

Abstract
Regulatory T (Treg) cells suppress abnormal/excessive immune responses to self‐ and nonself‐antigens to maintain immune homeostasis. In tumor immunity, Treg cells are involved in tumor development and progression by inhibiting antitumor immunity. There are several Treg cell immune suppressive mechanisms: inhibition of costimulatory signals by CD80 and CD86 expressed by dendritic cells through cytotoxic T‐lymphocyte antigen‐4, interleukin (IL)‐2 consumption by high‐affinity IL‐2 receptors with high CD25 (IL‐2 receptor α‐chain) expression, secretion of inhibitory cytokines, metabolic modulation of tryptophan and adenosine, and direct killing of effector T cells. Infiltration of Treg cells into the tumor microenvironment (TME) occurs in multiple murine and human tumors. Regulatory T cells are chemoattracted to the TME by chemokine gradients such as CCR4‐CCL17/22, CCR8‐CCL1, CCR10‐CCL28, and CXCR3‐CCL9/10/11. Regulatory T cells are then activated and inhibit antitumor immune responses. A high infiltration by Treg cells is associated with poor survival in various types of cancer. Therefore, strategies to deplete Treg cells and control of Treg cell functions to increase antitumor immune responses are urgently required in the cancer immunotherapy field. Various molecules that are highly expressed by Treg cells, such as immune checkpoint molecules, chemokine receptors, and metabolites, have been targeted by Abs or small molecules, but additional strategies are needed to fine‐tune and optimize for augmenting antitumor effects restricted in the TME while avoiding systemic autoimmunity. Here, we provide a brief synopsis of these cells in cancer and how they can be controlled to achieve therapeutic outcomes.
Keywords: immune checkpoint, immune suppression, tolerance, Treg, tumor
1. INTRODUCTION
The identification of tumor antigens has led to various manipulations of the host immune system to treat cancer.1 However, only a limited number of patients show antitumor effects, such as tumor regression, after multiple vaccinations despite the development of measurable humoral and cellular immune responses against tumor antigens. The immunosuppressive tumor microenvironment (TME) is a critical barrier for the development and augmentation of effective antitumor immune responses. The immunosuppressive TME is filled with immunosuppressive cells, such as regulatory T (Treg) cells,2 myeloid‐derived suppressor cells (MDSCs),3 and tumor‐associated macrophages,4 and increased expression of immunosuppressive molecules such as programmed cell death‐1 (PD‐1)5 and PD‐ligand 1 (PD‐L1).6 Therefore, the focus of cancer immunology to develop effective cancer immunotherapies has shifted to understanding and controlling these immune suppressive networks in the TME.
Meth A (a 3′‐methylcholanthrene‐induced sarcoma)‐bearing mice harbor immune suppressive CD4+ T cells, which are peripherally induced Treg cells.7 There are also thymus‐derived immune suppressive lymphocytes, which appear in autoimmune diseases caused by neonatal thymectomy.8 These lymphocytes were identified by Sakaguchi et al2 as CD25+CD4+ T cells and are currently known as thymus‐derived natural Treg cells. Regulatory T cells have central roles in the maintenance of self‐tolerance: they protect hosts from developing autoimmune diseases and allergies, whereas in malignancies, they hinder immune surveillance against cancer in healthy individuals and prevent the development of effective antitumor immunity in tumor‐bearing patients. Two Japanese groups found that antitumor immunity was inhibited by Treg cells by demonstrating tumor rejection and decreased tumor growth in mice given anti‐CD25 mAb to deplete Treg cells or nude (T cell‐deficient) mice transplanted with CD25+ cell‐deleted splenocytes.9, 10 Therefore, Treg cell depletion or control of immune suppression by Treg cells evokes/augments antitumor immune responses, which has led to the development of Treg cell‐targeted cancer immunotherapies.
Here, we discuss the roles of Treg cells in cancer and the development of Treg cell‐targeted cancer immunotherapies for immune precision medicine.
2. CHARACTERIZATION OF TREG CELLS
Regulatory T cells were initially defined as CD4+ T cells with a high expression of CD25 (interleukin [IL]‐2 receptor α‐chain). The Foxp3 gene, a member of the Forkhead/winged‐helix family of transcriptional regulators, was then discovered as a master regulator in developing Treg cells based on the following findings: Scurfy mice with a frameshift mutation in the Foxp3 gene have T cell inflammation in multiple organs and a lethal autoimmune disease because of effector T cell activation and increased cytokine production caused by the lack of Treg cells.11 In addition, mutation of the Foxp3 gene in humans leads to IPEX syndrome (X‐linked immune dysregulation, polyendocrinopathy, and enteropathy).12 Furthermore, the forced expression of FoxP3 in naive T cells results in an immune suppressive function. CD4+CD25− naive T cells that are transfected with Foxp3 can convert to CD4+CD25+ Treg‐like cells that produce inhibitory cytokines and express typical Treg‐cell molecules such as CD25, cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4), and glucocorticoid‐induced tumor necrosis factor (TNF) receptor‐related protein (GITR).13 Thus, FoxP3 is a lineage‐specific marker and a master regulatory gene in the generation, maintenance, and immune suppressive functions of Treg cells.
Regulatory T cells are classified into natural/thymic and peripherally induced Treg cells based on where they develop.14 FoxP3+ natural Treg cells are generated in the thymus as the functionally mature T‐cell subpopulation specialized for immune suppression (natural/thymic Treg cells). Some Treg cells are converted from conventional T cells following in vitro T‐cell receptor (TCR) stimulation with transforming growth factor (TGF)‐β or retinoic acid (peripherally induced Treg cells).15, 16 In humans, FoxP3+ T cells are readily induced from conventional T cells by TCR stimulation, but produce inflammatory cytokines rather than gain an immune suppressive function; however, several cytokines or specific microbiota environments induce Treg cells with an immune suppressive function from CD4+CD25− T cells.17 Currently, the in vivo function and stability of peripherally induced Treg cells, such as TGF‐β‐induced Treg cells, are unclear, particularly in humans.
Because human T cells transiently express Foxp3 in conventional T cells following TCR stimulation, FoxP3+ T cells in humans are heterogeneous in function and phenotype. CD25+CD4+ Treg cells express low levels of CD127 (the α‐chain of the IL‐7 receptor); thus, CD4+CD25+CD127lo T cells are considered to be Treg cells with suppressive activity.18 However, naive T cells stimulated by TCR signaling transiently increase FoxP3 expression and downregulate expression of CD127, which suggests that there is possible contamination of some activated non‐Treg cells in the CD4+CD25+CD127lo T‐cell fraction.
Therefore, it is necessary to distinguish Treg cells from FoxP3‐expressing conventional T cells in humans. We previously proposed that human Treg cells can be classified by the expression levels of FoxP3 (and/or CD25) and a naive marker CD45RA: (a) Fraction (Fr.) 1, naive/resting Treg cells, defined by FoxP3loCD45RA+CD25lo cells; (b) Fr. 2, effector/activated Treg (eTreg) cells, defined by FoxP3hiCD45RA−CD25hi cells; and (c) Fr. 3, non‐Treg cells, defined by FoxP3loCD45RA−CD25lo cells (see Table 1 and Figure 1).19 Naive Treg cells that have recently left the thymus but have not been activated in the periphery possess weak suppressive activity. After TCR stimulation in the draining lymph node, naive Treg cells vigorously proliferate and differentiate into highly suppressive and terminally differentiated eTreg cells. These eTreg cells then inhibit the maturation of antigen‐presenting cells (APCs) such as dendritic cells (DCs) in an antigen‐specific manner. In contrast, eTreg cells show their suppressive activity through consumption of IL‐2 by high affinity IL‐2 receptor, secretion of inhibitory cytokines including IL‐10, TGF‐β, and IL‐35 and degradation of ATP, an important cellular energy. These suppressive mechanisms act through an antigen‐nonspecific manner. In fact, in a TCR‐transgenic animal model, antigen‐specific Treg cells show a superior immune suppressive function compared with antigen‐nonspecific Treg cells, although the latter also have an immune suppressive activity.20 Therefore, although Treg cell suppression is partially antigen‐nonspecific, antigen‐specific Treg cells show a far stronger immune suppressive function.
Table 1.
Classification of FoxP3+CD4+ T cells in humans
| Fraction | Classification | Definition | Phenotype/cytokines | Characteristics |
|---|---|---|---|---|
| Fr. 1 | rTreg | FoxP3loCD45RA+CD25lo | CTLA‐4lo CD127lo/− Ki‐67− |
|
| Fr. 2 | eTreg | FoxP3hiCD45RA−CD25hi | CTLA‐4hi PD‐1+, ICOS+, GITR+, OX40+, CD15s+ CCR4+, CCR8+, IL‐10+, TGF‐β+ |
|
| Fr. 3 | Non‐Treg | FoxP3loCD45RA−CD25lo | IL‐2+, IFN‐γ+, IL‐17+ |
|
CCR4, CC chemokine receptor 4; CCR8, chemokine receptor 8; CTLA‐4, cytotoxic T‐lymphocyte antigen‐4; eTreg, effector/activated Treg; GITR, glucocorticoid‐induced tumor necrosis factor receptor‐related protein ICOS, inducible T‐cell costimulator; IFN‐γ, γ‐interferon; IL, interleukin; PD‐1, programmed cell death 1; rTreg, resting/naïve Treg; TCR, T‐cell receptor; TGF‐β, transforming growth factor‐β; Treg, regulatory T cell.
Figure 1.
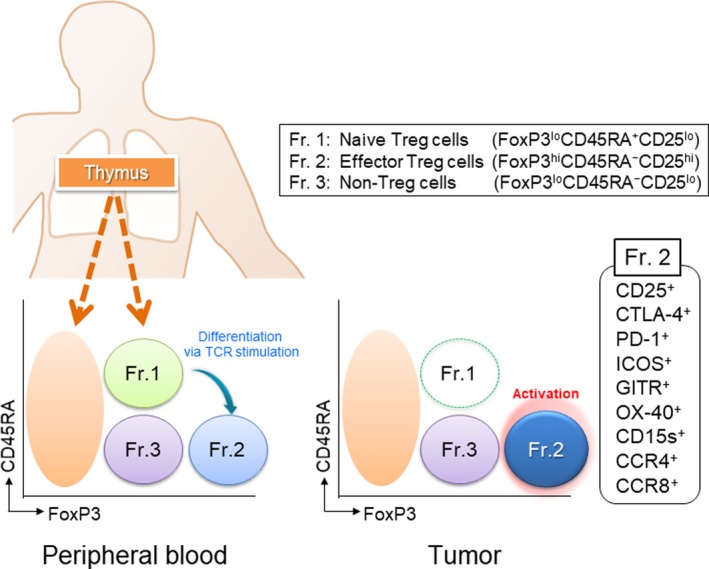
Classification of human regulatory T (Treg) cells. Human Treg cells can be classified into the following 3 subfractions: Fraction (Fr.) 1, naive/resting Treg cells, defined by FoxP3loCD45RA+CD25lo cells; Fr. 2, effector/activated Treg (eTreg) cells, defined by FoxP3hiCD45RA−CD25hi cells; and Fr. 3, non‐Treg cells, defined by FoxP3loCD45RA−CD25lo cells. Naive/resting Treg cells that have just left the thymus have a weak immune suppressive function and differentiate into effector/activated Treg cells following T‐cell receptor (TCR) stimulation. eTreg cells are the terminal differentiation state and harbor strong immune suppressive activity. Non‐Treg cells do not possess immune suppressive activity, but produce inflammatory cytokines. Typical staining pattern of CD4+ T cells in peripheral blood and lung cancer tissue. In general, the frequency of eTreg cells in humans is 1%‐5% in peripheral blood but approximately 10%‐50% in the TME. eTreg cells predominantly express various activation cell surface markers including cytotoxic T‐lymphocyte antigen‐4 (CTLA)‐4, programmed cell death (PD)‐1, inducible T‐cell costimulator (ICOS), glucocorticoid‐induced tumor necrosis factor receptor‐related protein (GITR), OX40, CD15s, CCR4, and CCR8. Naive/resting Treg cells are hardly detected in the tumor microenvironment
It is widely accepted that naive conventional T cells, including CD8+ T cells and CD4+FoxP3− T cells, are primed by APCs in the lymph nodes. In addition, these primed T cells are required to recognize cognate antigens through APCs for their optimal activation in the TME.21 Thus, T cells need to be activated by APCs not only in the lymph node but also in the TME. Given that one important suppressive mechanism by Treg cells is inhibiting the maturation of APCs, Treg cells control the priming and the activation of conventional T cells both in the lymph node and in the TME, respectively.
For the cognate antigens for Treg cells, particularly in the cancer setting, a recent study analyzed the TCR repertoire of tumor‐infiltrating Treg cells in patients with metastatic melanoma, gastrointestinal, and ovarian cancers. Regulatory T cells in the TME possessed a unique TCR repertoire distinct from other conventional CD4+ T cells, and the TCRs were specific for tumor‐specific antigens, meaning that Treg cells in the TME recognize tumor‐specific antigens. These Treg cells with tumor‐specific TCRs were also detected in the periphery.22 These findings suggest that tumor‐specific activation and the clonal expansion of Treg cells are promoted locally and systemically.
Non‐Treg cells that are immune stimulatory T cells produce inflammatory cytokines including γ‐interferon and IL‐17. Analysis of DNA methylation of the 5′‐flanking region in each fraction revealed that CpG methylation sites crucial for safeguarding the lineage stability of proliferating Treg cells are completely demethylated in Fr. 2 cells, but less demethylated in Fr. 3,19 which indicates that Fr. 2 cells stably transcribe Foxp3. The frequency of eTreg cells (Fr. 2) in humans is 2%‐5% in CD4+ T cells but increases to 10%‐50% in the TME.23
3. TREG CELLS IN THE TME AND THE CLINICAL RELEVANCE OF TREG CELLS
Infiltration of Treg cells into the TME occurs in various murine and human tumors.24 Cancer cells with inherent genetic instability form abnormal proteins that have not been previously recognized by the immune system, and these proteins become immunogenic antigens (neoantigens) that can spontaneously trigger CD8+ T‐cell responses. Cancer cells that present extremely immunogenic neoantigens are eliminated from the host through the process of immune surveillance.25 Cancers then promote an immune suppressive TME in which immune‐suppressing cells including Treg cells, MDSCs, and tumor‐associated macrophages and multiple immune checkpoint molecules are abundant (cancer immunoediting). In various types of cancer, the presence of high Treg cells and a low ratio of CD8+ T cells to Treg cells in the TME are associated with unfavorable prognosis.24 However, there are some exceptions, such as in colorectal cancer in which the presence of a high number of FoxP3+ T cells corresponds to a better prognosis. The accumulation of FoxP3+ non‐Treg cells in the TME of a subfraction of colorectal cancers with a high level of inflammatory cytokines, such as TGF‐β and IL‐12, is correlated with a favorable prognosis. By contrast, a high number of bona fide Treg cells, particularly eTreg cells, in the TME is correlated with a poor prognosis, as reported for other types of cancer in which FoxP3+ non‐Treg cells are hardly detected.26
What is the significance of Treg cell accumulation in the TME? Treg cells are chemoattracted by chemokine gradients.27 CCR4, CCR8, CCR10, and CXCR3 are chemokine receptors responsible for Treg cell migration to the TME in response to CC and CXC chemokines: CCR4 is bound by CCL17 and CCL22,28 CCR8 is bound by CCL1,29 CCR10 is bound by CCL28,30 and CXCR3 is activated by CXCL9/10/11.31 Thymus‐derived Treg cells preferentially recognize self‐antigens by high‐affinity TCR and clonally expand in the TME. In the TME, there are many tumor‐associated self‐antigens from dying tumor cells that are recognized by Treg cells rather than by effector and memory T cells.32 Therefore, one can envisage that Treg cells are chemoattracted and recognize cognate antigens abundant in the TME, which leads to Treg cell activation and proliferation, contributing to the development of an immunosuppressive TME. Immunosuppressive cytokines, such as TGF‐β and IL‐10, produced by tumor cells and immune cells in the TME also increase Treg cells.33 In addition, inducible T‐cell costimulator (ICOS), a T cell costimulatory molecule belonging to the CTLA‐4/PD‐1/CD28 family and expressed by Treg cells, is involved in the proliferation of activated Treg cells through binding to an ICOS ligand expressed by plasmacytoid DCs.34, 35
4. SUPPRESSIVE MECHANISMS OF TREG CELLS
Regulatory T cells suppress immune functions through various mechanisms such as CTLA‐4‐mediated suppression of APC function, consumption of IL‐2, production of immunosuppressive cytokines, and production of immune suppressive metabolites (Figure 2).
Figure 2.
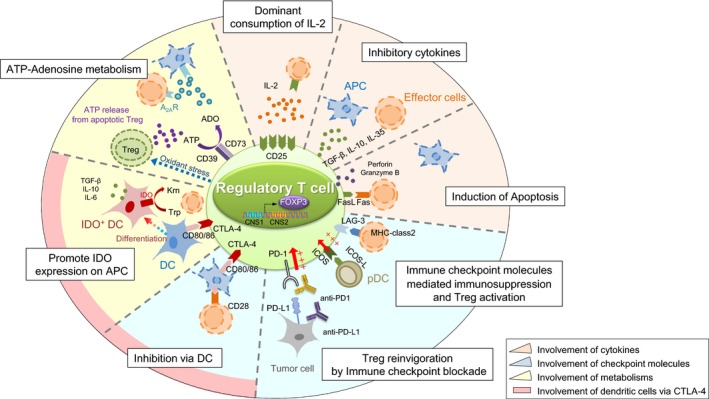
Suppressive mechanism of regulatory T (Treg) cells. Treg cells exert their immunosuppressive function through several mechanisms. The first immunosuppressive mechanisms involving cytokines include consumption of interleukin (IL)‐2 by Treg cells highly expressing CD25 (IL‐2 receptor α‐chain), suppression by inhibitory cytokines, such as transforming growth factor (TGF)‐β, IL‐10, and IL‐35, and direct killing of effector or antigen‐presenting cells (APC) by perforin, granzyme B, or Fas/Fas ligand (FasL) interaction. The second immunosuppressive mechanisms involving immune checkpoint molecules include inhibition of effector T cells by the lymphocyte activation gene‐3 (LAG‐3)‐MHC class II pathway and Treg activation through the inducible T‐cell costimulator (ICOS)‐ICOS ligand (ICOSL) and programmed cell death (PD)‐1/PD‐ligand (PD‐L)1 pathways. The third immunosuppressive mechanisms include metabolic modulation by indoleamine 2,3‐dioxygenase (IDO) expression in dendritic cells (DC), which exhausts T cells because critical amino acids for survival are depleted. Furthermore, the generation of adenosine from ATP, which is metabolized by CD39 and CD73 expressed in activated Treg cells, results in T cell suppression from the induction of negative signaling to effector T cells and APCs. The fourth immunosuppressive mechanism involves DCs through cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) and decreased CD80/86 expression by APCs by binding to CTLA‐4 expressed by activated eTreg cells, which causes impairment of APC maturation, downregulation of CD80/86 molecules on APCs, and attenuation of T cell stimulation. A2AR, A2A receptor; CNS, conserved noncoding sequence; Krn, kyneurenine; pDC, plasmacytoid dendritic cell; Trp, tryptophan
4.1. Involvement of DCs through CTLA‐4
Cytotoxic T‐lymphocyte antigen‐4 expressed by Treg cells impairs maturation of APCs, such as DCs, by binding to CD80/86.36 Antigen stimulation by immature DCs or DCs with a low expression of CD80/86 induces T cells with relatively low‐affinity TCRs for tumor‐antigens derived from self‐components to enter an anergic state characterized by hypoproliferation and hypoproduction of cytokines following antigen restimulation.37 The CTLA‐4 binds to CD80/86 on APCs with a higher affinity than CD28, thereby inhibiting costimulatory signals. In addition, CD80/86 bound to CTLA‐4 can be physically transferred from APCs to the surface or the cytoplasm of Treg cells by trogocytosis.38
4.2. Involvement of cytokines
Regulatory T cells highly express CD25 (IL‐2 receptor α‐chain), dominantly consume IL‐2 through high‐affinity IL‐2 receptors, and hardly produce IL‐2, thereby limiting the amount of IL‐2 for effector T cell proliferation/activation. Administration of a high dose of IL‐2 neutralizes Treg‐cell suppressive functions.39 Treg cells also produce inhibitory cytokines, such as TGF‐β, IL‐10, and IL‐35, to inhibit effector T‐cell activation.40, 41 Additionally, cytotoxic substances produced by Treg cells, such as perforin and granzyme, kill effector T cells.42
4.3. Involvement of immune checkpoint molecules
Well‐known immune checkpoint molecules CTLA‐4, ICOS, and lymphocyte activation gene‐3 (LAG‐3)43 are expressed by activated eTreg cells, which inhibits the cytotoxic function and proliferation of effector T cells.44 Programmed cell death‐1 is expressed by activated eTreg cells as well as effector T cells. Yet, the effects of PD‐1 inhibition on eTreg cells remain to be determined. Programmed cell death‐1 inhibits excessive activation of conventional T cells by suppressing TCR and costimulatory CD28 signaling and renders them dysfunctional or exhausted. Considering the similar expression level of PD‐1 by Treg cells in the TME and the similar dependency of TCR and CD28 signaling for their survival and function, PD‐1 inhibition could potentiate the activation and immunosuppressive function of Treg cells (see 5.2).45
4.4. Involvement of immune suppressive metabolites
Indoleamine 2, 3‐dioxygenase (IDO), an essential enzyme in the kynurenine pathway of tryptophan metabolism, and tryptophan 2, 3‐dioxygenase (TDO) deplete tryptophan in the TME and cause T cell dysfunction.46 The interaction between CTLA‐4 expressed by Treg cells and CD80/86 on APCs promotes IDO secretion.47 Additionally, Treg cells are more sensitive to oxidative stress than effector T cells because Treg cells have a lower expression of NRF2, which is a key transcription factor for antioxidant responses. Therefore, oxidative stress induces Treg cell apoptosis, and apoptotic Treg cells release a large amount of ATP. Subsequently, ATP is metabolized to adenosine by CD39 and CD73, which are highly expressed by Treg cells, and adenosine binds to the A2A receptor (A2AR), which inhibits effector T cells.48
5. TARGETING TREG CELLS IN CANCER IMMUNOTHERAPY
The presence of a high number of Treg cells and a low ratio of CD8+ T cells to Treg cells in the TME are correlated with poor prognosis, which suggests that Treg cells suppress tumor antigen‐specific T cell responses.24 Therefore, depletion of Treg cells or the control of Treg cell functions could be promising immunotherapies.
Depletion of Treg cells increases antitumor immune responses and contributes to tumor eradication in mouse models. Importantly, an initial study revealed that Treg cell depletion induced tumor regression in some tumor cell lines, such as Meth A and RL‐male 1, but not in others, such as AKSL2 and RL‐female 8.10 Therefore, Treg cell‐targeted therapy is unlikely to be effective against all tumors, as observed in current cancer immunotherapy in the clinic. Indeed, a phase I clinical trial of Treg cell depletion by the administration of anti‐CCR4 mAb in patients with solid tumors resulted in increased antitumor immune responses in several patients, but clinical responses were not observed in most patients.49 Another concern is that systemic Treg cell depletion might increase the risk of immune‐related adverse events (irAE), such as autoimmunity‐related toxicities. To ensure the safety of Treg cell‐targeted therapy, selective depletion of eTreg cells in the TME, rather than systemic Treg cell depletion, should be used to increase antitumor effects without inducing detrimental irAE. Thus, we need to identify biomarker(s) that distinguish tumors in which Treg cells are essential for tumor growth by clarifying the immune suppressive network in the TME.
Another approach for targeting Treg cells is to control or modulate Treg cell function and infiltration. In particular, manipulating the chemokine and/or cytokine axis, cell‐intrinsic signaling, or metabolites in Treg cells in the TME could cause relative changes in Treg cell function and infiltration. The presence of specific metabolites in the TME, such as IDO and adenosine, markedly affects Treg cell function and lineage stability. Fatty acid metabolism also promotes Treg cell development. Accelerated glycolytic metabolism by cancer cells results in the consumption of glucose and increase in the lactic and fatty acids in the TME. FoxP3 promotes oxidative phosphorylation and increasing nicotinamide adenine dinucleotide oxidation by decreasing glycolysis through suppressing Myc expression.50 Treg cells then uptake the lactic acid and fatty acid utilizing oxidative phosphorylation and fatty acid β‐oxidation, for maintaining their survival and immune suppressive function in the TME. Furthermore, modulation of PI3K changes FoxP3 expression.51 The clinical significance of these signaling pathways and metabolites in Treg cells is still unclear, but the regulation of Treg cell‐intrinsic signaling or metabolism, including transcriptional signaling, could be promising therapeutic targets in humans, and further studies are needed.
5.1. Regulatory T cell depletion
Regulatory T cells in the TME express several cell surface markers including CD25, CTLA‐4, PD‐1, ICOS, GITR, OX40, CD15s,52 CCR4, and CCR8, and these markers can be used to deplete Treg cells.
5.1.1. CD25
Because Treg cells were originally identified as CD25+CD4+ T cells, several studies explored the effects of targeting CD25 with Abs or a recombinant protein composed of IL‐2 and the active domain of the diphtheria toxin to deplete Treg cells.53 Treg cell depletion by anti‐CD25 mAb was evaluated in clinical trials; vaccination with multiple tumor‐associated peptides resulted in stable disease in 6 of 10 patients with a median progression‐free survival of 4.8 months when followed by Treg cell depletion with an anti‐CD25‐depleting mAb daclizumab in breast cancer patients.54 In contrast, another study showed that daclizumab depleted both effector T cells and Treg cells, but neither an antitumor immune response nor Ab production was observed. Because CD25 expression is induced following activation of effector T cells, CD25‐targeted Treg cell depletion could be accompanied by a reduction of effector cells, which suggests that there is a limited window for Treg cell depletion by targeting CD25 to increase antitumor T cell responses.
5.1.2. Cytotoxic T‐lymphocyte antigen‐4
Cytotoxic T‐lymphocyte antigen‐4 is constitutively expressed by FoxP3+CD4+ Treg cells and is upregulated by CD4+ and CD8+ effector T cells after activation. It was originally thought that anti‐CTLA‐4 mAb antagonized an inhibitory signal on activated CD4+ and CD8+ effector T cells and reinvigorated them to regain antitumor activity.55 However, recent studies have shown that the antitumor effects of anti‐CTLA‐4 mAb is dependent on depletion of CTLA‐4‐expressing Treg cells in the TME through Ab‐dependent cytotoxic activity (ADCC); depletion of the Fc function completely abrogates the antitumor effect of anti‐CTLA‐4 mAb.56, 57, 58 In addition, mice lacking CTLA‐4 only in Treg cells have increased antitumor immunity. Therefore, the increased antitumor effects by anti‐CTLA‐4 mAb are mainly caused by the suppression of Treg cell function and elimination of Treg cells in the TME, and further analyses to address the roles of CTLA‐4 in effector T cells and Treg cells in cancer settings are needed, particularly in humans.
5.1.3. OX40 and GITR
Other molecules, such as OX40,59 GITR,13 and LAG‐3,43 predominantly expressed by Treg cells, can also be candidates for Treg cell depletion and functional manipulation. The GITR protein is a costimulatory molecule expressed at low levels by resting CD4+ and CD8+ T cells and constitutively at high levels by FoxP3+CD4+ Treg cells. Activation of GITR signaling with an agonistic anti‐GITR mAb or GITR ligands inhibits the suppressive activity of FoxP3+CD4+ Treg cells and makes effector T cells resistant to FoxP3+CD4+ Treg‐mediated suppression.60 Another candidate is OX40, a costimulatory molecule of the TNF receptor superfamily. OX40 expression is transiently induced by the activation of effector T cells and constitutively expressed by FoxP3+CD4+ Treg cells. Agonistic anti‐OX40 mAb mediates antitumor effects by attenuating FoxP3+CD4+ Treg cell‐mediated immune suppression and activating effector T cell function.59 Therapies targeting GITR and OX40 are currently being tested in clinical trials with promising results.61
5.1.4. Chemokine and chemokine receptors
Regulatory T cell chemotaxis through CCL28‐CCR10, CCL1‐CCR8, and CCL22‐CCR4 into the TME has been studied. Blocking chemokine and chemokine receptor interactions attenuates Treg cell accumulation into the TME, which increases antitumor immune responses. For example, hypoxia in the TME induces CCL28 expression and chemoattracts CCR10+ Treg cells. Intratumoral administration of anti‐CCR10 immunotoxin, which blocks the interaction of CCL28 and CCR10, reduces Treg cell accumulation in the TME and increases the antitumor immune response in a mouse model.62
CCR4 is highly expressed by eTreg cells but not by naive Treg cells or most effector T cells, except for some Th2 and Th17 cells in peripheral blood.28 Using PBMCs from melanoma patients, increased tumor antigen (NY‐ESO‐1)‐specific CD4+ and CD8+ T cell responses are observed after depletion of CCR4+ eTreg cells.63 Anti‐CCR4 mAb is therefore instrumental in evoking and augmenting antitumor immunity in cancer patients by selectively depleting eTreg cells, and a phase Ia/Ib multicenter clinical trial has been carried out.49 Administration of anti‐CCR4 mAb (mogamulizumab) for advanced or recurrent solid tumor patients significantly reduces eTreg cells in peripheral blood. In addition, humoral responses against NY‐ESO‐1 and XAGE1 antigens were observed in 3 of 5 and 5 of 5 patients, respectively, in those with NY‐ESO‐1‐ or XAGE1‐positive antigens.49 Further clinical trials in combination with an immune checkpoint blockade are now ongoing.
The secretion of CCL1 by CD11b+CD14+ myeloid cells is involved in Treg cell infiltration.64 Increased expression of CCR8, a receptor for CCL1, is observed in Treg cells and NKT cells, particularly in cancer patients, and faint expression is observed in CD8+ T cells or Th1 cells. Interactions between CCL1 and CCR8 enhance the expression of FoxP3 through the STAT3 pathway, and activated CCR8+ Treg cells strongly suppress antitumor immunity by promoting ATP‐adenosine metabolism by CD39 and secretion of IL‐10 and granzyme B.29 The overall survival of breast cancer patients with a high infiltration of CCR8+FoxP3+ Treg cells is significantly shorter than that of patients with low infiltration.64 Therefore, therapies that target CCL1‐CCR8 molecules warrant testing in the clinic.
5.1.5. Chemotherapy
Low doses of cyclophosphamide selectively inhibit Treg cell proliferation and induce Treg apoptosis.65 In a phase II clinical trial, patients with advanced renal cell cancer received IMA901 vaccination consisting of multiple tumor‐associated peptides and GM‐CSF with or without prior treatment with cyclophosphamide. Addition of cyclophosphamide reduced Treg cells and increased antitumor immune responses. However, a phase III trial investigating the addition of IMA901, GM‐CSF, and cyclophosphamide to the standard care of sunitinib against renal cell carcinoma failed to show survival benefits. In addition to cyclophosphamide, cyclosporine A and tacrolimus also inhibit IL‐2 production, an essential cytokine for Treg cell activation and proliferation, and decrease Treg cells.66
5.2. Immune checkpoint inhibitors
Immune checkpoints, such as CTLA‐4 and PD‐1, are highly expressed by activated Treg cells. The role of Treg cells in anti‐CTLA‐4 mAb is discussed above. The effect of PD‐1 blockade on Treg cells harboring comparable expression levels of PD‐1 and effector T cells remains unclear. Programmed cell death‐1 inhibits excessive activation of effector T cells by suppressing TCR and CD28 signals and rendering them dysfunctional (so‐called exhausted T cells).67 Because Treg cells show comparable expression of PD‐1 with that of effector T cells, particularly in the TME, and are dependent on TCR and CD28 for their survival and function, PD‐1 blockade might activate the immune suppressive function of Treg cells. In line with this hypothesis, PD‐1‐deficient Treg cells possess strong immune suppressive activity and rescue autoimmune phenotypes.69 Anti‐PD‐1 mAb increases Treg cell‐mediated immune suppressive activity in some patients with gastric cancer, which contributes to hyperprogression during PD‐1 blockade therapy.45 However, another study reported that PD‐1‐blocked Treg cells show low immune suppressive activity, and further analyses to examine the roles of PD‐1 in effector T cells and Treg cells in cancer settings are needed.68
5.3. Regulatory T cell modulation
5.3.1. Cytokines
Mutations in the TGF‐β pathway are often observed in human cancers, and overactivation of this pathway is associated with tumor progression by stimulating angiogenesis and suppressing the innate and adaptive antitumor immune responses.70 Transforming growth factor‐β directly suppresses the function of effector T cells and natural killer cells. Glycoprotein A repetitions predominant (GARP) is a transmembrane protein containing leucine‐rich repeats that promotes the activation and secretion of TGF‐β.71 Increased GARP expression is observed in activated Treg cells,72 and GARP could be a candidate for Treg cell control in preclinical studies.
5.3.2. Tyrosine kinase inhibitors
Tyrosine kinase inhibitors, including imatinib and dasatinib, inhibit TCR signaling for Treg cell survival and function through off‐target effects. For example, dasatinib induces G0/G1 arrest of Treg cells and inhibits Treg cell function.73 In a clinical trial for dasatinib discontinuation, Treg cell reduction was observed and was associated with a favorable clinical outcome in patients with chronic myeloid leukemia.74
5.3.3. Phosphatidylinositol 3‐kinase‐PTEN‐MTOR axis
Inhibitors of P13K also effectively control immune suppression by Treg cells in mouse models.75 Analysis of PI3Kδ‐deficient mice revealed that PI3Kδ signaling attenuates immune suppression by Treg cells by reducing TCR and IL‐2 signals crucial for Treg cell development.76 The PI3K‐PTEN‐MTOR axis, downstream of TCR and costimulatory signaling, has a pivotal role in the development, function, and metabolism of T cells, particularly Treg cells. Regulatory T cell‐specific ablation of PTEN, the primary negative regulator of PI3K, impairs mitochondrial fitness, upregulates glycolysis, causes loss of FoxP3 expression in Treg cells, and induces effector T cells.77, 78 Furthermore, a cancer vaccine co‐administered with a PI3Kδ inhibitor reduces Treg cells and increases effector T cells, efficiently inhibiting tumor growth.79 In addition, Treg cell‐specific deletion of Atg7 or Atg5, two essential genes involved in autophagy, breaks self‐tolerance and facilitates tumor eradication because of increased MTOR complex 1 activity, c‐Myc expression, and glycolytic metabolism characteristic of anabolic upregulation.80
Heat shock protein (HSP) inhibitor induces PI3K/AKT phosphorylation and increases suppressive activity in Treg cells.81 Regulatory T cells treated with HSP70 significantly inhibit the proliferation and production of CD25−CD4+ T cells, which produce effector cytokines such as γ‐interferon and TNF‐α. An HSP90 inhibitor strongly induces cancer antigen‐specific effector T cells by decreasing Treg cells and MDSCs in the TME.
5.3.4. CD39 and CD73
CD39 and CD73 metabolize extracellular ATP to adenosine, which binds to A2AR and inhibits effector T‐cell activation. Adenosine negatively signals to the APCs and attenuates activation of effector T cells. Regulatory T cells express both CD39 and CD73 at high levels, especially in the TME.48 Thus, CD39 and CD73, which are critical to adenosine metabolism, are promising therapeutic targets and are currently under investigation in clinical trials.
5.3.5. Vascular endothelial growth factor‐vascular endothelial growth factor receptor axis
The vascular endothelial growth factor (VEGF)‐VEGF receptor 2 (VEGFR2) pathway is involved in the accumulation of immature DCs, MDSCs, and Treg cells, and the attenuation of T cell infiltration.82 Increased PD‐L1 expression and increased CD8+ T cell infiltration are observed after treatment with ramucirumab, a fully humanized IgG1 anti‐VEGFR2 mAb. In addition, a reduction of eTreg cell infiltration and PD‐1 expression by CD8+ T cells is observed in TILs compared with that of PBMCs after ramucirumab‐containing therapies. Furthermore, VEGFA promotes VEGFR2+ eTreg cell proliferation, and this effect is inhibited by Ramucirumab.23 Thus, targeting VEGFR2 molecules expressed by activated Treg cells or blocking the VEGF‐VEGFR2 axis might contribute to tumor‐shrinking through Treg cell inhibition.
6. CONCLUSIONS
Regulatory T cells have strong immune suppressive activity and inhibit antitumor immune response in tumor‐bearing hosts. High Treg cell infiltration in the TME is involved in unfavorable prognosis in patients with various types of cancer. Depletion of Treg cells and control Treg cell function have been tested in the clinic, but most of these therapies fail to selectively deplete or inhibit Treg cells. One obstacle to overcome is the lack of specific targets for the depletion and functional impairment of Treg cells, particularly tumor‐infiltrating Treg cells. In addition, because systemic depletion of Treg cells could increase a patient's risk of irAE, strategies that can selectively impair Treg cells in the TME are needed. The biology of Treg cells is complicated, but addressing these questions could lead to new therapeutic methods and immune precision medicine for each patient's cancer.
DISCLOSURE
No potential conflict of interest was disclosed by YO. HN has received honoraria and grants from Bristol‐Myers Squibb, Chugai, and Ono and grants from Astellas, BD Japan, Daiichi Sankyo, Kyowa Hakko Kirin, Sysmex, Taiho, Asahikasei, and Zenyaku Kogyo.
Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019;110:2080–2089. 10.1111/cas.14069
REFERENCES
- 1. Carey TE, Takahashi T, Resnick LA, Oettgen HF, Old LJ. Cell surface antigens of human malignant melanoma: mixed hemadsorption assays for humoral immunity to cultured autologous melanoma cells. Proc Natl Acad Sci USA. 1976;73:3278‐3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sakaguchi S. Regulatory T cells: key controllers of immunologic self‐tolerance. Cell. 2000;101:455‐458. [DOI] [PubMed] [Google Scholar]
- 3. Gabrilovich DI. Myeloid‐derived suppressor cells. Cancer Immunol Res. 2017;5:3‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD‐1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887‐3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293‐12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. North RJ, Bursuker I. Generation and decay of the immune response to a progressive fibrosarcoma. I. Ly‐1+ 2‐ suppressor T cells down‐regulate the generation of Ly‐1‐2+ effector T cells. J Exp Med. 1984;159:1295‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishizuka Y, Sakakura T. Thymus and reproduction: sex‐linked dysgenesia of the gonad after neonatal thymectomy in mice. Science. 1969;166:753‐755. [DOI] [PubMed] [Google Scholar]
- 9. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+ CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211‐5218. [PubMed] [Google Scholar]
- 10. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti‐CD25 (interleukin‐2 receptor alpha) monoclonal antibody. Can Res. 1999;59:3128‐3133. [PubMed] [Google Scholar]
- 11. Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged‐helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68‐73. [DOI] [PubMed] [Google Scholar]
- 12. Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20‐21. [DOI] [PubMed] [Google Scholar]
- 13. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self‐tolerance. Nat Immunol. 2002;3:135‐142. [DOI] [PubMed] [Google Scholar]
- 14. Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol. 2013;4:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25‐ naive T cells to CD4+CD25+ regulatory T cells by TGF‐beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875‐1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mucida D, Park Y, Kim G, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256‐260. [DOI] [PubMed] [Google Scholar]
- 17. Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232‐236. [DOI] [PubMed] [Google Scholar]
- 18. Liu W, Putnam AL, Xu‐Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701‐1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899‐911. [DOI] [PubMed] [Google Scholar]
- 20. Tang Q, Adams JY, Tooley AJ, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Investig. 2014;124:2425‐2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ahmadzadeh M, Pasetto A, Jia L, et al. Tumor‐infiltrating human CD4(+) regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol. 2019;4:pii: eaao4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tada Y, Togashi Y, Kotani D, et al. Targeting VEGFR2 with Ramucirumab strongly impacts effector/activated regulatory T cells and CD8(+) T cells in the tumor microenvironment. J Immunother Cancer. 2018;6:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fridman WH, Pages F, Sautes‐Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298‐306. [DOI] [PubMed] [Google Scholar]
- 25. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565‐1570. [DOI] [PubMed] [Google Scholar]
- 26. Saito T, Nishikawa H, Wada H, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22:679‐684. [DOI] [PubMed] [Google Scholar]
- 27. Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol. 2008;9:970‐980. [DOI] [PubMed] [Google Scholar]
- 28. Kurose K, Ohue Y, Sato E, et al. Increase in activated Treg in TIL in lung cancer and in vitro depletion of Treg by ADCC using an antihuman CCR28 mAb (KM2760). J Thorac Oncol. 2015;10:74‐83. [DOI] [PubMed] [Google Scholar]
- 29. Barsheshet Y, Wildbaum G, Levy E, et al. CCR29(+)FOXp3(+) Treg cells as master drivers of immune regulation. Proc Natl Acad Sci USA. 2017;114:6086‐6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eksteen B, Miles A, Curbishley SM, et al. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR30. J Immunol. 2006;177:593‐603. [DOI] [PubMed] [Google Scholar]
- 31. Muller M, Carter SL, Hofer MJ, et al. CXCR31 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol. 2007;179:2774‐2786. [DOI] [PubMed] [Google Scholar]
- 32. Nishikawa H, Kato T, Tanida K, et al. CD4+CD25+ T cells responding to serologically defined autoantigens suppress antitumor immune responses. Proc Natl Acad Sci USA. 2003;100:10902‐10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237‐1240. [DOI] [PubMed] [Google Scholar]
- 34. Dong C, Juedes AE, Temann UA, et al. ICOS co‐stimulatory receptor is essential for T‐cell activation and function. Nature. 2001;409:97‐101. [DOI] [PubMed] [Google Scholar]
- 35. Nagase H, Takeoka T, Urakawa S, et al. ICOS(+) Foxp3(+) TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with Helicobacter pylori. Int J Cancer. 2017;140:686‐695. [DOI] [PubMed] [Google Scholar]
- 36. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell‐extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11:852‐863. [DOI] [PubMed] [Google Scholar]
- 37. Maeda Y, Nishikawa H, Sugiyama D, et al. Detection of self‐reactive CD8(+) T cells with an anergic phenotype in healthy individuals. Science. 2014;346:1536‐1540. [DOI] [PubMed] [Google Scholar]
- 38. Qureshi OS, Zheng Y, Nakamura K, et al. Trans‐endocytosis of CD80 and CD86: a molecular basis for the cell‐extrinsic function of CTLA‐4. Science. 2011;332:600‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)‐2 and induction of autoimmune disease by IL‐2 neutralization. J Exp Med. 2005;201:723‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takahashi T, Kuniyasu Y, Toda M, et al. Immunologic self‐tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10:1969‐1980. [DOI] [PubMed] [Google Scholar]
- 41. Collison LW, Workman CJ, Kuo TT, et al. The inhibitory cytokine IL‐35 contributes to regulatory T‐cell function. Nature. 2007;450:566‐569. [DOI] [PubMed] [Google Scholar]
- 42. Cao X, Cai SF, Fehniger TA, et al. Granzyme B and perforin are important for regulatory T cell‐mediated suppression of tumor clearance. Immunity. 2007;27:635‐646. [DOI] [PubMed] [Google Scholar]
- 43. Camisaschi C, Casati C, Rini F, et al. LAG‐3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol. 2010;184:6545‐6551. [DOI] [PubMed] [Google Scholar]
- 44. Huang CT, Workman CJ, Flies D, et al. Role of LAG‐3 in regulatory T cells. Immunity. 2004;21:503‐513. [DOI] [PubMed] [Google Scholar]
- 45. Kamada T, Togashi Y, Tay C, et al. PD‐1(+) regulatory T cells amplified by PD‐1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci USA. 2019;116:9999‐10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uyttenhove C, Pilotte L, Theate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3‐dioxygenase. Nat Med. 2003;9:1269‐1274. [DOI] [PubMed] [Google Scholar]
- 47. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762‐774. [DOI] [PubMed] [Google Scholar]
- 48. Maj T, Wang W, Crespo J, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD‐L1‐blockade resistance in tumor. Nat Immunol. 2017;18:1332‐1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kurose K, Ohue Y, Wada H, et al. Phase Ia study of FoxP3+ CD4 treg depletion by infusion of a humanized anti‐CCR49 antibody, KW‐0761, in cancer patients. Clin Cancer Res. 2015;21:4327‐4336. [DOI] [PubMed] [Google Scholar]
- 50. Angelin A, Gil‐de‐Gómez L, Dahiya S, et al. Foxp3 reprograms T cell metabolism to function in low‐glucose, high‐lactate environments. Cell Metab. 2017;25:1282‐1293. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sauer S, Bruno L, Hertweck A, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA. 2008;105:7797‐7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miyara M, Chader D, Sage E, et al. Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc Natl Acad Sci USA. 2015;112:7225‐7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Foss F. Clinical experience with denileukin diftitox (ONTAK). Semin Oncol. 2006;33:S11‐S16. [DOI] [PubMed] [Google Scholar]
- 54. Rech AJ, Mick R, Martin S, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4:134ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ribas A. Tumor immunotherapy directed at PD‐1. N Engl J Med. 2012;366:2517‐2519. [DOI] [PubMed] [Google Scholar]
- 56. Bulliard Y, Jolicoeur R, Windman M, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor‐targeting antibodies. J Exp Med. 2013;210:1685‐1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Selby MJ, Engelhardt JJ, Quigley M, et al. Anti‐CTLA‐4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32‐42. [DOI] [PubMed] [Google Scholar]
- 58. Simpson TR, Li F, Montalvo‐Ortiz W, et al. Fc‐dependent depletion of tumor‐infiltrating regulatory T cells co‐defines the efficacy of anti‐CTLA‐4 therapy against melanoma. J Exp Med. 2013;210:1695‐1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jensen SM, Maston LD, Gough MJ, et al. Signaling through OX40 enhances antitumor immunity. Semin Oncol. 2010;37:524‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nishikawa H, Kato T, Hirayama M, et al. Regulatory T cell‐resistant CD8+ T cells induced by glucocorticoid‐induced tumor necrosis factor receptor signaling. Can Res. 2008;68:5948‐5954. [DOI] [PubMed] [Google Scholar]
- 61. Zappasodi R, Sirard C, Li Y, et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019;25:759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Facciabene A, Peng X, Hagemann IS, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226‐230. [DOI] [PubMed] [Google Scholar]
- 63. Sugiyama D, Nishikawa H, Maeda Y, et al. Anti‐CCR1 mAb selectively depletes effector‐type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA. 2013;110:17945‐17950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Plitas G, Konopacki C, Wu K, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45:1122‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336‐344. [DOI] [PubMed] [Google Scholar]
- 66. Shibutani S, Inoue F, Aramaki O, et al. Effects of immunosuppressants on induction of regulatory cells after intratracheal delivery of alloantigen. Transplantation. 2005;79:904‐913. [DOI] [PubMed] [Google Scholar]
- 67. Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. 2018;33:547‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: the role of PD-1 in Treg development and their involvement in autoimmunity onset and cancer progression. Front Immunol. 2018;9:2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD‐1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci USA. 2016;113:8490‐8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Colak S, Ten Dijke P. Targeting TGF‐beta signaling in cancer. Trends Cancer. 2017;3:56‐71. [DOI] [PubMed] [Google Scholar]
- 71. Roubin R, Pizette S, Ollendorff V, Planche J, Birnbaum D, Delapeyriere O. Structure and developmental expression of mouse Garp, a gene encoding a new leucine‐rich repeat‐containing protein. Int J Dev Biol. 1996;40:545‐555. [PubMed] [Google Scholar]
- 72. Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS ONE. 2008;3:e2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yamazaki S, Dudziak D, Heidkamp GF, et al. CD8+CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol. 2008;181:6923‐6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Imagawa J, Tanaka H, Okada M, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol. 2015;2:e528‐e535. [DOI] [PubMed] [Google Scholar]
- 75. Ali K, Soond DR, Pineiro R, et al. Inactivation of PI(3)K p110delta breaks regulatory T‐cell‐mediated immune tolerance to cancer. Nature. 2014;510:407‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Patton DT, Garden OA, Pearce WP, et al. Cutting edge: the phosphoinositide 3‐kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177:6598‐6602. [DOI] [PubMed] [Google Scholar]
- 77. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16:178‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Huynh A, DuPage M, Priyadharshini B, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16:188‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Luo CT, Liao W, Dadi S, Toure A, Li MO. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature. 2016;529:532‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wei J, Long L, Yang K, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. 2016;17:277‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fuhrmann‐Stroissnigg H, Ling YY, Zhao J, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Terme M, Pernot S, Marcheteau E, et al. VEGFA‐VEGFR pathway blockade inhibits tumor‐induced regulatory T‐cell proliferation in colorectal cancer. Can Res. 2013;73:539‐549. [DOI] [PubMed] [Google Scholar]