

Abstract
Background and Purpose
A cholecystokinin (CCK) system has been identified in white adipose tissue (WAT). Nevertheless, the endocrine actions of CCK on WAT remain unknown. Our goal was to investigate the role of CCK in regulating the production of adiponectin, an adipokine expressed in WAT, which is pivotal in preserving energy homeostasis.
Experimental Approach
The effect of the bioactive CCK fragment CCK‐8 on adiponectin production was studied both in vivo and in vitro. CCK‐8 effects were characterized in rats treated with selective CCK1 and CCK2 receptor antagonists as well as in pre‐adipocytes carrying the selective silencing of either CCK1 or CCK2 receptors. The influence of insulin on CCK‐8 responses was also analysed.
Key Results
In WAT, CCK‐8 increased plasma adiponectin levels and the expression of the adiponectin gene (Adipoq). In pre‐adipocytes, CCK‐8 up‐regulated adiponectin production. CCK‐8 effects were abolished by L‐365,260, a selective CCK2 receptor antagonist. CCK2 receptor knockdown also abolished the effects of CCK‐8 in pre‐adipocytes. Moreover, in vitro CCK‐8 effects were blocked by triciribine, a specific inhibitor of protein kinase B (Akt) and by the PPARγ antagonist T0070907. Silencing the expression of the insulin receptor inhibited CCK‐8‐induced Adipoq expression in pre‐adipocytes. Furthermore, insulin potentiated the effect of CCK‐8.
Conclusion and Implications
CCK‐8 stimulates adiponectin production in WAT by acting on CCK2 receptors, through a mechanism involving both Akt and PPARγ. Moreover, CCK‐8 actions are only observed in the presence of insulin. Our results could have translational value in the design of new insulin‐sensitizing therapies.
Abbreviations
- CCK
cholecystokinin
- Sc‐WAT
subcutaneous white adipose tissue
- Vis‐WAT
visceral white adipose tissues
- WAT
white adipose tissue
What is already known
Both CCK1 and CCK2 receptors are expressed in white adipocytes.
CCK receptors share signalling pathways with insulin receptors.
What this study adds
Stimulation of CCK2 receptors induces adiponectin gene (Adipoq) expression in adipocytes and promotes adiponectin production.
The activation of CCK2 receptors is ineffective in the absence of insulin.
What is the clinical significance
CCK2 receptor agonists could have translational value in the design of new insulin‐sensitizing therapies.
1. INTRODUCTION
Cholecystokinin (CCK) is a postprandial gut hormone that stimulates the activity of the exocrine pancreas (Singer, 1987), inhibits gastric emptying (Debas, Farooq, & Grossman, 1975), and promotes short‐term satiety by acting on CCK1 receptors located both in abdominal vagal afferents and in brainstem areas (Beglinger & Degen, 2004). The CCK2 receptors (also previously called gastrin receptors) are widely expressed in the CNS (Dufresne, Seva, & Fourmy, 2006) and, in the periphery; these receptors play a major role in maintaining the normal function of gastric mucosa (Chen et al., 2002). A recent study carried out in our laboratory has shown that activation of CCK2 receptors facilitates the uptake of dietary triglycerides by white adipocytes, suggesting that CCK is involved in regulating homeostasis in white adipose tissue (WAT) (Plaza, Merino, Cano, et al., 2018; Plaza, Merino, Sánchez‐Pernaute, et al., 2018). Related with that, studies carried out by Flatt's group have provided evidence of the effect of CCK in alleviating symptoms of insulin resistance (Irwin et al., 2012; Irwin, Montgomery, O'harte, Frizelle, & Flatt, 2013), which supports the concept of an insulin/CCK drive of energy metabolism.
Adiponectin, which is the most abundant adipokine, plays a relevant role in glucose and lipid metabolism (Havel et al., 1996). In particular, plasma concentration of this hormone positively correlates with insulin sensitivity (Stern, Rutkowski, & Scherer, 2016), especially in obese individuals (Kadowaki et al., 2006; Kantartzis et al., 2005). Moreover, low adiponectin levels are often found in individuals with insulin resistance as well as in patients with Type 2 diabetes mellitus (Yatagai et al., 2003) and are now considered as an independent risk factor for diabetes and insulin resistance as well as for cardiovascular disease (Kumada et al., 2003). Also, the adiponectin/leptin ratio has been proposed as a valuable index to estimate WAT dysfunction and obesity‐associated cardiometabolic risk (Frühbeck, Catalán, Rodríguez, & Gómez‐Ambrosi, 2018). Mechanisms regulating adiponectin production continue to be a matter of debate (Fang & Judd, 2018). Thus, some studies have shown that insulin induces adiponectin gene expression and secretion (Pereira & Draznin, 2005), while others have reported an inhibition (Fasshauer, Klein, Neumann, Eszlinger, & Paschke, 2002). Glucocorticoids and TNF‐α have been shown to inhibit adiponectin expression (Degawa‐Yamauchi et al., 2005). In the case of leptin, this hormone has also been shown to increase adiponectin mRNA and protein (Singh et al., 2016). Nevertheless, the meaning of this finding is difficult to interpret as, unlike other adipokines, both adiponectin production and plasma concentration are often inversely associated with blood leptin levels (B. Lee & Shao, 2014). Hence, mechanisms regulating adiponectin production are a topic of research of increasing interest.
Taking into account (a) the role of CCK in regulating WAT homeostasis (Plaza, Merino, Cano, et al., 2018), (b) the existence of a complete CCK system in WAT (Plaza, Merino, Sánchez‐Pernaute, et al., 2018), and (c) the already reported interaction between CCK and insulin in driving energy metabolism (Irwin et al., 2012; Irwin et al., 2013; Pathak, Flatt, & Irwin, 2018), we aimed at investigating the effect of the C‐terminal bioactive fragment of CCK, CCK‐8, on both adiponectin gene (Adipoq) expression and adiponectin production in rat WAT.
2. METHODS
2.1. In vivo studies
2.1.1. Animals
All animal care and experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals (European Communities Council Directive 86/609/EEC) and were approved by the Ethics Committee of the Universidad CEU—San Pablo (refs Py103‐15 and PROEX035/16). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. The in vivo studies were carried out in adult male Sprague–Dawley rats during the nocturnal period of the circadian cycle, under similar experimental conditions to those used in our laboratory to investigate the role of CCK‐8 on other aspects of energy metabolism (Merino, Cano, Guzman, Somoza, & Ruiz‐Gayo, 2008; Merino, Somoza, Ruiz‐Gayo, & Cano, 2008; Plaza, Merino, Cano, et al., 2018). The dose of CCK‐8 (10 μg·kg−1) was chosen on the basis of previous studies (Merino, Cano, et al., 2008; Plaza, Merino, Cano, et al., 2018). This dose did not significantly increase total CCK immunoreactivity in plasma and was able to activate CCK receptors in adipocytes (Plaza, Merino, Cano, et al., 2018). The time of CCK‐8 administration (10 p.m.) was selected because CCK‐8 has been shown to be more effective when administered earlier in the night (Kraly, 1981). Both plasma and WAT samples used in the current research were obtained from rats used in a recently published study (Plaza, Merino, Cano, et al., 2018) in which group sizes (n = 5–6) were calculated on the basis of an expected 20% reduction of food intake at the dose of 10‐μg·kg−1 CCK‐8, a statistical power >85%, and P < .05. The details of both the acute and chronic treatments with CCK‐8 have been provided in an earlier publication (Plaza, Merino, Cano, et al., 2018).
2.1.2. Pharmacological treatment
Treatment protocols are illustrated in Figure 1. Briefly, for acute treatment with CCK‐8, 14‐week‐old male Sprague–Dawley rats (Harlan, Spain; 400–450 g) were individually housed under 12 hr light/12 hr dark (22°C; lights on at 08:00hr) with standard rodent chow (Teklad; 14% of energy from fat; 3.1 kcal·g−1; Harlan, Spain) and water ad libitum. The day of the experiment, rats were fasted from 09:00 until 18:00 and then fed ad libitum. In order to minimize the number of rats, the experiment was organized in sequential assays that were carried out at 1‐week interval in the same animals, which were randomly assigned in each assay to either intervention or control groups. Operators were blinded to treatment assignment. Sulfated CCK‐8 was selected as this CCK fragment has been shown to be the most abundant form of CCK in rat and human plasma (Izzo, Brugge, & Praissman, 1984) that preserves the biological properties of full‐size CCK (Miller & Gao, 2008). (1) The first assay aimed at identifying the CCK receptor subtype involved in the effect of 10‐μg·kg−1 CCK‐8 on plasma adiponectin levels. This dose of CCK‐8 was chosen on the basis of previous studies aimed at identifying the role of CCK on the regulation of both feeding behaviour and energy balance (Cano, Merino, Ezquerra, Somoza, & Ruiz‐Gayo, 2008; Merino, Cano, et al., 2008; Plaza, Merino, Cano, et al., 2018). Animals received at 21:30hr either saline (n = 12), L‐365,260 (a CCK2 receptor antagonist; 1 mg·kg−1; n = 12), or SR‐27,897 (a CCK1 receptor antagonist; 0.3 mg·kg−1; n = 12) subcutaneously. Thirty minutes later, rats were treated intraperitoneally with either saline (saline + saline, n = 6; L‐365,260 + saline, n = 6; SR‐27,897 + saline, n = 6) or 10‐μg·kg−1 CCK‐8 (saline + CCK‐8, n = 6; L‐365,260 + CCK‐8, n = 6; SR‐27,897 + CCK‐8, n = 6). At 24:00hr, a sample of blood was obtained from the caudal vein after application of a mixture of lidocaine and prilocaine (EMLA; Figure 1a). (2) The second assay was designed to quantify adiponectin gene expression in adipose tissues and was performed in rats that were randomly assigned to receive either saline (n = 10) or L‐365,260 (1 mg·kg−1; n = 10) at 21:30hr. Five animals of each group received either saline or CCK‐8 (10 μg·kg−1) at 22:00hr. Animals were killed by decapitation, under inhalational anaesthesia with isoflurane, at 24:00hr, and visceral (Vis‐WAT) and subcutaneous adipose tissues (Sc‐WAT) were dissected and frozen in liquid nitrogen. Treatment with SR‐27,897 was omitted in this case as this antagonist failed to modify plasma adiponectin levels in Experiment 1 (Figure 1b).
Figure 1.
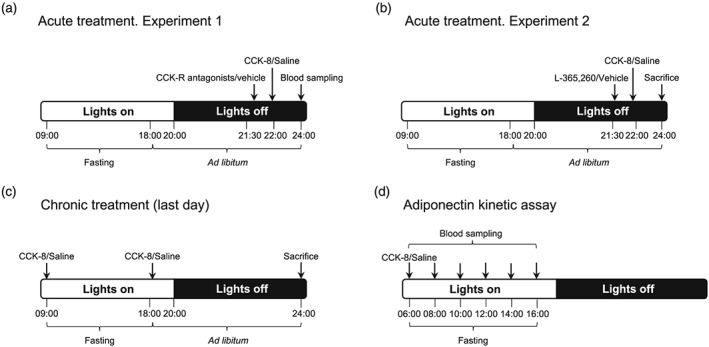
Experimental protocols used to analyse the effects of CCK‐8 on adiponectin production (a–c), and the time course of the effect of CCK‐8 on plasma adiponectin (d)
For chronic treatment, 12‐week‐old male Sprague–Dawley rats underwent chronic treatment (12 days; twice daily, at 09:00hr and 18:00hr) with either saline (n = 6) or CCK‐8 (n = 6, 10 μg·kg−1). On Day 12 of the treatment, animals were decapitated at 24:00hr, under inhalational anaesthesia with isoflurane (Figure 1c).
2.2. Plasma adiponectin kinetics
Another group of 14‐week‐old male Sprague–Dawley rats was used to characterize the time course of CCK‐8 on plasma adiponectin levels. Rats were treated with either saline (n = 6) or CCK‐8 (n = 6, 10 μg·kg−1) at 06:00 hr, food was removed, and blood samples were obtained by incision of the tail vein at 2‐hr intervals until 16:00 hr (Figure 1d). Sampling was performed under local anaesthesia with EMLA.
2.3. Plasma biochemistry
Glucose (GTM, Roche, Spain), triglycerides (Biolabo, France), and non‐esterified free‐fatty acids (NEFA; Wako Bioproducts, USA) were measured by spectrophotometric methods. Both plasma leptin and insulin were quantified by means of EIA. Data corresponding to these analyses have been already published by us (Plaza, Merino, Cano, et al., 2018) and are provided as Supporting Information. Plasma adiponectin (Abcam, UK) was analysed by EIA.
2.4. Isolation of adipose stromal vascular cells and differentiation of pre‐adipocytes
The procedure was carried out as previously described (Gil‐Ortega et al., 2013; Plaza, Merino, Cano, et al., 2018). Briefly, Vis‐WAT and Sc‐WAT samples from 12‐week‐old untreated Sprague–Dawley rats, provided by the animal facility of the Universidad CEU—San Pablo, were chopped and then incubated for 1 hr in α‐MEM containing 13.6‐U·ml−1 type NB4 collagenase (Serva, Germany) and 20‐μg·ml−1 type I DNAse (Roche, Spain). After enzymic digestion, samples were centrifuged at 300 x g (10 min, 25°C) and supernatants containing adipocytes discarded. The pellets containing the stromal vascular fraction were resuspended in 1‐ml α‐MEM, then filtered through a 37‐μm mesh, and centrifuged at 300 x g (10 min, 25°C). Pellets were treated with erythrocyte lysis buffer (155‐mM NH4Cl, 5.7‐mM K2HPO4, 0.1‐mM EDTA, pH = 7.3), then re‐centrifuged under identical conditions, resuspended in α‐MEM supplemented with 10% newborn calf serum (NCS, Sigma, Spain), 10‐U·ml−1 penicillin–streptomycin (Life Technologies, Spain), and 0.25‐μg·ml−1 amphotericin B (Life Technologies), plated in 12‐well plates (40,000 cells·cm−2), and cultured during 5–7 days. Differentiation of adipose‐derived stem cells into pre‐adipocytes was induced in differentiation medium (α‐MEM, containing 2% NCS, 66‐nM insulin, 1‐nM triiodothyronine, 1‐mM dexamethasone, 0.3‐mM rosiglitazone, and 10‐mg·ml−1 apotransferrin) for 7–9 days.
2.5. Determination of adiponectin levels in pre‐adipocytes treated with CCK‐8
Visceral and subcutaneous pre‐adipocytes were treated with CCK‐8 (10−6 M) during 4 hr. After this time, the medium was replaced by CCK‐free differentiation medium, and cells were collected immediately (CCK‐8 group, n = 5), as well as after 1 hr (CCK‐8 + 1 hr group, n = 5) or 2 hr (CCK‐8 + 2 hr group, n = 5). A control group (n = 5) without CCK‐8 treatment was used. Cells were homogenized in ice‐cold buffer containing 0.42‐M NaCl, 20‐mM HEPES (pH 7.9), 1‐mM Na4P2O7, 1‐mM EDTA, 1‐mM EGTA, 1‐mM DTT, 20% glycerol, 1‐mg·ml−1 aprotinin, 1‐mg·ml−1 leupeptin, 20‐mM sodium fluoride, 1‐mM trisodium orthovanadate, and 2‐mM phenylmethylsulfonyl fluoride. The homogenates were frozen at −80°C, thawed at 37°C three times, and then centrifuged for 10 min at 4°C. Equal amounts of protein (50 μg) were mixed with Laemmli buffer (50‐mM Tris pH = 6.8, 10% sodium dodecyl sulfate, 10% glycerol, 5% mercaptoethanol, and 2‐mg·ml−1 blue bromophenol), then loaded on an SDS‐PAGE gel, and submitted to electrophoresis. Proteins were transferred to nitrocellulose membranes (GE Healthcare, Spain) by using a Transblot apparatus (Bio‐Rad, Spain). The membranes were blocked with 5% dried skimmed milk powder in Tween‐PBS for 1 hr.
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Primary mouse monoclonal antibody against full‐length human adiponectin (ab22554; Abcam) was applied (at a dilution of 1:1,000) overnight at 4°C. After washing, an appropriate anti‐mouse secondary antibody (sc‐516102; Santa Cruz Biotechnologies, USA; dilution 1/5,000) was added for 1 hr at room temperature. Blots were washed, incubated in chemoluminescence reagents (ECL Prime; GE Healthcare), and bands detected using the ChemiDoc XRS+ Imaging System (Bio‐Rad). In order to check the equal loading of samples, blots were re‐incubated with β‐actin monoclonal antibody (Affinity Bioreagents, USA). All immunoblotting procedures follow the editorial on immunoblotting and immunohistochemistry (Alexander et al., 2018).
2.6. Analysis of Adipoq expression by RT‐qPCR
Visceral and subcutaneous pre‐adipocytes were incubated with CCK‐8 (10−7–10−5 M) for 2 hr, then washed with PBS, and preserved with Trizol (n = 5; samples were run in duplicate). Total RNA was extracted by using the Tri‐Reagent protocol (Life Technologies). cDNA was then synthesized from 1‐μg total mRNA by using a high‐capacity cDNA RT kit (Bio‐Rad). Quantitative RT‐PCR was performed by using designed primer pairs (Integrated DNA Technologies, USA; see Table 1). SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad) was used for amplification according to the manufacturer's protocols in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, USA). All gene values were normalized to the housekeeping gene 18s and β‐Actin. The ∆∆C(T) method was used to determine relative expression levels of rat genes. Statistics were performed using the ∆∆C(T) method (Livak & Schmittgen, 2001).
Table 1.
Designed primer pairs used in this study
| mRNA | Forward | Reverse |
|---|---|---|
| Rat Adipoq | 5′‐CCCTCCACCCAAGGAAACTT‐3′ | 5′‐CTTCCGCTCCTGTCATTCCA‐3′ |
| Rat Pparg | 5′‐CGTTCACAAGAGCTGACCCAATGG‐3′ | 5′‐ATAGTGGAAGCCTGATGCTTTATCCC‐3′ |
| Rat Il6 | 5′‐AGAGACTTCCAGCCAGTTGC‐3′ | 5′‐ACTCCAGAAGACCAGAGCAGA‐3′ |
| Rat Il1b | 5′‐TCACTCATTGTGGCTGTGGA‐3′ | 5′‐TGTCGTTGCTTGTCTCTCCT‐3′ |
| Rat Il10 | 5′‐CAGGACTTTAAGGGTTACTTGGGTTGC‐3′ | 5′‐AAACTCATTCATGGCCTTGTAGACACC‐3′ |
| Rat Tnfa | 5′‐TTCTCATTCCTGCTCGTGGC‐3′ | 5′‐CTCCGCTTGGTGGTTTGCTA‐3′ |
| Rat 18s | 5′‐ACTCAACACGGGAAACCTCA‐3′ | 5′‐AATCGCTCCACCAACTAAGA‐3′ |
| Rat Actin | 5′‐CCCTCTGAACCCTAAGGCCAACCG‐3′ | 5′‐GTGGTGGTGAAGCTGTAGCCACGC‐3′ |
2.7. Silencing of CCK and insulin receptors in pre‐adipocytes
Differentiated rat pre‐adipocytes were transfected with siRNAs for Cckar, Cckbr, and Insr (s234619, s218025, and s128753, respectively; Life Technologies), as previously described (Plaza, Merino, Cano, et al., 2018). Briefly, for each well, siRNAs (40 nmol·L−1) were diluted in 50‐μl Opti‐MEM (Life Technologies), incubated for 15 min (25°C), and then added to a solution containing 3‐μl lipofectamine RNAiMAX (Life Technologies) diluted in 50‐μl Opti‐MEM. After 15 additional min (25°C), 100 μl of the mixture were added drop‐wise to wells containing 400‐μl Opti‐MEM. The medium was replaced by differentiation medium after overnight incubation. Two days after transfection, the medium was changed to α‐MEM containing 0.1% BSA; then CCK‐8 (10−6 mol·L−1) was added and incubated for 2 hr (37°C, 5% CO2). The medium was removed, and mRNA was extracted as detailed above. Positive controls for transfection were performed by using Block‐iT Alexa Fluor Red (Life Technologies; n = 6; samples were run in duplicate). A Silencer Select negative control (4390843, Thermo Fisher Scientific, Spain) was used.
2.8. Inhibition of Akt with triciribine
Sc‐WAT and Vis‐WAT derived pre‐adipocytes were pre‐incubated with 10−5‐M triciribine in differentiation medium for 30 min and then incubated for 2 additional hr with 10−6‐M CCK‐8 (n = 6; samples were run in duplicate). After this time, mRNA was extracted as previously described. The dose of triciribine was selected accordingly to previous studies describing Akt inhibition with triciribine in adipocytes (C.‐L. Chang et al., 2010).
2.9. Inhibition of PPARγ with T0070907
Sc‐WAT and Vis‐WAT derived pre‐adipocytes were pre‐incubated with 10−5‐M T0070907 in differentiation medium for 30 min and then incubated for 2 additional hr with 10−6‐M CCK‐8, and mRNA was extracted (n = 6; samples were run in duplicate). The dose of T0070907 was selected on the basis of published data describing PPARγ antagonism by T0070907 in adipocytes (Bosco et al., 2017).
2.10. Combined treatment of pre‐adipocytes with CCK‐8 and insulin
Sc‐WAT and Vis‐WAT derived pre‐adipocytes were pre‐incubated with differentiation medium without insulin for 3 hr and then incubated for 2 additional hr with insulin (10−9 or 10−7 M) and/or CCK‐8 (10−7–10−5 M; n = 6; samples were run in duplicate).
2.11. Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Individual effects within a given group were analysed by using either a t test or a one‐way ANOVA, followed by Bonferroni post hoc test when F ANOVA achieved P < .05, and there was no significant variance in homogeneity. Individual group comparisons were made by using a two‐way ANOVA followed by Bonferroni post hoc test when appropriate. Statistical significance was set at P < .05. Normal distribution and variance homogeneity were assessed by using the Brown–Forsythe test. Outliers were identified by using the ROUT method (Q = 1%; GraphPad Prism software). All statistics were performed using GraphPad Prism software (GraphPad Software Inc., USA).
2.12. Materials
The CCK1 receptor antagonist, SR‐27,897 (1‐[2‐4‐(2‐chlorophenyl)thiazol‐2‐yl‐aminocarbonyl]‐indolylacetic acid), was kindly provided by Sanofi‐Synthelabo, France (Poncelet et al., 1993). The CCK2 receptor antagonist, L‐365,260 [(3R‐(+)‐2,3‐dihydro‐1‐methyl‐2‐oxo‐5‐phenyl‐1H‐1,4‐benzodiazepin‐3‐yl)‐N′‐(3‐methylphenyl)urea], was a gift of Merck Sharp and Dohme Research Laboratories, USA (R. Chang et al., 1989). Other chemicals were from Sigma (USA).
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to Pharmacology (Harding et al., 2017), and are permanently archived in the Concise Guide to Pharmacology 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Cidlowski et al., 2017; Alexander, Fabbro et al., 2017).
3. RESULTS
3.1. CCK‐8 enhanced adiponectin gene expression in both visceral and subcutaneous adipose tissues as well as in pre‐adipocytes
Figure 2a,c illustrates the effect of CCK‐8 on Adipoq expression in subcutaneous and visceral WAT, respectively. Adipoq expression was induced by acute CCK‐8 (10 μg·kg−1) only in Sc‐WAT, (Figure 2a), but was up‐regulated by chronic treatment in both Sc‐WAT and Vis‐WAT. In terms of Pparg, CCK‐8 induced its expression after chronic treatment in Sc‐WAT (Figure 2b) and was without effect in Vis‐WAT (Figure 2d).
Figure 2.
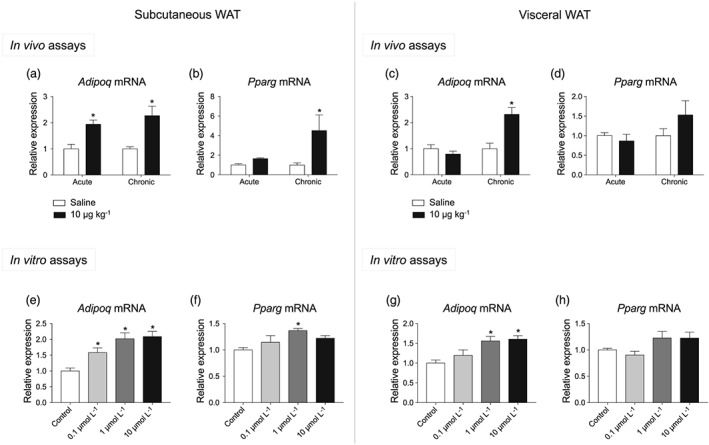
Effect of CCK‐8 on adiponectin and PPARγ gene expression. Effect of acute and chronic administration of CCK‐8 (10 μg·kg−1) on adiponectin (Adipoq) and PPARγ gene (Pparg) expression in subcutaneous (a, b) and visceral WAT (c, e). The effect of CCK‐8 was also assayed in pre‐adipocytes. Panels (e) and (g) show the effect of CCK‐8 on Adipoq expression in subcutaneous and visceral pre‐adipocytes. Panels (f) and (h) illustrate the effect of CCK‐8 on Pparg expression in subcutaneous and visceral pre‐adipocytes. Values are means ± SEM (n = 5 for acute in vivo, n = 6 for chronic in vivo, and n = 6 for in vitro assays). *P < .05, significantly different from their respective controls; ANOVA with Bonferroni's test
The effect of CCK‐8 on visceral and subcutaneous pre‐adipocytes appears illustrated in Figure 2e–h. In these assays, CCK‐8 induced Adipoq expression both in Sc‐WAT (Figure 2e), and Vis‐WAT (Figure 2g). Pparg expression was only induced by CCK‐8 in Sc‐WAT (Figure 2f), but not in visceral pre‐adipocytes (Figure 2h).
The robust effect of CCK‐8 on Adipoq expression led us to investigate the effect of CCK‐8 on adiponectin production both in vivo and in vitro. Otherwise, the similar qualitative pattern of Adipoq and Pparg gene expression suggested the involvement of PPARγ on adiponectin regulation, and therefore, the effect of PPARγ antagonists on Adipoq expression was further investigated.
3.2. CCK‐8 increased plasma adiponectin levels as well as adiponectin production in pre‐adipocytes
As shown in Figure 3a, plasma levels of adiponectin were increased by both acute and chronic treatments with CCK‐8. Figure 3b illustrates the time course effect of CCK‐8 on plasma adiponectin levels. Two‐way ANOVA revealed an effect of CCK‐8, which was found to be maximal 4 hr after CCK‐8 administration and reached basal values 2 hr later. However, adiponectin remained unaltered over the duration of the assay in saline‐treated rats. Accordingly, the AUC of plasma adiponectin levels was significantly enhanced by CCK‐8 (Figure 3c).
Figure 3.
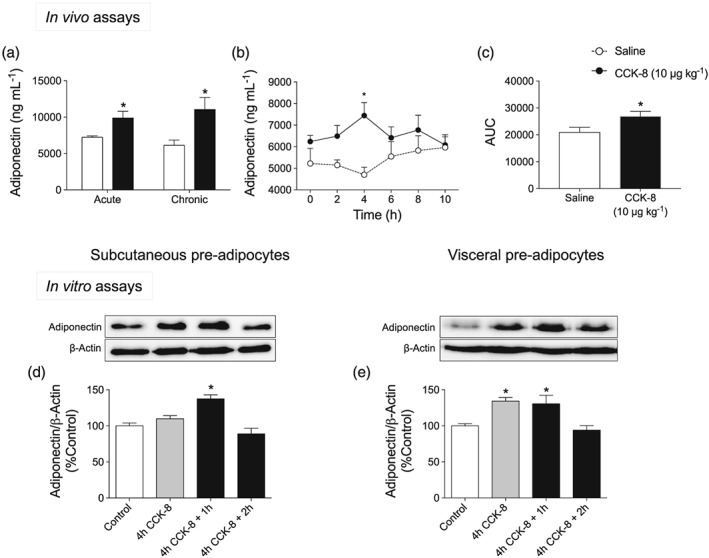
Effect of CCK‐8 on adiponectin levels. (a) Effect of both acute and chronic administration of CCK‐8 (10 μg·kg−1) on plasma adiponectin concentration. (b) Time course of plasma adiponectin levels after acute treatment with CCK‐8 (10 μg·kg−1). This assay was carried out between 06:00 hr, when CCK‐8 was administered, and 16:00 hr. (c) AUC corresponding to the time course of plasma adiponectin levels plotted in panel (b). Panels (d) and (e) illustrate the effect of 10‐μM CCK‐8 on adiponectin release in subcutaneous and visceral pre‐adipocytes, respectively. Values are means ± SEM (n = 6 for in vivo and n = 5 for in vitro assays). *P < .05, significantly different from their respective controls; ANOVA with Bonferroni's test
Figure 3d illustrates the time course of adiponectin content in subcutaneous pre‐adipocytes incubated with 1‐μM CCK‐8. The effect of CCK‐8 was maximal 1‐hr after 4‐hr incubation with CCK‐8. In this case, adiponectin production remained unchanged in control cells (see Figure S1). Similar results were detected in visceral pre‐adipocytes (Figure 3e), although in this case, CCK‐8 effects were already significant immediately after 4‐hr incubation with CCK‐8. In both cases, the effect of CCK‐8 was not observed in the CCK‐8 + 2 hr group.
The results illustrated in Figure 3, taken together, show that CCK‐8 triggers a transient increase of adiponectin production both in vivo and in vitro that seems to involve both Sc‐WAT and Vis‐WAT.
3.3. The effect of CCK‐8 on adiponectin production was mediated by CCK2 receptors
In order to identify the CCK receptor subtype involved in regulating adiponectin production, in vivo (effect of CCK1 and CCK2 receptor antagonists) and in vitro assays (effect of Cckar and Cckbr silencing) were carried out. As illustrated in Figure 4a, one‐way ANOVA revealed that the increase of plasma adiponectin triggered by CCK‐8 (10 μg·kg−1) was specifically abolished by the CCK2 receptor antagonist, L‐365,260, . Figure 4b shows the effect of the CCK2 receptor antagonist L‐365,260 on the up‐regulation of Adipoq triggered by CCK‐8. In this case, two‐way ANOVA indicated a significant effect of CCK‐8 and of L‐365,260, as well as a significant interaction for CCK‐8 × L‐365,260.
Figure 4.
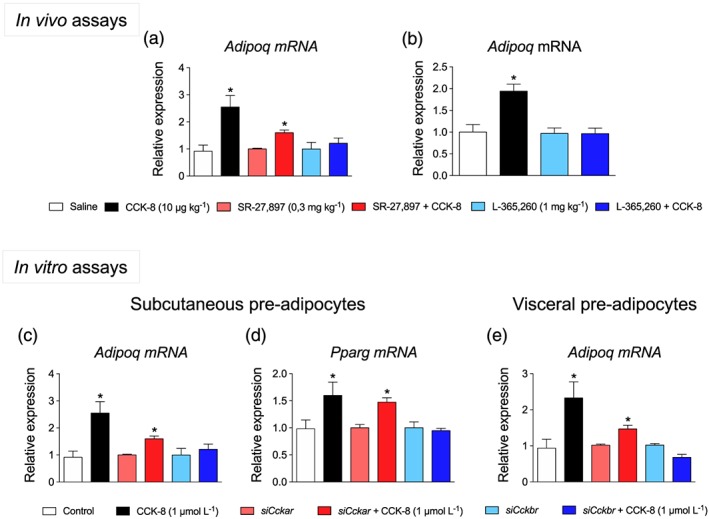
Effect of CCK receptor antagonists and silencing of CCK receptor genes on adiponectin production. (a) Effect of antagonists of CCK1 receptors (SR‐27,897) or CCK2 receptors (L‐365,260) on plasma adiponectin concentration. (b) Effect of L‐365,260 on Adipoq expression in Sc‐WAT from rats treated with CCK‐8. (c, d) Effect of Cckar and Cckbr silencing on Adipoq and Pparg expression in subcutaneous pre‐adipocytes. (e) Effect of Cckar and Cckbr silencing on Adipoq expression in visceral pre‐adipocytes. Values are means ± SEM (n = 6). *P < .05, significantly different from their respective controls; ANOVA with Bonferroni's test
Figure 4c,d illustrates the effects of silencing Cckar/Cckbr on Adipoq and Pparg expression in subcutaneous pre‐adipocytes. In this assay, the effect of CCK‐8 was specifically abolished in pre‐adipocytes carrying the Cckbr deletion, for Adipoq and Pparg expression. In visceral pre‐adipocytes, silencing Cckbr expression also abolished CCK‐8 effects on Adipoq expression (Figure 4e).
3.4. The inhibition of PKB by triciribine as well as the inhibition of PPARγ by T0070907 abolished the effect of CCK‐8 on adiponectin gene expression
In order to investigate the involvement of the Akt signalling pathway on CCK‐8‐induced expression of Adipoq and Pparg, the effect of the Akt inhibitor triciribine was investigated. Triciribine abolished the effect of CCK‐8 on Adipoq (Figure 5a) as well as on Pparg (Figure 5b) expression in subcutaneous pre‐adipocytes. In visceral pre‐adipocytes, triciribine also blocked the effect of CCK‐8 on Adipoq expression (Figure 5c).
Figure 5.
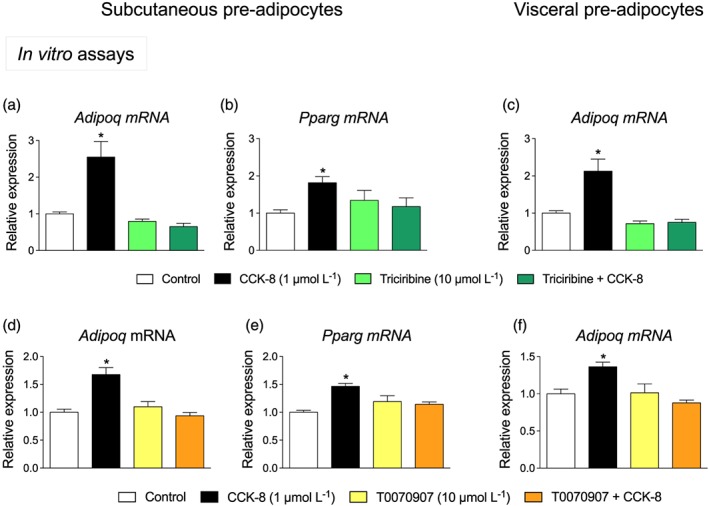
Effect of triciribine (an Akt inhibitor) and T0070907 (a PPARγ antagonist) on adiponectin gene (Adipoq) expression. Inhibition by triciribine of Adipoq (a) and Pparg expression (b) induced by CCK‐8 in subcutaneous pre‐adipocytes. Adipoq expression was also abolished by triciribine in visceral pre‐adipocytes (c). T0070907 also blocked the effect of CCK‐8 on Adipoq (d, f) and Pparg expression (e). Values are means ± SEM (n = 6). *P < .05, significantly different from their respective controls; ANOVA with Bonferroni's test
The involvement of PPARγ on Adipoq expression was assessed by using the selective PPARγ antagonist T0070907. As illustrated in Figure 5d,e, T0070907 antagonized the effect of CCK‐8 on Adipoq expression in subcutaneous, as well as in visceral pre‐adipocytes (Figure 5f; one outlier value was removed).
3.5. The effect of CCK‐8 on adiponectin gene expression requires the activation of the insulin receptor
The effect of CCK‐8 was analysed in pre‐adipocytes after silencing the expression of the insulin receptor gene (Insr) as well as in the presence of different concentrations of insulin. As illustrated in Figure 6a,b, the effect of CCK‐8 on Adipoq expression was abolished by Insr silencing in both subcutaneous and visceral pre‐adipocytes. Figure 6c and d shows the CCK‐8 concentration‐effect plots in the presence of different concentrations of insulin. In subcutaneous pre‐adipocytes (Figure 6c), two‐way ANOVA revealed a significant effect of insulin and of CCK‐8, as well as a significant interaction for insulin × CCK‐8. In visceral pre‐adipocytes (Figure 6d), two‐way ANOVA also revealed significant effects of insulin, and CCK‐8, as well as a significant interaction for insulin × CCK‐8.
Figure 6.
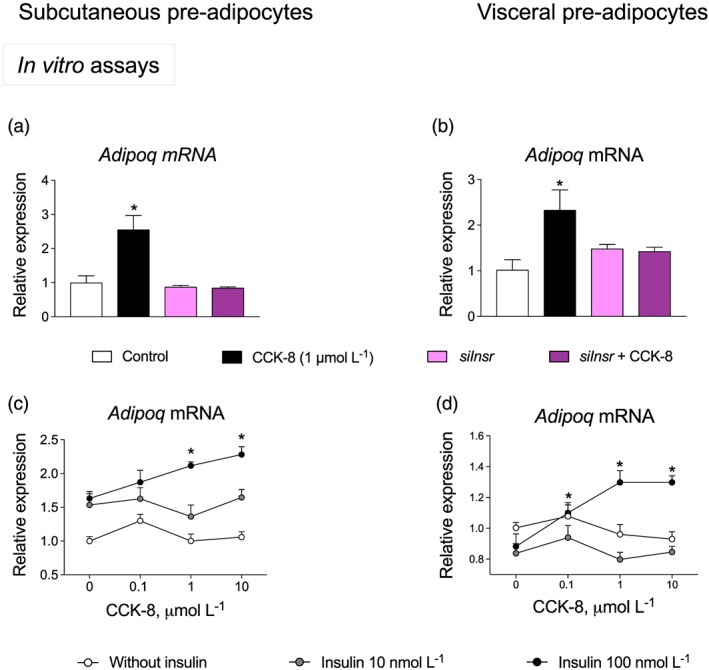
Insulin and insulin receptors are involved in the adiponectin gene (Adipoq) expression induced by CCK‐8. Effects of silencing of insulin receptors on Adipoq expression in subcutaneous (a) and visceral pre‐adipocytes (b). Concentration‐effect plots of CCK‐8 on Adipoq expression. Experiments were carried out in either the presence or absence of insulin in subcutaneous (c) and visceral pre‐adipocytes (d). Values are means ± SEM (n = 6). *P < .05, significantly different from their respective controls; ANOVA with Bonferroni's test
4. DISCUSSION
Our study showed that CCK‐8, by acting on CCK2 receptors, promoted the expression of Adipoq in subcutaneous and visceral WAT and, consequently, CCK‐8 increased plasma adiponectin levels. Moreover, CCK‐8 effects appeared to be insulin‐dependent and, in the case of subcutaneous WAT, linked to the activation of PPARγ. Taken together, our findings support the concept, already suggested by previous studies carried out by our group and others (King, Yang, Huesman, Rider, & Lo, 2015; Plaza, Merino, Cano, et al., 2018; Plaza, Merino, Sánchez‐Pernaute, et al., 2018), that both CCK and CCK binding sites have a relevant role in modulating WAT homeostasis, particularly in the case of Sc‐WAT.
The main finding of our study deals with the increase of plasma adiponectin triggered by CCK‐8. Such an effect was due to the activation of CCK‐2 receptors in adipocytes, since (a) CCK‐8 induced Adipoq expression in WAT as well as in pre‐adipocytes, (b) CCK‐8 increased adiponectin immunoreactivity in pre‐adipocytes, and (c) CCK‐8 effects were abolished by both genetic and pharmacological inactivation of CCK2 receptors. Considering that CCK is physiologically released after meals, our data suggest that the effect of CCK in regulating adiponectin synthesis would be perceptible during postprandial periods and would contribute to maintenance of plasma adiponectin levels. This hypothesis is compatible with the circadian oscillation of plasma adiponectin in humans, characterized by a nocturnal decline reaching a nadir in the early morning, and a further rise coincident with the first meal of the day, which tends to stabilize until the following nocturnal decrease (Gavrila et al., 2003; Scheer et al., 2010). In our experiments, plasma adiponectin concentration reached the highest level at 4 hr after CCK‐8 administration, which supports the concept of a long‐lasting effect of CCK‐8 in regulating adiponectinaemia. Nonetheless, it has to be noted that the kinetic assay illustrated in Figure 3b was carried out during the low activity period of the rat circadian cycle, a circumstance that limits the meaning of this result, and would explain the fact that plasma adiponectin reached lower values in this assay than when CCK‐8 was administered at 10 p.m.
Another interesting finding deals with the preservation of the effect of CCK‐8 on adiponectin production observed in chronically treated rats, which suggests that CCK‐8 did not induce tolerance in adipocytes. In fact, chronic CCK‐8 is more effective in promoting Adipoq expression in Vis‐WAT than acute CCK‐8. In any case, it has to be noted that the inhibition of food intake, together with the decrease of body weight triggered by CCK‐8 (Plaza, Merino, Cano, et al., 2018), might contribute to the increase of adiponectin plasma levels (Reinehr, Roth, Menke, & Andler, 2004). At this point, it has to be highlighted that chronic CCK‐8 led to a decrease of leptin plasma levels (Plaza, Merino, Cano, et al., 2018) that would rather limit the effect of CCK‐8 on Adipoq expression, as leptin has been shown to induce the expression of adiponectin (Frühbeck et al., 2017; Singh et al., 2016). A relevant circumstance that deserves attention is the fact that the effect of CCK‐8 on Adipoq expression seems to be slightly more intense in Sc‐WAT than in Vis‐WAT. This finding is of interest as subcutaneous pre‐adipocytes produce and release more adiponectin than visceral pre‐adipocytes (Baglioni et al., 2012), and, therefore, CCK2 receptor agonists might be useful drugs to stimulate adiponectin production in physiopathological situations, such as obesity, characterized by a decrease of plasma adiponectin. Some studies have already identified a decrease of adiponectin expression in isolated subcutaneous pre‐adipocytes from obese individuals (Degawa‐Yamauchi et al., 2005). This topic has been reviewed by Lafontan and Girard (2008). Otherwise, the similar pattern of expression of Adipoq and Pparg genes observed in the current study is compatible with the well‐characterized link between Adipoq expression and PPARγ activity (Banga et al., 2009), and with the role of adiponectin on insulin‐sensitizing therapies based on PPARγ agonists (Li et al., 2018; Tripathy et al., 2013). In this respect, our finding suggests that a pathway involving CCK receptors, PPARγ, and adiponectin could be interesting to investigate in developing of new insulin‐sensitizing therapies. Interestingly, CCK‐8 apparently promoted an anti‐inflammatory response as it tended to repress the expression of the pro‐inflammatory cytokines IL‐6, IL‐1β, and TNF‐α in both Sc‐WAT and Vis‐WAT (see Figure S2). This effect would be consistent with the known anti‐inflammatory effect of adiponectin (Frühbeck et al., 2017) and could contribute to an improvement in insulin sensitivity (Blüher, 2016).
The finding that triciribine blocks the effect of CCK‐8 on adiponectin expression further confirms the importance of Akt in the CCK2 receptor‐mediated responses in WAT (Plaza, Merino, Sánchez‐Pernaute, et al., 2018). The role of Akt in regulating adiponectin gene expression has not yet been fully elucidated, although Akt‐mediated phosphorylation of FoxO1 has been proposed to mediate the nuclear exclusion of this transcription factor. FoxO1 is a member of the forkhead box O transcription factor family that reduces the activity of the PPARγ‐responsive element, which is necessary for the adiponectin gene transcription (Fan et al., 2009; Tzivion, Dobson, & Ramakrishnan, 2011). Thus, Akt activation might indirectly promote PPARγ‐responsive element activity. The plausibility of this mechanism is stressed by the finding that CCK‐8 also induced Pparg expression in subcutaneous WAT. Moreover, Adipoq expression was abolished by the PPARγ antagonist, T0070907 (G. Lee et al., 2002). All these findings are consistent with the pivotal role assigned to PPARγ as an upstream master regulator of Adipoq expression (Shao et al., 2016). As triciribine also antagonized the effect of CCK‐8 on Pparg expression, one could hypothesize that both Akt and PPARγ would be integral to the mechanism that account for the regulation of CCK2 receptor‐mediated expression of adiponectin in subcutaneous adipocytes. Moreover, CCK2 receptors can also activate the ERK1/2 pathway (Höcker, 2004), which regulates the PPARγ–adiponectin axis (Maeda et al., 2001; Yamauchi et al., 2001). Nevertheless, in the case of visceral adipocytes, molecular mechanisms governing Pparg expression seem to be independent of CCK2 receptors.
In an effort to further identify the mechanism of action of CCK‐8, the relationship between CCK2 receptors and insulin receptors was explored. The finding that Adipoq expression was not induced by CCK‐8 in pre‐adipocytes carrying a deletion of the insulin receptor gene suggests a functional interaction between CCK2 receptors and insulin receptors. This possibility was further strengthened by the synergism observed between CCK‐8 and sub‐threshold concentrations of insulin. From our data, we cannot identify the molecular mechanism that could eventually account for such an interaction. Although in vivo studies suggest that insulin acts as a negative regulator of adiponectin synthesis and/or secretion (Basu, Pajvani, Rizza, & Scherer, 2007; Semple et al., 2006), insulin also positively regulated in vitro Adipoq expression by activating PPARγ, via suppressing FoxO1 activity (Fan et al., 2009). Hence, molecular mechanisms downstream of insulin receptors and CCK receptors could converge at this point to regulate Adipoq expression. Our findings are therefore consistent with the close relationship between plasma levels of adiponectin and insulin responsiveness (Cnop et al., 2003), as well as with the hypoadiponectinaemia observed in obese, insulin‐resistant patients (Annuzzi et al., 2010; Weyer et al., 2001). Moreover, postprandial plasma levels of CCK are lower in patients with Type 2 diabetes, than in control subjects (Rushakoff et al., 1993), proving a close relationship between CCK, insulin sensitivity, and adiponectin regulation.
Notably, the loss of effect of CCK‐8 in pre‐adipocytes lacking expression of insulin receptors (Figure 6a–d) supports the idea that both CCK and insulin may co‐operate in modulating adiponectin production by WAT. This concept is further supported by the study showing that sub‐threshold doses of insulin potentiate the effect of CCK‐8 in promoting Adipoq expression in both visceral and subcutaneous WAT. As Akt is a common signalling pathway element for both CCK2 receptors and insulin receptors, all these findings suggest that Akt might account for the interaction between CCK and insulin.
In conclusion, this study showed that CCK‐8 stimulated the synthesis of adiponectin by rat adipocytes. Such an effect depended on the activation of PKB (Akt) and involved PPARγ, suggesting that agonists of CCK2 receptors could have translational value in the design of new insulin‐sensitizing therapies. Further, our findings suggest that the insulin‐sensitizing effects of CCK agonists already reported by other authors (Irwin et al., 2012) might be linked to the effect of CCK on adiponectin production.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
A.P., B.M. and M.R.‐G. designed the research. A.P. and B.M. performed the experiments. A.P., B.M. and M.R.‐G. analyzed the data. A.P. and M.R.‐G. wrote the manuscript. N.D.O. revised the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Table S1. Effect of acute and chronic CCK‐8 on plasma parameters
Figure S1. Effect of CCK‐8 on adiponectin production in subcutaneous preadipocytes
Figure S2: Effect of CCK‐8 on the expression of IL1b, IL6, IL10 and TNFα
ACKNOWLEDGEMENTS
This study was supported by Ministerio de Ciencia, Innovación y Universidades (BFU2012‐35353 and BFU2016‐78556‐R), European Regional Development Fund, and Fundación Universitaria San Pablo CEU. A.P. is supported by a grant from Ministerio de Ciencia, Innovación y Universidades, Spain (BES‐2013‐063773).
Plaza A, Merino B, Del Olmo N, Ruiz‐Gayo M. The cholecystokinin receptor agonist, CCK‐8, induces adiponectin production in rat white adipose tissue. Br J Pharmacol. 2019;176:2678–2690. 10.1111/bph.14690
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174, S208–S224. 10.1111/bph.13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annuzzi, G. , Bozzetto, L. , Patti, L. , Santangelo, C. , Giacco, R. , Di Marino, L. , … Rivellese, A. A. (2010). Type 2 diabetes mellitus is characterized by reduced postprandial adiponectin response: A possible link with diabetic postprandial dyslipidemia. Metabolism‐Clinical and Experimental, 59(4), 567–574. 10.1016/j.metabol.2009.08.020 [DOI] [PubMed] [Google Scholar]
- Baglioni, S. , Cantini, G. , Poli, G. , Francalanci, M. , Squecco, R. , Di Franco, A. , … Luconi, M. (2012). Functional differences in visceral and subcutaneous fat pads originate from differences in the adipose stem cell. PLoS ONE, 7(5), e36569 10.1371/journal.pone.0036569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banga, A. , Unal, R. , Tripathi, P. , Pokrovskaya, I. , Owens, R. J. , Kern, P. A. , & Ranganathan, G. (2009). Adiponectin translation is increased by the PPARγ agonists pioglitazone and ω‐3 fatty acids. American Journal of Physiology‐Endocrinology and Metabolism, 296(3), E480–E489. 10.1152/ajpendo.90892.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, R. , Pajvani, U. B. , Rizza, R. A. , & Scherer, P. E. (2007). Selective downregulation of the high–molecular weight form of adiponectin in hyperinsulinemia and in type 2 diabetes: Differential regulation from nondiabetic subjects. Diabetes, 56(8), 2174–2177. 10.2337/db07-0185 [DOI] [PubMed] [Google Scholar]
- Beglinger, C. , & Degen, L. (2004). Fat in the intestine as a regulator of appetite—Role of CCK. Physiology & Behavior, 83(4), 617–621. 10.1016/j.physbeh.2004.07.031 [DOI] [PubMed] [Google Scholar]
- Blüher, M. (2016). Adipose tissue inflammation: A cause or consequence of obesity‐related insulin resistance? Clinical Science, 130(18), 1603–1614. 10.1042/CS20160005 [DOI] [PubMed] [Google Scholar]
- Bosco, D. B. , Roycik, M. D. , Jin, Y. , Schwartz, M. A. , Lively, T. J. , Zorio, D. A. , & Sang, Q. A. (2017). A new synthetic matrix metalloproteinase inhibitor reduces human mesenchymal stem cell adipogenesis. PLoS ONE, 12(2), e0172925 10.1371/journal.pone.0172925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano, V. , Merino, B. , Ezquerra, L. , Somoza, B. , & Ruiz‐Gayo, M. (2008). A cholecystokinin‐1 receptor agonist (CCK‐8) mediates increased permeability of brain barriers to leptin. British Journal of Pharmacology, 154(5), 1009–1015. 10.1038/bjp.2008.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C.‐L. , Au, L.‐C. , Huang, S.‐W. , Fai Kwok, C. , Ho, L.‐T. , & Juan, C.‐C. (2010). Insulin up‐regulates heme oxygenase‐1 expression in 3T3‐L1 adipocytes via PI3‐kinase‐and PKC‐dependent pathways and heme oxygenase‐1–associated microRNA downregulation. Endocrinology, 152(2), 384–393. [DOI] [PubMed] [Google Scholar]
- Chang, R. , Chen, T. , Bock, M. , Freidinger, R. , Chen, R. , Rosegay, A. , & Lotti, V. J. (1989). Characterization of the binding of [3H] L‐365,260: A new potent and selective brain cholecystokinin (CCK‐B) and gastrin receptor antagonist radioligand. Molecular Pharmacology, 35(6), 803–808. [PubMed] [Google Scholar]
- Chen, D. , Zhao, C. M. , Al–Haider, W. , Håkanson, R. , Rehfeld, J. F. , & Kopin, A. S. (2002). Differentiation of gastric ECL cells is altered in CCK2 receptor–deficient mice. Gastroenterology, 123(2), 577–585. 10.1053/gast.2002.34746 [DOI] [PubMed] [Google Scholar]
- Cnop, M. , Havel, P. , Utzschneider, K. , Carr, D. , Sinha, M. , Boyko, E. , … Kahn, S. E. (2003). Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: Evidence for independent roles of age and sex. Diabetologia, 46(4), 459–469. 10.1007/s00125-003-1074-z [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debas, H. T. , Farooq, O. , & Grossman, M. I. (1975). Inhibition of gastric emptying is a physiological action of cholecystokinin. Gastroenterology, 68(5), 1211–1217. [PubMed] [Google Scholar]
- Degawa‐Yamauchi, M. , Moss, K. A. , Bovenkerk, J. E. , Shankar, S. S. , Morrison, C. L. , Lelliott, C. J. , … Considine, R. V. (2005). Regulation of adiponectin expression in human adipocytes: Effects of adiposity, glucocorticoids, and tumor necrosis factor α. Obesity Research, 13(4), 662–669. 10.1038/oby.2005.74 [DOI] [PubMed] [Google Scholar]
- Dufresne, M. , Seva, C. , & Fourmy, D. (2006). Cholecystokinin and gastrin receptors. Physiological Reviews, 86(3), 805–847. 10.1152/physrev.00014.2005 [DOI] [PubMed] [Google Scholar]
- Fan, W. , Imamura, T. , Sonoda, N. , Sears, D. D. , Patsouris, D. , Kim, J. J. , & Olefsky, J. M. (2009). FOXO1 transrepresses peroxisome proliferator‐activated receptor γ transactivation, coordinating an insulin‐induced feed‐forward response in adipocytes. Journal of Biological Chemistry, 284(18), 12188–12197. 10.1074/jbc.M808915200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, H. , & Judd, R. L. (2018). Adiponectin regulation and function. Comprehensive Physiology, 8(3), 1031–1063. [DOI] [PubMed] [Google Scholar]
- Fasshauer, M. , Klein, J. , Neumann, S. , Eszlinger, M. , & Paschke, R. (2002). Hormonal regulation of adiponectin gene expression in 3T3‐L1 adipocytes. Biochemical and Biophysical Research Communications, 290(3), 1084–1089. 10.1006/bbrc.2001.6307 [DOI] [PubMed] [Google Scholar]
- Frühbeck, G. , Catalán, V. , Rodríguez, A. , & Gómez‐Ambrosi, J. (2018). Adiponectin‐leptin ratio: A promising index to estimate adipose tissue dysfunction. Relation with obesity‐associated cardiometabolic risk. Adipocytes, 7(1), 57–62. 10.1080/21623945.2017.1402151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frühbeck, G. , Catalán, V. , Rodríguez, A. , Ramírez, B. , Becerril, S. , Portincasa, P. , & Gómez‐Ambrosi, J. (2017). Normalization of adiponectin concentrations by leptin replacement in ob/ob mice is accompanied by reductions in systemic oxidative stress and inflammation. Scientific Reports, 7(1), 2752 10.1038/s41598-017-02848-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrila, A. , Peng, C.‐K. , Chan, J. L. , Mietus, J. E. , Goldberger, A. L. , & Mantzoros, C. S. (2003). Diurnal and ultradian dynamics of serum adiponectin in healthy men: Comparison with leptin, circulating soluble leptin receptor, and cortisol patterns. The Journal of Clinical Endocrinology & Metabolism, 88(6), 2838–2843. 10.1210/jc.2002-021721 [DOI] [PubMed] [Google Scholar]
- Gil‐Ortega, M. , Garidou, L. , Barreau, C. , Maumus, M. , Breasson, L. , Tavernier, G. , … Sengenès, C. (2013). Native adipose stromal cells egress from adipose tissue in vivo: Evidence during lymph node activation. Stem Cells, 31(7), 1309–1320. 10.1002/stem.1375 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2017). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havel, P. J. , Kasim‐Karakas, S. , Mueller, W. , Johnson, P. R. , Gingerich, R. L. , & Stern, J. S. (1996). Relationship of plasma leptin to plasma insulin and adiposity in normal weight and overweight women: Effects of dietary fat content and sustained weight loss. The Journal of Clinical Endocrinology & Metabolism, 81(12), 4406–4413. [DOI] [PubMed] [Google Scholar]
- Höcker, M. (2004). Molecular mechanisms of gastrin‐dependent gene regulation. Annals of the new York Academy of Sciences, 1014(1), 97–109. [DOI] [PubMed] [Google Scholar]
- Irwin, N. , Frizelle, P. , Montgomery, I. , Moffett, R. , O'Harte, F. , & Flatt, P. (2012). Beneficial effects of the novel cholecystokinin agonist (pGlu‐Gln)‐CCK‐8 in mouse models of obesity/diabetes. Diabetologia, 55(10), 2747–2758. 10.1007/s00125-012-2654-6 [DOI] [PubMed] [Google Scholar]
- Irwin, N. , Montgomery, I. , O'harte, F. , Frizelle, P. , & Flatt, P. (2013). Comparison of the independent and combined metabolic effects of subchronic modulation of CCK and GIP receptor action in obesity‐related diabetes. International Journal of Obesity, 37(8), 1058–1063. 10.1038/ijo.2012.179 [DOI] [PubMed] [Google Scholar]
- Izzo, R. S. , Brugge, W. R. , & Praissman, M. (1984). Immunoreactive cholecystokinin in human and rat plasma: Correlation of pancreatic secretion in response to CCK. Regulatory Peptides, 9(1–2), 21–34. 10.1016/0167-0115(84)90004-1 [DOI] [PubMed] [Google Scholar]
- Kadowaki, T. , Yamauchi, T. , Kubota, N. , Hara, K. , Ueki, K. , & Tobe, K. (2006). Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. The Journal of Clinical Investigation, 116(7), 1784–1792. 10.1172/JCI29126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantartzis, K. , Fritsche, A. , Tschritter, O. , Thamer, C. , Haap, M. , Schäfer, S. , … Stefan, N. (2005). The association between plasma adiponectin and insulin sensitivity in humans depends on obesity. Obesity, 13(10), 1683–1691. 10.1038/oby.2005.206 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, A. , Yang, Q. , Huesman, S. , Rider, T. , & Lo, C. C. (2015). Lipid transport in cholecystokinin knockout mice. Physiology & Behavior, 151, 198–206. 10.1016/j.physbeh.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraly, F. S. (1981). A diurnal variation in the satiating potency of cholecystokinin in the rat. Appetite, 2(3), 177–191. 10.1016/S0195-6663(81)80041-4 [DOI] [PubMed] [Google Scholar]
- Kumada, M. , Kihara, S. , Sumitsuji, S. , Kawamoto, T. , Matsumoto, S. , Ouchi, N. , … Matsuzawa, Y. (2003). Association of hypoadiponectinemia with coronary artery disease in men. Arteriosclerosis, Thrombosis, and Vascular Biology, 23(1), 85–89. 10.1161/01.ATV.0000048856.22331.50 [DOI] [PubMed] [Google Scholar]
- Lafontan, M. , & Girard, J. (2008). Impact of visceral adipose tissue on liver metabolism: Part I: Heterogeneity of adipose tissue and functional properties of visceral adipose tissue. Diabetes & Metabolism, 34(4), 317–327. 10.1016/j.diabet.2008.04.001 [DOI] [PubMed] [Google Scholar]
- Lee, B. , & Shao, J. (2014). Adiponectin and energy homeostasis. Reviews in Endocrine and Metabolic Disorders, 15(2), 149–156. 10.1007/s11154-013-9283-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, G. , Elwood, F. , McNally, J. , Weiszmann, J. , Lindstrom, M. , Amaral, K. , … Li, Y. (2002). T0070907, a selective ligand for peroxisome proliferator‐activated receptor γ, functions as an antagonist of biochemical and cellular activities. Journal of Biological Chemistry, 277(22), 19649–19657. 10.1074/jbc.M200743200 [DOI] [PubMed] [Google Scholar]
- Li, J. , Xue, Y.‐M. , Zhu, B. , Pan, Y.‐H. , Zhang, Y. , Wang, C. , & Li, Y. (2018). Rosiglitazone elicits an adiponectin‐mediated insulin‐sensitizing action at the adipose tissue‐liver axis in Otsuka Long‐Evans Tokushima Fatty rats. Journal of Diabetes Research, 2018, 1–12. 10.1155/2018/4627842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods, 25(4), 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Maeda, N. , Takahashi, M. , Funahashi, T. , Kihara, S. , Nishizawa, H. , Kishida, K. , … Matsuzawa, Y. (2001). PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose‐derived protein. Diabetes, 50(9), 2094–2099. 10.2337/diabetes.50.9.2094 [DOI] [PubMed] [Google Scholar]
- Merino, B. , Cano, V. , Guzman, R. , Somoza, B. , & Ruiz‐Gayo, M. (2008). Leptin‐mediated hypothalamic pathway of cholecystokinin (CCK‐8) to regulate body weight in free‐feeding rats. Endocrinology, 149(4), 1994–2000. 10.1210/en.2007-1286 [DOI] [PubMed] [Google Scholar]
- Merino, B. , Somoza, B. , Ruiz‐Gayo, M. , & Cano, V. (2008). Circadian rhythm drives the responsiveness of leptin‐mediated hypothalamic pathway of cholecystokinin‐8. Neuroscience Letters, 442(2), 165–168. 10.1016/j.neulet.2008.07.009 [DOI] [PubMed] [Google Scholar]
- Miller, L. J. , & Gao, F. (2008). Structural basis of cholecystokinin receptor binding and regulation. Pharmacology & Therapeutics, 119(1), 83–95. 10.1016/j.pharmthera.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak, V. , Flatt, P. R. , & Irwin, N. (2018). Cholecystokinin (CCK) and related adjunct peptide therapies for the treatment of obesity and type 2 diabetes. Peptides, 100, 229–235. [DOI] [PubMed] [Google Scholar]
- Pereira, R. I. , & Draznin, B. (2005). Inhibition of the phosphatidylinositol 3′‐kinase signaling pathway leads to decreased insulin‐stimulated adiponectin secretion from 3T3‐L1 adipocytes. Metabolism, 54(12), 1636–1643. 10.1016/j.metabol.2005.07.002 [DOI] [PubMed] [Google Scholar]
- Plaza, A. , Merino, B. , Cano, V. , Domínguez, G. , Pérez‐Castells, J. , Fernández‐Alfonso, M. S. , … Ruiz‐Gayo, M. (2018). Cholecystokinin is involved in triglyceride fatty acid uptake by rat adipose tissue. Journal of Endocrinology, 236(3), 137–150. 10.1530/JOE-17-0580 [DOI] [PubMed] [Google Scholar]
- Plaza, A. , Merino, B. , Sánchez‐Pernaute, A. , Torres‐García, A. J. , Rubio‐Herrera, M. A. , & Ruiz‐Gayo, M. (2018). Expression analysis of a cholecystokinin system in human and rat white adipose tissue. Life Sciences, 206, 98–105. 10.1016/j.lfs.2018.05.036 [DOI] [PubMed] [Google Scholar]
- Poncelet, M. , Arnone, M. , Heaulme, M. , Gonalons, N. , Gueudet, C. , Santucci, V. , … Soubrié, P. (1993). Neurobehavioural effects of SR 27897, a selective cholecystokinin type A (CCK‐A) receptor antagonist. Naunyn‐Schmiedeberg's Archives of Pharmacology, 348(1), 102–107. 10.1007/BF00168544 [DOI] [PubMed] [Google Scholar]
- Reinehr, T. , Roth, C. , Menke, T. , & Andler, W. (2004). Adiponectin before and after weight loss in obese children. The Journal of Clinical Endocrinology & Metabolism, 89(8), 3790–3794. 10.1210/jc.2003-031925 [DOI] [PubMed] [Google Scholar]
- Rushakoff, R. A. , Goldfine, I. , Beccaria, L. J. , Mathur, A. , Brand, R. J. , & Liddle, R. A. (1993). Reduced postprandial cholecystokinin (CCK) secretion in patients with noninsulin‐dependent diabetes mellitus: Evidence for a role for CCK in regulating postprandial hyperglycemia. The Journal of Clinical Endocrinology & Metabolism, 76(2), 489–493. [DOI] [PubMed] [Google Scholar]
- Scheer, F. , Chan, J. , Fargnoli, J. , Chamberland, J. , Arampatzi, K. , Shea, S. , … Mantzoros, C. S. (2010). Day/night variations of high‐molecular‐weight adiponectin and lipocalin‐2 in healthy men studied under fed and fasted conditions. Diabetologia, 53(11), 2401–2405. 10.1007/s00125-010-1869-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple, R. , Soos, M. , Luan, J. , Mitchell, C. , Wilson, J. , Gurnell, M. , … O'Rahilly, S. (2006). Elevated plasma adiponectin in humans with genetically defective insulin receptors. The Journal of Clinical Endocrinology & Metabolism, 91(8), 3219–3223. 10.1210/jc.2006-0166 [DOI] [PubMed] [Google Scholar]
- Shao, X. , Wang, M. , Wei, X. , Deng, S. , Fu, N. , Peng, Q. , … Lin, Y. (2016). Peroxisome proliferator‐activated receptor‐γ: master regulator of adipogenesis and obesity. Current Stem Cell Research & Therapy, 11(3), 282–289. 10.2174/1574888X10666150528144905 [DOI] [PubMed] [Google Scholar]
- Singer, M. (1987). Pancreatic secretory response to intestinal stimulants: A review. Scandinavian Journal of Gastroenterology, 22(sup139), 1–13. 10.3109/00365528709089768 [DOI] [PubMed] [Google Scholar]
- Singh, P. , Sharma, P. , Sahakyan, K. R. , Davison, D. E. , Sert‐Kuniyoshi, F. H. , Romero‐Corral, A. , … Somers, V. K. (2016). Differential effects of leptin on adiponectin expression with weight gain versus obesity. International Journal of Obesity, 40(2), 266–274. 10.1038/ijo.2015.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern, J. H. , Rutkowski, J. M. , & Scherer, P. E. (2016). Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metabolism, 23(5), 770–784. 10.1016/j.cmet.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy, D. , Daniele, G. , Fiorentino, T. V. , Perez‐Cadena, Z. , Chavez‐Velasquez, A. , Kamath, S. , … Folli, F. (2013). Pioglitazone improves glucose metabolism and modulates skeletal muscle TIMP‐3–TACE dyad in type 2 diabetes mellitus: A randomised, double‐blind, placebo‐controlled, mechanistic study. Diabetologia, 56(10), 2153–2163. 10.1007/s00125-013-2976-z [DOI] [PubMed] [Google Scholar]
- Tzivion, G. , Dobson, M. , & Ramakrishnan, G. (2011). FoxO transcription factors; regulation by AKT and 14‐3‐3 proteins. Biochimica et Biophysica Acta (BBA)‐Molecular Cell Research, 1813(11), 1938–1945. 10.1016/j.bbamcr.2011.06.002 [DOI] [PubMed] [Google Scholar]
- Weyer, C. , Funahashi, T. , Tanaka, S. , Hotta, K. , Matsuzawa, Y. , Pratley, R. E. , & Tataranni, P. A. (2001). Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. The Journal of Clinical Endocrinology & Metabolism, 86(5), 1930–1935. 10.1210/jcem.86.5.7463 [DOI] [PubMed] [Google Scholar]
- Yamauchi, T. , Kamon, J. , Waki, H. , Terauchi, Y. , Kubota, N. , Hara, K. , … Kadowaki, T. (2001). The fat‐derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nature Medicine, 7(8), 941–946. 10.1038/90984 [DOI] [PubMed] [Google Scholar]
- Yatagai, T. , Nagasaka, S. , Taniguchi, A. , Fukushima, M. , Nakamura, T. , Kuroe, A. , … Ishibashi, S. (2003). Hypoadiponectinemia is associated with visceral fat accumulation and insulin resistance in Japanese men with type 2 diabetes mellitus. Metabolism‐Clinical and Experimental, 52(10), 1274–1278. 10.1016/S0026-0495(03)00195-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Effect of acute and chronic CCK‐8 on plasma parameters
Figure S1. Effect of CCK‐8 on adiponectin production in subcutaneous preadipocytes
Figure S2: Effect of CCK‐8 on the expression of IL1b, IL6, IL10 and TNFα