

Abstract
Over the past decade, human host genome-wide association studies (GWASs) have contributed greatly to our understanding of the impact of host genetics on phenotypes. Recently, the microbiome has been recognized as a complex trait in host genetic variation, leading to microbiome GWAS (mGWASs). For these, many different statistical methods and software tools have been developed for association mapping. Applications of these methods and tools have revealed several important findings; however, the establishment of causal factors and the direction of causality in the interactive role between human genetic polymorphisms, the microbiome and the host phenotypes are still a huge challenge. Here, we review disease scoring approaches in host and mGWAS and their underlying statistical methods and tools. We highlight the challenges in pinpointing the genetic-associated causal factors in host and mGWAS and discuss the role of multi-omic approach in disease scoring statistics that may provide a better understanding of human phenotypic variation by enabling further system biological experiment to establish causality.
Keywords: genome-wide association study, microbiome, multi-omics, microbiome GWAS
Introduction
Our understanding of the diversity of the human genome has improved considerably in the past decade with the advances in high-throughput sequencing technology [1]. These high-throughput technologies have also revolutionized disease scoring statistics (DSS) approaches. DSS refer to the application of statistical methods to any ‘omic’ data to identify and characterize the factors underlying human phenotypic variation (Figure 1). Leveraging on the huge technological developments, DSS have led to the identification of various factors that shape the diversity of the human genome at individual, family and population levels. DSS provided deeper insights into the basis of phenotypic variation, including human appearance, disease susceptibility/resistance, disease severity and response to treatment [2]. In recent years, studies have also corroborated the role of the microbiome on human phenotypic variation, and the microbiome has emerged as a complex trait in human variation [3, 4]. These studies have shown that the microbiome is intimately involved in the interplay between health and disease [5, 6]. For example, alterations in the composition of the gut microbiome, also known as dysbiosis, are now known to be associated with many complex diseases such as inflammatory bowel disease, cancer and autoimmune disorders [7]. Altogether, these remarkable discoveries from human genome-wide association study (GWAS) and microbiome GWAS (mGWAS) have raised enthusiasm among researchers to conduct research to obtain a broader understanding of human genetic architecture, particularly as each new DSS approach continues to reveal novel biomarkers for disease phenotypes.
Figure 1.
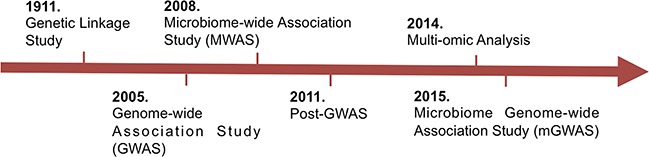
Development of various DSS.
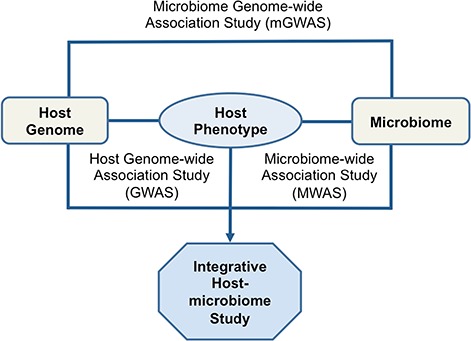
Although we know that the taxonomic composition and relative abundance of the microbiome is associated with host genetics and host phenotypes, little is known about the associated causative genetic factor; the precise molecular mechanisms underlying the expression of a given phenotype, is at best, poorly understood [3, 5]. Recent studies have suggested that the microbiome impacts host phenotype through alteration of the host gene regulation in the interfacing host epithelial cells or modification of open chromatin status in the intraepithelial lymphocytes [8]. The genetic risk factors in the observed relationship between host genetic polymorphisms, the host’s phenotypic expression and the microbiome composition are of fundamental biological and medical interest as a 1st step toward causality. DSS can play major roles toward dissecting associated genetic factors by helping to identify and prioritize likely associated variants in linkage disequilibrium (LD) with causal variant from the set of statistically associated genetic polymorphisms. This will enable further biological experiment to reveal their precise molecular mode of action. While DSS have enabled identification of novel genes, pathways and networks that harbor genetic variations responsible for a horde of phenotypes, current human host and mGWASs do not clearly provide a mechanistic understanding of how the consortium of host genetic variation, the microbiome and the environment cooperate to influence traits/disease. Furthermore, we even delve into the challenges host or mGWASs face in discerning true risk variants and the perplexity in understanding how these risk variants exert their effects [9]. However, these constitute an important step toward a global understanding of what and how human variation contributes to phenotypic differences ranging from development, physiology and behavior to pathogenesis of many human diseases. With this global view, it has today become critical to integrate different factors that potentially play roles in the human phenotypic variation. Consequently, the integration of multi-omic data within integrative DSS approaches (Figure 2) will greatly improve our understanding and unravel the complex interaction between host genetics, microbiome and environment that are pertinent to human health and disease. Such integrative DSS approaches will facilitate the understanding of causality and subsequently translate to clinical and medical applications, including proper diagnosis, prevention and treatment. It is important to note that the integration of such high-dimensional and diverse multi-omic data is itself challenging [10].
Figure 2.
Illustrative representation of host GWAS, mGWAS and the integrative host-microbiome study approach.
In this paper, we (1) discuss genetic risk in the observed associations between host genetics, the microbiome and the complex diseases using DSS, (2) outline the role of integrative multi-omic approaches to unravel causality and (3) conclude by highlighting some research areas where further work on DSS is needed to establish integrative genetic risk factors of complex traits and diseases.
