

Abstract
Glioblastoma is one of the most devastating human malignancies for which a novel efficient treatment is urgently required. This pre–clinical study shows that eribulin, a specific inhibitor of telomerase reverse transcriptase (TERT)‐RNA‐dependent RNA polymerase, is an effective anticancer agent against glioblastoma. Eribulin inhibited the growth of 4 TERT promoter mutation‐harboring glioblastoma cell lines in vitro at subnanomolar concentrations. In addition, it suppressed the growth of glioblastoma cells transplanted subcutaneously or intracerebrally into mice, and significantly prolonged the survival of mice harboring brain tumors at a clinically equivalent dose. A pharmacokinetics study showed that eribulin quickly penetrated brain tumors and remained at a high concentration even when it was washed away from plasma, kidney or liver 24 hours after intravenous injection. Moreover, a matrix‐assisted laser desorption/ionization mass spectrometry imaging analysis revealed that intraperitoneally injected eribulin penetrated the brain tumor and was distributed evenly within the tumor mass at 1 hour after the injection whereas only very low levels of eribulin were detected in surrounding normal brain. Eribulin is an FDA‐approved drug for refractory breast cancer and can be safely repositioned for treatment of glioblastoma patients. Thus, our results suggest that eribulin may serve as a novel therapeutic option for glioblastoma. Based on these data, an investigator‐initiated registration‐directed clinical trial to evaluate the safety and efficacy of eribulin in patients with recurrent GBM (UMIN000030359) has been initiated.
Keywords: eribulin, glioblastoma, mass spectrometry imaging, RdRP, TERT
1. INTRODUCTION
Glioblastoma (GBM) is one of the most devastating brain tumors with limited therapeutic options. The median survival is only 14 months even with multimodal therapies with surgery, radiation therapy and chemotherapy with temozolomide.1, 2 Glioblastoma patients may respond to temozolomide when MGMT promoter in the tumor cells is hypermethylated; however, the development of temozolomide resistance is inevitable. Temozolomide is ineffective when MGMT is hypomethylated.3 Although a number of molecular targeting agents have been tested for their efficacy against glioblastoma in clinical trials, none has so far been proved to prolong overall survival of GBM patients.4, 5 Development of a new effective drug for GBM is urgently needed.2, 6
Among the numerous genetic mutations,7, 8, 9 telomerase reverse transcriptase (TERT) promoter mutations, which are found in 60%–80% of glioblastomas, are the most common mutational events.10, 11 TERT is a reverse transcriptase subunit of telomerase that maintains telomere length by utilizing TERC as an RNA subunit.12 Telomere length is shortened at each cell division, and the cells undergo replicative senescence when telomeres attain a critical length. Most cancers, including gliomas, activate telomerase to evade this process.13 The promoter mutations of TERT occur at 2 hotspots (−124C > T or −146C > T) in a mutually exclusive manner. Either of these mutations creates a de novo binding site for GABPA, which activates the promoter and upregulates TERT expression.14, 15 The introduction of TERT promoter mutation in induced pluripotent stem cells abrogates telomerase silencing upon differentiation, and these cells overcome replicative senescence and infinitely multiply upon acquisition of proliferating mutations.16 Thus, it appears that TERT promoter mutations are the most common driver oncogenic event in GBM, making it an attractive therapeutic target.
Telomerase targeting therapies have been a subject of interest for several decades. Although telomerase is activated in most cancer cells, the great majority of normal cells exhibit only a low level of telomerase, making it an ideal cancer‐specific target. However, this strategy has not yet materialized in clinical settings.17 Imetelstat, an antisense oligonucleotide that inhibits telomerase by binding to TERC, is the only telomerase‐targeting drug that has been evaluated in phase II clinical trials of brain tumors.18 Although telomerase inhibition was observed, no objective response of imetelstat was achieved in refractory pediatric brain tumors.19 An inherent problem of targeting telomerase is that the inhibition of telomerase in a cancer cell already having elongated telomeres would not yield an immediate anti–proliferative effect. Telomerase inhibition may also lead to the activation of alternative lengthening of telomeres (ALT), a homologous recombination‐based telomerase‐independent telomere elongation mechanism, which would abrogate the effects of telomerase‐targeting anticancer therapy.17
Growing evidence indicates that TERT may have a telomerase‐independent activity (“non–canonical function”).20 One of the non–canonical functions of TERT is RNA‐dependent RNA polymerase (RdRP) activity, which catalyzes the synthesis of an RNA strand complementary to a template RNA.17, 21 Although TERT is well known as a reverse transcriptase, it has phylogenetic and structural similarities to viral RdRP, such as those present in hepatitis C virus, poliovirus and influenza virus.17 In addition, TERT has been shown to be involved in the production of siRNA,21 heterochromatin maintenance22 and de novo synthesis of RNA.23 Thus, TERT appears to function as an RdRP in mammals. Moreover, TERT forms a complex with Brahma‐related gene 1 (BRG1) and nucleostemin (NS), which contributes to heterochromatin maintenance, mitotic progression22 and maintenance of tumor‐initiating cell phenotypes.24 The suppression of TERT, BRG1 or NS disrupted heterochromatin formation and induced mitotic arrest.22 These data suggest that the inhibition of RdRP activity instead of TERT activity may serve as a novel TERT‐targeted anticancer strategy.17
We have recently identified eribulin as a specific inhibitor of TERT‐RdRP through drug screening.25 Eribulin mesylate (Halaven, Eisai, Tokyo, Japan) is a fully synthetic analog of halichondrin B, a natural product isolated from marine sponge and originally developed as a microtubule inhibitor.26 Eribulin is currently approved in more than 60 countries for the treatment of refractory breast cancers and liposarcomas. In a series of preliminary preclinical experiments, eribulin showed anticancer activities in diverse cancer cell lines, including a GBM cell line.27, 28, 29 In this study, we investigated the efficacy of eribulin in inhibiting GBM cell proliferation in vitro and in vivo, as well as the pharmacokinetics of eribulin in a mouse harboring intracerebrally‐transplanted human GBM cells. Our results showed that eribulin is active against GBM with TERT promoter mutations and penetrated mouse brain tumors. These data strongly suggest that eribulin can serve as an effective anticancer drug against GBM.
2. MATERIALS AND METHODS
2.1. Cell lines, vector, WST assay, pyrosequencing and ion proton analysis for targeted sequencing
Details of cell lines, vector, WST assay, pyrosequencing and ion proton analysis are described in a supporting document (Doc S1). Genotypes of cell lines used in this study and ion proton analysis data are shown in the supporting information (Tables S1 and S2).
2.2. Studies on brain tumor xenografts
Female BALB/c nu/nu athymic mice or SCID‐beige (6‐week to 8‐week‐old, Charles River Japan, Tokyo, Japan) were housed under specific pathogen‐free conditions. To establish a mouse brain tumor xenograft, U87MG, U87MG‐Fluc2, GSC23, GS‐Y03 (1 × 105 cells in 2 μL PBS) or LN229 (5 × 105 cells in 2 μL PBS) cells were stereotactically inoculated into the right cerebral hemisphere of immunodeficient mice by using a Hamilton syringe and stereotactic micro‐injector (Narishige, Tokyo, Japan). Saline (control) or eribulin (0.5 mg/kg) was administered to the mice 3 times per week (q2d × 3 per week). A 1‐week treatment (3 injections) period was defined as 1 cycle, and an appropriate number of therapy cycles was applied to each study. All animal studies were conducted under the protocols approved by the Committee for Ethics of Animal Experimentation of National Cancer Center, and the experiments were carried out in accordance with the Guidelines for Animal Experiments.
2.3. Subcutaneous tumor xenografts
U87MG cells (2 × 106 cells in 100 μL PBS) were implanted into the right flank of female BALB/c nu/nu athymic mice to establish subcutaneous tumor xenografts. Saline (control) or eribulin (0.125, 0.25, or 0.5 mg/kg) were intraperitoneally administered 3 times per week (q2d × 3 per week), and 1 week of therapy was defined as 1 cycle. Two cycles of therapy were applied to each group. The tumor volume was measured 3 times per week, compared between groups, and statistically analyzed. For RdRP assay, eribulin was applied to an independent set of subcutaneous tumor xenografts at the same dose level (see below). On the final day of saline or eribulin (day 20) administration, all the mice were killed, and the tumor specimens were harvested and quickly frozen in liquid nitrogen to measure RdRP activity as described below.
2.4. Bioluminescence study
For in vivo bioluminescence study, eribulin or saline was injected into mice harboring U87MG‐Fluc2 intracerebral tumors until the first mouse of the control group died. Bioluminescence signals were monitored on the same day of receiving injections, and the intensities of bioluminescence signals were statistically compared between the groups.
2.5. Survival study
For in vivo survival study, eribulin or saline was intraperitoneally injected into mice harboring intracranial U87MG, LN229, GSC23 or GS‐Y03 tumors. A total of 5 (U87MG), 9 (LN229), 2 (GSC23) or 3 (GS‐Y03) cycles of treatment were applied to each group. The survival of mice was observed and analyzed using the Kaplan‐Meier method.
2.6. Pharmacokinetics study
Eribulin (0.5 mg/kg) was intravenously injected via the tail vein to mice bearing intracerebral U87MG xenografts. The mice were killed at several time points (15 minutes, and 1, 2, 4, 6 and 24 hours after injection; n = 3 for each time point). Normal brain tissue, brain tumor tissue and plasma were collected from each mouse at each time point and quickly frozen for further analysis. Plasma and tissue concentrations of eribulin were measured by using liquid chromatography‐tandem mass spectrometry analysis (LC‐MS/MS). The method is described in detail in a supporting document (Doc S1).
2.7. Immunoprecipitation‐immunoblotting
Immunoprecipitation‐immunoblotting (IP‐IB) was performed as previously described.23 Briefly, 1 × 107 cells were lysed in 1 mL of lysis buffer A (0.5% NP‐40, 20 mM Tris‐HCl [pH 7.4] and 150 mM NaCl). After sonication, the lysate was pre–absorbed with Pierce Protein A Plus Agarose (Thermo Fisher Scientific, Tokyo, Japan) for 30 minutes at 4°C. The pre–absorbed lysate was mixed with 10 μg of an anti–human TERT mAb (clone 10E9‐2, MBL) and Pierce Protein A Plus Agarose, and incubated overnight at 4°C. After washing the beads, the immune complexes were subjected to SDS‐PAGE. An anti–human TERT mAb (clone 2E4‐2) and MouseTrueBlot ULTRA (Rockland) were used for immunoblotting.
Molecular imaging of the distribution of eribulin in the brain tumor tissue was performed using matrix‐assisted laser desorption/ionization mass spectrometry imaging (MALDI‐MSI). The method is described in detail in a supporting document (Doc S1). Briefly, frozen sections of 8 μm were thaw‐mounted onto microscopic glass slides and were covered with 2,5‐dihydroxybenzoic acid by using a sublimation apparatus (SVC‐700TMSG/7PS80, SANYU Electron, Tokyo, Japan) and an ImagePrep (Bruker Daltonics, Tokyo, Japan) in 2 steps.30 The distribution of eribulin in the brain tumor tissue was visualized at a high resolution using an atmospheric pressure imaging ion source (AP‐SMALDI 10, TransMIT) coupled to a quadrupole orbitrap mass spectrometer (Q Exactive, Thermo Fisher Scientific).
2.8. Immunoprecipitation‐RdRP assay
The immunoprecipitation‐RdRP (IP‐RdRP) assay was performed as described previously with some modifications.23 For all cell lines, 1 × 107 cells were lysed in 1 mL of lysis buffer A. After sonication, the cell lysates were cleared by centrifugation at 21 000 g at 4°C for 15 minutes. For xenografts, tissue pieces were homogenized in lysis buffer A by using the MagNA Lyser Instrument (Roche Diagnostics) at 6500 r/min for 30 seconds. The tissue lysates were cleared by centrifugation twice at 21 000 g at 4°C and assayed for protein concentration. Cell lysate (1 mL) or protein of tissue lysate (650 μg) was pre–absorbed with Pierce Protein A Plus Agarose and incubated overnight with 10 μg of anti–human TERT mAb (clone 10E9‐2) and Pierce Protein A Plus Agarose at 4°C. After washing, the bead suspension was treated with 20 units of Micrococcal Nuclease at 25°C for 15 minutes and washed again. The immune complexes were mixed with a synthetic RNA template (RNA #1, 5′‐GGGAUCAUGUGGGUCCUAUUACAUUUUAAACCCA‐3′), ribonucleotides and [α‐32P]UTP, and incubated at 32°C for 2 hours for RdRP reaction. The RdRP products were treated with RNase I and electrophoresed in a polyacrylamide gel containing 7 M urea. Images were captured by using a Typhoon FLA 7000 Phosphorimager and the signal intensities of the bands were quantified using ImageQuant TL software (GE Healthcare Japan).
2.9. Liquid chromatography‐tandem mass spectrometry analysis of eribulin concentration in brain tissue sections
The concentrations of eribulin in the targeted tissues/plasma were measured using a triple‐quadrupole mass spectrometer (QTRAP 5500, AB SCIEX) coupled to a Shimadzu HPLC system (Nexera X2, Shimadzu). Chromatographic separation was performed on the Unison UK‐C18 column (3 μm, 250 × 3 mm; Imtakt). The method is described in detail in a supporting document (Doc S1).
2.10. Statistical analysis
The IC50 values of the different cell lines obtained from the in vitro cytotoxicity assay were compared by 2‐way ANOVA. Comparison of bioluminescence signals obtained from the in vivo bioluminescence assay was performed by ANOVA. The results of in vivo survival assays were compared by log‐rank test. A P‐value <0.05 was considered significant. All analyses were performed using GraphPad Prism 5 software (GraphPad Software, San Diego, CA, USA).
3. RESULTS
3.1. Eribulin inhibits growth of glioblastoma cell lines with telomerase reverse transcriptase promoter mutations
The cytotoxic effect of eribulin on the TERT‐mutated GBM cell lines U87MG, U251MG, U118MG and LN229 and patient‐derived sphere‐cultured cells GSC23 and GS‐Y03 was examined in vitro using the WST assay (Figure 1). All 6 cell lines had TERT promoter mutations (Table S1). ES‐2 and TOV‐21G (OCCC with wild‐type TERT promoter) were used for comparison. The results showed that eribulin suppressed the growth of all GBM cells tested in a dose‐dependent manner (Figure 1A). The IC50 values of the TERT‐mutated cells U87MG, U251MG, U118MG, LN 229, GSC23 and GS‐Y03 (mean 0.32, 0.15, 0.36, 2.12, 0.21 and 1.10 nmol/L, respectively) were significantly lower than those of TERT‐wild‐type ES‐2 (17.16 nmol/L) and TOV‐21G (IC50 not reached) cells (P < 0.001) (Figure 1B). Thus, GBM cell lines that harbored TERT promoter mutations appeared to be more susceptible to eribulin than TERT‐wild‐type ES‐2 and TOV‐21G cells.
Figure 1.
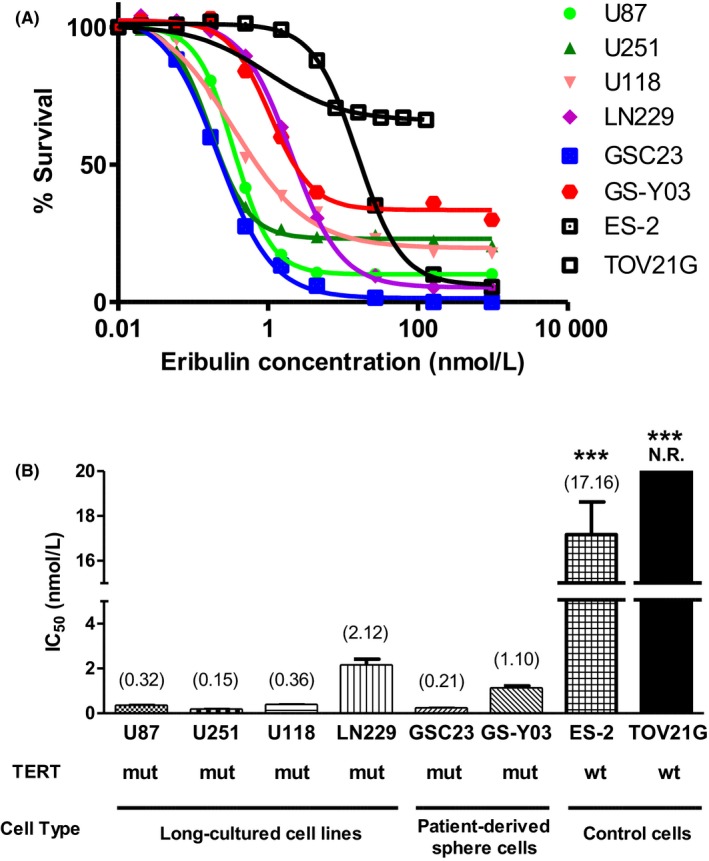
Effects of eribulin on proliferation of human glioblastoma cell lines examined by WST assay. A, The survival rates of U87MG, U251MG, U118MG, LN229 cells (established adherent‐cultured human GBM cell lines), GSC23, GS‐Y03 (patient‐derived sphere‐cultured human GBM cells) as well as ES‐2 and TOV‐21G cells (human ovarian cancer cell lines) were plotted against increasing concentrations of eribulin. Each assay was performed in triplicate. B, The IC 50 value of each cell line (A) is shown in a bar graph. Mean IC 50 value is plotted at the top of each bar. GBM cells with TERT mutation were significantly more sensitive to eribulin than the control ovarian cancer cells with wildtype TERT. Error bars indicate standard error. N.R., not reached; mut, mutated; wt, wildtype; ***P < 0.001
3.2. Eribulin significantly inhibited intracranial tumor growth
Next, we evaluated whether eribulin inhibited the growth of intracerebrally xenografted luciferase‐expressing U87MG cells (U87MG‐Fluc2) by using an in vivo imaging system. Transduction with a conventional replication‐defective lentiviral vector expressing firefly luciferase resulted in a comparable level of bioluminescence from U87‐Fluc2 cells exposed to luciferin. A highly robust quantitative correlation between cell count and bioluminescent signal intensity was confirmed by optical imaging in vitro (Figure S1).
Following stereotactic intracerebral implantation of U87MG‐Fluc2 cells into athymic mouse (105 cells/mouse) on day 0, all mice were intraperitoneally administered saline or eribulin (1.25 mg/kg) on days 5, 7, 9 and 12 (Figure 2A, black triangle). The bioluminescence signals in brain tumors were monitored by in vivo optical imaging at sequential time points (days 4, 6, 9 and 13; Figure 2). The initial bioluminescence signal intensities of both the groups were virtually the same at the first time‐point (day 4). Thereafter, the bioluminescence signals from mice treated with saline increased continually, indicating tumor growth, until 1 mouse in the saline group died on day 14 (Figure 2A, blue line). In contrast, the bioluminescence signals from mice treated with eribulin decreased (Figure 2A, red line). On day 13, the signal intensities in eribulin‐treated mice were significantly lower than those in the saline group (P < 0.001). These results indicated that eribulin inhibited the growth of intracerebrally transplanted U87MG‐Fluc2 tumors in vivo.
Figure 2.
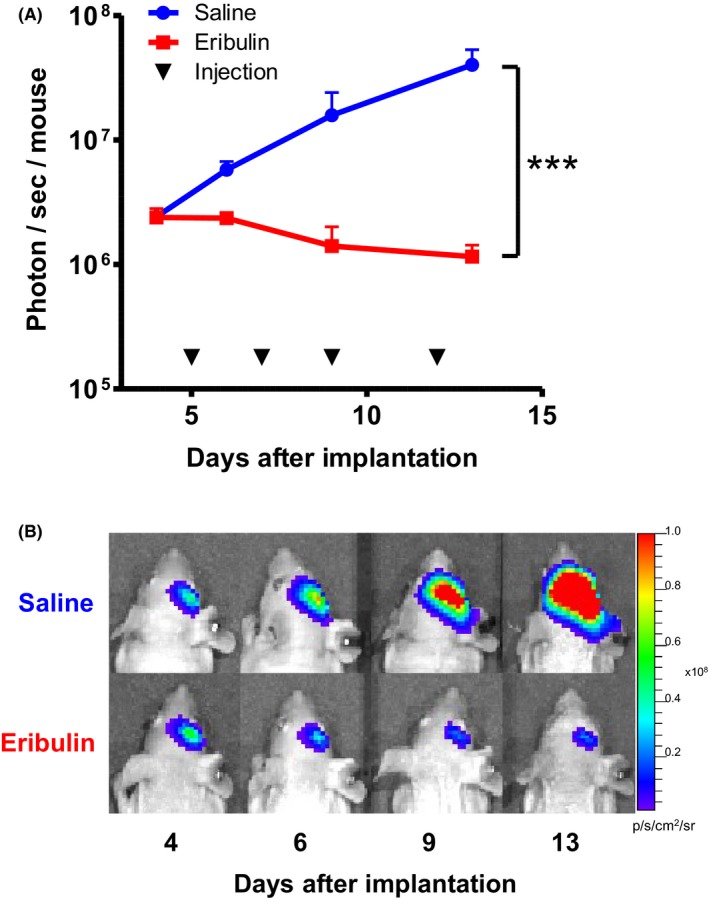
Anticancer effect of eribulin against intracerebrally transplanted U87MG‐Fluc2 tumors monitored by bioluminescent study at sequential time points. Mice injected with luciferase‐expressing U87MG cells (U87MG‐Fluc2) were subjected to intraperitoneal injections of saline or eribulin (0.5 mg/kg; q2d). A, The mean signal intensities in each group were plotted. Error bars indicate standard error. Mice treated with eribulin showed significantly low bioluminescent signal intensity. ***P < 0.001. B, Representative bioluminescent images of brain tumor xenografts in each group on days 4, 6, 9 and 13
3.3. Intraperitoneal eribulin administration significantly prolonged survival of mice with intracerebral tumors
We next evaluated the efficacy of eribulin in prolonging survival of mice harboring intracerebrally transplanted glioblastoma cells, U87MG, LN229 (established glioblastoma cell lines), GSC23 and GS‐Y03 (patient‐derived glioblastoma cells), which were highly susceptible to eribulin in vitro with an IC50 of 0.32 nmol/L. U87MG, GSC23 or GS‐Y03 cells (105 cells/mouse) were stereotactically implanted into the brain of BALB/c nu/nu athymic mice on day 0. LN229 cells (2 × 105 cells/mouse) were also implanted into the brain of SCID‐beige mice. Treatment schedules were modified for each cell line according to each survival time of the model. Saline or eribulin was intraperitoneally administered q2d × 3 per week for 5 (U87MG), 9 (LN229), 2 (GSC23) or 3 (GS‐Y03) cycles (weeks) and the survival of mice was observed. Eribulin treatment significantly prolonged survival of mice harboring intracerebral xenografts of established cell lines U87MG or LN229 tumor (median survival; 49 days vs 28 days, P < 0.001 or 81 days vs 76 days, P < 0.01, respectively, Figure 3A,B), as well as patient‐derived glioblastoma cells GSC23 or GS‐Y03 (median survival; 32 days vs 26 days, P < 0.001 or 34 days vs 28 days, P < 0.001, respectively, Figure 3C,D). All mice treated with eribulin in this survival study developed no neurological symptoms, including seizure, gait disturbance or abnormal behavior.
Figure 3.
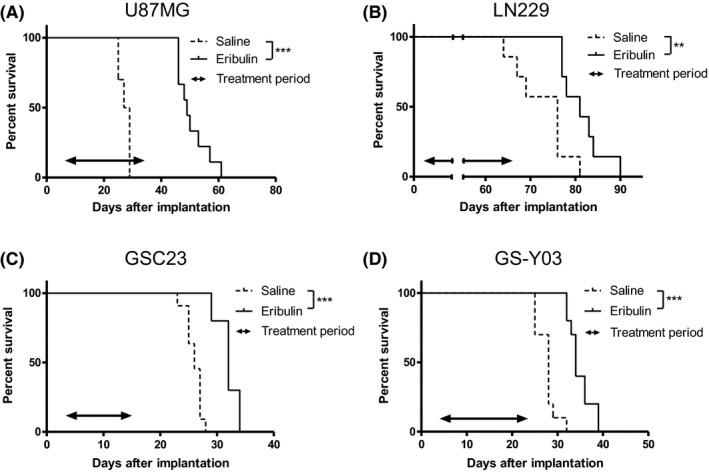
Kaplan‐Meier survival curves of mice with intracerebral glioblastoma treated with saline or eribulin. The log‐rank test was performed for statistical analysis. Mice harboring intracerebrally transplanted U87MG, LN229 (established glioblastoma cell lines, A and B), GSC23 or GS‐Y03 (patient‐derived glioblastoma cells, C and D) cells were intraperitoneally injected with saline or eribulin (0.5 mg/kg; q2d × 3 per week) during a treatment period (indicated by a double‐headed arrow). The survival of eribulin‐treated mice was significantly prolonged than that of untreated mice in all 4 cell lines. ***P < 0.001, **P < 0.01
3.4. Intravenously injected eribulin efficiently penetrated brain tumors and remained in the tumor tissue for more than 24 hours
We examined the pharmacokinetics of eribulin in mice with intracerebral tumors. U87MG cells were intracerebrally transplanted into athymic mice 21 days prior to the administration of eribulin. On day 21, eribulin (0.5 mg/kg) was injected via the tail vein, and plasma, normal brain tissues and intracerebral tumor tissues were harvested at different time points (0.25, 1, 2, 4, 6 and 24 hours after injection; n = 3 for each time point). As expected, the plasma concentration gradually decreased from 140 nmol/L (mean of 3 animals) at 0.25 hour after injection to 6.8 nmol/L at 6 hours after injection and was undetectable after 24 hours (Figure 4A, blue line). The concentration of eribulin in the normal brain tissue remained very low (maximum 2.16 nmol/L at 1 hour after injection) and quickly plunged below the detection limit after 2 hours. In contrast, the concentration of eribulin in U87MG brain tumor was equivalent to that in the plasma (127 nmol/L at 0.25 hours after injection) and remained high even after 24 hours of injection (91.7 nmol/L, Figure 4A, red line). Accordingly, the tumor/plasma ratio of eribulin increased with time up to 6 hours after intravenous injection (Figure 4B). A similar pharmacokinetic study was performed in subcutaneous U87MG xenografts by intravenously injecting eribulin (0.5 mg/kg). Then, the concentration of eribulin in the kidney, liver, subcutaneous tumor and plasma at 1, 4, and 24 hours after injection (n = 3 for each time point, Figure S2) was measured. Although eribulin concentration rapidly decreased in the kidney, liver and plasma and was undetectable after 24 hours, its concentration in the subcutaneous tumor remained high for 24 hours.
Figure 4.
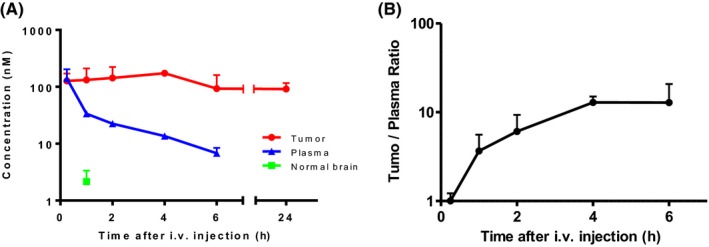
Pharmacokinetic study of eribulin in a mouse brain tumor model. Eribulin concentrations in plasma, transplanted brain tumor tissue and contralateral normal brain tissue from mice with intracerebral U87MG tumors. A, Mice with U87MG brain tumors were killed at 15 min, 1, 2, 4, 6 and 24 h after a single intravenous injection of eribulin (0.5 mg/kg, n = 3 for each time point). Plasma, tumor tissue and contralateral normal brain tissue were harvested and immediately frozen for measurement of eribulin concentration. Eribulin concentration in plasma gradually decreased after injection and became undetectable after 24 h. The concentration of eribulin in non–tumoral brain tissue was very low and undetectable after 2 h. In contrast, eribulin concentration in the brain tumor tissue was almost as high as that in plasma 15 minutes after injection,and remained high up to 24 h. B, The tumor/plasma ratio of eribulin gradually increased for up to 6 h (plasma concentration undetectable after 24 h). See supplementary data for tissue/plasma concentration of eribulin (Figure S2)
Upon observing the unexpectedly high concentration of eribulin in the brain tumor tissue, we investigated the distribution of eribulin within the tissue further by using MALDI‐MSI. A U87MG brain tumor model was developed and tumor‐bearing brain tissues were obtained at 1 and 24 hours after a single intravenous injection of eribulin (0.5 mg/kg). MALDI‐MSI analysis revealed that intraperitoneally injected eribulin penetrated the brain tumor and was distributed evenly within the tumor mass at 1 h after the injection (Figure 5). Only very low levels of eribulin were detected in normal brain parenchyma. To validate these results, the concentrations of eribulin in microdissected tumor lesion or contralateral normal brain lesion were measured by LC‐MS/MS at either 1 or 24 hours after the injection. The concentrations of eribulin in the tumor lesion were 113.86 and 74.19 ng/cm3 at 1 and 24 hours after the injection, respectively, whereas the concentration in the normal brain parenchyma was below the lower limit of quantitation, at both 1 and 24 hours after the injection (Figure S3).
Figure 5.
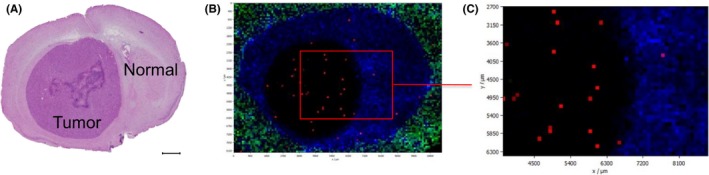
The whole brain was collected 1 h after intravenous administration of a single dose of eribulin (0.5 mg/kg). A, H&E staining of brain tumor tissue from mice. Scale bar, 1 mm. B, A fusion image of matrix‐assisted laser desorption/ionization mass spectrometry imaging in the same brain tissue section. Spatial resolution: 90 μm. Red signals, which indicates eribulin, were predominantly localized and evenly distributed in the tumor lesion. Blue and green signals indicate arbitrary molecules located outside the tumor. C, Enlarged image of a part of Figure 5B. Red and blue signals indicate eribulin and normal brain tissue, respectively
3.5. RdRP activity decreased in subcutaneous tumors after eribulin treatment
Finally, we evaluated the effect of eribulin on RdRP activity in the xenografted tumor tissues. The cell lines used in this study showed varying degrees of RdRP activities (Figure S4). U87MG cells (2 × 106 cells) were implanted into the right flank of athymic mice on day 0 (n = 6 per group). When the tumors reached an average size of 5 mm in diameter, eribulin (0.125, 0.25 or 0.5 mg/kg) or saline was injected intraperitoneally 3 times per week for 2 weeks (days 8, 11, 13, 15, 18 and 20), and the sizes of the subcutaneous tumors were measured. The growth of tumors treated with eribulin at all the 3 doses tested was significantly suppressed compared with that of the untreated control (P < 0.0001) (Figure 6A). The experiment was repeated (n = 3 per group) and all mice were killed 1 hour after the final administration of eribulin or saline on day 20. Then, the subcutaneous tumors were harvested. The tumor specimens were quickly frozen and RdRP activities analyzed. The results showed that eribulin treatment decreased RdRP activities in the subcutaneous tumors in a dose‐dependent manner (Figure 6B). The intracerebral tumor model also demonstrated a similar tendency (Figure S5). Of note, TERT messenger RNA expression was upregulated in all glioma cell lines tested (Figure S6).
Figure 6.
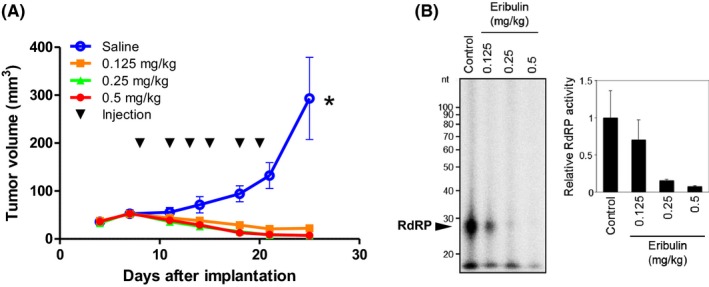
Effects of eribulin in a mouse subcutaneous tumor model. Mice with U87MG subcutaneous tumors were administered an intraperitoneal injection of saline or eribulin (0.125, 0.25 or 0.5 mg/kg; q2d × 3 per week) for 2 weeks, and the tumor size was recorded at several time points. A, Tumor size of each group on days 4, 7, 11, 14, 18, 21 and 25 were plotted. Eribulin treatment significantly suppressed subcutaneous tumor growth in all groups tested when compared with the saline group. Error bars indicate standard error. *P < 0.05. B, For RdRP activity analysis, the same experiment was repeated and subcutaneous tumors were harvested 1 h after eribulin injection on day 20. The RdRP activities of eribulin treated‐tumors decreased in a dose‐dependent manner. Error bars indicate standard error
4. DISCUSSION
In this study, we showed that eribulin has an anticancer efficacy against human glioblastoma cells in vitro and in vivo. The results of in vitro growth inhibition assay showed that the growth of all 4 glioma cell lines tested was strongly suppressed by eribulin, with most of the IC50 values being below 1 nM (Figure 1). Because most glioma cell lines harbored TERT promoter mutation, 2 ovarian cancer cell lines without TERT promoter hotspot mutations were tested as TERT promoter wild‐type cancer cells for comparison. It was previously reported25 that the IC50 values of these TERT‐wild‐type cells were much higher, with one of them (TOV‐21G) exhibiting a value over 100 nM. Although the correlation between sensitivity to eribulin and TERT status needs to be further investigated, all cell lines tested in this study and the cell lines with TERT promoter hotspot mutations tested in a previous study were highly sensitive to eribulin.25 The growth‐suppressing effect of eribulin in glioma cells was further demonstrated by real‐time in vivo bioluminescence monitoring of tumor growth by using mouse intracerebral xenografts (Figure 2). Moreover, continual administration of eribulin at a clinically equivalent dose (0.5 mg/kg, 3 times per week) to mice harboring brain tumors, both with established glioblastoma cell lines and patient‐derived glioblastoma sphere‐cultured cells, significantly prolonged their survival compared with untreated control mice (Figure 3). These results strongly suggested that eribulin may serve as a clinically applicable novel agent for glioblastoma.
In a previous preclinical study, glioblastoma was among the several cancer types that showed sensitivity to eribulin.27, 31 However, glioblastoma was not considered a target for eribulin because the drug could not penetrate the blood‐brain barrier (BBB).31 This was most likely because eribulin is a substrate of P‐glycoprotein (P‐gp), an efflux transporter that plays an important role in elimination of drugs at the BBB.32, 33 To explain the unexpectedly high efficacy of eribulin against intracerebral tumor, we hypothesized that although eribulin could not penetrate the BBB of normal brain, it may penetrate the brain tumor tissues. Large brain tumors (>0.25 mm in diameter) may develop abnormal tumor vessels with disrupt BBB, which eventually allows intravenously injected drugs to reach the brain tumor tissue.34, 35 Therefore, we studied the pharmacokinetics of eribulin in brain tumor‐bearing mice by chronologically measuring the concentrations of eribulin in the plasma, brain tumor tissue, and non–neoplastic brain tissues after a single intravenous injection. Notably, we found that eribulin concentration in the brain tumor tissue was almost as high as that in plasma within 15 minutes of injection (Figure 4). Although the plasma concentration gradually decreased and was below the detection level after 24 hours, the concentration of eribulin in the brain tumor tissue remained high for at least 24 hours. The concentration of eribulin in the normal brain tissue was very low and undetectable 2 hours after administration, confirming the previous findings. These results indicated that the high efficacy of eribulin against mouse brain tumor xenografts resulted from the efficient transfer of eribulin to brain tumor tissues. This transfer most likely occurred because the BBB in the brain tumor tissue was at least partially disrupted, which would otherwise have prevented eribulin from entering the brain. In support of this, it has been reported that eribulin reduced the size of metastatic brain tumors developed from breast cancers, suggesting that eribulin penetrated intracerebral tumors in these cases.36, 37, 38
The most intriguing finding in our study was that the concentration of eribulin in the U87MG brain xenograft remained high even 24 hours after the injection (91.7 ± 35.0 nM), although its concentration in the plasma decreased below the detectable level (Figure 4). MSI showed that eribulin was evenly distributed within the tumor tissue (Figures 5 and 6). These data confirmed that eribulin penetrated the brain tumor tissue and remained in the tumor hours after the injection. A similar phenomenon was observed in subcutaneous U87MG xenograft, where eribulin concentration remained stable for 24 hours after intravenous injection, while it gradually decreased and became undetectable after 24 hours in the plasma, kidney and liver (Figure S2). Furthermore, it was reported recently that eribulin quickly penetrated LOX human melanoma xenografts subcutaneously transplanted into mice.39 The concentration of eribulin in the tumor was high and remained stable for at least 36 hours after single or multiple (q2d × 3) intravenous injection, while plasma eribulin was cleared after 12‐24 hours. Thus, the above findings and the results of our study demonstrated that eribulin is retained in the tumor tissue for a long period of time (>24 hours) even after it is cleared from the plasma, regardless of the tumor type or location.
Although the reason for the paradoxical retention of eribulin in the tumor tissue compared to other organs, including the normal brain or plasma, is currently unknown, several underlying mechanisms can be proposed. Eribulin has been shown to bind to microtubule plus ends with high affinity40 and induce irreversible mitotic blockade, suggesting that the binding of eribulin to microtubules may be stable.41 Therefore, it is possible that eribulin bound to growing microtubules may be retained within replicating cells such as tumor cells, while quickly cleared from non–replicating cells. These findings are supported by the high distribution of eribulin in the bone marrow as documented by Eisai (http://medical.eisai.jp/products/di/IF/HAL_V_IF/HAL_V_IF.pdf). The high rate of retention in tumors may be a property specific to eribulin, because paclitaxel, another tubulin‐binding drug, shows reversible activity and is not retained in colon tumor xenografts.41, 42 Although elucidating the mechanism underlying eribulin retention in tumors is beyond the scope of this paper, it warrants further investigation. In addition, it remains unclear whether eribulin impairs self‐renewal ability in sphere‐forming glioblastoma cells. It is important to clarify this aspect of eribulin in the future because glioblastoma is a heterogeneous tumor that contains tumor‐initiating cells that cause recurrence of the tumor. Further study is warranted.
Eribulin was originally developed as a microtubule inhibitor.43, 44 However, it has recently been shown to have specific inhibitory activity against TERT‐RdRP as well.25 TERT‐RdRP appears to be involved in M‐phase progression through the promotion of heterochromatin assembly and the maintenance of stem‐cell property.21, 22, 24 Inhibition of TERT using shRNA immediately induced mitotic arrest.22 A previous study reported that the suppression of ovarian cancer cell growth by eribulin was dependent on the TERT status.25 In the present study, we showed that TERT‐RdRP activity in subcutaneous U87MG xenografts treated with eribulin decreased in a dose‐dependent manner (Figure 6). A similar tendency was observed in an intracerebral U87 MG xenograft model (Figure S5), although the difference was not statistically significant, most likely because the mouse brain tumor specimen treated with eribulin was too small. These results suggested that eribulin may exert anticancer effect through its function as a TERT‐RdRP inhibitor in addition to microtubule inhibitor activity. Further investigation assessing the exact mechanism by which eribulin suppresses tumor cell growth is underway.
A few studies have been conducted to detect the biomarker of eribulin in breast cancer and liposarcoma;45, 46, 47 however, no robust biomarker for eribulin mesylate confirmed in clinical settings has been identified yet. No paper has ever described molecular profiles of glioblastoma in association with eribulin treatment. In fact, although all TERT‐mutated cells used in this study exhibited high sensitivity to eribulin, there were small differences in IC50 (0.15‐2.12 nmol/L) among these cell lines. However, no distinct genetic changes were observed among the 93 genes examined for mutations (Table S2). We are currently conducting a multicenter, phase 2, investigator‐initiated clinical trial using eribulin in patients with recurrent glioblastoma and surgical specimens are being collected for molecular analysis for biomarkers to predict response to eribulin.
In conclusion, we showed that eribulin exerted anticancer activity against glioblastoma in vitro and in vivo, thereby significantly prolonging survival of mice harboring intracerebral glioblastoma xenografts. Eribulin penetrated brain tumor tissues and remained at a high concentration for more than 24 hours after its administration. Molecular imaging confirmed that eribulin was evenly distributed within the brain tumor tissues. In addition to microtubule inhibition, TERT‐RdRP inhibition may account for the highly efficient anti–GBM activity of eribulin. Our results thus suggested that eribulin may serve as an effective drug against GBM. Based on these data, an investigator‐initiated registration‐directed clinical trial to evaluate the safety and efficacy of eribulin in patients with recurrent GBM (UMIN000030359, https://upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000034631) has been initiated and is currently enrolling patients.
DISCLOSURE
Raku Shinkyo and Kiyomi Kikuchi are employees of Eisai. Ryo Nishikawa received honoraria and research funds which is not associated with this manuscript from Eisai. Akitake Mukasa received research funds which are not associated with this manuscript from Eisai. Yoshitaka Narita received research funds which are not associated with this manuscript from Abbvie, Ono Pharmaceutical, SBI Pharma, Stella‐Pharma, Daiichi‐Sankyo and Eisai. Akinobu Hamada received research funding which is not associated with this manuscript from Eisai. Koichi Ichimura received research funding which is not associated with this manuscript from EPS Corporation, Chugai Pharmaceutical, Daiichi‐Sankyo and Eisai. Other authors have no conflict of interest to declare regarding this manuscript. Part of the pharmacokinetics study (LC‐MS/MS) was performed in collaboration with Eisai.
Supporting information
ACKNOWLEDGMENTS
This research was supported by the Practical Research for Innovative Cancer Control from Japan Agency for Medical Research and Development, AMED (K.I.), the Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT) (K.M.) and the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) (K.M.) from Japan Agency for Medical Research and Development, AMED.
Takahashi M, Miki S, Fujimoto K, et al. Eribulin penetrates brain tumor tissue and prolongs survival of mice harboring intracerebral glioblastoma xenografts. Cancer Sci. 2019;110:2247‐2257. 10.1111/cas.14067
Takahashi and Miki contributed equally to this work.
REFERENCES
- 1. Dubrow R, Darefsky AS, Jacobs DI, et al. Time trends in glioblastoma multiforme survival: the role of temozolomide. Neuro Oncol. 2013;15:1750‐1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol. 2009;10:459‐466. [DOI] [PubMed] [Google Scholar]
- 3. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997‐1003. [DOI] [PubMed] [Google Scholar]
- 4. Chen R, Cohen AL, Colman H. Targeted therapeutics in patients with high‐grade gliomas: past, present, and future. Curr Treat Options Oncol. 2016;17:42. [DOI] [PubMed] [Google Scholar]
- 5. Prados MD, Byron SA, Tran NL, et al. Toward precision medicine in glioblastoma: the promise and the challenges. Neuro Oncol. 2015;17:1051‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987‐996. [DOI] [PubMed] [Google Scholar]
- 7. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48:768‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII‐expressing glioblastoma (ACT IV): a randomised, double‐blind, international phase 3 trial. Lancet Oncol. 2017. [DOI] [PubMed] [Google Scholar]
- 10. Arita H, Narita Y, Fukushima S, et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013;126:267‐276. [DOI] [PubMed] [Google Scholar]
- 11. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self‐renewal. Proc Natl Acad Sci U S A. 2013;110:6021‐6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masutomi K, Hahn WC. Telomerase and tumorigenesis. Cancer Lett. 2003;194:163‐172. [DOI] [PubMed] [Google Scholar]
- 13. Barthel FP, Wei W, Tang M, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49:349‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bell RJ, Rube HT, Kreig A, et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015;348:1036‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akincilar SC, Khattar E, Boon PL, Unal B, Fullwood MJ, Tergaonkar V. Long‐range chromatin interactions drive mutant TERT promoter activation. Cancer Discov. 2016;6:1276‐1291. [DOI] [PubMed] [Google Scholar]
- 16. Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer‐associated TERT promoter mutations abrogate telomerase silencing. Elife. 2015;4:e07918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maida Y, Masutomi K. Telomerase reverse transcriptase moonlights: therapeutic targets beyond telomerase. Cancer Sci. 2015;106:1486‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Man RJ, Chen LW, Zhu HL. Telomerase inhibitors: a patent review (2010‐2015). Expert Opin Ther Pat. 2016;26:679‐688. [DOI] [PubMed] [Google Scholar]
- 19. Salloum R, Hummel TR, Kumar SS, et al. A molecular biology and phase II study of imetelstat (GRN163L) in children with recurrent or refractory central nervous system malignancies: a pediatric brain tumor consortium study. J Neurooncol. 2016;129:443‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ozturk MB, Li Y, Tergaonkar V. Current insights to regulation and role of telomerase in human diseases. Antioxidants (Basel). 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maida Y, Yasukawa M, Furuuchi M, et al. An RNA‐dependent RNA polymerase formed by TERT and the RMRP RNA. Nature. 2009;461:230‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maida Y, Yasukawa M, Okamoto N, et al. Involvement of telomerase reverse transcriptase in heterochromatin maintenance. Mol Cell Biol. 2014;34:1576‐1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maida Y, Yasukawa M, Masutomi K. De novo RNA synthesis by RNA‐dependent RNA polymerase activity of telomerase reverse transcriptase. Mol Cell Biol. 2016;36:1248‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okamoto N, Yasukawa M, Nguyen C, et al. Maintenance of tumor initiating cells of defined genetic composition by nucleostemin. Proc Natl Acad Sci U S A. 2011;108:20388‐20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamaguchi S, Maida Y, Yasukawa M, Kato T, Yoshida M, Masutomi K. Eribulin mesylate targets human telomerase reverse transcriptase in ovarian cancer cells. PLoS ONE. 2014;9:e112438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dybdal‐Hargreaves NF, Risinger AL, Mooberry SL. Eribulin mesylate: mechanism of action of a unique microtubule‐targeting agent. Clin Cancer Res. 2015;21:2445‐2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Towle MJ, Nomoto K, Asano M, Kishi Y, Yu MJ, Littlefield BA. Broad spectrum preclinical antitumor activity of eribulin (Halaven(R)): optimal effectiveness under intermittent dosing conditions. Anticancer Res. 2012;32:1611‐1619. [PubMed] [Google Scholar]
- 28. Kolb EA, Gorlick R, Reynolds CP, et al. Initial testing (stage 1) of eribulin, a novel tubulin binding agent, by the pediatric preclinical testing program. Pediatr Blood Cancer. 2013;60:1325‐1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Swami U, Shah U, Goel S. Eribulin in cancer treatment. Marine Drugs. 2015;13:5016‐5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aikawa H, Hayashi M, Ryu S, et al. Visualizing spatial distribution of alectinib in murine brain using quantitative mass spectrometry imaging. Sci Rep. 2016;6:23749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Narayan S, Carlson EM, Cheng H, et al. Novel second generation analogs of eribulin. Part III: blood‐brain barrier permeability and in vivo activity in a brain tumor model. Bioorg Med Chem Lett. 2011;21:1639‐1643. [DOI] [PubMed] [Google Scholar]
- 32. Taur JS, DesJardins CS, Schuck EL, Wong YN. Interactions between the chemotherapeutic agent eribulin mesylate (E7389) and P‐glycoprotein in CF‐1 abcb1a‐deficient mice and Caco‐2 cells. Xenobiotica. 2011;41:320‐326. [DOI] [PubMed] [Google Scholar]
- 33. Goasdoue K, Miller SM, Colditz PB, Bjorkman ST. Review: the blood‐brain barrier; protecting the developing fetal brain. Placenta. 2017;54:111‐116. [DOI] [PubMed] [Google Scholar]
- 34. Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 2002;3:53‐57. [DOI] [PubMed] [Google Scholar]
- 35. Kim WY, Lee HY. Brain angiogenesis in developmental and pathological processes: mechanism and therapeutic intervention in brain tumors. FEBS J. 2009;276:4653‐4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang AY, Ying XX. Brain metastases from breast cancer and response to treatment with eribulin: a case series. Breast Cancer (Auckl). 2015;9:19‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsuoka H, Tsurutani J, Tanizaki J, et al. Regression of brain metastases from breast cancer with eribulin: a case report. BMC Res Notes. 2013;6:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Okita Y, Narita Y, Suzuki T, et al. Extended trastuzumab therapy improves the survival of HER2‐positive breast cancer patients following surgery and radiotherapy for brain metastases. Mol Clin Oncol. 2013;1:995‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugawara M, Condon K, Liang E, et al. Eribulin shows high concentration and long retention in xenograft tumor tissues. Cancer Chemother Pharmacol. 2017;80:377‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith JA, Wilson L, Azarenko O, et al. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry. 2010;49:1331‐1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Towle MJ, Salvato KA, Wels BF, et al. Eribulin induces irreversible mitotic blockade: implications of cell‐based pharmacodynamics for in vivo efficacy under intermittent dosing conditions. Cancer Res. 2011;71:496‐505. [DOI] [PubMed] [Google Scholar]
- 42. Hamaguchi T, Matsumura Y, Suzuki M, et al. NK105, a paclitaxel‐incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br J Cancer. 2005;92:1240‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Towle MJ, Salvato KA, Budrow J, et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61:1013‐1021. [PubMed] [Google Scholar]
- 44. Jordan MA, Kamath K, Manna T, et al. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol Cancer Ther. 2005;4:1086‐1095. [DOI] [PubMed] [Google Scholar]
- 45. Kashiwagi S, Fukushima W, Asano Y, et al. Identification of predictive markers of the therapeutic effect of eribulin chemotherapy for locally advanced or metastatic breast cancer. BMC Cancer. 2017;17:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oba T, Izumi H, Ito KI. ABCB1 and ABCC11 confer resistance to eribulin in breast cancer cell lines. Oncotarget. 2016;7:70011‐70027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wiemer EAC, Wozniak A, Burger H, et al. Identification of microRNA biomarkers for response of advanced soft tissue sarcomas to eribulin: translational results of the EORTC 62052 trial. Eur J Cancer. 2017;75:33‐40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials