

Abstract
It has been well established that microRNA (miR)‐143 is downregulated in human bladder cancer (BC). Recent precision medicine has shown that mutations in BC are frequently observed in FGFR3, RAS and PIK3CA genes, all of which correlate with RAS signaling networks. We have previously shown that miR‐143 suppresses cell growth by inhibiting RAS signaling networks in several cancers including BC. In the present study, we showed that synthetic miR‐143 negatively regulated the RNA‐binding protein Musashi‐2 (MSI2) in BC cell lines. MSI2 is an RNA‐binding protein that regulates the stability of certain mRNAs and their translation by binding to the target sequences of the mRNAs. Of note, the present study clarified that MSI2 positively regulated KRAS expression through directly binding to the target sequence of KRAS mRNA and promoting its translation, thus contributing to the maintenance of KRAS expression. Thus, miR‐143 silenced KRAS and MSI2, which further downregulated KRAS expression through perturbation of the MSI2/KRAS cascade.
Keywords: bladder cancer, KRAS, miR‐143, Musashi‐2, RNA‐binding protein
1. INTRODUCTION
Bladder cancer (BC) is the most common malignancy of the urinary tract. According to the American Cancer Society, approximately 79 000 new cases of BC and over 18 000 deaths were estimated to have occurred in the USA alone in 2017.1 Recent precision medicine showed that gene alternations in BC were frequently observed in FGFR3, RAS and PIK3CA,2, 3 all of which are correlated with RAS signaling networks. Among these networks, that of KRAS, in particular, is extremely complicated. Moreover, KRAS regulates more than 10 effector signaling pathways, and its expression is promoted mainly by receptor tyrosine kinases (RTK), including FGFR3.4, 5 Previous studies reported on the networks of KRAS.6, 7 In addition, microRNAs (miR) that directly target KRAS signaling impede KRAS‐driven tumorigenesis.7 Previous studies including ours demonstrated that miR‐143 suppresses KRAS‐mediated tumorigenesis.8, 9, 10 Moreover, miR‐143 is strongly downregulated in several cancers,9, 11, 12, 13, 14 including BC;15, 16 and it inhibits cell proliferation by suppressing both signaling pathways of PI3K/AKT and MAPK, which are downstream of KRAS effector signaling pathways, as well as KRAS in BC.17
The Musashi gene is a consequence of earlier gene duplication, and humans have two related genes, Musashi‐1 (MSI1) and Musashi‐2 (MSI2). MSI1 and MSI2 share approximately 75% amino acid identity in their overall structure and belong to a family of RNA‐binding proteins.18 MSI2 post‐transcriptionally regulates mRNA processing by binding to the recognition motifs located at the 3′UTR of target mRNAs, similar to MSI1. MSI2 preferentially interacts with an ACCUUUUUAGAA motif and other poly‐U sequences,19 UAG motifs, and UAG‐containing motifs ± additional flanking nucleotides.20, 21 The Musashi proteins were first linked to cancer based on studies showing elevated expression of MSI1 in gliomas,22 medulloblastomas,23 and hepatomas.24 MSI2 was identified as part of a translocation event with HoxA9 in chronic myeloid leukemias that preserved MSI2 RNA‐binding motifs,25 also implicating MSI2 in cancer development. The past several years have been marked by a surge of reports elucidating the frequency and mechanisms of involvement of MSI2, in particular, in multiple forms of human cancer,19, 26, 27, 28 including BC.29 Like MSI1, moreover, Dong et al30 reported that MSI2 is directly regulated in a negative way by miR‐143.
In the present study, we clarified the correlation between KRAS and MSI2, both of which are targets of miR‐143. Notably, knockdown of MSI2 induced downregulation of KRAS, and overexpression of MSI2 upregulated KRAS without causing an increase in the level of KRAS mRNA. These results indicated that MSI2 post‐transcriptionally regulated KRAS expression. Furthermore, by using a luciferase reporter assay and surface plasmon resonance (SPR), we demonstrated that MSI2 positively regulated KRAS expression through directly binding to the target sequence UAGUA in the 3′UTR region of KRAS mRNA. Taken together, our findings indicated the extremely potent anticancer activity of synthetic miR‐143 (syn‐miR‐143), and it enabled us to clarify and better understand the role of the novel miR‐143/MSI2/KRAS cascade in human BC.
2. MATERIALS AND METHODS
2.1. RNA immunoprecipitation
RNA immunoprecipitation (RIP) was carried out with a RIP‐assay Kit (Medical & Biological Laboratories Co., Ltd., Aichi, Japan) according to the manufacturer's instructions.
2.2. RNA‐stability measurements
The RNA polymerase II transcriptional inhibitor 5,6‐dichlorobenzimidazole riboside (DRB) was procured from Tokyo Chemical Industry (Tokyo, Japan). T24 cells were seeded on the day prior to transfection with the cDNA plasmid encoding MSI2 or control vector. The cells were treated with DRB at 24 hours after transfection. Cellular RNA was harvested at time 0, 2, 4, 6 and 8 hours and used for qRT‐PCR analysis of KRAS mRNA. RNA half‐lives were calculated from linear regression of log‐transformed expression values.31 ANCOVA was carried out on the resulting regression lines to assess statistical significance.
2.3. Human tumor xenograft model
Animal experimental protocols were approved by the Committee for Animal Research and Welfare of Gifu University (approval no. H30‐42). BALB/cSLC‐nu/nu (nude) mice were obtained from Japan SLC (Shizuoka, Japan). Human bladder cancer T24 cells were inoculated into the back of each mouse. At 7 days after the inoculation, we confirmed engraftment of the tumors. When the tumor size had reached approximately 100 mm3, treatment was started. siRNA or miRNA carried by Lipofectamine RNAi MAX ( Invitrogen, Carlsbad, CA, USA) was injected into the tumor every 2 days for a total of three times. Each group contained three mice. Tumor volume was calculated by the formula: 0.5236 L1 (L2)2, where L1 is the long axis and L2 is the short axis.
Other methods are shown in Data S1.
3. RESULTS
3.1. Impact of KRAS on proliferation of bladder cancer cell lines
To investigate the function of KRAS as an oncogene in human BC, we first assessed the association between cell growth and KRAS and that between it and HRAS in BC cell lines T24 and 253JB‐V. Knockdown of HRAS by use of siRNA significantly suppressed cell proliferation, and knockdown of KRAS resulted in a more potent growth inhibition than that obtained with knockdown of HRAS (Figure 1A). In addition, KRAS effector signaling proteins, AKT and ERK1/2, were downregulated by both knockdowns (Figure 1B). Of note, this knockdown was more prominent in T24 cells, which have an HRAS mutation, not a KRAS one. These results suggested that KRAS contributed considerably to cell proliferation in BC, as did HRAS.
Figure 1.
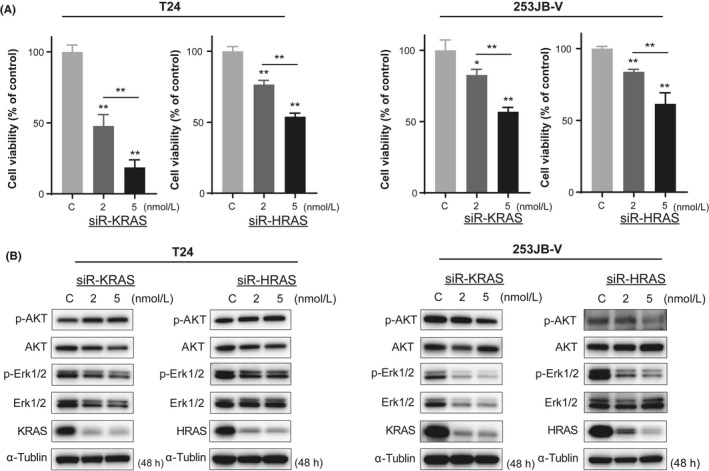
KRAS strongly contributes to cell growth in bladder cancer (BC) cell lines. Cell growth inhibition (A) and protein expression (B) with siR‐KRAS or siR‐HRAS in T24 and 253JB‐V cells. *P < .05; **P < .01. Means + SD indicated by error bars are shown
3.2. Syn‐miR‐143 directly silences the key genes of KRAS networks and MSI2
Previously, we reported that miR‐143 inhibited cell proliferation with apoptosis through silencing PI3K/AKT and MAPK signaling pathways, which are major growth‐related effector signal pathways in KRAS networks in BC.17, 32 As shown in Figure 2A, the expression levels of miR‐143 were extremely downregulated in both T24 and 253JB‐V cells. Recently, we developed a chemically modified miR‐143 that has potent RNase‐resistant anticancer activity (Figure S1). This syn‐miR‐143 silences not only KRAS but also KRAS effector signaling molecules, AKT and ERK.10
Figure 2.
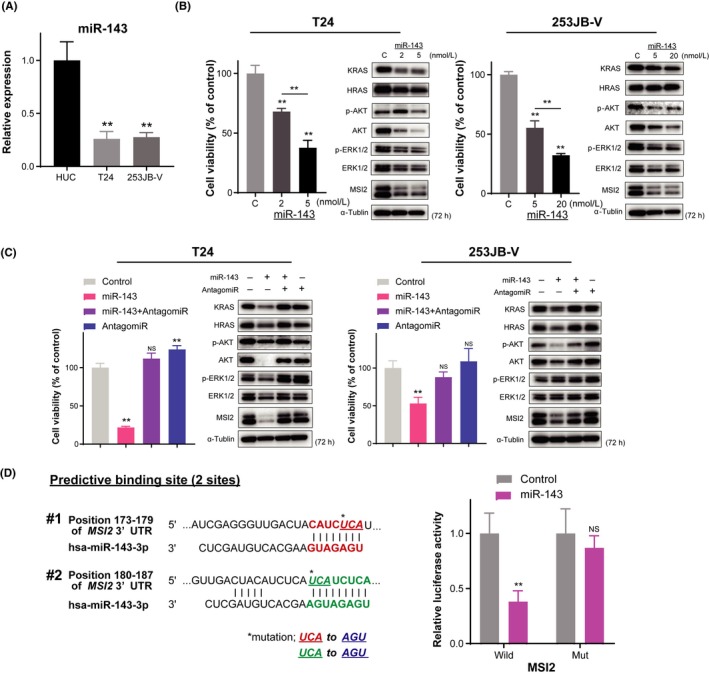
Ectopic expression of microRNA (miR)‐143 induces significant downregulation of KRAS and Musahi‐2 (MSI2) through RNA interference. A, Relative expression levels of miR‐143 in T24 and 253JB‐V cells. B, Dose‐dependent effect of miR‐143 on cell viability and protein expression levels of the target genes. C, Above effects of miR‐143 were verified in cells treated with antagomiR‐143. D, Luciferase activities after cotransfection with control or miR‐143 and wild‐type or mutant‐type pMIR vectors having the predictive miR‐143 binding site in the 3′UTR of MSI2. Left panel shows complementation in the regions of the 3′UTR of MSI2 mRNA (positions 173‐179: #1 and 180‐187: #2) to the mature miR‐143. Colored (red and green) sequences of two sites indicate the predicted binding sites for miR‐143. The nucleotide sequence of the mutated site is shown in blue. **P < .01. Means + SD indicated by error bars are shown. NS, not significant
To clarify how KRAS networks contribute to carcinogenesis and cell growth in BC, we introduced syn‐miR‐143 into T24 cells, which induced apoptosis to a greater extent than that obtained with Ambion miR‐143 (Ambion, Carlsbad, CA, USA), probably as a result of extreme silencing of KRAS networks (Figure S2).10 As shown in Figure 2B, ectopic expression of syn‐miR‐143 led to significant growth inhibition in both cell lines. Western blot analysis indicated that syn‐miR‐143 strongly decreased the expression of KRAS protein and its effector signaling proteins AKT and ERK1/2. Interestingly, RNA‐binding protein MSI2 was also downregulated. MSI2 was recently reported as a target of miR‐143 in cervical cancer30 and has the fourth‐most frequent genetic alterations in BC across almost all major cancers as assessed in The Cancer Genome Atlas (TCGA) BC cohort using the cBioPortal for Cancer Genomics (cBioPortal; Figure S3), and has specific target sequences recognized by miR‐143 according to in silico prediction tools in TargetScan. In addition, treatment with antagomiR‐143 reversed the growth inhibition and lowered protein levels of both KRAS and MSI2 elicited by syn‐miR‐143 (Figure 2C). To examine whether miR‐143 directly bound to the 3′UTR region of MSI2 mRNA, we cloned the 3′UTR of MSI2 mRNA containing the possible miR‐143 binding site in a reporter plasmid. As a result, luciferase activity of wild‐type pMIR‐MSI2 was inhibited after cotransfection with miR‐143 and the reporter plasmid DNA in T24 cells (Figure 2D). In contrast, decrease in luciferase activity was abrogated in the case of mutated binding sites. Together, these results indicated that miR‐143 could silence MSI2 expression at the translation step and inhibit BC cell proliferation, in part, through suppression of MSI2 expression. Hence, next we focused on the oncogenic function of MSI2 and the interaction between MSI2 and KRAS.
3.3. Musashi‐2 is upregulated in clinical tumor samples of BC
We examined the expression of MSI2 in 10 samples from BC patients by western blot analysis. Clinicopathological findings of the patients are shown in Table S1. As shown in Figure 3A, MSI2 was upregulated in six of the 10 clinical BC samples examined compared with its level in normal bladder tissues in the same patients, and expression of miR‐143 was downregulated in all cases according to qRT‐PCR results (Figure 3B). Therefore, all cases of MSI2 overexpression corresponded to downregulation of miR‐143. In addition, qRT‐PCR analysis of MSI2 mRNA showed upregulation in T24 and 253JB‐V cells, and the increase was more prominent in T24 cells than in 253JB‐V cells (Figure 3C). Based on these results, we focused on T24 cells in the following experiments.
Figure 3.
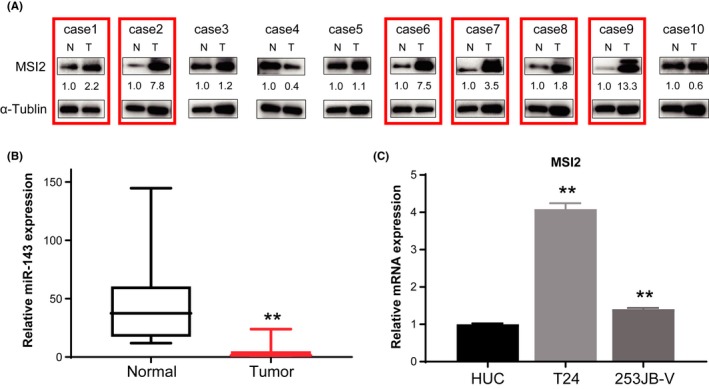
Expression of Musahi‐2 (MSI2) in clinical bladder cancer (BC) samples and cell lines. A, Protein expression levels of MSI2 in 10 BC tumor tissue samples from BC patients. Overexpression of MSI2 in tumor samples is highlighted by red‐colored boxes. N, normal; T, tumor in the same patient. B, Relative expression levels of MSI2 in T24 and 253JB‐V cells. C, Relative mRNA expression level of MSI2 in BC cell lines compared with that in HUC. **P < .01. Means + SD indicated by error bars are shown. HUC, human urothelial cell; miR, microRNA
3.4. Relationship between MSI2 and KRAS or HRAS in T24 cells
To examine the interaction between MSI2 and KRAS, we carried out knockdown and overexpression of MSI2 by using siRNA (Figure S4) and MSI2 expression vector (pF5A‐MSI2), respectively. Knockdown of MSI2 by siR‐MSI2 induced cell growth inhibition along with downregulation of KRAS (Figure 4A). In contrast, overexpression of MSI2 promoted cell proliferation and upregulated KRAS (Figure 4B). Importantly, no significant change in KRAS mRNA levels was observed in the case of either silencing or overexpression of MSI2 (Figure 4A,B). On the contrary, expression levels of HRAS mRNA corresponded to those of MSI2. Taken together, these data suggested that KRAS and HRAS were downstream of MSI2 and that MSI2 may have post‐transcriptionally regulated the transcripts of KRAS and HRAS. Notably, the expression of MSI2 was also affected by KRAS or HRAS, because either knockdown of KRAS or HRAS caused a decrease in the expression of MSI2 protein (Figure 4C). Also, silencing of either KRAS or HRAS caused the expression of HRAS or KRAS, respectively, to decrease. These results indicated that MSI2 and KRAS or HRAS are coordinated with each other, although it is difficult to clarify how MSI2 interacts with KRAS and HRAS, given the complicated nature of RAS signaling networks.
Figure 4.
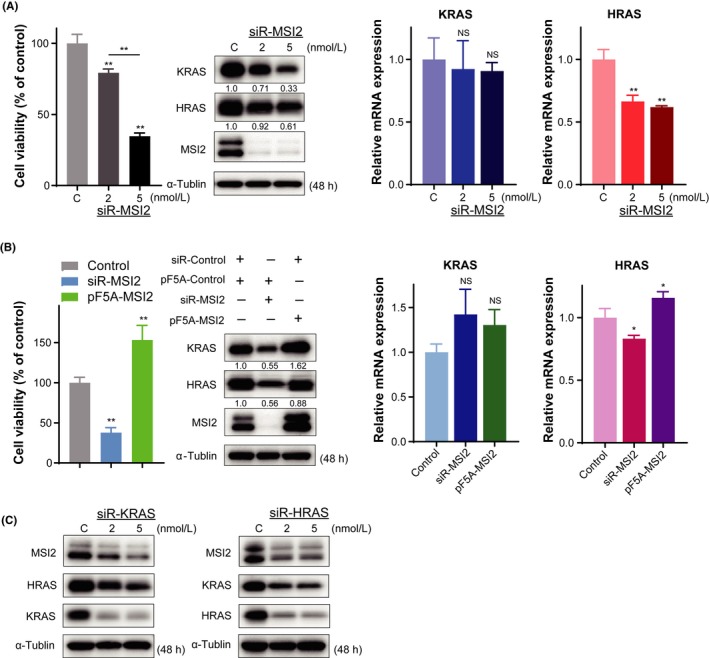
Relationship between Musahi‐2 (MSI2) and KRAS or HRAS in expression profiles of T24 cells. A, Effects of MSI2 knockdown using siRNA on cell growth (left panel). Protein and mRNA expression levels of KRAS and HRAS after siR‐MSI2 transfection (middle and right panels). B, Effects of MSI2 knockdown and overexpression on cell growth (left panel). Protein and mRNA expression levels of KRAS and HRAS in MSI2‐silenced and ‐overexpressed cells (middle and right panels). C, MSI2 and RAS protein profiles after transfection with siR‐KRAS or siR‐HRAS. *P < .05; **P <0.01. Means + SD indicated by error bars are shown. NS, not significant
3.5. Musashi‐2 directly binds to mRNA of KRAS
To clarify whether or not MSI2 bound directly to mRNAs of KRAS and HRAS, using BC cells, we first carried out MSI2‐immunoprecipitation (IP) followed by qRT‐PCR. Western blot analysis, carried out as a quality check, showed that MSI2 was detected in input and MSI2‐IP samples only, not in the control IgG‐IP sample. qRT‐PCR findings showed that the MSI2‐immunoprecipitated RNA fraction was significantly enriched in KRAS mRNA (Figure 5A), whereas there was no enrichment of HRAS mRNA in IgG or MSI2 fractions. These data suggested that MSI2 directly bound to KRAS but not to HRAS. Therefore, the data on HRAS observed in Figure 4A,B were supposedly as a result of an indirect impact of MSI2.
Figure 5.
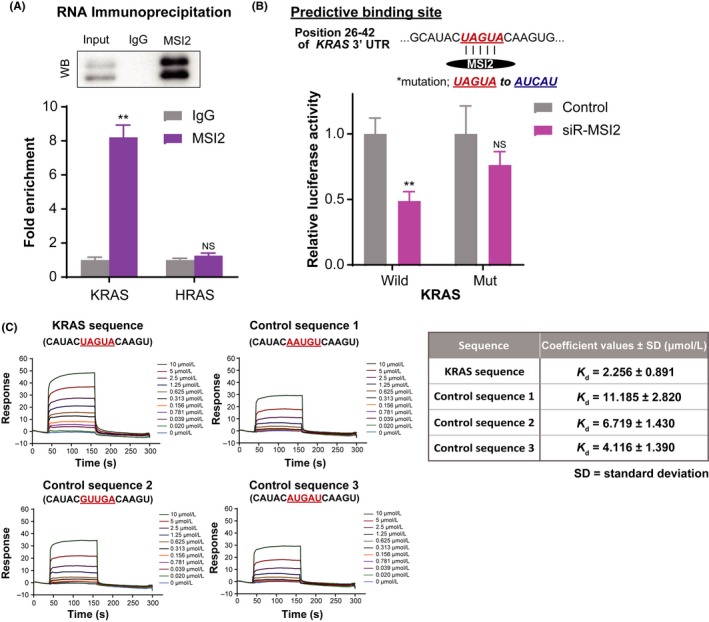
Musashi‐2 (MSI2) directly binds to UAGUA in 3′UTR of KRAS mRNA. A, RNA‐immunoprecipitation of MSI2 complexes with anti‐MSI2 or control antirabbit IgG antibody, followed by qRT‐PCR of KRAS and HRAS. Relative enrichment was calculated by enrichment over the control. Western blot analysis as a quality check of immunoprecipitated MSI2 is also shown. B, Luciferase activities after cotransfection with control or siR‐MSi2 and wild‐type or mutant‐type pMIR vectors having the predictive MSI2 binding site in the 3′UTR of KRAS. Upper panel shows the position 26‐42 of the 3′UTR of KRAS mRNA. Colored sequences indicate the predicted binding site for MSI2. Site of the mutated sequence is shown in blue. **P <0.01. Means + SD indicated by error bars are shown. NS, not significant. C, Representative surface plasmon resonance (SPR) sensorgrams for binding of MSI2 to synthetic RNAs containing KRAS mRNA (UAGUA) or its scrambled sequences (AAUGU, GUUGA, and AUGAU) (left panel). Dissociation constants between MSI2 protein and RNAs shown in the left panel. Average ± SD from three experiments (right panel)
In addition, to determine the direct interaction between MSI2 and KRAS mRNA, we cloned the predicted MSI2 binding site UAGUA in the 3′UTR region of KRAS mRNA in a reporter plasmid vector (Figure 5B). Results of the luciferase reporter assay indicated that activity of wild‐type pMIR‐KRAS was decreased by siR‐MSI2 compared with that obtained with control siRNA, indicating that the activity paralleled the level of MSI2 protein expression. On the contrary, decrease in the activity of the pMIR vector was almost canceled when the mutated MSI2 binding site AUCAU was used. Thus, these data clearly showed the promoting roles of MSI2 in the translation step of luciferase mRNAs.
To examine direct interaction between MSI2 and UAGUA, we next used the SPR assay. To this end, a recombinant MSI2 protein containing the two RNA‐binding domains was expressed (Figure S5) and immobilized on a sensor‐chip surface. A synthetic 15‐mer KRAS mRNA containing UAGUA or its scrambled sequence as a control was injected over the sensor chip. As shown in the left panel of Figure 5C, the UAGUA sequence gave the highest binding response to the immobilized MSI2 (reaching 50 resonance units at 10 μmol/L). In addition, a dissociation constant (Kd) of UAGUA for MSI2 was 2‐5‐fold lower than that for the control RNA (right panel of Figure 5C), suggesting that MSI2 preferentially bound to the UAGUA sequence.
Collectively, these data showed that MSI2 directly interacted with KRAS mRNA by recognizing and binding to one of the specific UAGUA sequences.
3.6. Musashi‐2 post‐transcriptionally enhances translation of KRAS
To investigate how MSI2 regulates the processing of KRAS mRNAs after transcription, we first examined the localization of MSI2 in cells by using immunofluorescence. MSI2 was located mainly in the cytoplasm in T24 and 253JB‐V cells (Figure 6A) in agreement with the reports in a public database (The Human Protein Atlas).
Figure 6.
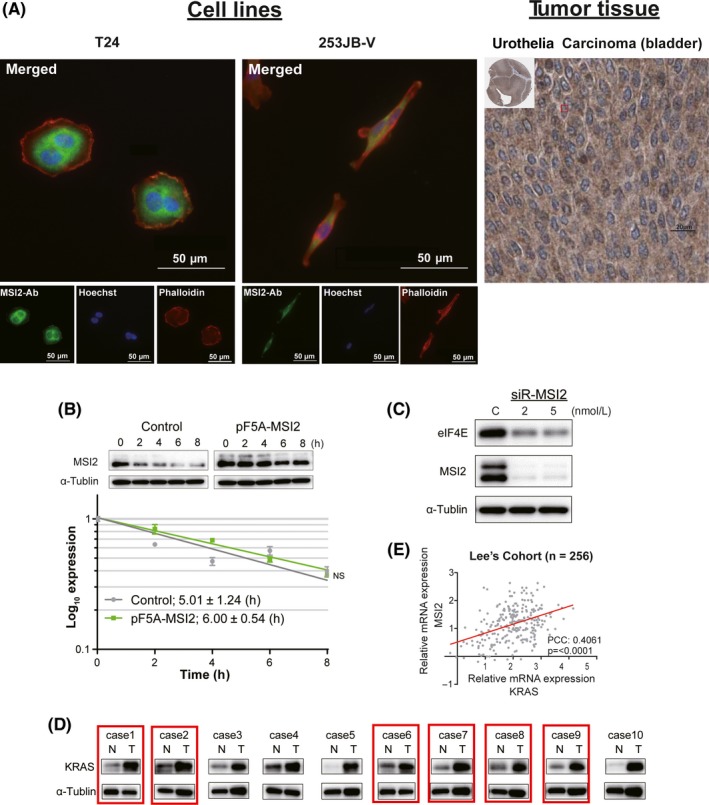
Cell localization of Musashi‐2 (MSI2) and functions against KRAS mRNA. A, Immunofluorescence of MSI2 (green), a nucleic marker, Hoechst 33342 (blue), and an actin marker, Phalloidin (red) in T24 and 253JB‐V cells (left panel). Immunohistochemistry images as obtained from the Human Protein Atlas also show MSI2 in bladder carcinoma (BC) tissue (right panel). B, KRAS mRNA stability curves were plotted as qRT‐PCR expression with time. ANCOVA was used for determining statistical significance. Mean standard error is indicated for each time point. Half‐life in hours was calculated from the stability curves. NS, not significant. C, Protein expression of translational initiator eIF4E in MSI2‐silenced cells. D, Protein expression levels of MSI2 and KRAS in 10 tumor tissue samples from BC patients. Cases with co‐upregulated MSI2 and KRAS in the tumor samples are highlighted by red boxes. The samples are the same as in Figure 3A. N, normal; T, tumor in the same patient. E, Correlation of mRNA expression levels between MSI2 and KRAS in human BC samples from Lee's cohort (n = 256) estimated by bioinformatics. PCC, Pearson's correlation coefficient
Cellular localization of MSI2 and the findings in the current study suggest that MSI2 might have the ability to regulate the stability or translation of target mRNAs.33 To determine whether MSI2 could regulate the stability of KRAS mRNA, we estimated the rate of mRNA decay after treatment with DRB. Time‐course RNA decay curves for KRAS mRNA were prepared from qRT‐PCR data after DRB treatment of cells transfected with pF5A‐control or pF5A‐MSI2. As a result, the half‐life of KRAS mRNA was not significantly changed in either case, whereas overexpression of MSI2 was achieved in the case of pF5A‐MSI2 transfection (Figure 6B). These data thus showed that MSI2 functioned to enhance the translation of KRAS mRNA rather than to stabilize the mRNA, the finding of which is well supported by the results given in Figure 4A,B. To further validate that MSI2 regulated the translation, we assessed the expression of translational initiator eIF4E by western blot analysis. Notably, knockdown of MSI2 induced the downregulation of eIF4E (Figure 6C). These data suggested that MSI2 played a role in enhancing translation. Furthermore, western blot analysis showed that MSI2 and KRAS were co‐upregulated in six cases of 10 clinical BC samples compared with their expression in normal bladder tissues (Figure 6D). In Lee's cohort,34 there was a significant positive correlation of mRNA expression levels between MSI2 and KRAS in human BC (Figure 6E). These data suggested that the patients, which had abundant MSI2 mRNA, could have increment of KRAS protein expression through efficient translation by MSI2. Furthermore, we showed that the silencing effect of KRAS by ectopic expression with syn‐miR‐143 was certainly canceled by overexpression of MSI2 (Figure S6). This finding suggested that downregulated expression of MSI2 by miR‐143 was significant in the growth of BC cells.
Collectively, these data indicated that MSI2 functioned to accelerate the translation of KRAS mRNA in the cytoplasm and had crucial roles as a KRAS enhancer in BC cells. This machinery was closely correlated with KRAS networks, in which there was a positive circuit for enhancement of KRAS mRNA expression by KRAS effector signaling (AKT and ERK)10 (Figures 7, S7 and S8).
Figure 7.
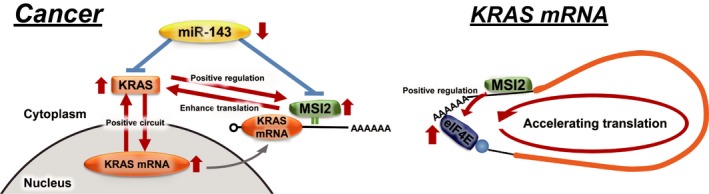
Schematic diagrams showing the roles of Musashi‐2 (MSI2) in KRAS networks and the association of MSI2 with KRAS mRNA. MicroRNA (miR)‐143/MSI2/KRAS cascade (left panel) and possible machinery for MSI2‐mediated enhancement of the translation of KRAS mRNA (right panel)
3.7. MicroRNA‐143/MSI2/KRAS cascade on T24 cell‐xenografted tumors in nude mice
We have clarified the novel miR‐143/MSI2/KRAS cascade in vitro. To further validate the cascade between miR‐143, MSI2 and KRAS, we examined the antitumor effect by using syn‐miR‐143 and siR‐MSI2 in vivo. As shown in Figure 8A, growth suppression of tumors was observed in the groups treated with siR‐MSI2 or syn‐miR‐143. In addition, the tumor‐suppressive effect of syn‐miR‐143 was greater than that of siR‐MSI2. Western blot analysis of the tissue samples from grafted tumors showed that MSI2 was significantly silenced in both treated groups, Furthermore, decreased expression of KRAS protein was also shown in both groups as it had been found in vitro (Figures 2B and 4A), and the KRAS‐silencing effect of syn‐miR‐143 was greater than that of siR‐MSI2 (Figure 8B). These findings suggested that MSI2 contributed to the growth of the engrafted tumor through upregulation of KRAS, and that the miR‐143/MSI2/KRAS cascade could exist even in an in vivo experiment.
Figure 8.
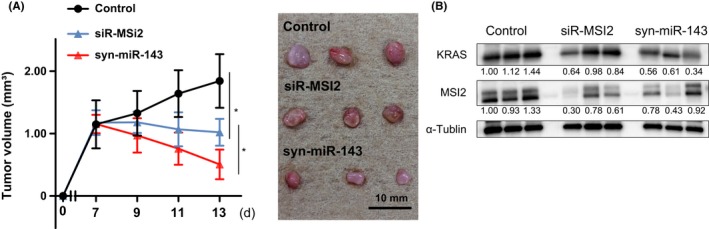
Antitumor activity of siR‐MSI2 and syn‐miR‐143 in T24 cell‐xenografted mice. A, Changes in tumor size of mice treated with control siRNA, siR‐MSI2 or syn‐miR‐143 (n = 3; left panel). (Right panel) Representative photograph of tumors. Upper, middle and lower photos show the tumors from control and treated mice, respectively. B, Protein expression levels of MSI2 and KRAS in control, siR‐MSI2 or syn‐miR‐143‐treated tumor tissues. *P < .05. Means ± SD indicated by error bars are shown. miR, microRNA; MSI2, Musashi‐2
4. DISCUSSION
In the current study, we clarified a novel network operating miR‐143, MSI2 and KRAS (Figure 7). We were also able to show that the expression of KRAS was affected by MSI2 through binding of the latter to KRAS mRNA, the finding of which was validated by RNA‐IP, SPR, and the expression profiles of the genes involved. Previously, we showed that miR‐143 directly silences KRAS signaling networks;10, 17 and Dong et al30 reported that miR‐143 also targets RNA‐binding protein MSI2. However, the association between these targets of miR‐143, KRAS and MSI2 had not been previously reported. Given the earlier reports that MSI2 has been suggested to interact preferentially with the UAG‐containing motifs in the 3′UTR region of its target RNAs,21, 35, 36, 37 we predicted the binding site in KRAS mRNA to be UAGUA and showed by a using luciferase reporter assay and the SPR technique (Figure 5B,C) that MSI2 protein preferentially binds to the sequence. In addition, it was earlier reported that MSI2 has functions to affect the stabilization or translation of its target mRNAs.21, 30, 38 Based on our results, MSI2 did not impact the stability of KRAS mRNA despite its direct binding to it (Figure 6B). Given that MSI2 positively regulated the translational initiator eIF4E (Figure 6C), we propose that MSI2 functioned to enhance the translation of KRAS mRNA rather than its stabilization. With regard to the role of translational regulation, MSI2 is regulated by site‐specific phosphorylation, which converts MSI2 from a repressor to an activator of target mRNA translation, and MAPK and Ringo/CDK contribute to MSI2 regulatory phosphorylation, as does MSI1.39, 40 MAPK also contributes to the translational machinery including eIF4E.41 MSI2 may be included in the cascades, and MAPK positively regulates eIF4E through phosphorylation of MSI2. In addition, given the RAS signaling pathways, KRAS and HRAS could regulate MSI2 through MAPK indirectly. Indeed, it is reasonable that MSI2 was downregulated in cells by knockdown of KRAS and HRAS, in which ERK1/2 was also inhibited (Figures 1B and 4C).
We demonstrated an association between the specific sequence UAGUA and the ability of MSI2 to enhance the translation of its target mRNA. However, the SPR technique showed that the control sequences, despite the absence of a UAG motif, gave a weak binding response to MSI2 protein (Figure 5C). These data suggested that the specificity of MSI2 binding to transcripts may not be so high. The impact of MSI2 on its target RNAs could be due not only to binding ability, but also to other mechanisms. The two RNA recognition motif (RRM) of MSI2 are possibly involved in the mechanism. Biochemical and structural studies have suggested that RRM1 contributes the majority of the binding energy and specificity, whereas RRM2 has a more supportive role.37 In addition, Bennett et al21 reported that these two RRM may provide a mechanism for MSI2 to distinguish its veritable targets. However, this machinery is presently barely understood and further validation is warranted.
In the present study, we clarified that MSI2 directly targeted KRAS, promoting translation of its mRNA. The RTK/RAS pathway has been reported to be involved in the regulation of cell proliferation in several cancers.42, 43, 44, 45 Recent precision medicine studies showed that gene alterations in RTK/RAS pathways occurred in up to 60% of BC patients.46 Among these signaling pathways, in particular, up to 80% of non‐muscle invasive BC (NMIBC) harbor activating point mutations in FGFR3,47, 48 which activate the RAS/MAPK pathway.4, 5, 49 Moreover, the alteration of KRAS occurs more frequently than that of HRAS.2, 3 Thus, KRAS and KRAS signaling networks are dominant pathways in BC. Previously, we have clarified the signaling networks. The “positive circuit” through the constitutive KRAS activation‐stimulation of effector signaling pathways (PI3K/AKT and MAPK) occurs in colorectal cancer, resulting in enhanced nuclear KRAS transcription.10 As shown in Figure S7, this cascade was also seen in BC cell lines and, again, the “positive circuit” also occurred in HRAS signaling networks. These data suggested that KRAS and HRAS interacted with each other, indicating that either MSI2 or miR‐143 indirectly affected the expression of HRAS by regulating KRAS. Indeed, inhibition of the signals of ERK and AKT occurred, resulting in indirect regulation of HRAS expression in the case of treatment with siR‐MSI2 or syn‐miR‐143 (Figures 2B and S8).
Recently, it was reported that miR‐143 has a significant antitumor role in BC. Lin et al15 reported that transfection of BC cells with miR‐143 significantly inhibited cell proliferation through decreased expression of RAS protein. Wang et al50 showed that overexpression of miR‐143 inhibited cell proliferation in BC. Furthermore, we demonstrated previously that syn‐miR‐143 functions as a tumor suppressor in BC cells.17, 32 In an earlier study, we also showed that miR‐143 directly targets KRAS signaling networks.10 Furthermore, it was reported that miR‐143 also directly targets MSI2 in cervical cancer.30 In the present study, we clarified the novel association between MSI2 and KRAS, both of which are targets of miR‐143, validating the interaction between MSI2 and the UAGUA sequence of the KRAS transcript in vitro (Figures 4 and 5, S6). Previously, genome‐wide analyses demonstrated that MSI2 binds to a multitude of target genes.37 Fox et al51 validated targets C‐MET, BRD4, and HMGA2 in pancreatic cancer, and Park et al52 validated 48 genes in hematopoietic stem cells and 11 genes in leukemia stem cells.38 Thus, as MSI2 binds to a great number of targets, KRAS cannot be listed at the top in genome‐wide analysis of MSI2‐ribonucleoproteins. Syn‐miR‐143 has allowed us to propose the possibility of association between the two proteins, resulting in a better understanding of the novel miR‐143/MSI2/KRAS expression system (Figure 7).
Collectively, we showed that miR‐143 directly impacted MSI2 expression through its RNAi action, which also effectively inhibited KRAS networks as a novel mechanism in human BC. Moreover, this evidence was confirmed by the results of an in vivo experiment (Figure 8). Taken together, these findings indicated the complicated nature of KRAS networks and the tight control of their maintenance.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was carried out with the support of SHIONOGI. This work was supported by the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) from Japan Agency for Medical Research and Development (AMED) (16cm0106202 h0001 to YA).
Tsujino T, Sugito N, Taniguchi K, et al. MicroRNA‐143/Musashi‐2/KRAS cascade contributes positively to carcinogenesis in human bladder cancer. Cancer Sci. 2019;110:2189–2199. 10.1111/cas.14035
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Robertson AG, Kim J, Al‐Ahmadie H, et al. Comprehensive molecular characterization of muscle‐invasive bladder cancer. Cell. 2017;171:540‐556. e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Springer SU, Chen CH, Rodriguez Pena MDC, et al. Non‐invasive detection of urothelial cancer through the analysis of driver gene mutations and aneuploidy. Elife. 2018;7:e32143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oliveras‐Ferraros C, Cufi S, Queralt B, et al. Cross‐suppression of EGFR ligands amphiregulin and epiregulin and de‐repression of FGFR3 signalling contribute to cetuximab resistance in wild‐type KRAS tumour cells. Br J Cancer. 2012;106:1406‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Touat M, Ileana E, Postel‐Vinay S, Andre F, Soria JC. Targeting FGFR signaling in cancer. Clin Cancer Res. 2015;21:2684‐2694. [DOI] [PubMed] [Google Scholar]
- 6. Tsai FD, Lopes MS, Zhou M, et al. K‐Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane‐targeting motif. Proc Natl Acad Sci USA. 2015;112:779‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jinesh GG, Sambandam V, Vijayaraghavan S, Balaji K, Mukherjee S. Molecular genetics and cellular events of K‐Ras‐driven tumorigenesis. Oncogene. 2018;37:839‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen X, Guo X, Zhang H, et al. Role of miR‐143 targeting KRAS in colorectal tumorigenesis. Oncogene. 2009;28:1385‐1392. [DOI] [PubMed] [Google Scholar]
- 9. Akao Y, Nakagawa Y, Iio A, Naoe T. Role of microRNA‐143 in Fas‐mediated apoptosis in human T‐cell leukemia Jurkat cells. Leuk Res. 2009;33:1530‐1538. [DOI] [PubMed] [Google Scholar]
- 10. Akao Y, Kumazaki M, Shinohara H, et al. Impairment of K‐Ras signaling networks and increased efficacy of epidermal growth factor receptor inhibitors by a novel synthetic miR‐143. Cancer Sci. 2018;109:1455‐1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akao Y, Nakagawa Y, Naoe T. MicroRNAs 143 and 145 are possible common onco‐microRNAs in human cancers. Oncol Rep. 2006;16:845‐850. [PubMed] [Google Scholar]
- 12. Akao Y, Nakagawa Y, Kitade Y, Kinoshita T, Naoe T. Downregulation of microRNAs‐143 and ‐145 in B‐cell malignancies. Cancer Sci. 2007;98:1914‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akao Y, Nakagawa Y, Hirata I, et al. Role of anti‐oncomirs miR‐143 and ‐145 in human colorectal tumors. Cancer Gene Ther. 2010;17:398‐408. [DOI] [PubMed] [Google Scholar]
- 14. Takagi T, Iio A, Nakagawa Y, Naoe T, Tanigawa N, Akao Y. Decreased expression of microRNA‐143 and ‐145 in human gastric cancers. Oncology. 2009;77:12‐21. [DOI] [PubMed] [Google Scholar]
- 15. Lin T, Dong W, Huang J, et al. MicroRNA‐143 as a tumor suppressor for bladder cancer. J Urol. 2009;181:1372‐1380. [DOI] [PubMed] [Google Scholar]
- 16. Zhou H, Tang K, Xiao H, et al. A panel of eight‐miRNA signature as a potential biomarker for predicting survival in bladder cancer. J Exp Clin Cancer Res. 2015;34:53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noguchi S, Yasui Y, Iwasaki J, et al. Replacement treatment with microRNA‐143 and ‐145 induces synergistic inhibition of the growth of human bladder cancer cells by regulating PI3K/Akt and MAPK signaling pathways. Cancer Lett. 2013;328:353‐361. [DOI] [PubMed] [Google Scholar]
- 18. Sakakibara S, Nakamura Y, Satoh H, Okano H. Rna‐binding protein Musashi2: developmentally regulated expression in neural precursor cells and subpopulations of neurons in mammalian CNS. J Neurosci. 2001;21:8091‐8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang S, Li N, Yousefi M, et al. Transformation of the intestinal epithelium by the MSI2 RNA‐binding protein. Nat Commun. 2015;6:6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zearfoss NR, Deveau LM, Clingman CC, et al. A conserved three‐nucleotide core motif defines Musashi RNA binding specificity. J Biol Chem. 2014;289:35530‐35541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bennett CG, Riemondy K, Chapnick DA, et al. Genome‐wide analysis of Musashi‐2 targets reveals novel functions in governing epithelial cell migration. Nucleic Acids Res. 2016;44:3788‐3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kanemura Y, Mori K, Sakakibara S, et al. Musashi1, an evolutionarily conserved neural RNA‐binding protein, is a versatile marker of human glioma cells in determining their cellular origin, malignancy, and proliferative activity. Differentiation. 2001;68:141‐152. [DOI] [PubMed] [Google Scholar]
- 23. Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178‐15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shu HJ, Saito T, Watanabe H, et al. Expression of the Musashi1 gene encoding the RNA‐binding protein in human hepatoma cell lines. Biochem Biophys Res Comm. 2002;293:150‐154. [DOI] [PubMed] [Google Scholar]
- 25. Barbouti A, Hoglund M, Johansson B, et al. A novel gene, MSI2, encoding a putative RNA‐binding protein is recurrently rearranged at disease progression of chronic myeloid leukemia and forms a fusion gene with HOXA9 as a result of the cryptic t(7;17)(p15;q23). Can Res. 2003;63:1202‐1206. [PubMed] [Google Scholar]
- 26. Kharas MG, Lengner CJ, Al‐Shahrour F, et al. Musashi‐2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. 2010;16:903‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kudinov AE, Deneka A, Nikonova AS, et al. Musashi‐2 (MSI2) supports TGF‐beta signaling and inhibits claudins to promote non‐small cell lung cancer (NSCLC) metastasis. Proc Natl Acad Sci USA. 2016;113:6955‐6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo K, Cui J, Quan M, et al. The novel KLF4/MSI2 signaling pathway regulates growth and metastasis of pancreatic cancer. Clin Cancer Res. 2017;23:687‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang C, Zhang W, Wang L, et al. Musashi‐2 promotes migration and invasion in bladder cancer via activation of the JAK2/STAT3 pathway. Lab Invest. 2016;96:950‐958. [DOI] [PubMed] [Google Scholar]
- 30. Dong P, Xiong Y, Hanley SJB, Yue J, Watari H. Musashi‐2, a novel oncoprotein promoting cervical cancer cell growth and invasion, is negatively regulated by p53‐induced miR‐143 and miR‐107 activation. J Exp Clin Cancer Res. 2017;36:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen CY, Ezzeddine N, Shyu AB. Messenger RNA half‐life measurements in mammalian cells. Methods Enzymol. 2008;448:335‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noguchi S, Mori T, Hoshino Y, et al. MicroRNA‐143 functions as a tumor suppressor in human bladder cancer T24 cells. Cancer Lett. 2011;307:211‐220. [DOI] [PubMed] [Google Scholar]
- 33. Gerstberger S, Hafner M, Tuschl T. A census of human RNA‐binding proteins. Nat Rev Genet. 2014;15:829‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee J‐S, Leem S‐H, Lee S‐Y, et al. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J Clin Oncol. 2010;28:2660‐2667. [DOI] [PubMed] [Google Scholar]
- 35. Katz Y, Li F, Lambert NJ, et al. Musashi proteins are post‐transcriptional regulators of the epithelial‐luminal cell state. eLife. 2014;3:e03915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lan L, Xing M, Douglas JT, Gao P, Hanzlik RP, Xu L. Human oncoprotein Musashi‐2 N‐terminal RNA recognition motif backbone assignment and identification of RNA‐binding pocket. Oncotarget. 2017;8:106587‐106597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kudinov AE, Karanicolas J, Golemis EA, Boumber Y. Musashi RNA‐binding proteins as cancer drivers and novel therapeutic targets. Clin Cancer Res. 2017;23:2143‐2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park SM, Gonen M, Vu L, et al. Musashi2 sustains the mixed‐lineage leukemia‐driven stem cell regulatory program. J Clin Invest. 2015;125:1286‐1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arumugam K, MacNicol MC, Wang Y, et al. Ringo/cyclin‐dependent kinase and mitogen‐activated protein kinase signaling pathways regulate the activity of the cell fate determinant Musashi to promote cell cycle re‐entry in Xenopus oocytes. J Biol Chem. 2012;287:10639‐10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. MacNicol MC, Cragle CE, McDaniel FK, et al. Evasion of regulatory phosphorylation by an alternatively spliced isoform of Musashi2. Sci Rep. 2017;7:11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roux PP, Topisirovic I. Signaling pathways involved in the regulation of mRNA translation. Mol Cell Biol. 2018;38:e00070-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111:817‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the Wnt/beta‐catenin pathway through LRP6 in colorectal cancer. Oncogene. 2015;34:4914‐4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wong GS, Zhou J, Liu JB, et al. Targeting wild‐type KRAS‐amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. 2018;24:968‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Regad T, Targeting RTK. Signaling pathways in cancer. Cancers. 2015;7:1758‐1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sanchez‐Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome Atlas. Cell. 2018;173:321‐337. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15:25‐41. [DOI] [PubMed] [Google Scholar]
- 48. Inamura K. Bladder cancer: new insights into its molecular pathology. Cancers. 2018;10:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yadav V, Zhang X, Liu J, et al. Reactivation of mitogen‐activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B‐RAF V600E mutant melanoma. J Biol Chem. 2012;287:28087‐28098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang H, Li Q, Niu X, et al. miR‐143 inhibits bladder cancer cell proliferation and enhances their sensitivity to gemcitabine by repressing IGF‐1R signaling. Oncol Lett. 2017;13:435‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fox RG, Lytle NK, Jaquish DV, et al. Image‐based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature. 2016;534:407‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Park SM, Deering RP, Lu Y, et al. Musashi‐2 controls cell fate, lineage bias, and TGF‐beta signaling in HSCs. J Exp Med. 2014;211:71‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials