

Abstract
Vasohibin‐2 (VASH2) is expressed in various cancers and promotes their progression. We recently reported that pancreatic cancer patients with higher VASH2 expression show poorer prognosis. Herein, we sought to characterize the role of VASH2 in pancreatic cancer. We used LSL‐Kras G12D ; LSL‐Trp53 R172H ; Pdx‐1‐Cre (KPC) mice, a mouse model of pancreatic ductal adenocarcinoma (PDAC), and cells isolated from them (KPC cells). Knockdown of Vash2 from PDAC cells did not affect their proliferation, but decreased their migration. When Vash2‐knockdown PDAC cells were orthotopically inoculated, liver metastasis and peritoneal dissemination were reduced, and the survival period was significantly prolonged. When KPC mice were crossed with Vash2‐deficient mice, metastasis was significantly decreased in Vash2‐deficient KPC mice. VASH2 was recently identified to have tubulin carboxypeptidase activity. VASH2 knockdown decreased, whereas VASH2 overexpression increased tubulin detyrosination of PDAC cells, and tubulin carboxypeptidase (TCP) inhibitor parthenolide inhibited VASH2‐induced cell migration. We next clarified its role in the tumor microenvironment. Tumor angiogenesis was significantly abrogated in vivo when VASH2 was knocked down or deleted. We further examined genes downregulated by Vash2 knockdown in KPC cells, and found chemokines and cytokines that were responsible for the recruitment of myeloid derived suppressor cells (MDSC). Indeed, MDSC were accumulated in PDAC of KPC mice, and they were significantly decreased in Vash2‐deficient KPC mice. These findings suggest that VASH2 plays an essential role in the metastasis of PDAC with multiple effects on both cancer cells and the tumor microenvironment, including tubulin detyrosination, tumor angiogenesis and evasion of tumor immunity.
Keywords: angiogenesis, MDSC, pancreatic ductal adenocarcinoma, tubulin detyrosination, vasohibin‐2
Abbreviations
- CAS
cancer‐associated fibroblasts
- EMT
epithelial‐to‐mesenchymal transition
- GM‐CSF
granulocyte‐macrophage colony‐stimulating factor
- G‐MDSC
granulocytic myeloid‐derived suppressor cells
- IHC
immunohistochemistry
- MDSC
myeloid derived suppressor cells
- NF‐κB
nuclear factor kappa B
- PDAC
pancreatic ductal adenocarcinoma
- TAM
tumor associated macrophages
- TCP
tubulin carboxypeptidase
- VASH
vasohibin
1. INTRODUCTION
Pancreatic cancer is an extremely malignant cancer, with a 5‐year survival rate of <5%. Although surgical resection is the only possible curative treatment, this cancer is often advanced and metastasized when diagnosed.1 Standard chemotherapies for pancreatic cancer provide only slightly increased survival periods owing to high resistance and aggressiveness that has been associated with the tumor microenvironment such as collagen deposition, high number of activated CAF and increased inflammatory cell infiltration.2, 3, 4, 5
We have isolated VASH1 as an endothelium‐derived angiogenesis inhibitor,6 and VASH2 as its homolog.7 Although VASH1 and VASH2 have no classical signal sequence for their secretion, they are efficiently secreted through binding with another newly discovered protein, SVBP (small vasohibin binding protein).8 The VASH gene is highly conserved between species. Lower organisms possess a single ancestral VASH gene, which has evolved into VASH1 and VASH2 genes in vertebrates. Whereas VASH1 is expressed in endothelial cells, VASH2, which is more similar to the ancestral VASH, is not expressed in normal tissues except in the testis after birth.9 However, VASH2 is overexpressed in various cancer cells, and promotes tumor angiogenesis, accumulation of CAF and EMT of cancer cells.10, 11, 12, 13, 14 VASH2 knockdown or blockade in cancer cells including human ovarian cancer and hepatocellular carcinoma showed significant anticancer effects,10, 11, 15, 16 and VASH2 has consequently been identified as a potential molecular target for anticancer therapy.
Microtubules (MT) are a major component of cytoskeletons, and show important functions ranging from intracellular transport to cell motility and cell division. MT are composed of repeating α‐ and β‐tubulin heterodimers, and they are subjected to a number of post‐translational modifications including the tyrosination‐detyrosination cycle. Tubulin tyrosination is mediated by tubulin tyrosine ligase whereas its detyrosination is mediated by TCP. One such modification, namely tubulin detyrosination, is reported to be accumulated in cancer cells during tumor development and is correlated with histological grade in breast cancer.17, 18 VASH have recently been identified to have TCP activity, which catalyzes the C‐terminus tyrosine residue of α‐tubulin.19, 20 Thus, VASH2 may influence cancer cell behavior by augmenting tubulin detyrosination.
We recently showed VASH2 expression in KPC cell lines and that its expression correlates with poor prognosis in PDAC patients.21 Furthermore, VASH2 amplification occurs frequently in primary PDAC (10%).22 Herein, we used LSL‐Kras G12D/+ ; LSL‐Trp53 R172H/+ ; Pdx‐1‐Cre (KPC) mice, a mouse model of PDAC, and a KPC‐derived pancreatic cancer cell line to examine the role of VASH2 in pancreatic cancer. We further crossed KPC and Vash2‐deficient mice, and examined the role of VASH2 in pancreatic cancer progression. The present study indicates that VASH2 plays an important role in the metastasis of pancreatic cancer. Its possible roles both in PDAC cells and in the tumor microenvironment are described herein.
2. MATERIALS AND METHODS
2.1. Genetically engineered mouse models
LSL‐Kras G12D/+; LSL‐Trp53 R172H/+ ; Pdx‐1‐Cre mice were generated as described previously.23 Vash2 LacZ/LacZ mice were maintained as described previously.24 To analyze the role of Vash2 in pancreatic cancer, we then generated K‐rasLSL G12D/+ ; Trp53LSL R172H/+ ; Vash2 LacZ/+ mice and these mice were crossed with Pdx‐1‐Cre; Vash2 LacZ/+ mice to generate LSL‐Kras G12D/+; LSL‐Trp53 R172H/+ ; Pdx‐1‐Cre; Vash2 LacZ/+ (KPC Vash2 LacZ/+) mice and LSL‐Kras G12D/+; LSL‐Trp53 R172H/+; Pdx‐1‐Cre; Vash2 LacZ/LacZ (KPC Vash2 LacZ/LacZ) mice. These mice were killed and used for examination at a humane endpoint. Survival endpoints included lack of responsiveness to manual stimulation, immobility, or inability to eat or drink. Mice were maintained and treated in accordance with the protocol approved by the Committee on Animal Experimentation of Tohoku University, Japan.
2.2. Cell culture
Murine pancreatic cancer cells (KPC cells) were isolated from the tumors of KPC mice as described previously.25 shRNA sequence for murine Vash2 expression suppression was 5′‐TTCGAAGATTCCTATAAGAAATA for KPC cells, and for human VASH2 expression suppression was 5′‐ GGGCTATCAAATCCGAATTTAGC for PANC‐1 cells. The respective sequence was inserted into a pBAsi‐mU6 Pur expression vector (Takara Bio, Kusatsu, Japan) or pBAsi‐hU6 Neo expression vector (Takara Bio) for mouse Vash2 shRNA or human VASH2 shRNA expression, respectively. KPC and PANC‐1 cells were transfected with the respective vector or empty vector for control by using FuGENE HD (Promega Corporation, Madison, WI, USA) transfection reagent. After transfection, cells were selected in puromycin or G418 (FUJIFILM Wako Pure Chemical Corporation, Tokyo, Japan)‐containing medium. Next, bulk cells were seeded at a density of 0.5 cells/well in 96‐well plates. After confirmation of gene expression by RT‐qPCR, mouse Vash2 knockdown clones from KPC cells and human VASH2 knockdown clones from PANC‐1 were established. Cells were maintained in DMEM (FUJIFILM Wako Pure Chemical Corporation) with 10% FBS (Sigma‐Aldrich, St Louis, MO, USA) and penicillin‐streptomycin (FUJIFILM Wako Pure Chemical Corporation).
2.3. Adenoviral infection
For VASH2 overexpression in SUIT‐2, we used adenovirus vectors encoding human VASH2 (AdVASH2).24 SUIT‐2 cells were plated in 6‐well plates at 3 × 105 cells/well and cultured overnight at 37°C in DMEM containing 10% FBS. On the following day, medium was replaced with DMEM containing 2% FBS and cells were infected with AdVASH2 or AdLacZ at a final MOI of 50. Cells were incubated for an additional 48 hours, followed by western blot analysis and Transwell migration assays were carried out.
2.4. Tail vein injection of mouse KPC cells
Athymic nude mice (Charles River Laboratories, Wilmington, MA, USA) were divided into two groups (KPC/shControl and KPC/shVash2 cells), and 3 × 105 cells suspended in 100 μL PBS were injected into the tail vein of the mice. Twenty days after, wet weight of lungs was measured.
2.5. Orthotopic implantation of KPC cells
Wild‐type mice of mixed 129/SvJae and C57BL/6 genetic background were injected with 5 × 105 cells suspended in 50 μL HBSS (FUJIFILM Wako Pure Chemical Corporation) into the pancreas. Four weeks later, visible metastatic lesions in liver and mesentery were analyzed and volumes of ascites were measured and specimens were used for IHC.
2.6. Cell proliferation assay
Cells were plated in 96 wells at 3 × 103 cells/well and cultured overnight at 37°C in DMEM containing 10% FBS. Subsequently, at different time points (0, 24, 48, 72 hours), 10 μL cell proliferation reagent WST‐1 (Sigma‐Aldrich) was added and cells were further incubated for 1 hour at 37°C. Absorbance was then determined at 450 nm.
2.7. Cell migration and invasion assays
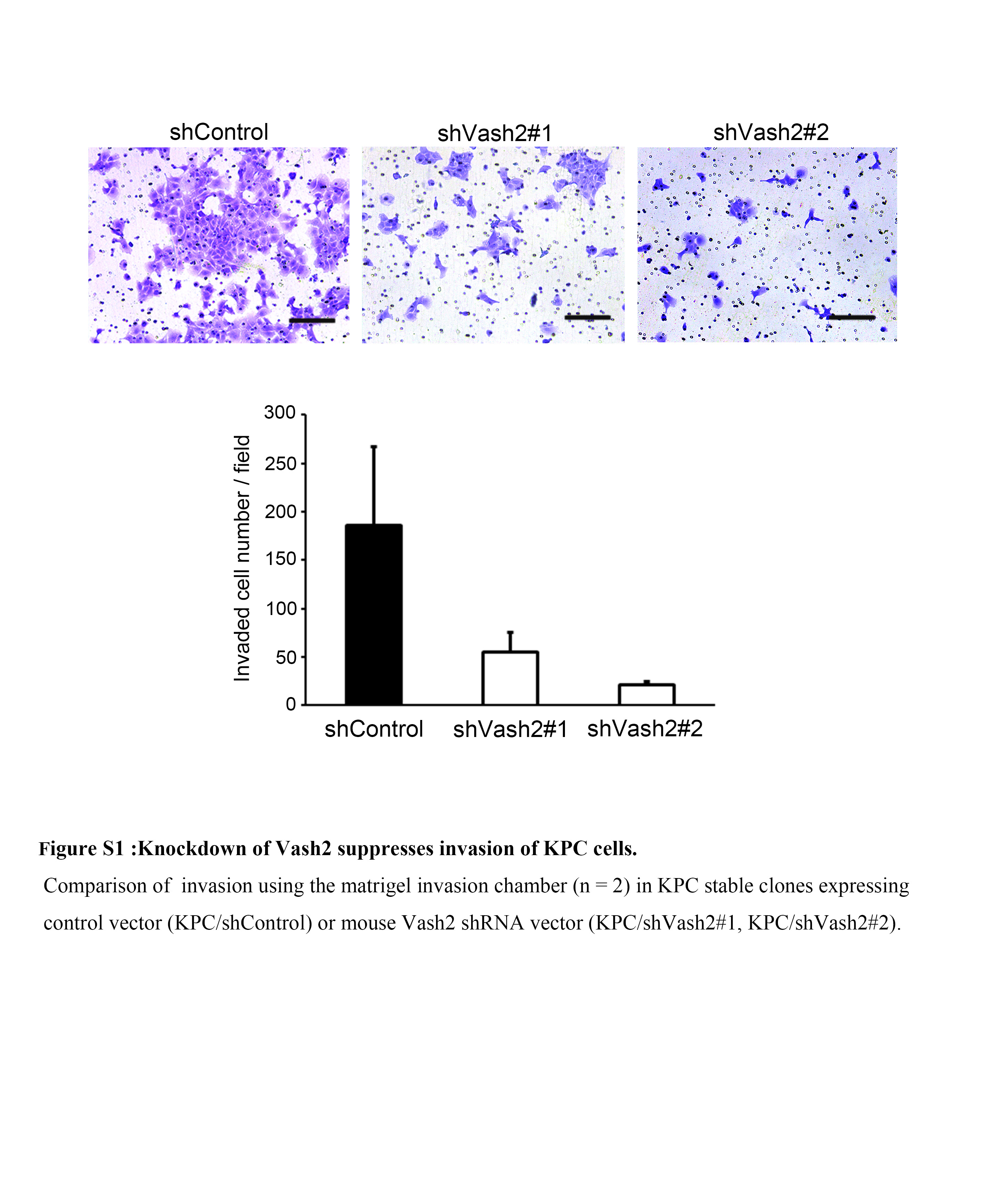
Cell migration was determined using a 24‐well Transwell with an 8.0 μm pore polycarbonate membrane insert (Corning Inc. Corning, NY, USA). Cells were added at 5 × 104 cells/well to the upper chamber (insert); and the lower chamber was filled with DMEM containing 10% FCS. The cells were allowed to migrate for 16 hours (KPC), 6 hours (PANC‐1), or 24 hours (SUIT‐2), and those that had migrated across the filter were fixed in methanol and stained with crystal violet. Total number of migrated cells was counted in at least five fields using ImageJ software. Invasion was determined by using a 24‐well Matrigel invasion chamber (Corning Inc.). Following incubation in DMEM 2% FCS, 5 × 104 cells were added to the upper chamber, after which the lower chamber was filled with DMEM 10% FCS. The cells were incubated for 24 hours, and invaded cells were counted.
2.8. Western blot analysis
Cells were harvested from the culture dish with a scraper in RIPA buffer containing 0.1% SDS (Nakalai Tesque, Kyoto, Japan). Equal amounts of proteins were separated by SDS‐PAGE, blotted onto PVDF membranes (Bio‐Rad, Hercules, CA, USA). Next, the membrane was incubated with anti‐detyrosinated tubulin (Abcam, Cambridge, UK), anti‐alpha‐tubulin (Abcam), anti‐human Vash2 mAb (5E3 clone)7 and the appropriate HRP‐conjugated secondary antibody. Bands were detected using Immobilon Western Chemiluminescent HRP substrate (Merck Millipore, Burlington, MA, USA) and LAS‐4000 (Fuji Photo Film; Fuji, Tokyo, Japan).
2.9. Quantitative real‐time reverse transcriptase‐polymerase chain reaction
For preparation of total RNA from cultured cells and mouse tissue, total RNA was isolated using an RNeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Complementary DNA was prepared from total RNA using ReverTra Ace (ToYoBo, Osaka, Japan). qRT‐PCR was carried out using a CFX96 real‐time PCR detection system (Bio‐Rad) according to the manufacturer's instructions. Each mRNA level was measured and normalized to β‐actin. Sequences of the primers are shown in Table S1.
2.10. Microarray analysis
KPC/shControl cells and KPC/shVash2 cells were harvested and total RNA was extracted as described above. RNA integrity was confirmed by Agilent 2100 bioanalyzer (Agilent Technologies, Santa, Clara, CA, USA). Briefly, total RNA was converted to cDNA, followed by in vitro transcription and labeling of Cy3‐CTP into nascent cRNA. After labeling, cRNA was subjected to Agilent SurePrint G3 Mouse GE 8 × 60K Microarrays (Agilent Technologies). Fluorescence signals were scanned as described in the manufacturer's protocol. Signal intensities were calculated by Feature Extraction Software (Agilent Technologies).
2.11. Transfection and luciferase assays
KPC cells were transiently cotransfected with pNFκB‐Met‐Luc reporter plasmid (Clontech Laboratories, Mountain View, CA, USA) and internal control pSV‐β‐Galactosidase Vector (Promega Corp., Madison, WI, USA) by FugeneHD (Promega Corp.). After 48 hours, the medium was replaced with DMEM containing 10% FCS, collected 8 hours later, and luciferase activities were quantified using a Ready‐To‐Glow Secreted Luciferase Reporter System (Clontech Laboratories). Values were normalized relative to β‐galactosidase activity detected by β‐Galactosidase Enzyme Assay System (Promega Corp.).
2.12. Immunohistochemical analysis
Tumor tissues were fixed with 4% formalin, dehydrated in ethanol, embedded in paraffin, cut on microtome and stained with H&E or antibodies according to standard protocols. Antibodies used in the analysis were detyrosinated tubulin (Abcam), alpha‐tubulin (Merck Millipore), E‐cadherin (Cell Signaling Technology, Danvers, MA, USA), CD3 (Nichirei Biosciences, Tokyo, Japan), CD4 (Cell Signaling Technology), CD8 (Cell Signaling Technology), Ly‐6G (Abcam), CD11b (Abcam), F4/80 (Bio‐Rad), Arginase‐1 (Cell Signaling Technology), and CXCL1 (Abcam). Staining signals were visualized using Histofine Simple Stain MAX PO (Nichirei Biosciences) or SignalStain Boost IHC Detection reagent (Cell Signaling Technology) followed by counterstaining with hematoxylin. For microscopic fluorescence analysis, secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 555 (Thermo Fisher Scientific, Waltham, MA, USA) were used. Nuclei were stained with DAPI. The images were captured with DM2000 LED (Leica Microsystems, Wetzlar, Germany) or KEYENCE BZ‐9000 microscope (Keyence Corporation, Osaka, Japan).
2.13. Statistical analysis
Data are expressed as mean and standard deviation (SD) or standard error of means (SEM). For determining differences between two groups, data were analyzed using the Student's t test. For a comparison of more than two groups, Holm‐Bonferroni test was used. For Kaplan‐Meier plots, log‐rank test was applied for statistical analyses. P‐value <.05 was considered statistically significant.
3. RESULTS
3.1. Vasohibin‐2 is expressed in PDAC cells and promotes their migration
To examine the role of Vash2 in pancreatic cancer, we used KPC mice in which oncogenic Kras G12D and TP53 R172H were conditionally expressed under the control of the pancreas‐specific Pdx‐1 promoter.23 We first analyzed Vash2 expression in samples from KPC mice at each stage, and found that Vash2 was significantly upregulated in pancreatic tissue during PDAC progression (Figure 1A). We then used a pancreatic cancer cell line derived from PDAC of a KPC mouse (KPC cells),25 and established two stable Vash2‐knockdown KPC cells (shVash2#1 and #2) (Figure 1B). Knockdown of Vash2 did not significantly change the proliferation of KPC cells in vitro (Figure 1C). We then carried out Transwell migration assay and found that knockdown of Vash2 significantly decreased the migration and invasion of KPC cells (Figures 1D, S1). The human pancreatic cancer cell line PANC‐1 also showed decreased cell migration when VASH2 was stably knocked down (Figure 1E). Conversely, VASH2 overexpression significantly increased migration of human pancreatic cancer cell line SUIT‐2 in which endogenous VASH2 was almost absent (Figure 1F).
Figure 1.
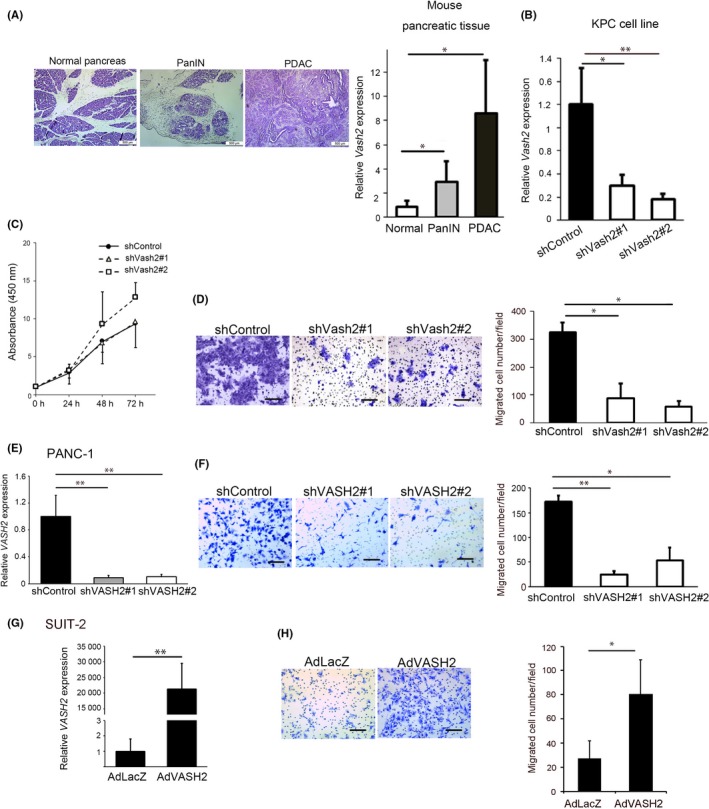
Vasohibin‐2 (Vash2) was upregulated during pancreatic ductal adenocarcinoma (PDAC) progression and promoted PDAC cell migration. A, Representative images of H&E staining and comparison of Vash2 mRNA expression using qRT‐PCR analysis in normal pancreatic tissue (n = 5), PanIN (n = 5) and PDAC (n = 7) from KPC mice. B‐D, Comparison of (B) Vash2 mRNA expression (n = 3), (C) proliferation using WST‐1 assay (n = 2) and (D) migration using the Boyden chamber (n = 3) in KPC stable clones expressing control vector (KPC/shControl) or mouse Vash2 shRNA vector (KPC/shVash2#1, KPC/shVash2#2). E, F, Comparison of (E) VASH2 mRNA expression, (F) migration in PANC‐1 cells stably transfected with control vector (PANC‐1/shControl) or human Vash2 shRNA vector (PANC‐1/shVASH2#1, PANC‐1/shVASH2#2) (n = 3). G, H, Comparison of (G) VASH2 mRNA expression and (H) migration of SUIT‐2 cells infected with LacZ adenovirus (AdLacZ) or human VASH2 adenovirus (AdVASH2) (n = 3). Error bars represent SD. Scale bars are 500 μm (A) and 100 μm (D, F, H). *P < .05, **P < .01
These data indicate that VASH2 expression is increased during PDAC progression, and promotes PDAC cell migration.
3.2. Vasohibin‐2 is required for PDAC progression
To determine whether Vash2 is involved in the metastasis of PDAC, we injected KPC cells into the tail vein of nude mice. Lung metastasis was significantly decreased in the mice injected with Vash2‐knockdown cells (Figure 2A). Next, we inoculated KPC cells orthotopically in the pancreas of mice. Weights of primary tumors formed by Vash2‐knockdown cells were significantly reduced in comparison to primary tumors formed by control cells (Figure 2B,C). Incidence of metastasis in the liver was not significantly different, but metastases were small in Vash2‐knockdown KPC cells (Figure 2D,E). Peritoneal dissemination and ascites were significantly reduced in Vash2‐knockdown KPC cells (Figure 2D,F). When overall survival was compared in the orthotopic mouse model, mice bearing Vash2‐knockdown cells survived longer than mice bearing control cells (Figure 2G).
Figure 2.
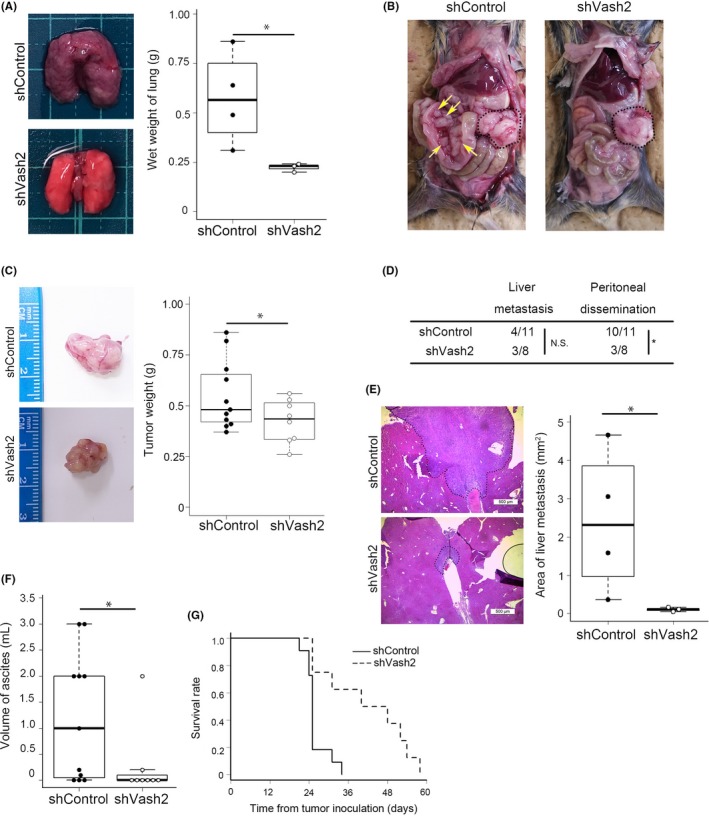
Knockdown of vasohibin‐2 (Vash2) expression inhibited pancreatic ductal adenocarcinoma progression in a mouse model. A, Representative lung images and wet weight quantification of lung in nude mice injected with KPC/shControl (n = 4) or KPC/shVash2 (n = 3) cells into the tail vein. B‐F, KPC/shControl (n = 11) or KPC/shVash2 (n = 8) cells were orthotopically injected into the pancreas of wild‐type mice. B, Representative images of dissected mice. Primary tumors are delimited by the dashed lines. Yellow arrows indicate mesentery metastases. C, Representative images of excised primary tumor specimens and tumor weights are shown. D, Incidence of metastasis or invasion confirmed by autopsy. Fisher's exact test was used in analysis. *P < .05. E, Representative data of H&E staining of liver metastasis and quantification of metastases (n = 3‐4). Scale bars are 500 μm. F, Measurements of volume of ascites. G, Kaplan‐Meier analysis of survival rates in the orthotopic model injected with KPC/shControl (n = 11) or KPC/shVash2 (n = 7). Log‐rank test was used in analysis. P = .002. Student's t test was used in (A‐C, E and F). *P < .05
To further clarify the role of Vash2 in PDAC formation in KPC mice, we crossed KPC mice with Vash2‐deficient mice to obtain Vash2‐heterodeficient KPC mice (KPC/Vash2 LacZ/+) or Vash2‐homodeficient KPC mice (KPC/Vash2 LacZ/LacZ), and compared their phenotypes. Incidences of PDAC were almost equal in all mice irrespective of their genotype. Importantly, however, metastasis was significantly decreased in Vash2‐deficient mice in a gene‐dosage‐sensitive method (Figure 3A). In these pancreatic tumors, Vash2 depletion did not affect differentiation (Figures 3B,C and S2).
Figure 3.
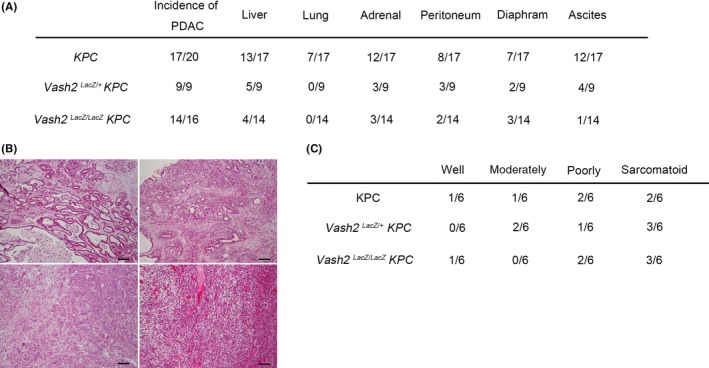
Deletion of vasohibin‐2 (Vash2) gene suppressed pancreatic ductal adenocarcinoma (PDAC) metastasis in KPC mice. A, Table shows incidence of primary tumor, invasion and metastasis in KPC,KPC/Vash2 LacZ/+ mice and KPC/Vash2 LacZ/LacZ mice. B, Representative histological images of PDAC in KPC mice showing well‐differentiated adenocarcinoma (upper left), moderately differentiated adenocarcinoma (upper right), poorly differentiated adenocarcinoma (bottom left), and sarcomatoid carcinoma (bottom right). H&E staining. Scale bars indicate 100 μm. C, Table shows raw number of each differentiation status of PDAC in KPC mice (n = 6), KPC/Vash2 LacZ/+ mice (n = 6), and KPC/Vash2 LacZ/LacZ mice (n = 6)
These results indicate that Vash2 plays an essential role in PDAC metastasis.
3.3. Vasohibin‐2 increases α‐tubulin detyrosination for cancer cell migration
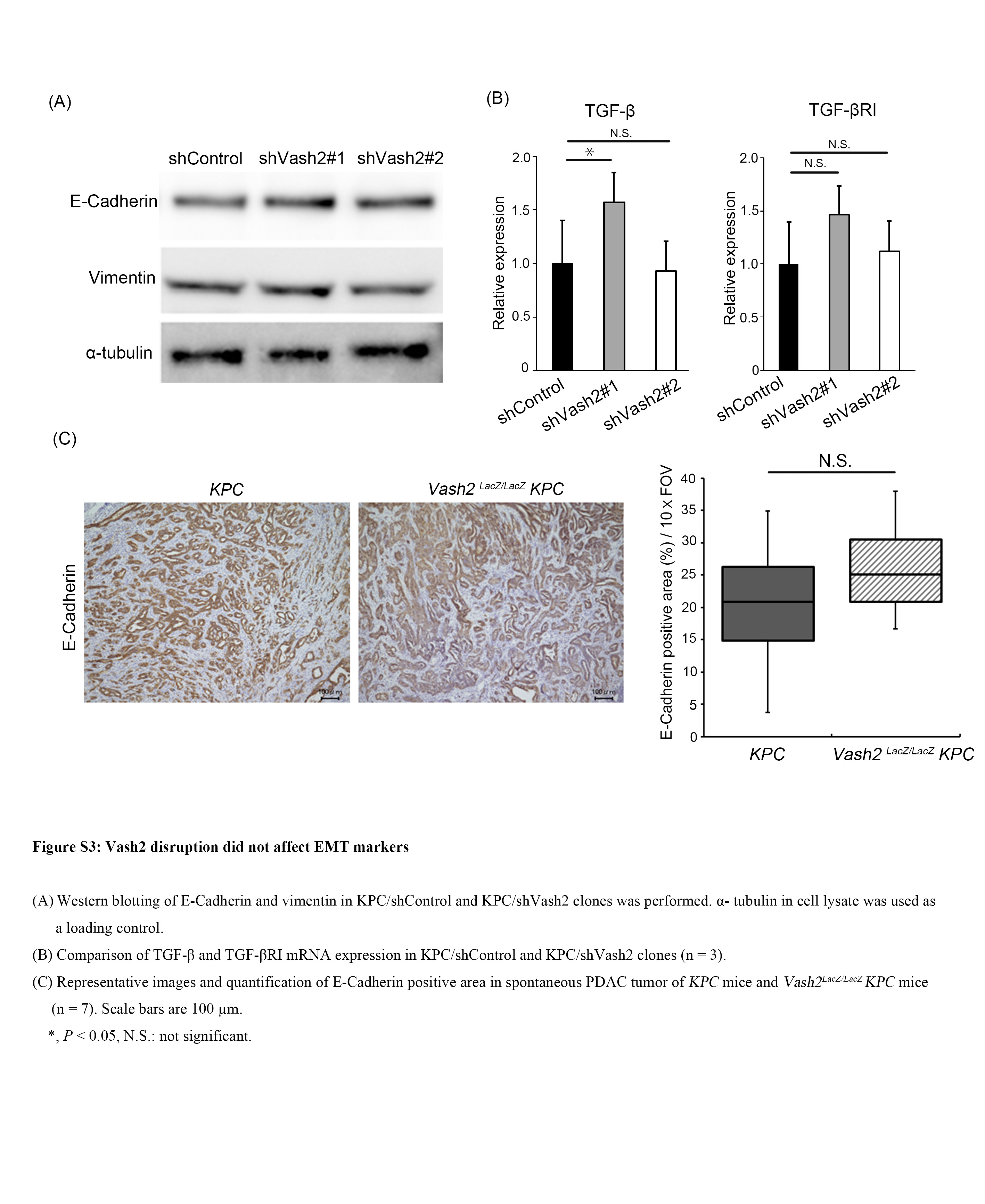
Our previous study showed that VASH2 modulates the metastatic potential of tumor cells through EMT by regulation of transforming growth factor beta (TGF‐β) signaling in ovarian cancer cells.13 We confirmed EMT markers in KPC cell lines and PDAC in KPC mice. Vash2‐knockdown did not affect E‐cadherin and vimentin protein levels nor TGF‐β and TGF‐βRI mRNA levels in the KPC cell line (Figure S3A,B). Furthermore, IHC analyses showed that no significant changes were observed in E‐cadherin level in KPC/Vash2 LacZ/LacZ mice (Figure S3C). These results indicate that EMT may not be the major cause of VASH2‐induced metastatic potential.
Vasohibins have recently been identified to have TCP activity, which deletes a tyrosine residue from the carboxy‐terminus of α‐tubulin.19, 20 We confirmed that detyrosinated tubulin was decreased in Vash2‐knockdown KPC cells and tissue of orthotopic tumors (Figure 4A‐C). Conversely, VASH2 overexpression strongly increased detyrosinated tubulin and cell migration of SUIT‐2 (Figure 4D). We then asked if this TCP activity was related to cell migration. When VASH2‐overexpressed SUIT‐2 cells were treated with a TCP inhibitor parthenolide,26 the increased cell migration was almost completely inhibited (Figure 4E). Thus, the TCP activity of VASH2 is required for cancer cell migration and metastasis.
Figure 4.
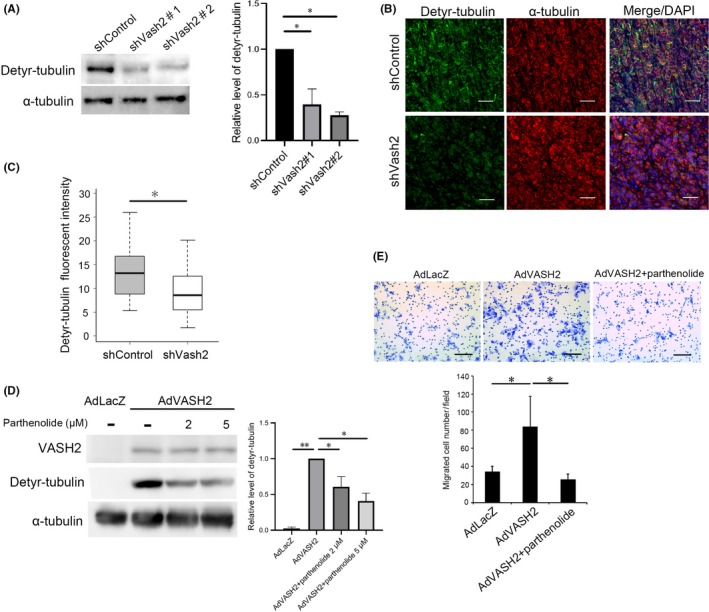
Vasohibin‐2 (Vash2) increased α‐tubulin detyrosination for cancer cell migration. A, Western blotting of detyrosinated α‐tubulin and total α‐tubulin in KPC/shControl and KPC/shVash2 clones was carried out (n = 3). Quantification of the western blotting is shown on the right. B, Representative images of double fluorescent immunohistochemical staining for detyrosinated α‐tubulin (green), total α‐tubulin (red) in the orthotopic tumors formed by KPC/shControl and KPC/shVash2 clones. C, Quantification of detyrosinated α‐tubulin immunofluorescence intensity (n = 4). D, Western blotting of VASH2, detyrosinated tubulin and α‐tubulin in SUIT‐2 cells infected with AdLacZ or AdVASH2 and treated with DMSO (‐) or indicated concentration of parthenolide for 48 h. Quantification data are shown on the right. E, SUIT‐2 cells were infected with AdLacZ or AdVASH2 and incubated for 48 h. Thereafter, migration assay was carried out in the presence of DMSO (‐) or 5 μmol/L parthenolide (n = 3). Quantification of the western blotting is shown on the right. Error bars represent standard deviation (SD). Scale bars are 100 μm. *P < .05, **P < .01
3.4. Role of VASH2 in tumor angiogenesis
As we previously noted that VASH2 accelerated tumor angiogenesis,10, 12 we examined the vasculature in the orthotopic tumors. Vascular area was significantly reduced in tumors formed by Vash2‐knockdown KPC cells (Figure 5A). We next examined tumor vasculature in KPC mice. Similar to the orthotopic model, tumor angiogenesis was significantly decreased in Vash2‐deficient mice (Figure 5B), confirming the proangiogenic role of VASH2 in PDAC.
Figure 5.
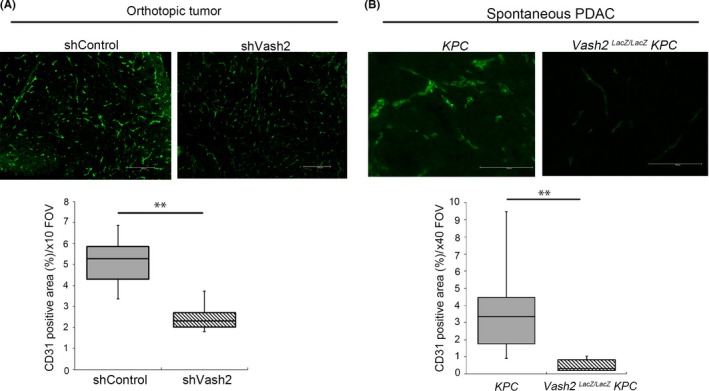
Vasohibin‐2 (Vash2) depletion suppressed tumor angiogenesis in pancreatic ductal adenocarcinoma (PDAC). Representative images and quantification of positive area of fluorescent immunohistochemical (IHC) staining for CD31 in (A) orthotopic KPC tumor (n = 4) and (B) KPC mice (n = 7). KPC cells were injected into 5‐8‐wk‐old WT mice, and mice were killed after 25‐28 d for IHC. KPC mice were killed at endpoint and used. At least three images per sample were analyzed. Scale bars are 200 μm (A) and 100 μm (B). **P < .01
3.5. Role of VASH2 in the tumor immune microenvironment
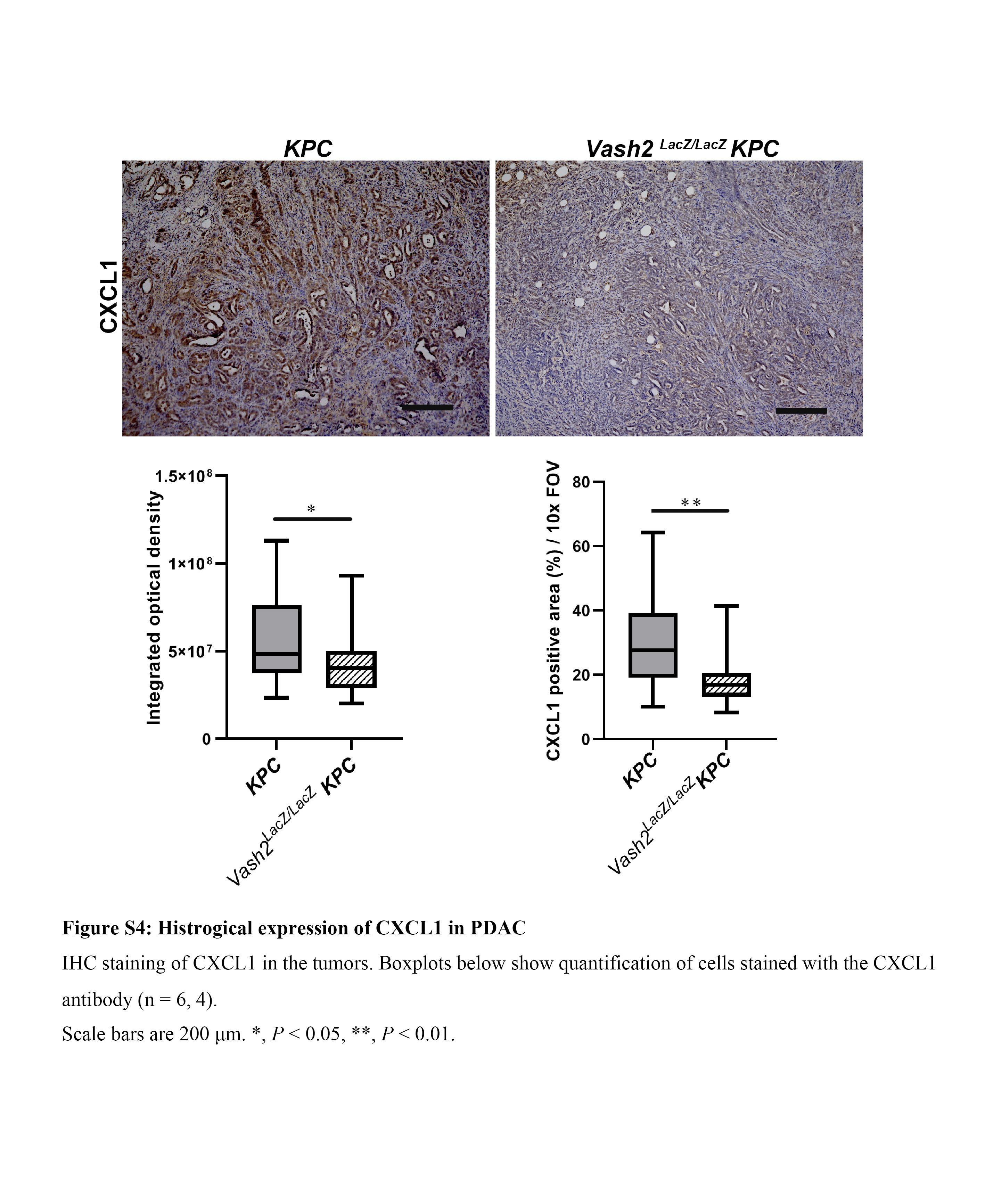
To further clarify VASH2 function, the downstream targets of VASH2 in KPC cells were analyzed using microarray to compare gene expression between control and Vash2‐knockdown KPC cells. Microarray showed that 911 genes were upregulated and 1695 genes were downregulated by Vash2 knockdown in KPC cells (<0.5 fold). Gene enrichment analysis by KEGG pathway showed that downregulated genes were related to inflammation, including cytokine‐cytokine receptor interaction, chemokine signaling and tumor necrosis factor (TNF) signaling (Figure 6A). NF‐κB signaling is a classical proinflammatory signaling pathway, and we therefore assessed NF‐κB activity using a pNF‐κB‐MetLuc2 reporter vector which contained multiple copies of the NF‐κB response element. Knockdown of Vash2 decreased NF‐κB promoter activity (Figure 6B). We further examined gene expression included in these pathways using RT‐qPCR, and downregulation of secreted proteins such as chemokines (Cxcl2, Cxcl5, Ccl2, Ccl5) and cytokines (Csf1, Csf2, Il‐1A) were observed (Figure 6C). CXCL2 and CXCL5 are ligands for CXCR2, which is expressed in G‐MDSC and neutrophils.27 These ligands recruit G‐MDSC, which enable evasion of tumor immunity in PDAC.4 Csf2 encodes the cytokine GM‐CSF, and tumor‐derived GM‐CSF drives differentiation of G‐MDSC.28 We confirmed downregulation of CXCR2 ligands and GM‐CSF mRNA expression (Figure 6D), and also CXCL1 protein levels (Figure S4).
Figure 6.
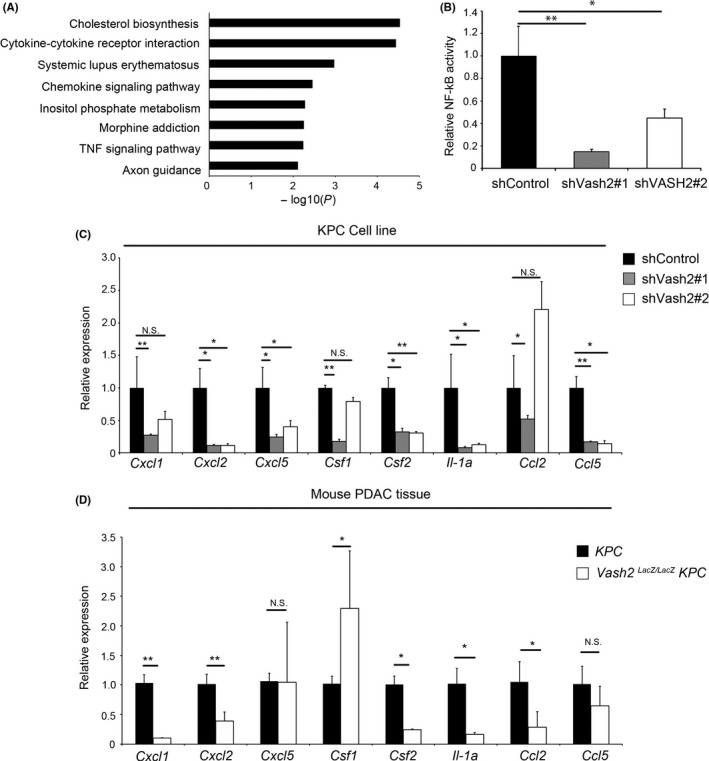
Vasohibin‐2 (Vash2) modulated the expression of inflammatory chemokines and cytokines in pancreatic ductal adenocarcinoma (PDAC) cells. A, Pathway enrichment analysis (by Metascape) of genes downregulated by Vash2 knockdown in KPC cells. B, Quantification of nuclear factor kappa B (NF‐kB) activity in KPC/shControl and KPC/shVash2 clones using luciferase reporter assay. (n = 3). C, Quantification of chemokine and cytokine mRNA expression in KPC/shControl and KPC/shVash2 clones (n = 3). D, Quantification of chemokine and cytokine mRNA expression in the PDAC of KPC mice and KPC/Vash2 LacZ/LacZ mice (n = 7). Error bars represent SEM (B, C) and SD (D). *P < .05, **P < .01. NS, not significant
CCL2 induces recruitment of CCR2+ inflammatory monocytes, which are precursors of TAM.29 The expression of Ccl2 was significantly reduced in PDAC of KPC/Vash2 LacZ/LacZ mice but not in Vash2‐knockdown KPC cells (Figure 6C,D), which indicated that Vash2 may control Ccl2 expression indirectly in mice.
Thus, VASH2 may regulate cytokine and chemokine expression, which controls the recruitment of G‐MDSC and TAM in the tumor microenvironment.
3.6. Vasohibin‐2 is involved in the evasion of tumor immunity
Accumulation of G‐MDSC prevents cytotoxic T‐cell infiltration into the tumor, and this supports tumor progression in PDAC.4 We speculated that VASH2 modulates G‐MDSC accumulation through the upregulation of CXCR2 ligands and GM‐CSF. We therefore examined the presence of CD11b+ Ly6G+ G‐MDSC by immunohistochemical double staining. Quantification of G‐MDSC in tumor showed that G‐MDSC accumulation was decreased in PDAC of KPC/Vash2 LacZ/LacZ mice (Figure 7A). We next investigated T‐cell infiltration in the tumor. IHC analysis showed that the number of CD3+ T cells significantly increased in PDAC of mice and was correlated with the number of cytotoxic CD8+ T cells (Figure 7B).
Figure 7.
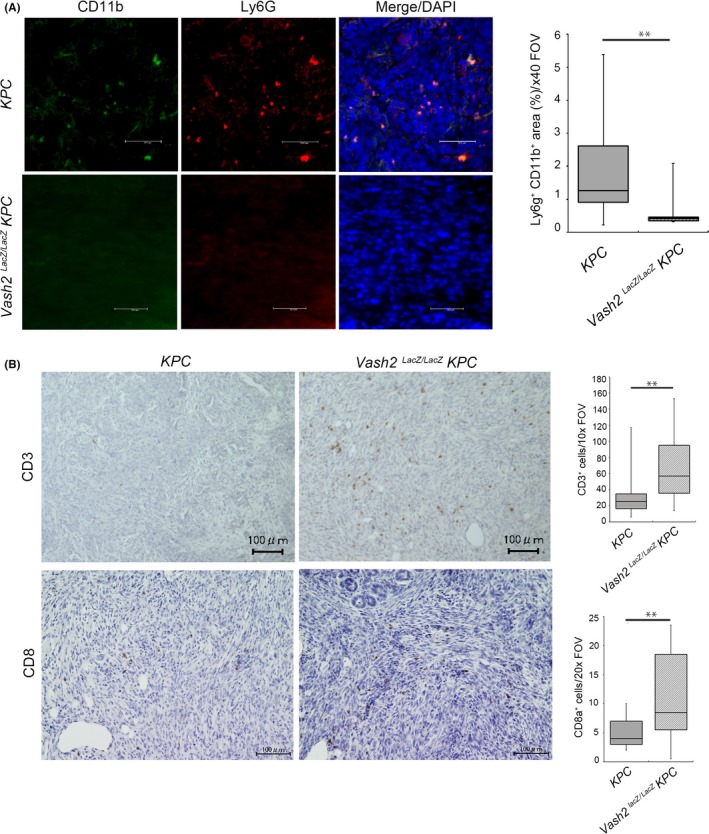
Vasohibin‐2 (Vash2) modulated myeloid derived suppressor cell accumulation and T‐cell infiltration in pancreatic ductal adenocarcinoma (PDAC). A, Representative images and quantification of CD11b and Ly6G double‐positive area in spontaneous PDAC tumor of KPC mice and Vash2 LacZ/LacZ KPC mice. (n = 7). B, Immunohistochemical (IHC) staining of CD3 and CD8 in the tumors. Boxplots on the right show quantification of cells stained with the indicated antibodies (n = 7). Mice were killed at endpoint and used for IHC. Scale bars are 100 μm. *P < .05, **P < .01
Macrophages are broadly divided into proinflammatory M1‐TAM and anti‐inflammatory M2‐TAM.30 We showed that deletion of the Vash2 gene caused significant reduction of Arginase‐1 positive M2‐TAM in PDAC (Figure S5).
These data suggest that VASH2 is involved in the evasion of tumor immunity in PDAC through the accumulation of G‐MDSC and M2‐TAM.
4. DISCUSSION
Pancreatic ductal adenocarcinoma is a highly metastatic cancer. Herein, we examined the significance of VASH2 in PDAC, and showed that VASH2 plays an essential role in the progression of PDAC by acting on both cancer cells and the tumor microenvironment.
VASH2 knockdown significantly reduced pancreatic cancer cell migration, whereas its overexpression increased their migration. Accordingly, the VASH2‐induced cell migration was due to its direct effect on PDAC cells. Although we and others have previously reported that VASH2 promotes EMT of various cancer cells, our present study indicated that EMT was not the primary cause of VASH2‐mediated metastasis of PDAC. In contrast, the present study indicates that cytoskeletal microtubule modification is critical for VASH2‐mediated metastasis.
Microtubules receive a variety of post‐translational modifications including tubulin detyrosination. VASH2 has recently been shown to have TCP activity.19, 20 In accordance with this evidence, the present study showed that VASH2 stimulated metastatic activity of PDAC cells, at least in part, through its TCP activity within the cell. Accumulation of detyrosinated tubulin is documented in breast cancers and neuroblastomas with poor prognosis.18, 31 In addition, detyrosinated tubulin is involved in the metastasis of breast cancer cells though the formation of cellular protrusions, so‐called microtentacles.26 We suggest that VASH2 may play a considerable role in the accumulation of detyrosinated tubulin in such circumstances.
When bound to SVBP, VASH2 is efficiently secreted,8, 32 and this may be responsible for the paracrine effect of VASH2 in the tumor microenvironment. We have previously reported that when VASH2 was knocked down in cancer cells, it prevented tumor angiogenesis in ovarian cancer cells and hepatocellular carcinoma.10, 11 In addition, neutralizing anti‐VASH2 monoclonal antibody can inhibit VASH2‐induced tumor angiogenesis.16 Thus, VASH2 secreted by cancer cells stimulates tumor angiogenesis, and our present study supports this theory. We evaluated and determined that cell proliferation in vitro was not affected by VASH2 expression levels, whereas tumor weights were decreased in vivo by VASH2 knockdown. As the reduction of vascular area was observed in the tumors of Vash2‐knockdown KPC cells, we speculated that the decrease of tumor size was caused, at least in part, by the inhibition of angiogenesis.
Besides tumor angiogenesis, our present study showed for the first time that VASH2 affects tumor immunity in the tumor microenvironment. This theory is derived from the analysis of VASH2‐regulated gene expression in cancer cells. We noticed that VASH2 regulated the expression of inflammatory chemokines and cytokines in cancer cells, which, in turn, controlled the recruitment of MDSC and M2‐TAM. Accordingly, these activities of VASH2 in the tumor immune microenvironment is indirectly mediated by VASH2‐regulated genes such as CXCR2 ligands and GM‐CSF in PDAC cells.
Myeloid‐derived suppressor cells and M2‐TAM have recently been recognized as critical players in the tumor immune microenvironment. MDSC represent a heterogeneous population of immature myeloid cells. Under normal conditions, these immature cells differentiate into macrophages, dendritic cells, and granulocytes. But, under pathological conditions such as cancer, they are arrested in their differentiation, resulting in the accumulation of MDSC. Increasing evidence indicates that MDSC not only provide evasion of tumor immunity but also augment the metastatic potential of tumor cells through promoting pre‐metastatic niche formation.33 Indeed, CXCR2 signaling is reported be responsible for the accumulation of MDSC in PDAC, and blockade of CXCR2 signaling contributes to improved tumor immunity and reduced metastasis of PDAC in mice.4, 34 The transition of M1‐TAM to M2‐TAM is another aspect of immune response during the progression of PDAC, as the accumulation of M2‐TAM is evident in PDAC.30 The present study indicates that VASH2 is responsible for the modulation of tumor immunity through accumulation of M2‐TAM as well.
One of the most significant advancements in recent anticancer therapy is immune checkpoint treatment. However, responses to such treatment are not uniform across tumor types. In particular, it is not effective for PDAC because of its immunosuppressive tumor microenvironment, as the accumulation of MDSC prevents the infiltration of cytotoxic T lymphocytes in PDAC. The present study suggests that the expression of VASH2 in PDAC cells is critical for this immunosuppressive tumor microenvironment of PDAC.
In conclusion, VASH2 plays an essential role in PDAC progression with multiple functions in both cancer cells and the tumor microenvironment. Its role in cancer cells is tubulin detyrosination which directly accelerates the metastatic activity of PDAC cells. Its role in the tumor microenvironment is tumor angiogenesis as a result of its paracrine activity and the evasion of tumor immunity due to altered gene expression of PDAC cells (Figure 8). We propose VASH2 to be a relevant target for the treatment of refractory PDAC.
Figure 8.
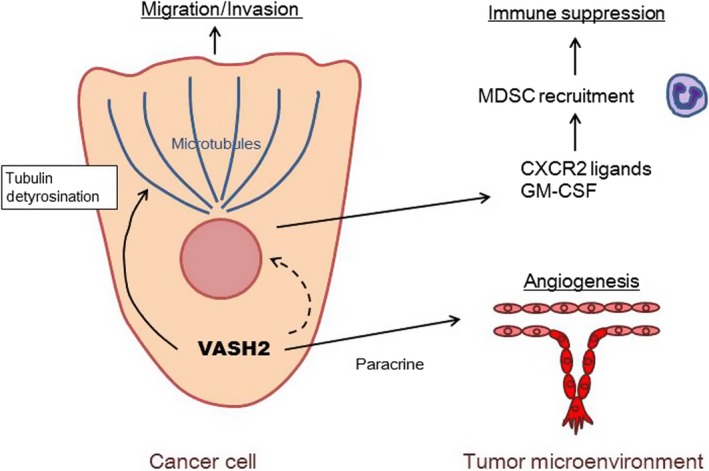
Schematic model illustrating the role of vasohibin‐2 (VASH2) in pancreatic ductal adenocarcinoma (PDAC). VASH2‐dependent increase in tubulin detyrosination directly accelerates the migration of PDAC cells. In the tumor microenvironment, VASH2 promotes tumor angiogenesis as a result of paracrine activity and the evasion of tumor immunity as a result of altered gene expression in PDAC cells. GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; MDSC, myeloid derived suppressor cells
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported by a Grant‐in‐Aid for Scientific Research (B) (16H04689) to Y. Sato and a Grant‐in‐Aid for Early‐Career Scientists (18K15205) to R. Norita‐Iida from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Iida‐Norita R, Kawamura M, Suzuki Y, et al. Vasohibin‐2 plays an essential role in metastasis of pancreatic ductal adenocarcinoma. Cancer Sci. 2019;110:2296–2308. 10.1111/cas.14041
Iida‐Norita and Kawamura contributed equally to this manuscript.
REFERENCES
- 1. Tuveson DA, Neoptolemos JP. Understanding metastasis in pancreatic cancer: a call for new clinical approaches. Cell. 2012;148(1–2):21‐23. [DOI] [PubMed] [Google Scholar]
- 2. Neesse A, Michl P, Frese KK, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861‐868. [DOI] [PubMed] [Google Scholar]
- 3. Ireland L, Santos A, Ahmed MS, et al. Chemoresistance in pancreatic cancer is driven by stroma‐derived insulin‐like growth factors. Cancer Res. 2016;76(23):6851‐6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steele CW, Karim SA, Leach JDG, et al. CXCR4 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29(6):832‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor‐infiltrating macrophages decreases tumor‐initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Watanabe K, Hasegawa Y, Yamashita H, et al. Vasohibin as an endothelium‐derived negative feedback regulator of angiogenesis. J Clin Invest. 2004;114(7):898‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shibuya T, Watanabe K, Yamashita H, et al. Isolation and characterization of vasohibin‐2 as a homologue of VEGF‐inducible endothelium‐derived angiogenesis inhibitor vasohibin. Arterioscler Thromb Vasc Biol. 2006;26(5):1051‐1057. [DOI] [PubMed] [Google Scholar]
- 8. Suzuki Y, Kobayashi M, Miyashita H, Ohta H, Sonoda H, Sato Y. Isolation of a small vasohibin‐binding protein (SVBP) and its role in vasohibin secretion. J Cell Sci. 2010;123(18):3094‐3101. [DOI] [PubMed] [Google Scholar]
- 9. Sato Y. The vasohibin family: a novel family for angiogenesis regulation. J Biochem. 2013;153(1):5‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takahashi Y, Koyanagi T, Suzuki Y, et al. Vasohibin‐2 expressed in human serous ovarian adenocarcinoma accelerates tumor growth by promoting angiogenesis. Mol Cancer Res. 2012;10(9):1135‐1146. [DOI] [PubMed] [Google Scholar]
- 11. Xue X, Gao W, Sun B, et al. Vasohibin 2 is transcriptionally activated and promotes angiogenesis in hepatocellular carcinoma. Oncogene. 2013;32(13):1724‐1734. [DOI] [PubMed] [Google Scholar]
- 12. Kitahara S, Suzuki Y, Morishima M, et al. Vasohibin‐2 modulates tumor onset in the gastrointestinal tract by normalizing tumor angiogenesis. Mol Cancer. 2014;32(10):1441‐1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Norita R, Suzuki Y, Furutani Y, et al. Vasohibin‐2 is required for epithelial–mesenchymal transition of ovarian cancer cells by modulating transforming growth factor‐β signaling. Cancer Sci. 2017;108(3):419‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzuki Y, Kitahara S, Suematsu T, Oshima M, Sato Y. Requisite role of vasohibin‐2 in spontaneous gastric cancer formation and accumulation of cancer‐associated fibroblasts. Cancer Sci. 2017;108(12):2342‐2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koyanagi T, Suzuki Y, Saga Y, et al. In vivo delivery of siRNA targeting vasohibin‐2 decreases tumor angiogenesis and suppresses tumor growth in ovarian cancer. Cancer Sci. 2013;104(12):1705‐1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koyanagi T, Suzuki Y, Komori K, et al. Targeting human vasohibin‐2 by a neutralizing monoclonal antibody for anti‐cancer treatment. Cancer Sci. 2017;108(3):512‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lafanechère L, Courtay‐Cahen C, Kawakami T, et al. Suppression of tubulin tyrosine ligase during tumor growth. J Cell Sci. 1998;111(Pt 2):171‐81. [DOI] [PubMed] [Google Scholar]
- 18. Mialhe A, Lafanechère L, Job D, et al. Tubulin detyrosination is a frequent occurrence in breast cancers of poor prognosis. Cancer Res. 2001;61(13):5024‐5027. [PubMed] [Google Scholar]
- 19. Aillaud C, Bosc C, Peris L, et al. Vasohibins/SVBP are tubulin carboxypeptidases (TCPs) that regulate neuron differentiation. Science. 2017;358(6469):1448‐1453. [DOI] [PubMed] [Google Scholar]
- 20. Nieuwenhuis J, Adamopoulos A, Bleijerveld OB, et al. Vasohibins encode tubulin detyrosinating activity. Science. 2017;358(6369):1453‐1456. [DOI] [PubMed] [Google Scholar]
- 21. Kim JC, Kim KT, Park JT, Kim HJ, Sato Y, Kim HS. Expression of vasohibin‐2 in pancreatic ductal adenocarcinoma promotes tumor progression and is associated with a poor clinical outcome. Hepatogastroenterology. 2015;62(138):251‐256. [PubMed] [Google Scholar]
- 22. Witkiewicz AK, McMillan EA, Balaji U, et al. Whole‐exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469‐483. [DOI] [PubMed] [Google Scholar]
- 24. Kimura H, Miyashita H, Suzuki Y, et al. Distinctive localization and opposed roles of vasohibin‐1 and vasohibin‐2 in the regulation of angiogenesis. Blood. 2009;113(19):4810‐4818. [DOI] [PubMed] [Google Scholar]
- 25. Hamada S, Taguchi K, Masamune A, Yamamoto M, Shimosegawa T. Nrf2 promotes mutant K‐ras/p53‐driven pancreatic carcinogenesis. Carcinogenesis. 2017;38(6):661‐670. [DOI] [PubMed] [Google Scholar]
- 26. Whipple RA, Vitolo MI, Boggs AE, Charpentier MS, Thompson K, Martin SS. Parthenolide and costunolide reduce microtentacles and tumor cell attachment by selectively targeting detyrosinated tubulin independent from NF‐κB inhibition. Breast Cancer Res. 2013;15(5):R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stromnes IM, Greenberg PD, Hingorani SR. Molecular pathways: myeloid complicity in cancer. Clin Cancer Res. 2014;20(20):5157‐5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bayne LJ, Beatty GL, Jhala N, et al. Tumor‐derived granulocyte‐macrophage colony‐stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanford DE, Belt BA, Panni RZ, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR1 axis. Clin Cancer Res. 2013;19(13):3404‐3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cui R, Yue W, Lattime EC, Stein MN, Xu Q, Tan XL. Targeting tumor‐associated macrophages to combat pancreatic cancer. Oncotarget. 2016;7(31):50735‐50754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kato C, Miyazaki K, Nakagawa A, et al. Low expression of human tubulin tyrosine ligase and suppressed tubulin tyrosination/detyrosination cycle are associated with impaired neuronal differentiation in neuroblastomas with poor prognosis. Int J Cancer. 2004;112(3):365‐375. [DOI] [PubMed] [Google Scholar]
- 32. Kadonosono T, Yimchuen W, Tsubaki T, et al. Domain architecture of vasohibins required for their chaperone‐dependent unconventional extracellular release. Protein Sci. 2017;26(3):452‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Safarzadeh E, Orangi M, Mohammadi H, Babaie F, Baradaran B. Myeloid‐derived suppressor cells: important contributors to tumor progression and metastasis. J Cell Physiol. 2018;233(4):3024‐3036. [DOI] [PubMed] [Google Scholar]
- 34. Sano M, Ijichi H, Takahashi R, et al. Blocking CXCLs–CXCR34 axis in tumor–stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune‐inflammatory microenvironment. Oncogenesis. 2019;8(2):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials