

Abstract
Background and Purpose
Microvascular barrier breakdown is a hallmark of sepsis that is associated with sepsis‐induced multiorgan failure. Histidine‐rich glycoprotein (HRG) is a 75‐kDa plasma protein that was demonstrated to improve the survival of septic mice through regulation of cell shape, spontaneous ROS production in neutrophils, and adhesion of neutrophils to vascular endothelial cells. We investigated HRG's role in the LPS/TNF‐α‐induced barrier dysfunction of endothelial cells in vitro and in vivo and the possible mechanism, to clarify the definitive roles of HRG in sepsis.
Experimental Approach
EA.hy 926 endothelial cells were pretreated with HRG or human serum albumin before stimulation with LPS/TNF‐α. A variety of biochemical assays were applied to explore the underlying molecular mechanisms on how HRG protected the barrier function of vascular endothelium.
Key Results
Immunostaining results showed that HRG maintains the endothelial monolayer integrity by inhibiting cytoskeleton reorganization, losses of VE‐cadherin and β‐catenin, focal adhesion kinase degradation, and cell detachment induced by LPS/TNF‐α. HRG also inhibited the cytokine secretion from endothelial cells induced by LPS/TNF‐α, which was associated with reduced NF‐κB activation. Moreover, HRG effectively prevented the LPS/TNF‐α‐induced increase in capillary permeability in vitro and in vivo. Finally, Western blot results demonstrated that HRG prevented the phosphorylation of MAPK family and RhoA activation, which are involved mainly in the regulation of cytoskeleton reorganization and barrier permeability.
Conclusions and Implications
Taken together, our results demonstrate that HRG has protective effects on vascular barrier function in vitro and in vivo, which may be due to the inhibition of MAPK family and Rho activation.
Abbreviations
- FAK
focal adhesion kinase
- HRG
histidine‐rich glycoprotein
- HSA
human serum albumin
- p‐MLC
phosphorylated myosin light chain
What is already known
HRG has an anti‐septic effect in mice through the maintenance of neutrophil quiescence.
What this study adds
HRG produced significant protective effects on vascular endothelial cells in vitro and in vivo.
What is the clinical significance
The rapid decrease in plasma HRG in sepsis contributes to the dysregulation of vascular endothelial cells.
1. INTRODUCTION
Sepsis is a leading cause of death with infection worldwide (Riedemann, Guo, & Ward, 2003). It is a life‐threatening organ dysfunction caused by a dysregulated host response to infection (Singer et al., 2016; Vincent, Opal, Marshall, & Tracey, 2013). Septic organ dysfunction is due mainly to an overwhelming systemic inflammatory process, characterized by the activation of inflammatory cells as well as the enhanced production and release of various cytokines (Gill, Taneja, Rohan, Wang, & Mehta, 2014). Endothelial dysfunction is one of key processes involved in the pathogenesis of sepsis, and it is reported to be associated with patient mortality in the clinic (Backer, Creteur, Preiser, Dubois, & Vincent, 2002; Trzeciak et al., 2007). It is generally agreed that endothelial dysfunction promotes the adhesion of neutrophils to vascular endothelial cells, coagulation abnormalities, microvascular leakage, and hypoperfusion leading to sepsis‐induced multiorgan failure (Aird, 2003). Thus, the recovery and maintenance of the body's endothelial barrier function must be critical to survival in cases of sepsis. These processes are likely to provide novel targets for the treatment of sepsis.
The endothelial barrier is maintained mostly by the balance between its intrinsic contractility and intercellular adhesive interactions (Hoelzle, Svitkina, & Blanchoin, 2011; Huveneers et al., 2012). The intrinsic contractility—which is produced by cytoskeleton rearrangement, the cortical actin dissolution, and stress fibre formation—can result in endothelial retraction and gap formation (Dejana, Corada, & Lampugnani, 1995; Jean et al., 2014; Nwariaku et al., 2003). The actin cytoskeleton of endothelial cells is tethered to adherent focal adhesions, which mediate cell–matrix interactions (Burridge, Fath, Kelly, Nuckolls, & Turner, 1988; Wu, 2005). Cell–cell contacts are mediated by adherent junctions among endothelial cells, and these contacts are composed mainly of VE‐cadherin. VE‐cadherin is an endothelium‐specific transmembrane adhesion molecule that regulates the cytoskeleton reorganization via an interaction with actin filaments through catenin binding, which also plays a critical role in modulating endothelial permeability (Gumbiner, 1996; Luscinskas & Shaw, 2001; Rodrigues & Granger, 2015).
Hyperpermeability is a hallmark of different inflammatory diseases such as acute respiratory distress syndrome and sepsis, and aberrant actin dynamics during sepsis leads to intercellular junction destabilization, vascular hyperpermeability, and immune cell recruitment. Therefore, the actomyosin contractility machinery has become the focus of many studies investigating sepsis (Schnoor et al., 2017). In this process, RhoA triggers the formation of stress fibres through the phosphorylation of myosin light chain (MLC) by Rho‐associated kinase (ROCK; García‐Ponce et al., 2016). Phosphorylated MLC (p‐MLC) enables the myosin molecule to change conformation, interact with actin, and slide along its filaments, leading to increased contractility. Members of the MAPK family also interact with the cytoskeleton, and in particular, activated ERK1/2 or p38 was shown to mediate changes in actin dynamics, leading to stress fibre formation, redistribution of VE‐cadherin junctions, and permeability changes in endothelial cells (Guay et al., 1997; Nwariaku et al., 2002).
Activation of the vascular endothelium in septic conditions results in the production of various pro‐inflammatory molecules including leukocyte adhesion molecules as well as soluble cytokines and chemokines (Pober & Cotran, 1990). The activation of NF‐κB in endothelial cells is required for the release of pro‐inflammatory cytokines, including IL‐1β, IL‐6, and TNF‐α (Baeuerle & Baltimore, 1996). Therefore, the inhibition of NF‐κB activation will control the overproduction of cytokines in sepsis conditions, which further protects the barrier function in the pathogenesis of sepsis.
Histidine‐rich glycoprotein (HRG) is a 75‐kDa plasma protein. Human HRG is synthesized in the liver and is present in the blood at a concentration range of 60–100 μg·ml−1 (Koide, Foster, Yoshitake, & Davie, 1986). The primary structure of human HRG is predicted to be a 507‐amino acid multidomain polypeptide consisting of two cystatin‐like regions at the N‐terminus, a histidine‐rich region flanked by proline‐rich regions, and a C‐terminal domain. HRG is an adaptor protein that interacts with many ligands, including Zn2+, haem, tropomyosin, heparin, heparan sulphate, plasminogen, and complement (Ronca & Raggi, 2015).
It has been shown that HRG can regulate many biological processes, such as angiogenesis, coagulation, fibrinolysis, and the phagocytosis of apoptotic cells (Poon, Patel, Davis, Parish, & Hulett, 2011). Shannon et al. (2010) demonstrated that HRG decreased the mortality of a septic mouse model with Streptococcus pyogenes‐induced abscesses by the killing and trapping of bacteria in the abscess sites. Wake et al. (2016) demonstrated that HRG prevents septic lethality through the regulation of neutrophils and the inhibition of the strong attachment of neutrophils to vascular endothelial cells. In recent clinical studies, HRG was further proposed as a new biomarker to predict the outcome of a sepsis patient (Kuroda et al., 2018; Nishibori, Wake, & Morimatsu, 2018). Based on these findings, our present research demonstrates for the first time the protective effects of HRG on the endothelium, especially the barrier dysfunction of endothelium in systemic septic conditions, and the possible mechanism. This will provide new evidence for the use of HRG as a supplementary therapy for the treatment of sepsis.
2. METHODS
2.1. Cell culture
EA.hy 926 cells (ATCC Cat# CRL‐2922, RRID:CVCL_3901), a hybridoma of HUVECs and the human epithelial cell line A549 were cultured using DMEM (Sigma) supplemented with 10% FBS (Gibco), 10% l‐glutamine (#G7513, Sigma), and 5% penicillin/streptomycin (Gibco) in 5% CO2 at 37°C. After reaching confluence, the endothelial cells were detached from culture flasks using 0.25% trypsin–EDTA (Gibco), washed, and resuspended in DMEM. These cells were passaged every 3–4 days, and all experiments were performed with cells kept in culture between three and six passages.
2.2. Purification of HRG from human plasma
HRG was purified from human plasma by our lab as described previously (Mori, Takahashi, Yamaoka, Okamoto, & Nishibori, 2003). Human plasma was supplied by the Japanese Red Cross Society from the healthy volunteer's donation. The study protocol complied with the principles outlined in the Declaration of Helsinki, and all subjects signed an informed consent. Briefly, human plasma was incubated with nickel–nitrilotriacetic acid (Ni–NTA) agarose (Qiagen, Hilden, Germany) for 2 hr at 4°C with gentle shaking. The gel was packed into a column and washed successively with 10 mmol·L−1 Tris‐buffered saline (pH 8.0) containing 10 mmol·L−1 imidazole and then 10 mmol·L−1 Tris buffer (pH 8.0) containing 1 mol·L−1 NaCl. Human HRG was eluted by 0.5 mol·L−1 imidazole in 10 mmol·L−1 Tris‐buffered saline (pH 8.0). The protein eluate from Ni–NTA was further purified by a Mono Q column (GE Healthcare, Little Chalfont, UK) with a NaCl gradient. Purified human HRG was identified by SDS‐PAGE and Western blotting with a human HRG‐specific antibody.
2.3. Immunostaining assay
EA.hy 926 cell suspensions (5 × 105 cells·ml−1) were cultured in 96‐well plates (Falcon, Tewksbury, MA) until confluent. The monolayer was then washed with PBS and pretreated with HRG or human serum albumin (HSA; 1 μmol·L−1) for 30 min before stimulation with LPS (Escherichia coli 0111:B4, Sigma) or TNF‐α (Sigma‐Aldrich; 100 ng·ml−1). After incubation, the cells were washed with PBS twice and fixed with 4% paraformaldehyde (Wako, Japan). Cell cytoskeleton F‐actin was stained with phalloidin–Alexa 568 (Invitrogen, Carlsbad, CA) at room temperature (RT) for 1 hr, and the intercellular junction proteins or NF‐κB were stained with primary antibodies for VE‐cadherin (Abcam, #ab33168, RRID:AB_870662), β‐catenin (Abcam, #ab16051), or NF‐κB p65 (Abcam, #ab16502, RRID:AB_443394) for 1 hr at 37°C, followed by Alexa Fluor 488 goat anti‐rabbit IgG (Invitrogen) secondary antibody for 1 hr at RT. Nuclei were stained with DAPI for 5 min. The samples were observed using a confocal microscope (LSM 780, Carl Zeiss). The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
2.4. Western blotting
Cells cultured in six‐well plates were collected with RIPA (50 mmol·L−1 Tris–HCl, 150 mmol·L−1 NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mmol·L−1 EDTA, 1 mmol·L−1 DTT, 20 mmol·L−1 β‐glycerophosphate, and protease/phosphatase inhibitors added immediately before use) and then electrophoresed on a polyacrylamide gel and transferred onto a PVDF membrane (Bio‐Rad, Hercules, CA). The membrane was blocked with 10% skimmed milk for 1 hr and incubated overnight at 4°C with rabbit anti‐VE‐cadherin Ab, anti‐p‐VE‐cadherin Ab (Abcam, #ab119785), anti‐FAK Ab (Abcam, #ab40794, RRID:AB_732300), anti‐p‐FAK Ab (Abcam, #ab81298), anti‐p38 Ab (Cell Signaling Technology, #9212S, RRID:AB_330713), anti‐ERK1/2 Ab (Cell Signaling Technology, #12940), anti‐p‐p38 (T180/Y183, Cell Signaling Technology, #4631SS, RRID:AB_331765) or anti‐p‐ERK1/2 Ab (T202/Y204, Cell Signaling Technology, Cat# 4370, RRID:AB_2315112), and anti‐MLC and anti‐p‐MLC Ab (Abcam, #ab157747) followed by goat anti‐rabbit IgG‐HRP (MBL, Nagoya, Japan) for 2 hr at RT. The signals were visualized by the luminal‐based enhanced chemiluminescence HRP substrate method (Thermo Fisher Scientific, MA). An Image Quant LAS4000 system was used for detection, and images were analysed with ImageJ software Version 1.51.
2.5. Cell detachment assay
First, 96‐well plates were coated with 5 μg per well extracellular matrix protein–laminin (Sigma‐Aldrich) at 37°C for 2 hr, then washed twice with PBS. EA.hy 926 endothelial cell suspensions were added into plain or laminin‐coated plates together with the mixture of HRG or HSA and LPS or TNF‐α (100 ng·ml−1). After incubation for 2 hr, the detached cells were washed out with PBS, and the adherent cells were fixed with 4% paraformaldehyde for 30 min and stained for 15 min with 1% crystal violet (Wako, Osaka, Japan). The cells were washed twice with PBS and air dried and then observed with an All‐in‐One Fluorescence Microscope (BZ‐X700, Keyence, Japan). After the microscopic observation, crystal violet was extracted from the cells with 100% methyl alcohol, and the OD was measured at 595‐nm wavelength with a multi‐detection reader Flex Station 3 (Molecular Devices, CA).
2.6. Cytometric bead array
We examined the cytokines secreted into the supernatant of cultured endothelial cells by performing a cytometric bead array using a Human Soluble Protein Master Buffer Kit and cytokine Flex Set (BD Biosciences, CA) following the manufacturer's instructions. Generally, multiple capture beads for IL‐6, IL‐8, IL‐1α, and IFN‐γ were mixed together. The mixed capture beads were co‐incubated with 50 μl of supernatant and detection reagent for 2 hr. The beads were then washed carefully and resuspended. Samples were analysed using a FACSCanto II system (BD Biosciences). The data were analysed with FCAP Array software.
2.7. RNA isolation and real‐time PCR
Cells were harvested, and mRNA was extracted using an RNeasy mini kit (Qiagen). cDNA was synthesized with a Takara RNA PCR kit Version 3.0 (Takara Bio, Nagahama, Japan) according to the manufacturer's instructions. Real‐time PCR was performed with a Light Cycler (Roche, Switzerland) according to the manufacturer's instructions. The primers shown in Table S1 were used to amplify specific cDNA fragments. β‐actin expression was used to normalize cDNA levels. The PCR products were analysed by a melting curve to ascertain the specificity of amplification, and the relative fold gene expression of samples was calculated with the Δ–Δ Ct method.
2.8. In vitro permeability assay
EA.hy 926 endothelial cells were seeded at a density of 5 × 105 cells·ml−1 onto 24‐well plates with 6.5‐mm‐diameter transwell inserts and a 0.4‐μm pore size polyester membrane (Corning, MA) and cultured until confluent. Cells were pre‐incubated for 30 min with HRG/HSA before stimulation with LPS/TNF‐α. After 12 hr, the culture medium was replaced with medium containing 0.5 mg·ml−1 FITC–dextran (70, 150, and 250 kDa, Sigma‐Aldrich) in the upper chamber. The fluorescence in the lower chamber, which represents the permeability of the monolayer of endothelial cells, was detected with the Flex Station 3 (Ex = 492 nm; Em = 518 nm) at different time points.
2.9. Animals
Adult male C57BL/6N mice (22 ± 3 g, 8 weeks, RRID:MGI_5658420) were purchased from SLC (Hamamatsu, Japan) and then housed in the Okayama University institutional animal units (12‐hr light cycle). The mice were maintained on a standard rodent diet with free access to water. Up to four mice were kept per plastic cage with aspen wood bedding material. All procedures involving animals were performed in accordance with guidelines of Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and approved by the Ethics Review Board of the Okayama University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology (McGrath and Lilley, 2015). Every effort was taken to minimize the number of animals used and their suffering (in line with the 3Rs).
2.10. Randomization and blinding
Animals were randomized for treatment. Data collection and evaluation of all experiments were performed blindly of the group identity.
2.11. Sepsis model
Two types of septic animal models were used in the present study: an LPS‐induced endotoxaemia model and a caecal ligation puncture (CLP) model. The LPS‐induced septic model was established with an i.v. injection of LPS (10 mg·kg−1) and was used for experiments 12 hr thereafter (London et al., 2010; Xu, Wu, Hack, Bao, & Cunningham, 2015). The CLP septic animal models were established as described in our previous study (Wake et al., 2016). Briefly, the mice were anaesthetized with 3% isoflurane in 48.5% N2O and 48.5% O2, and a ligature was set below the ileocaecal valve. The caecum was gently exteriorized from the peritoneal cavity and punctured twice with an 18‐gauge needle and was then returned back to the abdomen. The mice were killed 24 hr later with pentobarbital sodium (50 mg·kg−1, i.p.).
2.12. Vascular permeability in septic mice
We assessed the vascular permeability of different organs in endotoxaemic or septic mice by determining the Evans blue leakage from capillaries. First, HRG or HSA in vehicle (PBS) was administered through the tail vein immediately after the operation. Each mouse was given 20 mg·kg−1 HRG or HSA in a volume of 200 μl (i.v.). After 12 or 24 hr, mice were given an i.v. injection of Evans blue albumin (EBA, 20 mg·kg−1, Wako). EBA was allowed to circulate for 1 hr, and the mice were then anaesthetized and perfused with saline. The lung, liver, and kidney were excised, weighed, homogenized, and kept in 2 ml of formalin for 48 hr at 60°C. The extracted dye was then measured with a spectrometer at 610 nm. The absorbance value was converted to μg of Evans blue dye . g‐1 wet weight of lung, liver, or kidney respectively (London et al., 2010).
2.13. Rho GTPase pull‐down assay
RhoA regulates molecular events by cycling between an inactive GDP‐bound form and an active GTP‐bound form. In this study, RhoA GTPase activity was detected by a Rho GTPase pull‐down assay for the GTP‐bound active RhoA according to the manufacturer's instructions (STA‐403‐A‐T, Cell Biolabs, CA). EA.hy 926 cell suspensions (5 × 105 cells·ml−1) were cultured in six‐well plates until confluence. The cells were then washed with PBS and pretreated with HRG or HSA (1 μmol·L−1) for 30 min before the stimulation with LPS for 1 hr. At the end of the incubation, the culture medium was removed, and the cells were washed with ice‐cold PBS. Then, the cells were detached from the plate by adding ice‐cold 1× lysis buffer (Part No. 240102‐T) and scraping with a cell scraper. Cell lysates were then incubated with agarose beads containing rhotekin‐binding domain to pull down the activated RhoA. The beads were collected, and proteins were analysed by immunoblotting.
2.14. Data and statistical analysis
Data were analysed with GraphPad Prism statistic software (Version 6.01, San Diego, CA). All values are presented as the means ± SEM and analysed by ANOVA followed by Bonferroni's test or post hoc Fisher test when the F statistic was significant. P < .05 was considered statistically significant. At least five independent experiments were performed with all the assays. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018).
2.15. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
3. RESULTS
3.1. HRG inhibited stress fibre formation and cytoskeleton reorganization
Given the well‐known important functions of actin and actin‐binding proteins for endothelial barrier homeostasis (García‐Ponce, Citalán‐Madrid, Velázquez‐Avila, Vargas‐Robles, & Schnoor, 2015), we investigated the effects of HRG on the stress fibre formation and cytoskeleton remodelling induced by LPS/TNF‐α in endothelial cells. Under the resting condition, F‐actin predominated as a thin band along the cell boundaries, and no F‐actin bundles were present inside the cells (Figure 1a, left panel). We observed that 10 ng·ml−1 of LPS/TNF‐α stimulation for 2 hr induced the reorganization of the actin cytoskeleton, as evidenced by an increase in the thickness of the peripheral F‐actin band and the appearance of F‐actin bundles (stress fibres, indicated by arrows in Figure 1a, middle panel) inside the cell. Our quantitative analysis of these actin structures by determining the pixel intensities of these fibres inside the cells (Figure 1b, upper panel) revealed a significant increase in such F‐actin fibres compared with the control group (Figure 1c, left panel). Additionally, the fluorescence intensity of the horizontal diameter of the cell shown in Figure 1b (lower panel, red line) was measured as the central stress fibre density as described by Peacock et al. (2007). These measurements invariably revealed a significant increase in actin fibres crossing the cell centres of the cells stimulated with LPS/TNF‐α (Figure 1c, right panel).
Figure 1.
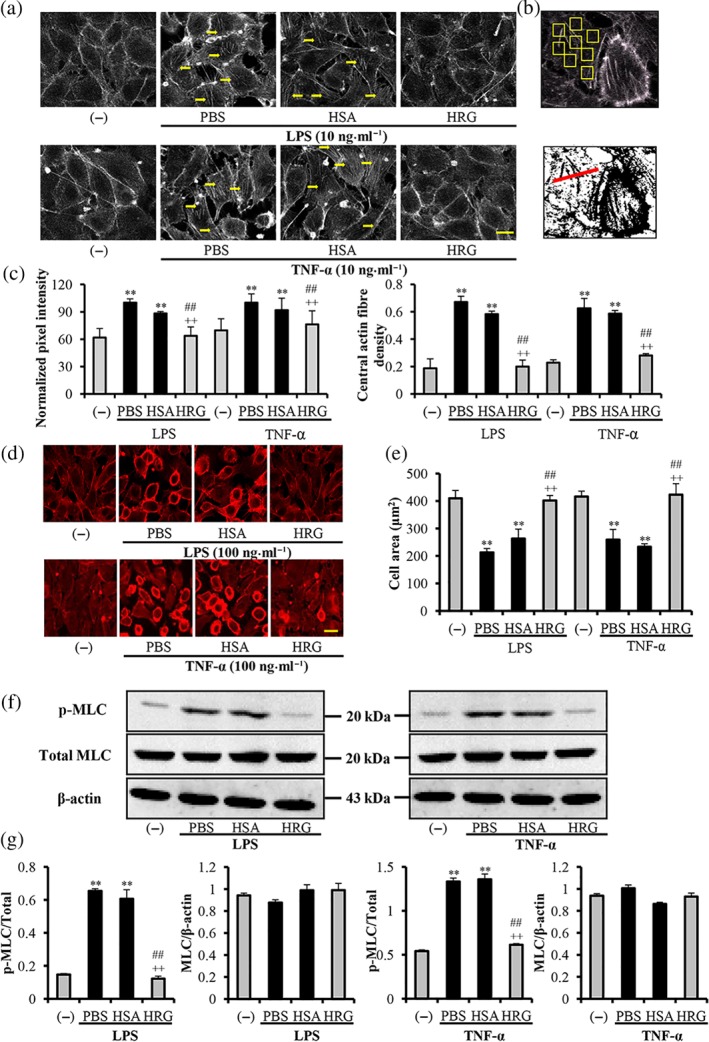
Effects of histidine‐rich glycoprotein (HRG) on LPS/TNF‐α‐induced stress fibre formation and cytoskeleton reorganization. EA.hy 926 endothelial cells were incubated with 1 μmol·L−1 of human serum albumin (HSA), HRG, or PBS for 30 min before stimulation with 10 ng·ml−1 LPS/TNF‐α for 2 hr (a) or 100 ng·ml−1 for 12 hr (d). The actin distribution and morphology of endothelial cells were analysed by fluorescence using Alexa 568–phalloidin. Actin staining is in grey (a) or red (d), and nucleus staining is blue. Images are representative of five independent experiments. Bar = 20 μm. (b) Upper: The method of actin fibre quantification is depicted as the yellow arrow shown in (a). Lower: the assessment of central actin fibre density, the results of which are shown in (c). Pixel intensities (c) and cell areas (e) were quantified using ImageJ software. (f)Western blot results of HRG on the LPS/TNF‐α‐induced phosphorylation of myosin light chain (MLC). (g) The quantification of Western blotting results in (f). The results in graphs are shown as the means ± SEM (n = 5 per group). One‐way ANOVA followed by the post hoc Fisher test, **P < .05 versus control, ## P < .05, and ++ P < .05 versus PBS and HSA respectively
After the cells were stimulated with LPS/TNF‐α (100 ng·ml−1) for 12 hr, we found that F‐actin had broken down and redistributed at the cell periphery accompanied by cell contraction towards the cell centre (Figure 1d, middle panel). The results were quantified with the cell area shown in Figure 1e. HRG (1 μmol·L−1) effectively inhibited this stress fibre formation (Figure 1a, right panel) and cell morphological changes (Figure 1d, right panel) and maintained the monolayer integrity. The results also demonstrated the concentration‐dependent effects of HRG on the morphology changes induced by LPS (Figure S1). HRG (1 μmol·L−1) significantly inhibited the phosphorylation of MLC induced by LPS/TNF‐α (Figure 1f). The quantification of Western blotting results is illustrated in Figure 1g. These results suggested that HRG inhibited the stress fibre formation and cytoskeleton reorganization through the regulation of MLC phosphorylation. However, HSA, a major plasma protein, did not show any inhibitory effects at the same concentration (1 μmol·L−1). The inhibitory effects of HRG on the LPS/TNF‐α induced cytoskeleton reorganization, and morphology changes are also true for the primary culture of human lung microvascular endothelial cells (HMVECs; Figure S6).
3.2. HRG prevented the loss of intercellular adherent junctions
VE‐cadherin is a major transmembrane adherent junction protein in vascular endothelial cells, and it represents a crucial determinant of endothelial barrier integrity. The immunostaining of VE‐cadherin and β‐catenin showed a linear staining pattern at the cell borders under control conditions in confluent endothelial monolayers (Figure 2a,b, left panel). Incubation with LPS/TNF‐α for 6 hr led to intercellular gap formation and a pronounced loss of VE‐cadherin and β‐catenin staining at the cell borders (middle panel). Pretreatment with HRG significantly prevented the loss of both VE‐cadherin and β‐catenin (Figure 2a,b, right panel). The inhibitory effects of HRG on the loss of VE‐cadherin also showed dose dependency (Figure S2). These protective effects are also true for the HMVECs (Figure S7). Scanning electron microscopy observations also confirmed the formation of intercellular gaps, thickness of the cell membrane, and microparticle‐like structure on the cell surface induced by LPS; this was also inhibited by HRG (Figure 2c). In parallel, the Western blotting results of VE‐cadherin also showed that HRG prevented the decrease in the expression of VE‐cadherin induced by LPS or TNF‐α (Figure 2d,e). The phosphorylation and dephosphorylation of intercellular tyrosine residues in the VE‐cadherin complex are responsible for the modulation of VE‐cadherin function (Weidert, Pohler, Gomez, & Dong, 2014). Wessel et al. (2014) showed that Tyr 685 and Tyr 731 of VE‐cadherin distinctly and selectively regulate the induction of vascular permeability or leukocyte extravasation. In this study, we investigated the effects of HRG on the Y‐685 phosphorylation in VE‐cadherin. The results showed that HRG effectively inhibited the LPS/TNF‐α‐stimulated phosphorylation of VE‐cadherin at Tyr 685 site (Figure 2d,e).
Figure 2.
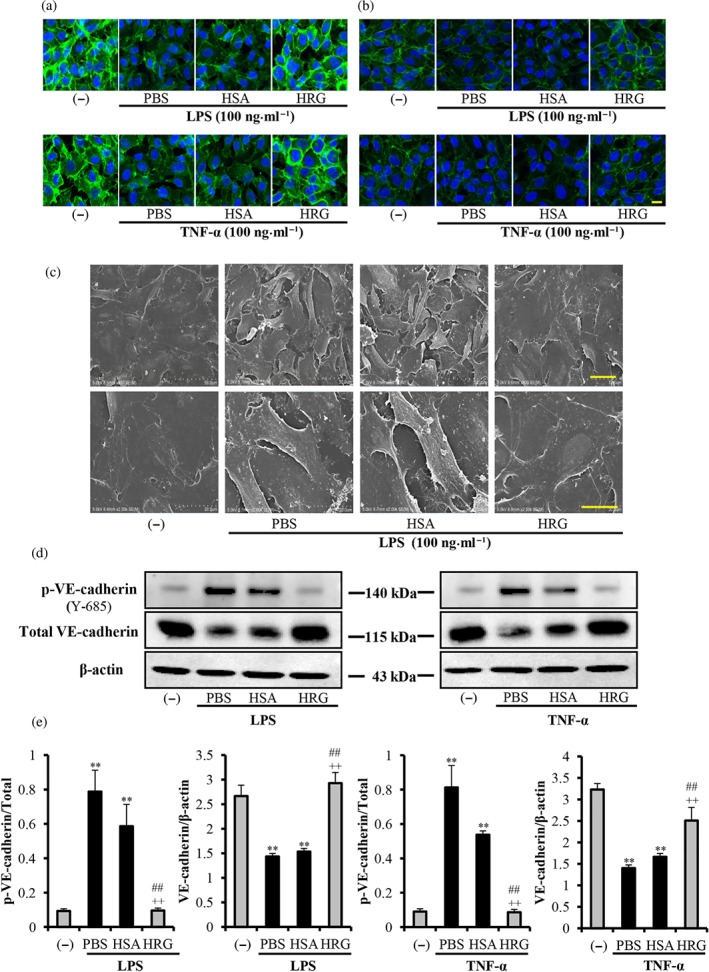
Histidine‐rich glycoprotein (HRG) prevents the loss of intercellular junction. Immunostaining results of VE‐cadherin/β‐catenin in endothelial cells stimulated with 100 ng·ml−1 LPS or TNF‐α for 6 hr after treatment with HRG or human serum albumin (HSA). (a, b) Cells were incubated with anti‐VE‐cadherin mAb or anti‐β‐catenin mAb for 2 hr and then stained with Alexa Fluor 488 (green) goat‐anti‐rabbit IgG. The cells were also stained with DAPI (blue) to visualize the nuclei. The pictures are representative of three independent experiments (n = 5 per group). Scale bar = 20 μm. (c) Scanning electron microscopy pictures of vascular endothelial cells. Scale bar = 50 μm (upper panel) or 20 μm (lower panel). (d, e) Quantification of results of the p‐VE‐cadherin and VE‐cadherin by Western blotting. One‐way ANOVA followed by the post hoc Fisher test, **P < .05 versus control, ## P < .05, and ++ P < .05 versus PBS and HSA respectively
Moreover, HRG prevented the LPS/TNF‐α‐induced loss of other junction molecules like occludin and ZO‐1, which were involved in the endothelium permeability control (Figure S3).
3.3. LPS/TNF‐α‐induced degradation of focal adhesion kinase and cell detachment were reduced by HRG
Focal adhesion kinase (FAK) is associated with actin stress fibres, which serve as holding points for cytoskeletal tension. FAK is responsible for the regulation of cell contractility and cell–matrix adhesion. LPS was reported to induce the stress fibre formation, associated with phosphorylation and degradation of FAK. The results in Figure 3a showed that LPS and TNF‐α induced slight FAK degradation and strongly stimulated phosphorylation of FAK, which were inhibited by the treatment with HRG. These results proved that HRG not only prevented the loss of FAK but also effectively regulated the FAK phosphorylation stimulated by LPS/TNF‐α. To further confirm the regulation effects of HRG on cell–matrix adhesion, the cell detachment assay was performed. Results showed that LPS/TNF‐α stimulation led to a large number of cells detaching from the plain plates (Figure 3b(A,C)) and laminin‐coated plates (Figure 3b(B,D)), whereas HRG effectively prevented the cell detachment under both conditions. The results also statistically analysed the measurement of OD of crystal violent remaining on the plates at 595 nm (Figure 3c).
Figure 3.
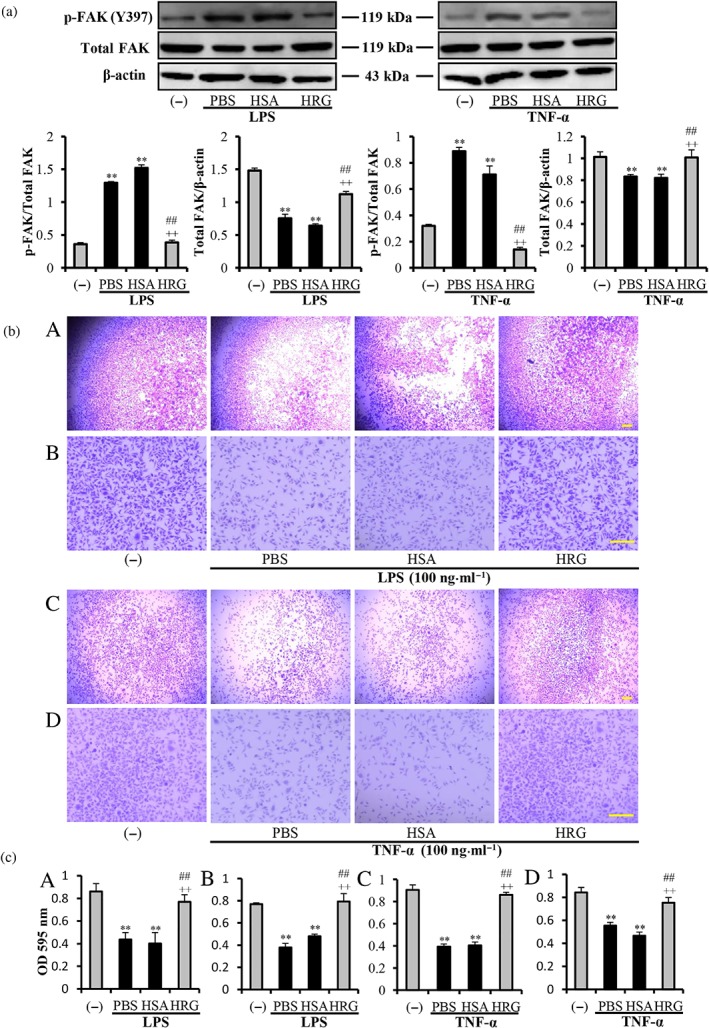
Effects of histidine‐rich glycoprotein (HRG) on the degradation of focal adhesion kinase (FAK) and cell detachment. (a) Western blot results and quantification of the phosphorylated FAK (p‐FAK) and degraded FAK induced by LPS/TNF‐α with ImageJ. (b) The cell suspensions were plated into 96‐well plain plates (A, C) or laminin‐coated plates (B, D). The cell detachment was evaluated by staining the remaining cells with 1% crystal violet. (c) Quantitative analysis of crystal violet extracted from the remaining cells. Panels A–D correspond to A–D in (b). Data are means ± SEM in the graph (n = 5 per group). One‐way ANOVA followed by the post hoc Fisher test, **P < .05 versus control, ## P < .05, and ++ P < .05 versus PBS and human serum albumin (HSA) respectively. Scale bar = 10 μm
3.4. HRG suppresses the cytokine secretion and mRNA expression of inflammation‐related molecules in endothelial cells
According to the results, a 12‐hr incubation of endothelial cells with LPS/TNF‐α resulted in a large increase in the secretion of IL‐6, IL‐8, IL‐1α, and IFN‐γ in the culture media compared with the non‐stimulated control group. HRG (1 μmol·L−1) almost completely inhibited all of the cytokine release induced by LPS, and the same concentration of HRG had less effect on the IL‐6 release induced by TNF‐α (Figure 4a). Consistent with above results, HRG also reduced the expression of IL‐6, IL‐8, IL‐1α, IFN‐γ, and IL‐10 at the mRNA levels (Figure 4b). From these results, we speculate that the reduced cytokine secretions from endothelial cells by HRG were due to the regulation of both secretory pathway and mRNA expression.
Figure 4.
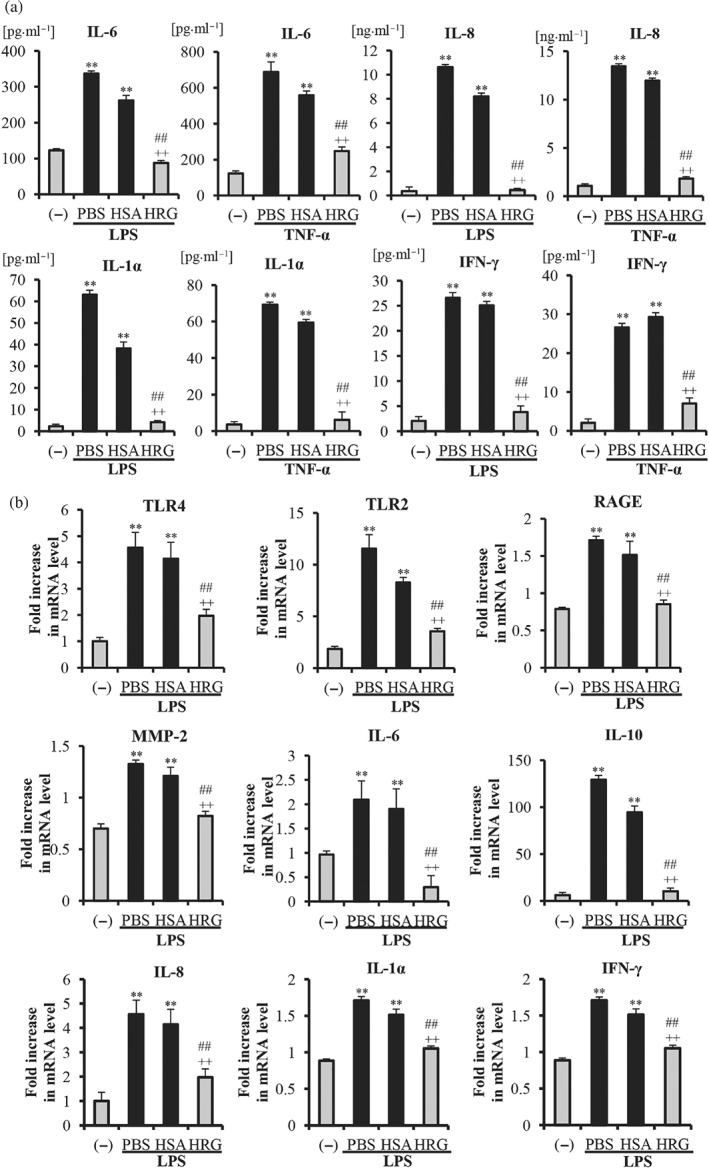
Histidine‐rich glycoprotein (HRG) inhibited the cytokine secretion and mRNA expressions of inflammation‐related molecules in endothelial cells. The cell culture media were collected for the determination of cytokines. (a) The levels of IL‐6, IL‐8, IL‐1α, and IFN‐γ in the culture media from each group are shown as the means ± SEM (n = 5 per group). (b) The expressions of TLR2, TLR4, RAGE, MMP‐2, IL‐6, IL‐8, IL‐1α, IFN‐γ, and IL‐10 at the mRNA level on endothelial cells were measured by quantitative RT‐PCR. The results were normalized to the expression of β‐actin and are expressed as the means ± SEM (n = 5 per group). One‐way ANOVA followed by the post hoc Fisher test, **P < .05 versus control, ## P < .05, and ++ P < .05 versus PBS and human serum albumin (HSA) respectively
Moreover, our previous study (Wake et al., 2016) already proved that the systemic injection of HRG significantly reduced the cytokine levels in the septic mice compared with control HSA‐injected group. In the present study, the effects of HRG on the serum cytokine levels in LPS‐injected mice were also determined with cytometric bead array. HRG significantly reduced the IL‐6, IL‐10, and TNF‐α levels in serum of LPS‐injected mice compared with control HSA‐injected group (Figure S4).
As presented in Figure 4b, LPS enhanced the expression of all three receptors (TLR2, TLR4, and RAGE) on endothelial cells. Interestingly, HRG partially but significantly inhibited the stimulatory effects of LPS on the expression of all three receptors. The stimulatory effect of LPS on the expression of TLR2 was higher than those of the other two receptors, and the inhibitory effect of HRG on the expression of this receptor was also more pronounced. MMP‐2 has been reported to be secreted from different cells (Alexander & Elrod John, 2002), leading to the degradation of tight junctions and adherens junctions, which results in an increase in the vascular permeability. The results also showed that HRG can effectively suppress the LPS‐induced up‐regulation of MMP‐2 mRNA.
3.5. LPS/TNF‐α‐induced NF‐κB activation was prevented by HRG in immunostaining
NF‐κB/p65 is a member of the NF‐κB protein family. It is transferred to the nucleus and bound to a specific sequence in the genome promoter regions, resulting in the regulation of proinflammatory responses and endothelial permeability when the cells receive inflammatory stimuli (Sprague & Khalil, 2009). We observed that NF‐κB/p65 was located in the cytoplasm in the non‐stimulated control group, whereas in the LPS/TNF‐α treated cells, NF‐κB/p65 was translocated into the nuclei. LPS‐induced changes in translocation occurred 12 hr after stimulation (Figure 5a), and the translocation by TNF‐α was evident 6 hr after stimulation (Figure 5b). However, pretreatment with HRG clearly suppressed the LPS/TNF‐α‐induced NF‐κB/p65 translocation to nuclei in endothelial cells (Figure 5a,b). The results of our quantitative analysis of the relative intensity of fluorescence in the nuclei/cytosol are provided in Figure 5c. The preventive effects of HRG on the LPS/TNF‐α‐induced NF‐κB activation were also observed in HMVECs (Figure S8).
Figure 5.
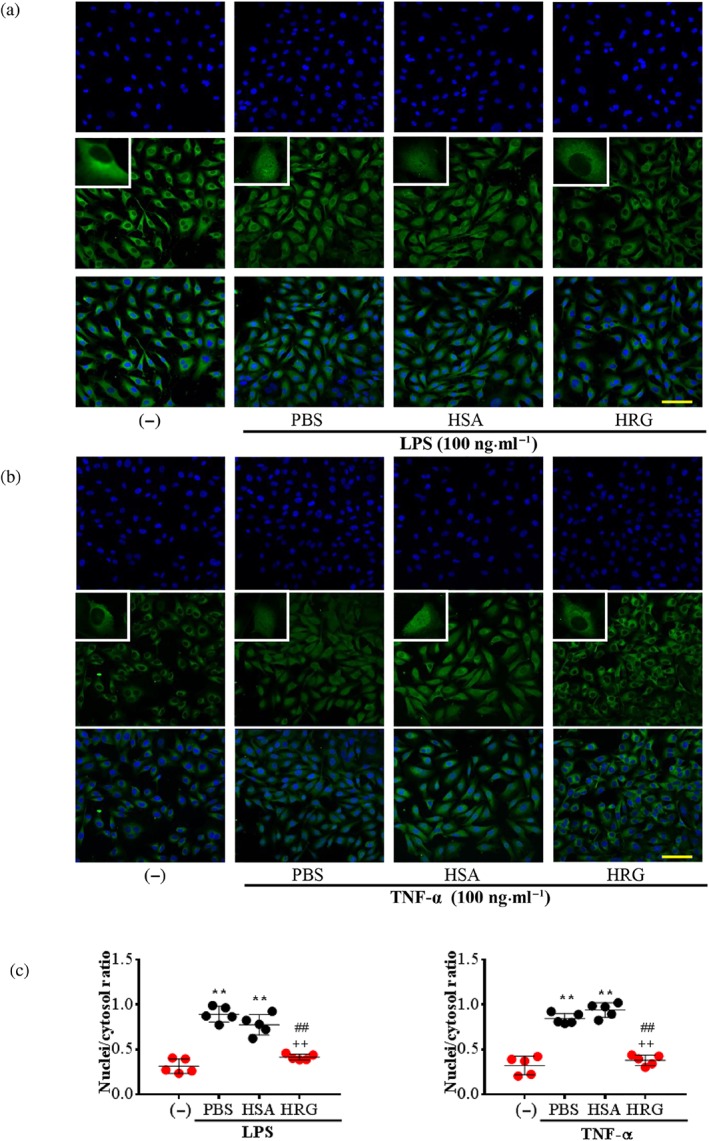
Histidine‐rich glycoprotein (HRG) suppresses the LPS/TNF‐α‐induced activation of NF‐κB in endothelial cells. Immunostaining results of NF‐κB in endothelial cells stimulated with 100 ng·ml−1 LPS for 12 hr or 100 ng·ml−1 TNF‐α for 6 hr after treatment with HRG or human serum albumin (HSA). (a, b) Cells were stained with anti‐NF‐κB/p65 mAb for 2 hr and then stained with Alexa Fluor 488 (green) goat‐anti‐rabbit IgG. Cells were also stained with DAPI (blue) to visualize the nucleus. The results shown are representative of ≥5 experiments. Scale bar = 10 μm. (c) The nuclei/cytosol fluorescence intensity ratio represents the translocation of NF‐κB from the cytoplasm to nucleus. Data are means ± SEM (n = 5 per group). One‐way ANOVA followed by the post hoc Fisher test, **P < .05 versus control, ## P < .05, and ++ P < .05 versus PBS and HSA
3.6. Protective role of HRG on the permeability of vascular endothelium in vitro and in vivo
In the FITC–dextran transwell assay as depicted in Figure 6a, LPS/TNF‐α induced significant leakage of 70‐kDa FITC–dextran from the endothelial monolayer compared with the control group, and pretreatment with HRG produced a significant inhibition of dextran leakage during the 12‐hr stimulation compared with the HSA treatment group. This effect was also true for LPS‐induced 150‐ or 250‐kDa FITC–dextran leakage (Figure 6b,c). Moreover, HRG also showed the inhibitory effects on the LPS/TNF‐α‐induced hyperpermeability in HMVEC monolayer in vitro (Figure S5).
Figure 6.
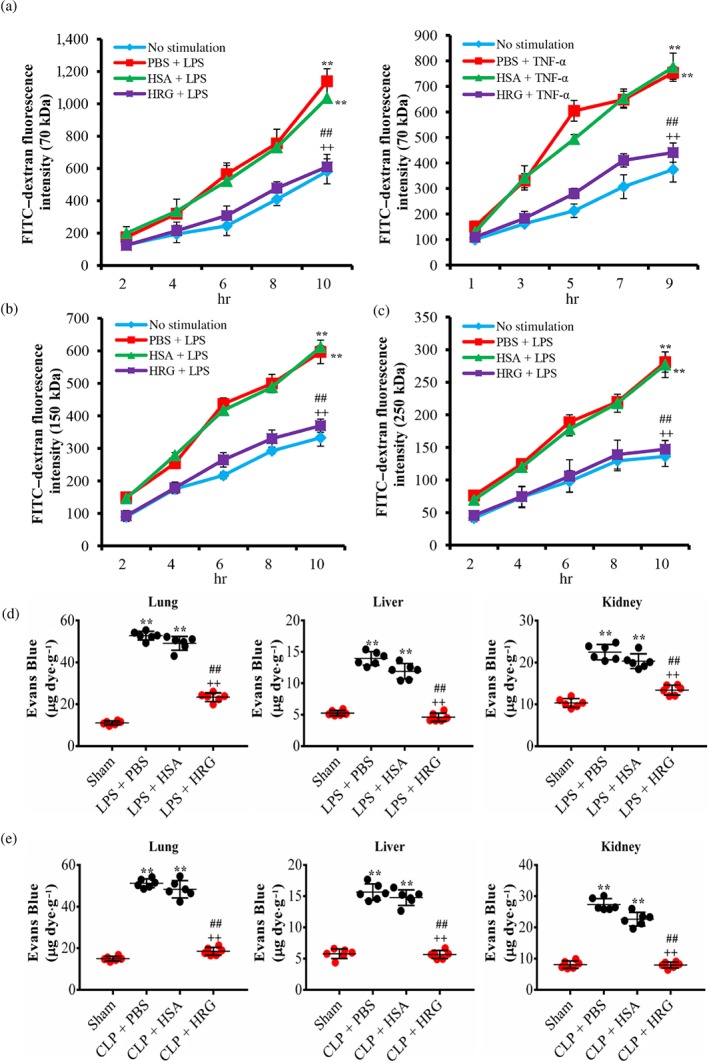
Protective effects of histidine‐rich glycoprotein (HRG) on endothelial permeability in vitro and in vivo. (a–c) Effects of HRG on LPS/TNF‐α‐induced hyperpermeability of the endothelial monolayer. The graph shows the fluorescence intensity of FITC–dextran leakage, at different sizes, from transwell filters to the lower chamber. Data are means ± SEM (n = 5 per group). One‐way ANOVA followed by the post hoc Fisher test, **P < .05 versus control, ## P < .05, and ++ P < .05 versus PBS and human serum albumin (HSA). (d) Mice were given an i.v. injection of LPS (10 mg·kg−1) and then treated with HRG or HSA (20 mg·kg−1) for 12 hr. (e) Mice were subjected to caecal ligation puncture (CLP) or a sham operation and then an immediate i.v. injection of HRG or HSA. At 12 or 24 hr later, the mice received an i.v. injection of Evans blue dye (EBA), and EBA extraction was measured in the lung, liver, and kidney to assess vascular permeability (n = 6 per group). The extracted dye was then measured with a spectrometer at 610 nm. The absorbance value was converted to μg Evans blue dye.g‐1 wet weight of lung, liver, or kidney respectively. Statistical analysis was performed by one‐way ANOVA with Bonferroni's post hoc test, **P < .05 versus sham, ## P < .05, and ++ P < .05 versus PBS and HSA respectively
To examine whether HRG reduces hyperpermeability under cytokine storm conditions in vivo, we used a bacterial endotoxin model of inflammation. LPS administration triggered a large increase in capillary permeability at 12 hr after the injection. Using EBA as a tracer, we observed that HRG significantly reduced the EBA leakage from capillaries in the lung, liver and kidney of LPS‐injected mice (Figure 6d). To test the physiological relevance of our findings and to determine whether the effect of HRG is limited to the administration of LPS, we used a model of polymicrobial sepsis known as CLP. HRG also significantly reduced vascular permeability in the lung, liver, and kidney after 24 hr in the sepsis‐induced barrier dysfunction model (Figure 6e).
3.7. HRG prevented the activation of MAPK and Rho signal pathway induced by LPS
ROCK and MAPK cascades, which include p38 and ERK1/2, play critical roles in the regulation of VE‐cadherin redistribution and cytoskeletal polymerization in endothelial permeability control (Yuan, 2002). The results show that LPS stimulation induced p38 and ERK1/2 phosphorylation in a time‐dependent manner, as the activation of p38 and ERK1/2 was biphasic with an initial increase (peaking at 1 hr) followed by a slight drop in the phosphorylation levels (Figure 7a,b). HRG effectively inhibited the phosphorylation of p38 and ERK1/2 induced by LPS (Figure 7c). The inhibitory effect of HRG on the LPS‐induced activation of Rho A (GTP bound) was shown in Figure 7d. The quantification of Western blotting results is illustrated in Figure 7e.
Figure 7.
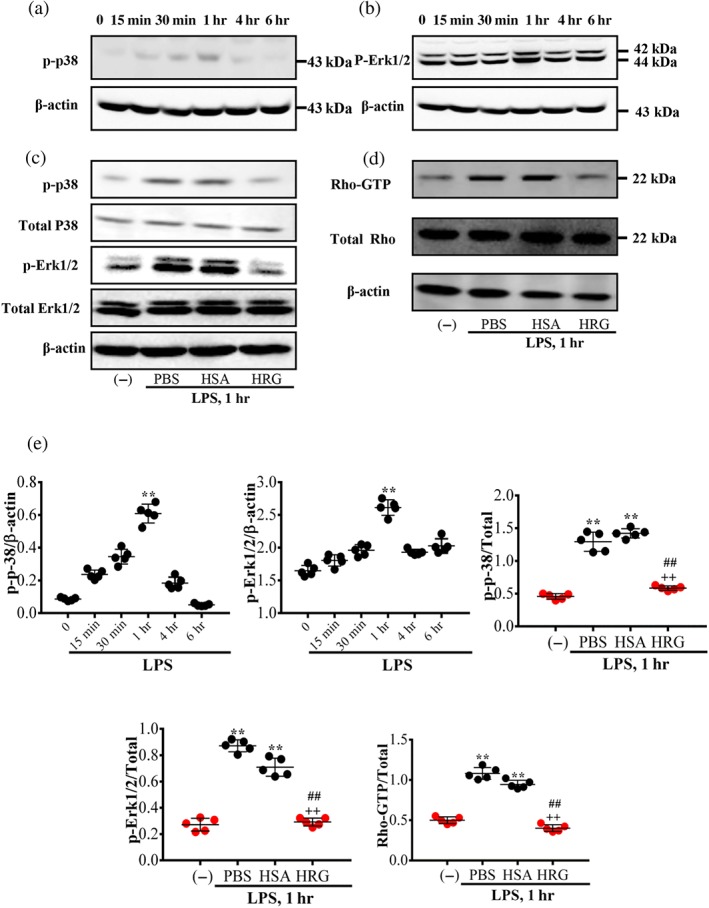
Effects of histidine‐rich glycoprotein (HRG) on the inhibition of MAPK and Rho activation. The cells were collected after stimulation with LPS at different time point, and a Western blot assay was then performed. (a, b) Time‐dependent phosphorylation of p38 and ERK1/2 induced by LPS. (c) Effects of HRG on the LPS‐induced phosphorylation of p38 and ERK1/2 at 1 hr. (d) The effect of HRG on the activation of RhoA was measured at 1 hr. (e) Quantification of the above Western blotting results. Data are means ± SEM in the graph (n = 5 per group). One‐way ANOVA followed by the post hoc Fisher test, **P < .05 versus control, ## P < .05, and ++ P < .05 versus PBS and human serum albumin (HSA) respectively
4. DISCUSSION
Endothelial barrier dysfunction and microvascular leakage critically contribute to the pathogenesis of organ failure and sepsis‐related complications (Goldenberg, Steinberg, Slutsky, & Lee, 2011). The endothelium is thus generally regarded as a target for the treatment of sepsis, especially with regard to endothelial barrier repair strategies. Currently, there is no appropriate therapy for recovery of the endothelial barrier dysfunction in sepsis. The present study extends our previous investigations and shows that plasma protein HRG has a potent protective effect on LPS‐ or septic‐induced barrier dysfunction of endothelium.
The function of the vascular barrier is maintained by a balance between endothelial cells' contractile forces and adhesive cell–cell and cell–matrix tethering forces; the former generate centripetal tension, and the latter regulate cell shape. Stress fibres composed of bundles of polymerized actin are the primary elements of the contractile machinery of endothelial cells, which assemble in a characteristic manner in response to permeability‐increasing mediators. In our experiments, stimulation with a low concentration (10 ng·ml−1) of LPS/TNF‐α for 2 hr induced stress fibre formation in endothelial cells (Figure 1a), whereas a higher concentration of LPS/TNF‐α (100 ng·ml−1) for 12 hr resulted in marked changes in cell morphology (Figure 1d). These results suggested a time‐ and dose‐dependent increase in cytoskeleton rearrangement and contractile forces. HRG effectively inhibited the stress fibre formation and cell rounding, thus maintaining the monolayer integrity in a dose‐dependent manner (Figure S1). Phosphorylated MLC (p‐MLC) enables the formation of stress fibre and leads to increased contractility in endothelial cells (García‐Ponce et al., 2016; Nwariaku et al., 2002). The results in Figure 1f,g show that HRG effectively reduced LPS/TNF‐α‐induced phosphorylation of MLC, indicating that HRG's protective effects on endothelial barrier function are related to the prevention of contractile stress fibre formation through the inhibition of the phosphorylation of MLC.
VE‐cadherin is the major transmembrane intercellular adherens junction that links to the actin cytoskeleton through α‐catenin and β‐catenin to provide both mechanical stability and the transduction of extracellular signals into the cell, and it thus represents a crucial determinant of endothelial barrier integrity (Dejana & Orsenigo, 2013). Soluble VE‐cadherin appears to play a role in sepsis and has the potential to be a clinical marker for the early detection of the loss of microvascular barrier functions in sepsis (Flemming et al., 2015). Focal adhesions provide additional adhesive forces in barrier regulation by forming a critical bridge for bidirectional signal transduction between the actin cytoskeleton and the cell–matrix interface. FAK is a major contributor to the regulation of focal adhesion formation and is a key determinant of vessel wall permeability through the regulation of cell–matrix adhesion. Our results demonstrated that HRG inhibited the loss of VE‐cadherin and β‐catenin at the cell border (Figure 2a,b) and also prevented the FAK degradation and phosphorylation induced by LPS/TNF‐α (Figure 3a). In addition, HRG enhanced the cell–matrix adhesion by inhibiting the cell detachment from laminin‐coated plates (Figure 3b,c). These results revealed that HRG protected the permeability of vascular endothelial cells through the regulation of intercellular junctions as well as cell matrix adhesion. Two phosphorylation sites of VE‐cadherin (Tyr 731 and Tyr 685) have been reported to be involved in controlling endothelial permeability and regulation of cell–cell adhesions (Weidert et al., 2014). In fact, it was found that HRG inhibited the phosphorylation of VE‐cadherin at Tyr 685. This mechanism may contribute to the maintenance of the intercellular junctions.
During the septic process, many pathogenic factors can stimulate the production of inflammatory mediators that lead to microvascular dysfunction. LPS acting on the endothelial cells can facilitate the release of secondary mediators such as TNF‐α, IL‐1α, and IL‐8 and the activation of toll‐like receptors. Thus, the inhibition of the production of proinflammatory cytokines may be a key factor in the prevention and therapy of sepsis. In present study, we examined the secretion of IL‐6, IL‐8, IL‐1α, and IFN‐γ and the expression of mRNAs for TLR2, TLR4, RAGE, MMP‐2, IL‐6, IL‐8, IL‐1α, IFN‐γ, and IL‐10 in LPS‐induced endothelial cells. The results presented in Figure 4 demonstrated that pretreatment with HRG can strongly inhibit the enhanced expression/secretion of cytokines as well as pattern recognition receptors. IL‐8 is a well‐known neutrophil chemotactic factor. The inhibitory effects of HRG on the IL‐8 secretion and expression also supported the previous finding that HRG can strongly suppress the adhesion of neutrophils to lung vasculatures in septic mice (Wake et al., 2016). Moreover, the suppressive effects of HRG on hypercytokinaemia in septic mice were also observed in the LPS‐injected mice (Figure S4). These findings support the hypothesis that HRG may exert anti‐septic effects through the regulation of cytokine production and barrier dysfunction in vascular endothelial cells, in addition to a strong regulator role for neutrophils (Wake et al., 2016).
LPS activates the release of these cytokines by a transcriptional mechanism involving the activation of NF‐κB (Cohen, 2002). NF‐κB is an important transcription factor involved mainly in inflammatory responses. Our immunofluorescence staining analysis revealed that HRG suppressed the translocation of NF‐κB (p65) from the cytoplasm into the nucleus induced by LPS/TNF‐α (Figure 5), which further explains the effective role of HRG on inhibition of cytokine secretion induced by LPS.
Our results showed that HRG prevented the LPS/TNF‐α‐induced FITC–dextran leakage with different size from the endothelial monolayer, even after 12‐hr stimulation (Figure 6a–c). The fluorescence intensity of 70‐kDa FITC–dextran in lower chamber of vascular endothelial monolayer stimulated by LPS is higher than that of 150 and 250 kDa. In fact, with the prolongation of the stimulation time, the endothelial monolayer seemed to spontaneously develop FITC–dextran leakage. This may be ascribed to the usage of FBS‐free medium for the cell culture. Even under such conditions, HRG had a significant inhibitory effect on the permeability compared with the HSA‐treated group, but HRG had no effect on the basal permeability.
HRG also prevented the hyperpermeability of different organs, the lung, liver, and kidney, in the LPS‐injected mice and septic mice (Figure 6d,e). Wake et al. (2016) demonstrated that HRG treatment inhibited the leukocyte infiltration and oedema in lung and glomerulus in CLP mice at 24 hr after the onset. Thus, the present results are consistent with the previous findings concerning the protection of capillary permeability by HRG.
In our experiment, EA.hy 926‐immortalized endothelial cell line was used for the study of the effects of HRG on the barrier dysfunction induced by LPS/TNF‐α. To further confirm the role of HRG on the endothelial permeability, we also performed several experiments with culture of primary HMVECs, which was often used for the permeability study. The results showed that HRG has potent inhibitory effects on the LPS/TNF‐α‐induced hyperpermeability, cytoskeleton reorganization, loss of VE‐cadherin, and NF‐κB activation (Figures S5–S8) in HMVECs, which provided critical evidence that HRG produces protective effects on the barrier function under inflammatory conditions in both EA.hy 926 endothelial cells and HMVECs.
To elucidate the potential mechanisms leading to the inhibition of NF‐κB activation by HRG, we explored LPS‐induced signalling cascades including two distinct types of MAPKs (p38 MAPK and ERK1/2). The two MAPKs are known to act as upstream intermediates providing a link between extracellular stimuli and cytokine production in various cell lines. Once activated, both p38 MAPK and ERK1/2 may activate transcription factors such as NF‐κB (Hippenstiel et al., 2000; Johnson & Lapadat, 2002). In addition, both p38 MAPK and ERK1/2 are involved in the regulation of cytoskeleton rearrangement and the permeability control of endothelial cells. We observed a time‐dependent activation of p38 and ERK1/2 by LPS with the maximal activation at 1 hr. This transient activation of MAPKs was inhibited by the treatment with HRG, whereas HSA had no such effects (Figure 7a–c). A key contractile event in endothelial cells is the phosphorylation of regulatory MLC catalysed by Ca2+/calmodulin‐dependent MLC kinase and/or through the activation of the Rho/Rho kinase pathway. It has been known for several years that RhoA and ROCK activation are present downstream of many permeability‐increasing mediators and contribute to increased permeability. Our results also demonstrated that HRG inhibited the activation of RhoA, which explains the manner in which HRG may inhibit the morphology changes and hyperpermeability of endothelial cells stimulated by LPS/TNF‐α (Figure 7d).
Clinical study was also performed to collect blood samples from patients within 24 hr of ICU admission, and HRG levels were measured with elisa. The results showed that plasma HRG levels of the septic patients (8.71 μg·mL−1) were significantly lower than in healthy volunteer (63.0 μg·mL−1) and also lower than in non‐infective systemic inflammatory response syndrome patients (33.3 μg·mL−1; Kuroda et al., 2018; Nishibori et al., 2018). HRG showed a high sensitivity and specificity for diagnosing sepsis. Moreover, plasma HRG levels were a good prognostic indicator, and the determination of plasma HRG alone could still predict mortality (Kuroda et al., 2018; Nishibori et al., 2018). Our present research proved that HRG has a potent protective effect on the vascular endothelium barrier under septic conditions, which indicates that HRG is suitable as a supplementary treatment for sepsis. However, the specific mechanism through which HRG mediates this effect on the endothelium needs to be further investigated.
5. CONCLUSION
The present study provides strong evidences that HRG efficiently protects vascular endothelial cells under inflammatory conditions. The effects may contribute to the anti‐septic effects of HRG administered systemically. Thus, the present study not only clarifies the effects of HRG on vascular endothelial cells but also facilitates our understanding of the pathophysiology of sepsis. As a whole, supplementary therapy with HRG should be encouraged for the treatment of sepsis.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
S.G. conceived the study, designed the experiments, analysed the data, and wrote the manuscript; M.N. and H.W. edited the manuscript; S.M. and H.W. contributed in the purification of HRG from human plasma; Y.G., D.W., and K.L. contributed in performing the experiments; and K.T. and H.T. contributed in critically reviewing the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Fig. S1.
Dose‐dependent effects of HRG on the LPS‐induced cell morphology changes.
Figure S2. Dose‐dependent effects of HRG on the LPS‐induced loss of VE‐cadherin.
Figure S3. HRG prevents the loss of intercellular junctions.
Figure S4. Effects of HRG on the cytokine levels in the serum of LPS‐injected mice.
Figure S5. Protection effects of HRG on the HMVECs permeability in vitro.
Figure S6. Effects of HRG on LPS/TNF‐α‐induced cytoskeleton reorganization in HMVECs.
Figure S7. HRG prevents the loss of intercellular junction in HMVECs.
Figure S8. HRG suppresses the LPS/TNF‐α‐induced NF‐κB activation in HMVECs.
Table S1. RT‐PCR Primer Sequence
ACKNOWLEDGEMENTS
This research was supported by grants from Japan Agency for Medical Research and Development (18im0210109h0002), the Secom Science and Technology Foundation to M.N., and the Grant‐in‐Aid for Scientific Research (15H04686 to M.N. and 16K08232 to K.T.) and a Grant‐in‐Aid for Young Scientists (17K15580 to H.W.) from the Japan Society for the Promotion of Science. The authors thank the Japanese Red Cross Society for providing the human fresh frozen plasma.
Gao S, Wake H, Gao Y, et al. Histidine‐rich glycoprotein ameliorates endothelial barrier dysfunction through regulation of NF‐κB and MAPK signal pathway. Br J Pharmacol. 2019;176:2808–2824. 10.1111/bph.14711
REFERENCES
- Aird, W. C. (2003). The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood, 101, 3765–3777. 10.1182/blood-2002-06-1887 [DOI] [PubMed] [Google Scholar]
- Alexander, J. S. , & Elrod John, W. (2002). Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. Journal of Anatomy, 200, 561–574. 10.1046/j.1469-7580.2002.00057.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174 (Suppl. 1, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174 (Suppl. 1, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. , (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide forsubmission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backer, D. , Creteur, J. , Preiser, J.‐C. , Dubois, M.‐J. , & Vincent, J.‐L. (2002). Microvascular blood flow is altered in patients with sepsis. American Journal of Respiratory and Critical Care Medicine, 166, 98–104. 10.1164/rccm.200109-016OC [DOI] [PubMed] [Google Scholar]
- Baeuerle, P. A. , & Baltimore, D. (1996). NF‐κB: Ten years after. Cell, 87, 13–20. 10.1016/S0092-8674(00)81318-5 [DOI] [PubMed] [Google Scholar]
- Burridge, K. , Fath, K. , Kelly, T. , Nuckolls, G. , & Turner, C. (1988). Focal adhesions: Transmembrane junctions between the extracellular matrix and the cytoskeleton. Annual Review of Cell Biology, 4, 487–525. 10.1146/annurev.cb.04.110188.002415 [DOI] [PubMed] [Google Scholar]
- Cohen, J. (2002). The immunopathogenesis of sepsis. Nature, 420, 885–891. 10.1038/nature01326 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana, E. , Corada, M. , & Lampugnani, M. G. (1995). Endothelial cell‐to‐cell junctions. The FASEB Journal, 9, 910–918. 10.1096/fasebj.9.10.7615160 [DOI] [PubMed] [Google Scholar]
- Dejana, E. , & Orsenigo, F. (2013). Endothelial adherens junctions at a glance. Journal of Cell Science, 126, 2545–2549. [DOI] [PubMed] [Google Scholar]
- Flemming, S. , Burkard, N. , Renschler, M. , Vielmuth, F. , Meir, M. , Schick, M. A. , … Schlegel, N. (2015). Soluble VE‐cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovascular Research, 107, 32–44. 10.1093/cvr/cvv144 [DOI] [PubMed] [Google Scholar]
- García‐Ponce, A. , Citalán‐Madrid, A. F. , Vargas‐Robles, H. , Chánez‐Paredes, S. , Nava, P. , Betanzos, A. , … Schnoor, M. (2016). Loss of cortactin causes endothelial barrier dysfunction via disturbed adrenomedullin secretion and actomyosin contractility. Scientific Reports, 6, 29003 10.1038/srep29003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Ponce, A. , Citalán‐Madrid, A. F. , Velázquez‐Avila, M. , Vargas‐Robles, H. , & Schnoor, M. (2015). The role of actin‐binding proteins in the control of endothelial barrier integrity. Thrombosis and Haemostasis, 113, 20–36. 10.1160/TH14-04-0298 [DOI] [PubMed] [Google Scholar]
- Gill, S. E. , Taneja, R. , Rohan, M. , Wang, L. , & Mehta, S. (2014). Pulmonary microvascular albumin leak is associated with endothelial cell death in murine sepsis‐induced lung injury in vivo. PLoS ONE, 9, e88501 10.1371/journal.pone.0088501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg, N. M. , Steinberg, B. E. , Slutsky, A. S. , & Lee, W. L. (2011). Broken barriers: A new take on sepsis pathogenesis. Science Translational Medicine, 3, 88ps25. [DOI] [PubMed] [Google Scholar]
- Guay, J. , Lambert, H. , Gingras‐Breton, G. , Lavoie, J. N. , Huot, J. , & Landry, J. (1997). Regulation of actin filament dynamics by p38 map kinase‐mediated phosphorylation of heat shock protein 27. Journal of Cell Science, 110, 357–368. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B. M. (1996). Cell adhesion: The molecular basis of tissue architecture and morphogenesis. Cell, 84, 345–357. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenstiel, S. , Soeth, S. , Kellas, B. , Fuhrmann, O. , Seybold, J. , Krüll, M. , … Suttorp, N. (2000). Rho proteins and the p38‐MAPK pathway are important mediators for LPS‐induced interleukin‐8 expression in human endothelial cells. Blood, 95, 3044–3051. [PubMed] [Google Scholar]
- Hoelzle, M. K. , Svitkina, T. , & Blanchoin, L. (2011). The cytoskeletal mechanisms of cell–cell junction formation in endothelial cells. Molecular Biology of the Cell, 23, 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers, S. , Oldenburg, J. , Spanjaard, E. , van der Krogt, G. , Grigoriev, I. , Akhmanova, A. , … de Rooij, J. (2012). Vinculin associates with endothelial VE‐cadherin junctions to control force‐dependent remodeling. The Journal of Cell Biology, 196, 641–652. 10.1083/jcb.201108120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean, C. , Chen, X. L. , Nam, J.‐O. , Tancioni, I. , Uryu, S. , Lawson, C. , … Schlaepfer, D. D. (2014). Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. The Journal of Cell Biology, 204, 247–263. 10.1083/jcb.201307067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, G. L. , & Lapadat, R. (2002). Mitogen‐activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science, 298, 1911–1912. 10.1126/science.1072682 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide, T. , Foster, D. , Yoshitake, S. , & Davie, E. W. (1986). Amino acid sequence of human histidine‐rich glycoprotein derived from the nucleotide sequence of its cDNA. Biochemistry, 25, 2220–2225. 10.1021/bi00356a055 [DOI] [PubMed] [Google Scholar]
- Kuroda, K. , Wake, H. , Mori, S. , Hinotsu, S. , Nishibori, M. , & Morimatsu, H. (2018). Decrease in histidine‐rich glycoprotein as a novel biomarker to predict sepsis among systemic inflammatory response syndrome. Critical Care Medicine, 46(4), 570–576. [DOI] [PubMed] [Google Scholar]
- London, N. R. , Zhu, W. , Bozza, F. A. , Smith, M. , Greif, D. M. , Sorensen, L. K. , … Li, D. Y. (2010). Targeting Robo4‐dependent slit signaling to survive the cytokine storm in sepsis and influenza. Science Translational Medicine, 23, 3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscinskas, F. W. , & Shaw, S. K. (2001). The biology of endothelial cell–cell lateral junctions. Microcirculation, 8, 141–142. [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, S. , Takahashi, H. K. , Yamaoka, K. , Okamoto, M. , & Nishibori, M. (2003). High affinity binding of serum histidine‐rich glycoprotein to nickel–nitrilotriacetic acid: The application to microquantification. Life Sciences, 73, 93–102. 10.1016/S0024-3205(03)00261-3 [DOI] [PubMed] [Google Scholar]
- Nishibori, M. , Wake, H. , & Morimatsu, H. (2018). Histidine‐rich glycoprotein as an excellent biomarker for sepsis and beyond. Critical Care, 22, 209 10.1186/s13054-018-2127-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwariaku, F. E. , Chang, J. , Zhu, X. , Liu, Z. , Duffy, S. L. , Halaihel, N. H. , … Turnage, R. H. (2002). The role of p38 MAP kinase in tumor necrosis factor‐induced redistribution of vascular endothelial cadherin and increased endothelial permeability. Shock, 18, 82–85. 10.1097/00024382-200207000-00015 [DOI] [PubMed] [Google Scholar]
- Nwariaku, F. E. , Rothenbach, P. , Liu, Z. , Zhu, X. , Turnage, R. H. , & Terada, L. S. (2003). Rho inhibition decreases TNF‐induced endothelial MAPK activation and monolayer permeability. Journal of Applied Physiology, 95, 1889–1895. 10.1152/japplphysiol.00225.2003 [DOI] [PubMed] [Google Scholar]
- Peacock, J. G. , Miller, A. L. , Bradley, W. D. , Rodriguez, O. C. , Webb, D. J. , & Koleske, A. J. (2007). The Abl‐related gene tyrosine kinase acts through p190RhoGAP to inhibit actomyosin contractility and regulate focal adhesion dynamics upon adhesion to fibronectin. Molecular Biology of the Cell, 18, 3860–3872. 10.1091/mbc.e07-01-0075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pober, J. S. , & Cotran, R. S. (1990). Cytokines and endothelial cell biology. Physiological Reviews, 70, 427–451. 10.1152/physrev.1990.70.2.427 [DOI] [PubMed] [Google Scholar]
- Poon, I. K. H. , Patel, K. K. , Davis, D. S. , Parish, C. R. , & Hulett, M. D. (2011). Histidine‐rich glycoprotein: The Swiss Army knife of mammalian plasma. Blood, 117, 2093–2101. 10.1182/blood-2010-09-303842 [DOI] [PubMed] [Google Scholar]
- Riedemann, N. C. , Guo, R.‐F. , & Ward, P. A. (2003). Novel strategies for the treatment of sepsis. Nature Medicine, 9, 517–524. 10.1038/nm0503-517 [DOI] [PubMed] [Google Scholar]
- Rodrigues, S. F. , & Granger, D. N. (2015). Blood cells and endothelial barrier function. Tissue Barriers, 3, e978720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronca, F. , & Raggi, A. (2015). Structure–function relationships in mammalian histidine–proline‐rich glycoprotein. Biochimie, 118, 207–220. [DOI] [PubMed] [Google Scholar]
- Schnoor, M. , García‐Ponce, A. , Vadillo, E. , Pelayo, R. , Rossaint, J. , & Zarbock, A. (2017). Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cellular and Molecular Life Sciences, 74, 1985–1997. 10.1007/s00018-016-2449-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, O. , Rydengård, V. , Schmidtchen, A. , Mörgelin, M. , Alm, P. , Sørensen, O. E. , & Björck, L. (2010). Histidine‐rich glycoprotein promotes bacterial entrapment in clots and decreases mortality in a mouse model of sepsis. Blood, 116, 2365–2372. 10.1182/blood-2010-02-271858 [DOI] [PubMed] [Google Scholar]
- Singer, M. , Deutschman, C. S. , Seymour, C. , Shankar‐Hari, M. , Annane, D. , Bauer, M. , … Angus, D. C. (2016). The third international consensus definitions for sepsis and septic shock (sepsis‐3). Jama, 315, 801–810. 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague, A. H. , & Khalil, R. A. (2009). Inflammatory cytokines in vascular dysfunction and vascular disease. Biochemical Pharmacology, 78, 539–552. 10.1016/j.bcp.2009.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciak, S. , Dellinger, R. P. , Parrillo, J. E. , Guglielmi, M. , Bajaj, J. , Abate, N. L. , … Hollenberg, S. M. (2007). Early microcirculatory perfusion derangements in patients with severe sepsis and septic shock: Relationship to hemodynamics, oxygen transport, and survival. Annals of Emergency Medicine, 49, 88–98. 10.1016/j.annemergmed.2006.08.021 [DOI] [PubMed] [Google Scholar]
- Vincent, J.‐L. , Opal, S. M. , Marshall, J. C. , & Tracey, K. J. (2013). Sepsis definitions: Time for change. Lancet, 381, 774–775. 10.1016/S0140-6736(12)61815-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake, H. , Mori, S. , Liu, K. , Morioka, Y. , Teshigawara, K. , Sakaguchi, M. , … Nishibori, M. (2016). Histidine‐rich glycoprotein prevents septic lethality through regulation of immunothrombosis and inflammation. eBioMedicine, 9, 180–194. 10.1016/j.ebiom.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidert, E. , Pohler, S. E. , Gomez, E. W. , & Dong, C. (2014). Actinomyosin contraction, phosphorylation of VE‐cadherin, and actin remodeling enable melanoma‐induced endothelial cell–cell junction disassembly. PLoS ONE, 9, e108092 10.1371/journal.pone.0108092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel, F. , Winderlich, M. , Holm, M. , Frye, M. , Rivera‐Galdos, R. , Vockel, M. , … Vestweber, D. (2014). Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE‐cadherin. Nature Immunology, 15, 223–230. 10.1038/ni.2824 [DOI] [PubMed] [Google Scholar]
- Wu, M. H. (2005). Endothelial focal adhesions and barrier function. The Journal of Physiology, 569, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C. , Wu, X. , Hack, B. K. , Bao, L. , & Cunningham, P. N. (2015). TNF causes changes in glomerular endothelial permeability and morphology through a Rho and myosin light chain kinase‐dependent mechanism. Physiological Reports, 3, e12636 10.14814/phy2.12636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S. Y. (2002). Protein kinase signaling in the modulation of microvascular permeability. Vascular Pharmacology, 39, 213–223. 10.1016/S1537-1891(03)00010-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1.
Dose‐dependent effects of HRG on the LPS‐induced cell morphology changes.
Figure S2. Dose‐dependent effects of HRG on the LPS‐induced loss of VE‐cadherin.
Figure S3. HRG prevents the loss of intercellular junctions.
Figure S4. Effects of HRG on the cytokine levels in the serum of LPS‐injected mice.
Figure S5. Protection effects of HRG on the HMVECs permeability in vitro.
Figure S6. Effects of HRG on LPS/TNF‐α‐induced cytoskeleton reorganization in HMVECs.
Figure S7. HRG prevents the loss of intercellular junction in HMVECs.
Figure S8. HRG suppresses the LPS/TNF‐α‐induced NF‐κB activation in HMVECs.
Table S1. RT‐PCR Primer Sequence