Human variation
Human populations differ in the distribution and frequency of their phenotypic expressions. These `human variations’ result from both their genetic components whose compositions are largely shaped by their genomic history and nongenetic components. The genomic history of human evolution is characterized by the exchange of genetic materials across individuals, resulting into individuals with unique genetic features. Consequently, the current pattern of human genetic diversity is hypothesized to be ‘ancestral’, most genetic variants having occurred once in human history and vertically spread across populations, rather than due to recurrent mutations [11]. This genetic mixing generated substantial genetic variation. It is estimated that the human genome contains between 4.1 and 5.0 million polymorphisms, at least 99% of them being Single Nucleotide Polymorphisms (SNPs) and short indels [12]. Despite the large number of genetic variants in the genome, only a minute fraction of them are hypothesized to be causal of disease [11], the majority being neutral (having no contribution to phenotypic variation) or near neutral.
In addition to genetic variation, the human microbiome, the collection of bacteria, archaea, fungi, protozoans and viruses that colonize our body surfaces and their respective genomes, plays an important role in health and disease variation [5, 8]. A long-standing goal in human genetic studies is elucidating the fraction of heritability and environment that may contribute to the variation in health and disease [11]. This goal is, however, challenging owing to the myriad of confounding factors that likely mask true and causative associations. To this end, various DSS methods for host and mGWAS have been developed to investigate the association between genetic variability and/or microbiome composition and disease susceptibility/resistance [12, 13].
It is worth to note that current developed omic technologies, particularly genotyping arrays and imputation panels, have widely been designed for populations of European descent with long-range patterns of LD [13]. Additionally, current microbial databases are built with genomes of European descent populations. The insufficient capture of some haplotypes from non-European populations limits the power to detect important associations. In addition, many new genetic associations to diseases that have been identified from both host and mGWASs have primarily been applied to samples from population of European ancestry [12, 13]. This has an implication that a substantial proportion of functionally important variations in other populations are not captured. This may partly explain the challenge in replicating variants identified in some populations let alone drawing a strong mechanistic link between the associated variants and disease phenotypes. Variants associated with diseases found in populations of European descent do not always replicate in non-European, particularly African populations [12, 14] for several reasons, including differences in allelic architecture, LD and confounding of environmental factors across populations. In addition, genetic determinants of disease and their effect sizes have also been shown to vary significantly between European and African populations [13, 14].
The high levels of genetic diversity and the burden of complex diseases in non-European, particularly in African populations, may further introduce both challenges and opportunities not only for host and mGWAS but also for the general omic analyses [9, 14]. The high genetic diversity, environmental heterogeneity and high burden of diseases that characterize the African population [15] make it clear that leveraging the African genomic data is pertinent to a robust analysis. Notwithstanding, current genome-wide DSS that leverage a single level of omic data may be limited for adequately gaining insights into the basis of observed phenotypic expressions.
Whereas the importance of increasing host GWAS to include samples from non-European ancestry has been long appreciated, the observation that the composition and relative abundance of the human microbiome is influenced by diet, environment and host genetics has demonstrated how studies on diverse backgrounds may provide valuable insights. For example, Smits et al. compared the gut microbiome of the Hadza hunter-gatherers of Western Tanzania to 18 others in 16 different countries with varying lifestyles [16]. They found the gut microbiome composition to be clearly differentiated between traditional and industrialized populations. With the inclusion of multiple genetically diverse populations in the analysis, it is hoped that it will be possible to (1) elucidate the genetic architecture of many complex traits, (2) more accurately reconstruct ancestral haplotype which cannot occur in non-Africans and (3) shed light on the role of the human microbiome in disease susceptibility and resistance given differing environment and the burden of communicable and noncommunicable diseases in Africa, South America and Asia. The disparity in omic research, in terms of genetic diversity of the study population, and technology capacity across the globe, may not favor the advancement of our understanding of the intricate pathogenesis of complex diseases. In addition, such disparity forms a major obstacle toward the full development of appropriate global prevention and treatment strategies particularly in light of precision medicine.
Methodologies underpinning host and microbiome DSS
To identify one or more genetic variants associated with a given disease, the most commonly used approach today is to perform a genetic association study. This goal is achieved by comparing the frequency of one or more genetic variants between cases and controls [17, 18]. This has led to the advent of the development of various host-based GWAS models, mostly based on linear mixed, random, mixed effects and Bayesian frameworks [19, 20]. Box 1 provides a brief genesis of statistical methods for host-based GWAS approaches. Although many of these approaches and tools have been effective at uncovering the genetic basis of many complex traits, they have potentially missed out many novel genetic variants and/or failed to disentangle true signals from spurious associations owing to limitations in the underlying statistical models. Methodologically, these association frameworks (Supplementary Table S1 ) can be classified into two broad categories: linear regression-based and linear mixed model (LMM)-based frameworks. Supplementary Table S1 displays the category of the tools and provides a brief description on each tool.
Box 1: statistical methods in host GWAS.
