the neural correlates of facial affect processing in bipolar disorder and schizophrenia: a meta-analysis of functional imaging studies. Psychol Med 2013; 43(3): 553–569. [DOI] [PubMed] [Google Scholar]
- 12.Linden DE. The challenges and promise of neuroimaging in psychiatry. Neuron 2012; 73(1): 8–22. [DOI] [PubMed] [Google Scholar]
- 13.Craddock N, Sklar P. Genetics of bipolar disorder. Lancet 2013; 381(9878): 1654–1662. [DOI] [PubMed] [Google Scholar]
- 14.McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry 2003; 60(5): 497–502. [DOI] [PubMed] [Google Scholar]
- 15.Kieseppä T, Partonen T, Haukka J, Kaprio J, Lönnqvist J. High concordance of bipolar I disorder in a nationwide sample of twins. Am J Psychiatry 2004; 161(10): 1814–1821. [DOI] [PubMed] [Google Scholar]
- 16.Edvardsen J, Torgersen S, Røysamb E, Lygren S, Skre I, Onstad S et al. Heritability of bipolar spectrum disorders. Unity or heterogeneity? J Affect Disord 2008; 106(3): 229–240. [DOI] [PubMed] [Google Scholar]
- 17.Craddock N, Sklar P. Genetics of bipolar disorder: successful start to a long journey. Trends Genet 2009; 25(2): 99–105. [DOI] [PubMed] [Google Scholar]
- 18.O’Donovan MC, Owen MJ. The implications of the shared genetics of psychiatric disorders. Nat Med 2016; 22(11): 1214–1219. [DOI] [PubMed] [Google Scholar]
- 19.Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45(9): 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460(7256): 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018; 359(6376): 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lichtenstein P, Yip BH, Björk C, Pawitan Y, Cannon TD, Sullivan PF et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 2009; 373(9659): 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Consortium C-DGotPG. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381(9875): 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 2008; 40(9): 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Psychiatric GCBDWG. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011; 43(10): 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green EK, Grozeva D, Forty L, Gordon-Smith K, Russell E, Farmer A et al. Association at SYNE1 in both bipolar disorder and recurrent major depression. Mol Psychiatry 2013; 18(5): 614–617. [DOI] [PubMed] [Google Scholar]
- 27.Green EK, Hamshere M, Forty L, Gordon-Smith K, Fraser C, Russell E et al. Replication of bipolar disorder susceptibility alleles and identification of two novel genome-wide significant associations in a new bipolar disorder case-control sample. Mol Psychiatry 2013; 18(12): 1302–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou L, Bergen SE, Akula N, Song J, Hultman CM, Landén M et al. Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum Mol Genet 2016; 25(15): 3383–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol 2015; 134: 36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iqbal Z, Vandeweyer G, van der Voet M, Waryah AM, Zahoor MY, Besseling JA et al. Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum Mol Genet 2013; 22(10): 1960–1970. [DOI] [PubMed] [Google Scholar]
- 31.Forstner AJ, Hecker J, Hofmann A, Maaser A, Reinbold CS, Mühleisen TW et al. Identification of shared risk loci and pathways for bipolar disorder and schizophrenia. PLoS One 2017; 12(2): e0171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381(9875): 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu W, Cohen-Woods S, Chen Q, Noor A, Knight J, Hosang G et al. Genome-wide association study of bipolar disorder in Canadian and UK populations corroborates disease loci including SYNE1 and CSMD1. BMC Med Genet 2014; 15: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 2008; 7(5): 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nurnberger JI Jr., Koller DL, Jung J, Edenberg HJ, Foroud T, Guella I et al. Identification of pathways for bipolar disorder: a meta-analysis. JAMA Psychiatry 2014; 71(6): 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machado-Vieira R, Ibrahim L, Henter ID, Zarate CA. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav 2012; 100(4): 678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perlis RH, Smoller JW, Ferreira MA, McQuillin A, Bass N, Lawrence J et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry 2009; 166(6): 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scarr E, Pavey G, Sundram S, MacKinnon A, Dean B. Decreased hippocampal NMDA, but not kainate or AMPA receptors in bipolar disorder. Bipolar Disord 2003; 5(4): 257–264. [DOI] [PubMed] [Google Scholar]
- 39.McCullumsmith RE, Kristiansen LV, Beneyto M, Scarr E, Dean B, Meador-Woodruff JH. Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res 2007; 1127(1): 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nudmamud-Thanoi S, Reynolds GP. The NR1 subunit of the glutamate/NMDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neurosci Lett 2004; 372(1–2): 173–177. [DOI] [PubMed] [Google Scholar]
- 41.Meador-Woodruff JH, Hogg AJ Jr., Smith RE. Striatal ionotropic glutamate receptor expression in schizophrenia, bipolar disorder, and major depressive disorder. Brain Res Bull 2001; 55(5): 631–640. [DOI] [PubMed] [Google Scholar]
- 42.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse 2006; 60(8): 585–598. [DOI] [PubMed] [Google Scholar]
- 43.Loebrich S, Rathje M, Hager E, Ataman B, Harmin DA, Greenberg ME et al. Genomic mapping and cellular expression of human CPG2 transcripts in the SYNE1 gene. Mol Cell Neurosci 2016; 71: 46–55. [DOI] [PubMed] [Google Scholar]
- 44.Puckelwartz MJ, Kessler E, Zhang Y, Hodzic D, Randles KN, Morris G et al. Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice. Hum Mol Genet 2009; 18(4): 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A et al. Nesprin-1 and −2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet 2007; 16(23): 2816–2833. [DOI] [PubMed] [Google Scholar]
- 46.Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 1993; 363(6431): 718–722. [DOI] [PubMed] [Google Scholar]
- 47.Cottrell JR, Borok E, Horvath TL, Nedivi E. CPG2: a brain- and synapse-specific protein that regulates the endocytosis of glutamate receptors. Neuron 2004; 44(4): 677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25(14): 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25(16): 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011; 27(21): 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38(16): e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A et al. Integrative analysis of 111 reference human epigenomes. Nature 2015; 518(7539): 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rathje M, Fang H, Bachman JL, Anggono V, Gether U, Huganir RL et al. AMPA receptor pHluorin-GluA2 reports NMDA receptor-induced intracellular acidification in hippocampal neurons. Proc Natl Acad Sci U S A 2013; 110(35): 14426–14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujino T, Lee WC, Nedivi E. Regulation of cpg15 by signaling pathways that mediate synaptic plasticity. Mol Cell Neurosci 2003; 24(3): 538–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Savitz JB, Price JL, Drevets WC. Neuropathological and neuromorphometric abnormalities in bipolar disorder: view from the medial prefrontal cortical network. Neurosci Biobehav Rev 2014; 42: 132–147. [DOI] [PubMed] [Google Scholar]
- 56.Selvaraj S, Arnone D, Job D, Stanfield A, Farrow TF, Nugent AC et al. Grey matter differences in bipolar disorder: a meta-analysis of voxel-based morphometry studies. Bipolar Disord 2012; 14(2): 135–145. [DOI] [PubMed] [Google Scholar]
- 57.Arnone D, Cavanagh J, Gerber D, Lawrie SM, Ebmeier KP, McIntosh AM. Magnetic resonance imaging studies in bipolar disorder and schizophrenia: meta-analysis. Br J Psychiatry 2009; 195(3): 194–201. [DOI] [PubMed] [Google Scholar]
- 58.Eker C, Simsek F, Yılmazer EE, Kitis O, Cinar C, Eker OD et al. Brain regions associated with risk and resistance for bipolar I disorder: a voxel-based MRI study of patients with bipolar disorder and their healthy siblings. Bipolar Disord 2014; 16(3): 249–261. [DOI] [PubMed] [Google Scholar]
- 59.Morris RW, Sparks A, Mitchell PB, Weickert CS, Green MJ. Lack of cortico-limbic coupling in bipolar disorder and schizophrenia during emotion regulation. Transl Psychiatry 2012; 2: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci 2011; 14(3): 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ikeda M, Takahashi A, Kamatani Y, Okahisa Y, Kunugi H, Mori N et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol Psychiatry 2018; 23(3): 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith EN, Koller DL, Panganiban C, Szelinger S, Zhang P, Badner JA et al. Genome-wide association of bipolar disorder suggests an enrichment of replicable associations in regions near genes. PLoS Genet 2011; 7(6): e1002134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ et al. Finding the missing heritability of complex diseases. Nature 2009; 461(7265): 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bomba L, Walter K, Soranzo N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol 2017; 18(1): 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Loebrich S, Djukic B, Tong ZJ, Cottrell JR, Turrigiano GG, Nedivi E. Regulation of glutamate receptor internalization by the spine cytoskeleton is mediated by its PKA-dependent association with CPG2. Proc Natl Acad Sci U S A 2013; 110(47): E4548–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCarroll SA, Hyman SE. Progress in the genetics of polygenic brain disorders: significant new challenges for neurobiology. Neuron 2013; 80(3): 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Consortium SWGotPG. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511(7510): 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Richards AL, Jones L, Moskvina V, Kirov G, Gejman PV, Levinson DF et al. Schizophrenia susceptibility alleles are enriched for alleles that affect gene expression in adult human brain. Mol Psychiatry 2012; 17(2): 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 2010; 6(4): e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012; 337(6099): 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459(7243): 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharp SI, Lange J, Kandaswamy R, Daher M, Anjorin A, Bass NJ et al. Identification of rare nonsynonymous variants in SYNE1/CPG2 in bipolar affective disorder. Psychiatr Genet 2017; 27(3): 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Loebrich S, Nedivi E. The function of activity-regulated genes in the nervous system. Physiol Rev 2009; 89(4): 1079–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.