The traditional statistical method for GWAS was the simple linear (1) or logistic (2) regression, which phenotype 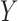 and the fixed effects
and the fixed effects 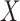 by the relation:
by the relation:
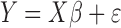 |
(1) |
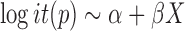 |
(2) |
where 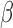 is the effect size,
is the effect size, 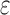 is random noise and
is random noise and 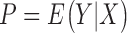 is the expected value of phenotype given genotype. LMM modifies (1) by introducing a random effects
is the expected value of phenotype given genotype. LMM modifies (1) by introducing a random effects  so that the model becomes
so that the model becomes
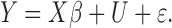 |
(3) |
While LMM represented a powerful methodology, three key issues remained at the forefront of its implementation: (i) computational cost in evaluating the variance parameters, (ii) strategy of modeling SNP effects (fixed SNP effect vs random SNP effect) and confounders and (iii) method of modeling genetic architecture of the phenotype (infinitesimal versus non-infinitesimal), leading to development of various statistical tools. Under an infinitesimal model, the χ2 [1 Degree of Freedom (d.o.f)] test statistic for testing association in equation (3), with the hypothesis  is
is
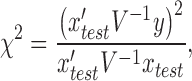 |
(4) |
where, 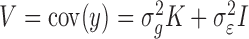 ;
; 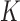 is the genetic relationship matrix (kinship matrix) that models sample structure and
is the genetic relationship matrix (kinship matrix) that models sample structure and 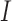 is
is 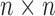 identity matrix. The variance parameters
identity matrix. The variance parameters 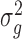 and
and 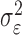 are typically unknown and to be computed. The computation of variance components in LMM is, however, expensive. Various approaches have been proposed to reduce computational cost. Kang et al. [22] proposed a direct estimation
are typically unknown and to be computed. The computation of variance components in LMM is, however, expensive. Various approaches have been proposed to reduce computational cost. Kang et al. [22] proposed a direct estimation 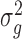 and
and 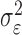 by maximizing the REML function, and then applying spectral decomposition [23] obviating more computationally intensive approach of determining the best linear unbiased prediction via Henderson’s iterative procedure [24].
by maximizing the REML function, and then applying spectral decomposition [23] obviating more computationally intensive approach of determining the best linear unbiased prediction via Henderson’s iterative procedure [24].
Computational efficiency is improved from other approaches that replace the usual relatedness matrix computed from all genome-wide SNPs by low-rank relatedness matrix [23]. In addition, the effective sample size of a random effect is reduced by clustering subjects into genetically similar groups. Moreover, logistic mixed model has recently been advocated for, in place of LMM, analysis of binary traits. This is because LMM is based on the assumption that the trait has constant residual variance—an assumption that is usually violated by binary traits in the presence of covariates [24]. In recent years, in light of trait polygenicity, LMM methods that simultaneously test the effect of multiple SNPs have been developed, for example, GCTA [25]. In these, the model is set as
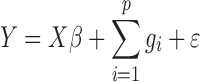 |
(6) |
where 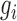 is a vector of genetic effects on whole genome. Bayesian modeling technique has since been adopted for joint analyses of multiple SNPs, because of their ability to increase power by leveraging known information on marker effects. Chen et al.
[24] proposed Hierarchical Bayesian model, in which the SNP random effects are assumed to follow the mixture distribution:
is a vector of genetic effects on whole genome. Bayesian modeling technique has since been adopted for joint analyses of multiple SNPs, because of their ability to increase power by leveraging known information on marker effects. Chen et al.
[24] proposed Hierarchical Bayesian model, in which the SNP random effects are assumed to follow the mixture distribution:
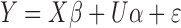 |
(7) |
where 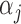 values are assumed to follow a Gaussian distribution.
values are assumed to follow a Gaussian distribution.
This improved power over other LMM-based methods, such as GCTA modeled [25] by equation (6). Meanwhile, Loh et al. [33], in the current popular BOLT-LMM tool, undertook a different Bayesian approach to capture non-infinitesinal genetic architecture, modeling SNP effect sizes by fitting non-Gaussian mixture prior distribution that better models small- and large-effect sizes. In this framework, the test statistic in (4) is derived to be
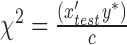 , where
, where 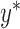 is a vector of residual phenotypes obtained by fitting a Gaussian mixture of priors to the standard LMM and
is a vector of residual phenotypes obtained by fitting a Gaussian mixture of priors to the standard LMM and 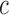 is calibration factor defined so that the LD score regression intercept of the χ2 test statistic under the non-infinitesimal model matches with that under the infinitesimal model (4).
is calibration factor defined so that the LD score regression intercept of the χ2 test statistic under the non-infinitesimal model matches with that under the infinitesimal model (4).
Linear regression-based approaches (Box 1) model the phenotype of an individual as function of fixed effects (which include genotype at the candidate marker, as well as optional covariates such as age, gender and other clinical information). In such models, the inflation in the test statistic can then be controlled by using different methods. These include genomic control, multidimensional scaling, structured association and principal component analysis. These methods are implemented in the GWAS software tools including STRAT [21] and the currently popular PLINK [22] (Supplementary Table S1). Although these methods are effective at controlling inflation in test statistic when the population has structures, they do not suffice in the presence of population substructure [19]. In particular, they do not account for the complete genealogy of all study subjects. LMM (Box 2), an extension of the standard linear regression, partitions the explanatory variables into two groups: fixed effects, which are modeled as parameters that are fixed but unknown, and random effects, which are modeled as being drawn from a random distribution. This provides a powerful method to simultaneously account for various levels of sample structure, including population stratification, family structure and cryptic relatedness. Principally, this is achieved by fitting population structure as a fixed effect and incorporating marker-based kinship information via the variance–covariance structure of the random effect for the individuals [23, 24]. To date, LMM remains the workhorse for association mapping and nearly all-current GWAS tools are based on it. Importantly, not only does LMM provide a control for confounding due to sample structure, but it also increases the statistical power to detect causal variants and enables estimation of heritability explained by genotyped markers [25]. This is achieved by applying a correction that is specific to a given type of sample structure [26, 27].
Box 2: statistical methods in mGWAS.
Diversity metrics (alpha diversity or beta diversity) can be leveraged as phenotype for mGWAS. In mGWAS [35] beta diversity is leveraged as phenotype in GWAS. The beta diversity analysis uses microbiome distance measures such as UniFrac and Bray-Curtis dissimilarity. Let 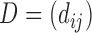 be a beta diversity distance matrix between subjects
be a beta diversity distance matrix between subjects 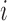 and
and 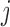 . Let
. Let 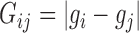 be the genetic distance. The main hypothesis here is that if a SNP is associated with the microbiome (through its distance matrix), then the microbiome distance measure should be smaller for pairs of subjects that have identical genotype at the SNP given SNP. Then, assuming a linear relationship,
be the genetic distance. The main hypothesis here is that if a SNP is associated with the microbiome (through its distance matrix), then the microbiome distance measure should be smaller for pairs of subjects that have identical genotype at the SNP given SNP. Then, assuming a linear relationship,
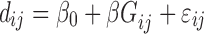 |
where 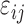 is the environmental effect. If
is the environmental effect. If 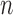 is the number of individuals, then
is the number of individuals, then 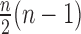 pairs of subjects can be clustered into three groups with genetic distance 0, 1 and 2. The hypothesis
pairs of subjects can be clustered into three groups with genetic distance 0, 1 and 2. The hypothesis 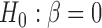 versus
versus 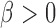 is tested using a score statistic,
is tested using a score statistic, 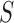 , derived by minimizing
, derived by minimizing 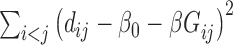 :
:
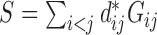 , where
, where 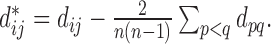 The conditional variance of
The conditional variance of 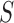 on
on 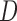 is
is 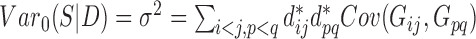 ,
, 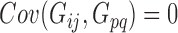 when
when 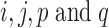 are distinct.
are distinct.
Then the variance-scaled score statistic for testing association of a microbiome distance with a genetic distance across pairs of subjects is then given by 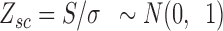 .
.
This model was subsequently extended to incorporate multiple distance matrices so as to improve statistical power.
Lynch et al. [36], proposed an mGWAS method that uses, as phenotype, relative abundance computed at a given taxonomic level. Owing to the high dimension of microbiome relative abundance data, their method applies regularization based on lasso regression, which results in a sparse solution. Host SNPs associated with microbiome are then determined via permutation test, while specific taxa correlated with host genetic variation identified using stability selection [37].
While LMMs has become the method of choice for GWAS, it presents a substantial computational challenge. Methods that compute the exact association test statistics from complete or imputed genotype data certainly provide ‘accurate’ P-values and thus maintain high power. Current software tools using exact computation of the test statistic include EMMA [28], FaST-LMM [29] and more recently GEMMA [23] (Supplementary Table S1). Although the P-values produced by GEMMA are identical to EMMA and FaST-LMM, the algorithm in GEMMA provides a higher efficiency, in terms of per-SNP computational time. It is important to note, however, that even with efficient implementation, the computation of variance component in these tools scale in time of O(mn3), where m is the number of markers and n is the number of individuals, and as such these tools are impractical for analysis of large samples (Box 2) [26]. To circumvent computational cost, several approximation methods have been proposed. These include (i) obviating repetitive estimation of variance components, as implemented in the software tools TASSEL [29] and EMMAX [30] (Supplementary Table S1) and (ii) step-wise implementation [31]: estimation of the residuals from the LMM under the null model and then using these residuals as phenotypes for analysis by the standard linear model (e.g. implemented in software GRAMMAR [32]. This substantially reduces the per-SNP computation time. Another important note is that these approximation-based tools show reduced power at SNPs with small effect sizes. Recently, Loh et al. [33] proposed an efficient approximation method that adapts the LMM by taking a Bayesian perspective and modeling non-infinitesimal genetic architectures via non-Gaussian mixture prior distributions, invoking the fast variational approximation to compute approximate phenotypic residuals. Their methods, implemented in BOLT-LMM and BOLT-REML (Supplementary Table S1), are the current state-of-the-art methods for host GWAS analysis not only in terms of computation time and memory requirement in large cohorts but also, importantly, in terms of the genetic architecture modeled. Apart from the methods proposed by Loh et al. [33], all currently existing tools are based on the infinitesimal model in which all variants are assumed causal with effect sizes following independent Gaussian distribution. In addition, all current tools, as far as we know (Supplementary Table S1), have computational times that scale with the square or cube of sample size, rendering them unfeasible in large data sets. Overall, all these state-of-the-art methods of host GWAS cannot distinguish confounding from polygenicity in the association test [19]. In contrast to host GWAS, methodological development for mGWAS has just begun to emerge. The past 3 years have seen the development of statistical methods for investigating host–microbiome interactions. These methods leverage microbiome features (relative abundance of microbial taxa, alpha diversity, beta diversity or microbial pathway) as a complex trait and determine their correlation with host genetics, by testing either multiple-distance matrices across pairs of subjects or taxa relative abundance [34]. Principally, GWAS can be performed between any given set of genotypes and phenotypes, and thus, although most of mGWAS carried out to date have been limited to the metagenomic level, the framework can similarly be performed at the metatranscriptomic, metaproteomic and metabolomic levels.
The two currently existing mGWAS tools are based on linear regression model as illustrated in Box 3 and Supplementary Table S2 summarizes the functionalities of these mGWAS tools. The 1st mGWAS tool is mGWAS [35]. It is a statistical framework for identifying host genetic variants associated with microbiome beta diversity with or without interacting with environmental factors and corrects for skewness and kurtosis. The 2nd mGWAS tool, HOMINID [36], which is based on Lasso regression, identifies associations between host SNPs and microbiome taxa. Additionally, by using Lasso regression plus stability selection with randomized Lasso, this tool enables identification of microbial taxa that are correlated with specific host SNPs.
Box 3: some unsolved challenges in host and microbiome GWAS.
Statistical methodologies for host-based and mGWAS for identifying causal factors in the observed associations between the environment, host genetics, the microbiome, and complex phenotypes
Determining the direction of causality in the interplay between host genetics and microbiome in complex traits
Determining the impact of host epigenetics on microbiome features
Accounting for interaction among microbial species, in identifying specific microbial taxa associated with host genetic variation
Correcting for confounders arising from the multiple factors that modulate the microbiome composition
Modelling the true genetic architecture of complex traits
Accounting for the missing heritability in host GWAS
Limitations in current methods and tools for host and mGWAS
Although substantial progress in the development of methods and tools for host genome-wide association mapping has occurred over the past decade and many new tools continue to be unveiled, many significant challenges need to be addressed before the gap between statistical association and biological association can be narrowed. Similarly, initial foray into mGWAS using custom-made mGWAS tools or ported host GWAS tools have illuminated several important microbiome-associated host genetic polymorphisms. Even so, however, several methodological limitations and pitfalls exit in using these tools. Box 1 provides some challenges in host and mGWAS.
Most current host GWAS tools are built on the infinitesimal genetic architecture, which makes the implicit assumption that all variants are causal with small-effect sizes independently drawn from Gaussian distributions. For complex traits, it is now known, however, that only a small proportion of the genetic variants are actually causal [17]. Thus, employing this assumption clearly limits power. Bayesian techniques have been invoked to incorporate non-infinitesimal genetic architecture [33, 34]. However, the prior on marker effect sizes is assumed to follow independent Gaussian distributions. The validity of the Gaussian assumption, according to the central limit theory, requires sufficiently large sample sizes that may not necessarily be the case in GWAS analyses. This concern can be even greater for the case of ascertained case–control studies because in such cases the distributions of genetic effects are no longer Gaussian [37] and the independence between genetic effects and environmental effects is lost due to disproportionate sampling of cases and controls [38]. In the same light, methods and tools so far developed to leverage the microbiome as a complex quantitative trait for association mapping assumes the infinitesimal genetic architecture and do not incorporate the effect of genetic interactions on the microbiome. Early insights gained from the mGWAS studies suggest that only a few host loci interact with the microbiome; thus, a more realistic non-infinitesimal genetic architecture could be modeled by taking a Bayesian approach and incorporating small and large effect size loci using suitable distributions. However, the question of how to choose the appropriate distribution and/or incorporate the (possibly) different distributions of marker-effect sizes (because not all markers were created equal) remains open for future research. In addition, existing methods [34] that leverages multiple microbiome beta-diversity distance matrices apply the strongest association, defined as the highest P-value from the set of P-values obtained from each distance matrix, to evaluate significance. Statistical techniques that allow to rescale the overall statistic threshold by effect of distance matrices should provide increased power. Here methods such as truncated product method [39] could be employed. Meanwhile, aptly approaches to tackle the complexity implied by the multi-dimensional interactions among genetic loci would perhaps include recursive partitioning method, multifactor dimensionality reduction or Bayesian technique.
It must be pointed out that another great limitation of current host and mGWAS methods alike is the absence of robust models for interactions of host genetics and the microbiome with the environment. While significant progress has been achieved in this direction for host GWAS, no appreciable stride has been made on the mGWAS side. The plasticity of microbiome data to a plethora of environmental factors makes realistic investigation of microbiome–environment interactions an extremely difficult problem. A 1st step toward addressing this issue will be to define a ‘gold standard’ for statistical methods that will be developed for such complex interactions. Of course, given that complexity of the microbiome, in terms of dimensionality and features (zero inflated, over dispersed and multiple outliers), any such models that incorporate environmental factors is likely to be computationally intractable. In such a case, likelihood free methods such as approximate Bayesian computation could be adopted. These limitations and challenges in current DSS frameworks consequently impact the progress toward a complete understanding of the nature association between host genetics, gene regulation and microbiome.
Dilemma in risk factors: host genetics, gene regulation and microbiome causality
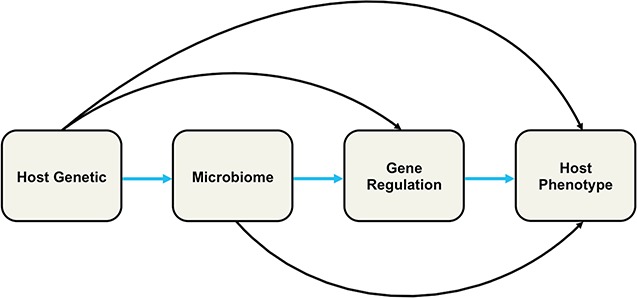
The results of several host GWAS conducted over the years have provided several insights into the biological processes underlying many diseases [40]. Several genes, pathways and regulatory networks have been identified for a number of complex diseases, including cardiovascular diseases, type 2 diabetes and cancer [41, 42]. While the pathogenesis and nature of the aberrant activities in these pathways and regulatory networks is coming into view for some complex diseases, the apparent molecular circuitry remains generally elusive for most traits. Meanwhile, initial forays into mGWAS have already demonstrated the intimate link between host genetic polymorphisms and microbiome attributes [43, 44]. For example, a pioneering study by Blekhman et al. using samples from 93 individuals identified multiple host loci associated with changes in abundance of microbial taxa [5]. Intriguingly, some of the loci detected in this and other mGWAS carried out to date overlap with several expression quantitative trait loci that have previously been identified across multiple tissues [38]. Moreover, this link is observed to be tissue specific [45] and enriched with specific human proteins [43]. These discoveries suggest the likely influence of host gene regulation on specific tissues that interact with the microbiome. However, little is known about the interplay between host genetic variation, microbiome composition and host gene expression and how this impacts host traits (Figure 3).
Figure 3.
Possible direction of interaction between host genetic, microbiome and gene regulation on host phenotype.
Several challenges remain toward establishing causality. These association results are often limited to correlations and may generally end up identifying consequential changes rather than the true risk factors that may lead to establish genetics causality [13, 2]. For example, the predictive power of disease risk remains poor because current identified variants account for only a small proportion of additive genetic variation [2]. Consequently, current findings from disease scoring approaches have not yet had major impact on therapeutic optimization for the majority of complex traits. Moreover, it has become apparent that changes in gene regulation are at the center stage of biological mechanisms underlying most associations, and yet our current knowledge of gene regulation is still limited [46]. Without a comprehensive knowledge of gene regulation and the paucity in available tools for studying regulation, the transition from statistical associations to biological insights (biological mechanisms, the particular genes involved and the direction of causality) remains a challenge. Nevertheless, significant advances in technology coupled with multi-omic approaches, which is able to simultaneously capture millions of data points, will enable system-wide examination of complex interactions in biological system [47].
Omic data integration
The multi-omic approach involves understanding the complexity of the living as a whole by incorporating data at multiple biological levels: from gene sequencing to protein expression and metabolic structures [48]. Therefore, these data cover wide range of the biological information involved in the variations that occur in the genes and cellular networks and influence the functioning of organic systems in their entirety [48]. This approach has mainly been driven by the surge in high-throughput sequencing efforts over the past two decades. Multi-omic approach will be able to dramatically improve our way of analyzing the biological system, which relies on a single-omic model, that offers limited insight into the complex and dynamic nature of biological networks and their association with environment factors. Integrating data from all these omic levels will be useful for identifying new biomarkers, generating new knowledge and/or developing new diagnostic tools. The integration of multi-omic data with GWAS will bring more insight in the causal factors and the interactions between the host, microbiome and environment. However, multi-omics is still in its infancy [47, 49] and requires large-scale studies that need state-of-the-art tools. So far, many integrative multi-omic tools have been developed [50]. Multi-omic method, however, poses significant challenges in terms of the analysis approach, the statistical methods and the interpretation of these numerous data [51]. One of the most important challenges is the difficulty of representing existing knowledge about the molecular processes involved in many complex diseases, given that biological systems are a myriad of complex connections and are closely linked to their evolutionary history. Another fundamental challenge is that of matching the heterogeneous data from different methods and platforms. It requires a synchronization of huge amount of data that vary in data format, thus potentially will add bias and noise to data integration processes. This step can use various strategies either merging the different data coming from the same subjects or trying to homogenize the data. However, it is very critical because matching the data must not only put the data in the same files but also bring a new meaningful knowledge. In addition, incorporating various omic data each of which typically contains many measurement errors raises the issue of quality control. This is compounded by the fact that the measured molecular data are prone to bias arising from samples preparation and processing. Another challenge is analyzing the data of huge size and of different classifications (molecular data, measurement features, technology used, biological samples, type of study, etc.). Therefore, bioinformatics approaches and pipelines and their associated mathematical models need to address the dimensionality and heterogeneity of the data. In doing so, machine-learning approaches may be more suitable for data integration and related modeling, as they provide a robust way of leveraging hidden knowledge from various omic data types to improve analyses. On the other hand, topological data analysis methods that examine the shape of data with geometric dimensional conversions [52] can also be suitable in finding hidden patterns compared to other standards methods such as correlation-based analysis [53, 54] and unsupervised data integration (matrix factorization methods, Bayesian methods, network-based methods, multiple kernel learning and multi-step analysis) [53, 55]. The other approach to explore is the complex network-based approaches, which may be worthy of exploration to efficiently handle the multi-omic data deluge [53].
Role of bioinformatics in the era of multi-omics
Considering the molecular variation of biomarkers, the rapid growth in large-scale omic technologies opens windows for global views of biological system in a holistic hypothesis-driven manner. Integrative approaches need to be designed and applied to various levels of biological information to comprehend the pathogenesis of complex diseases [49]. Bioinformatic approaches, resources and computational biology tools are at the center for the advancement in implementing real-time multi-omic integration and health care analytics. Bioinformatics is arguably a recent field but has substantially contributed to the advancement in the modernization of computational techniques and capacity to handle the amount of biological data generated by genome sequencing and variation studies [1, 10, 15]. Bioinformatics currently plays a critical role in deciphering omic data and organizing information in all aspects of independent omic layers. While bioinformatics and computational biology tackle challenges raised from each independent omic data type in the past decades [15, 49], today multi-omic era has raised further challenges in integrating various levels of biological information [47, 49]. Furthermore, analyzing and interpreting such integrative biological information demand outstrips supply and further bioinformatics and computational biology capacities are needed. This issue raises the need to strengthen the multidisciplinary nature of bioinformatics and education. In doing so, this will have a critical impact on the discovery of multi-omic diagnostics, biomarkers, clinical decision-making and data-driven medicine.
Conclusions and perspectives
The substantial role of host genetics on the microbiome and on host phenotypes has been identified using the wealth of available DSS tools. Although many of these methods and tools have been effective at uncovering the genetic basis of many complex traits, they have potentially missed out many novel genetic variants and/or failed to disentangle true signals from spurious associations owing to limitations in the underlying single-omic statistical models. Given the current experimental observations of the strong influence of host genetics on the microbiome and the role of the microbiome on host phenotypes, it has become increasingly apparent that integrating the microbiome in host GWAS will reveal many important insights and it will be a strong 1st step toward establishing causality. A critical challenge facing the host genetics and microbiome field is the identification of genetics risk factors with strong effect to allow the establishment of the direction of causality in the observed associations between the environment, host genetics, microbiome and complex diseases. A complete solution for this issue would be one of the major breakthroughs for a century-long problem on understanding human variation relevant.
The integrative analysis may be the key to better understand the role and mechanisms of host genetics, microbiome and environment in the manifestation of many complex diseases (Figure 2). This will ultimately revolutionize therapeutics, driving the era of precision medicine. The major challenge for this integration lies in the development of novel statistical techniques and multi-omic data integration. The integrative analyses will throw bioinformatics and human genetic studies into a brand-new era, quickening the pace of movement from population level to individual level understanding of complex human traits and diseases.
It will be interesting to perform simulations to examine the power of various current host and mGWAS methods and to investigate their performance under various important factors, such as sample size, marker effect size, host genetic correlations, population structure and microbial interactions. Such simulations will not only highlight current methodological limitations but also guide development and validation of future association tools. In brief, we discussed DSS approaches in host GWAS and in mGWAS, outlining their associated methods and tools. We further discussed the limitations of these methods and highlighted the dilemma in dissecting causality between host genetic, microbiome and environment. Finally, we underscored the importance of integrating multi-omic data with GWAS and outlined some of the challenges in this data integration. We believe that this paper may motivate the development of new methods for mGWAS.
Key Points
Discussing issues related to host and mGWAS
Outlining current methods and tools available for host and mGWAS
Discussing the importance of integrative multi-omic approaches in understanding causal factors and the direction of their effects on host phenotypes
Supplementary Material
Acknowledgements
We are grateful to Oyekanmi Nashiru and Hassan Ghazal for helpful discussions. Computations were performed using facilities provided by CHPC (https://www.chpc.ac.za/).
Awany Denis received his MSc degrees from Makerere University and African Institute for Mathematical Sciences. He is a PhD student at the University of Cape Town, South Africa.
Imane Allali received her PhD degree in bioinformatics from Mohammed V University in Rabat, Morocco. She is a postdoctoral fellow at the Computational Biology Division, University of Cape Town, South Africa.
Emile R. Chimusa received his PhD degree in bioinformatics from University of Cape Town. He is a senior lecturer at the Division of Human Genetics, Department of Pathology, University of Cape Town, South Africa.
Funding
This work was funded in part by DAAD, the German Academic Exchange Programme, under grant number 91653117. Some of the authors are also funded in part by the National Institutes of Health Common Fund under grant numbers 1U54HG009790–01(IFGeneRA), U01HG009716 (HI Genes Africa), U24HG006941 (H3ABioNet) and 1U01HG007459–01 (SADaCC) and by Wellcome Trust/AESA under grant number H3A/18/001. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
References
- 1. Zhang J, Chiodini R, Badr A, et al. The impact of next-generation sequencing on genomics. J Genet Genomics 2011;38(3):95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Visscher PM, Brown MA, McCarthy MI, et al. Five years of GWAS discovery. Am J Hum Genet 2012;90(1):7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goodrich JK, Davenport ER, Clark AG, et al. The relationship between the human genome and microbiome comes into view. Annu Rev Genet 2017;51:413–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sandoval-Motta S, Aldana M, Martínez-Romero E, et al. The human microbiome and the missing heritability problem. Front Genet 2017;8:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blekhman R, Goodrich JK, Huang K, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol 2015;16(1):191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cho, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet 2012;13(4):260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome. Nat Rev Microbiol 2016;14(8):508. [DOI] [PubMed] [Google Scholar]
- 8. Richards A, Muehlbauer L, Alazizi A, et al. Gut microbiota composition impacts host gene expression by changing chromatin accessibility. 2018bioRxiv. doi: 10.1101/210294 10.1101/210294. [DOI] [Google Scholar]
- 9. Hall B, Tolonen AC, Xavier RJ. Human genetic variation and the gut microbiome in disease. Nat Rev Genet 2017;18(11):690. [DOI] [PubMed] [Google Scholar]
- 10. Kohl M, Megger DA, Trippler M, et al. A practical data processing workflow for multi-OMICS projects. Biochim Biophys Acta 2014;1844(1):52–62. [DOI] [PubMed] [Google Scholar]
- 11. Frazer KA, Murray SS, Schork NJ, et al. Human genetic variation and its contribution to complex traits. Nat Rev Genet 2009;10(4):241. [DOI] [PubMed] [Google Scholar]
- 12. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017;101(1):5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peprah E, Xu H, Tekola-Ayele F, et al. Genome-wide association studies in Africans and African Americans: expanding the framework of the genomics of human traits and disease. Public Health Genomics 2015;18(1):40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet 2010;11(12):843. [DOI] [PubMed] [Google Scholar]
- 15. Shameer K, Badgeley MA, Miotto R, et al. Translational bioinformatics in the era of real-time biomedical, health care and wellness data streams. Brief Bioinform 2016;18(1):125–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smits SA, Leach J, Sonnenburg ED, et al. Seasonal cycling in the gut microbiome of the Hadza hunter—gatherers of Tanzania. Science 2017;357(6353):802–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zuk O, Hechter E, Sunyaev SR, et al. The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 2012;109(4):1193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am J Hum Genet 2010;86(1):6–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang J, Zaitlen NA, Goddard ME, et al. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet 2014;46(2):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pasaniuc, Price AL. Dissecting the genetics of complex traits using summary association statistics. Nat Rev Genet 2017;18(2):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Porras-Hurtado L, Ruiz Y, Santos C, et al. An overview of STRUCTURE: applications, parameter settings, and supporting software. Front Genet 2013;4:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Purcell S., Neale B., Todd-Brown K., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81(3): 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet 2012;44(7):821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen H, Wang C, Conomos MP, et al. Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. Am J Hum Genet 2016;98(4):653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang J, Lee SH, Goddard ME, et al. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011;88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Price L, Zaitlen NA, Reich NA, et al. New approaches to population stratification in genome-wide association studies. Nat Rev Genet 2010;11(7):459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Golan D, Rosset S. Mixed models for case-control genome-wide association studies: major challenges and partial solutions. In: Borgan ∅, Breslow N, Chatterjee N, et al. (1st edn). Handbook of Statistical Methods for Case-Control Studies. Boca Raton, FL: Chapman and Hall/CRC2018:495–514.
- 28. Yu J, Pressoir G, Briggs WH, Bi IV, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 2006;38(2):203. [DOI] [PubMed] [Google Scholar]
- 29. Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet 2012;44(7):821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aulchenko YS, De Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics 2007;177(1):577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kang HM, Sul JH, Service SK, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet 2010;42(4):348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Golan ES, Lander, Rosset S. Measuring missing heritability: inferring the contribution of common variants. Proc Nat Acad Sci U S A 2014;111(49)E5272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Loh P-R, Tucker G, Bulik-Sullivan BK, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet 2015;47(3):284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hua X, Song L, Yu G, et al. MicrobiomeGWAS: a tool for identifying host genetic variants associated with microbiome composition. 2015BioRxiv:031187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lynch J, Tang K, Priya S, et al. HOMINID: a framework for identifying associations between host genetic variation and microbiome composition. GigaScience 2017;6(12):1–7.doi: 10.1093/gigascience/gix107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao N, Chen J, Carroll IM, et al. Testing in microbiome-profiling studies with MiRKAT, the microbiome regression-based kernel association test. Am J Hum Genet 2015;96(5):797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Günther F, Wawro N, Bammann K. Neural networks for modeling gene–gene interactions in association studies. BMC Genet 2009;10(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koch L. Complex disease: a global view of regulatory networks. Nature Rev Genet 2016;17(5):252.27040485 [Google Scholar]
- 39. Shu L, Chan KHK, Zhang G, et al. Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet 2017;13(9):e1007040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gao L, Uzun Y, Gao P, et al. Identifying noncoding risk variants using disease-relevant gene regulatory networks. Nat Commun, 2018;9(1):702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goodrich JK, Davenport ER, Clark AG, et al. The relationship between the human genome and microbiome comes into view. Annu Rev Genet 2017;51:413–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davenport ER, Cusanovich DA, Michelini K, et al. Genome-wide association studies of the human gut microbiota. PLoS One 2015;10(11):e0140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huttenhower C, Gevers D, Knight R, et al. Structure, function and diversity of the healthy human microbiome. Nature 2012;486(7402):207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Price AL, Spencer CC, Donnelly P. Progress and promise in understanding the genetic basis of common diseases. Proc Biol Sci 2015;282(1821):20151684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Palsson B, Zengler K. The challenges of integrating multi-omic data sets. Nat Chem Biol 2010;6(11):787. [DOI] [PubMed] [Google Scholar]
- 46. Ritchie MD, Holzinger ER, Li R, et al. Methods of integrating data to uncover genotype–phenotype interactions. Nat Rev Genet 2015;16(2):85. [DOI] [PubMed] [Google Scholar]
- 47. Hasin Y, Seldin M and Lusis A. Multi-omics approaches to disease. Genome Biol 2017;18(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang S, Chaudhary K, Garmire LX. More is better: recent progress in multi-omics data integration methods. Front Genet 2017;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Palsson B, Zengler K. The challenges of integrating multi-omic data sets. Nat Chem Biol 2010;6(11):787. [DOI] [PubMed] [Google Scholar]
- 50. Zhong S, Jiang D, McPeek MS. CERAMIC: case–control association testing in samples with related individuals, based on retrospective mixed model analysis with adjustment for covariates. PLoS Genet 2016;12(10):e1006329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu G, Gail MH, Consonni D, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol 2016;17(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yoo S, Huang T, Campbell JD, et al. MODMatcher: multi-omics data matcher for integrative genomic analysis. PLoS Comput Biology 2014;10(8):e1003790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet 2010;42(4):355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lippert C, Listgarten J, Liu, et al. FaST linear mixed models for genome-wide association studies. Nat Methods 2011;8(10):833. [DOI] [PubMed] [Google Scholar]
- 55. Bradbury PJ, Zhang Z, Kroon DE, et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 2007;23(19)2633–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.