

Visual Abstract

Key Words: atrial cardiomyopathy, atrial fibrillation, ischemia/reperfusion, myocardial infarction, remote remodeling
Abbreviations and Acronyms: AF, atrial fibrillation; CatA, cathepsin A; Cx43, connexin 43; ECM, extracellular matrix; ICM, ischemic cardiomyopathy; I/R, ischemia/reperfusion; LA, left atrial; LAD, left anterior descending coronary artery; LV, left ventricular; MRI, magnetic resonance imaging; mRNA, messenger ribonucleic acid; PL, permanent left anterior descending ligation; SAR, (S)-3-{[1-(2-Fluoro-phenyl)-5-methoxy-1H-pyrazole-3-carbonyl]-amino}-3-o-tolyl-propionic-acid
Highlights
-
•
The role of the protease cathepsin A for the progression of left ventricular remote remodeling and atrial cardiomyopathy in ischemic cardiomyopathy is unknown.
-
•
In rats with ventricular ischemia and reperfusion, cathepsin A is up-regulated in the left ventricular and atrial tissue remote from the infarcted area.
-
•
Pharmacological inhibition of cathepsin A protease activity by SAR significantly reduces remote ventricular remodeling and atrial extracellular matrix remodeling, represented by fibrosis formation and connexin 43 lateralization.
-
•
Prevention of ventricular remote remodeling and atrial cardiomyopathy by SAR increased ventricular viable myocardium and atrial emptying function reducing susceptibility to atrial fibrillation.
-
•
Remote ventricular and atrial extracellular matrix remodeling may represent a promising target for pharmacological atrial fibrillation upstream therapy following myocardial infarction.
Summary
After myocardial infarction, remote ventricular remodeling and atrial cardiomyopathy progress despite successful revascularization. In a rat model of ventricular ischemia/reperfusion, pharmacological inhibition of the protease activity of cathepsin A initiated at the time point of reperfusion prevented extracellular matrix remodeling in the atrium and the ventricle remote from the infarcted area. This scenario was associated with preservation of more viable ventricular myocardium and the prevention of an arrhythmogenic and functional substrate for atrial fibrillation. Remote ventricular extracellular matrix remodeling and atrial cardiomyopathy may represent a promising target for pharmacological atrial fibrillation upstream therapy following myocardial infarction.
Long-term outcome after myocardial infarction is predicted by left ventricular (LV) dysfunction due to ischemic cardiomyopathy (ICM), which develops in ∼40% of post-myocardial infarct patients (1). Current clinical practice is focused on maximizing myocardial salvage by coronary revascularization (2). However, LV remodeling due to structural and functional changes in the extracellular matrix (ECM) often progresses even following successful revascularization and is the most common reason for heart failure after myocardial infarction 3, 4. Progressive ECM remodeling is characterized by scar formation and thinning of the infarcted ventricular wall as well as by maladaptive ECM alterations in the remote noninfarcted ventricular myocardium, which ultimately impairs LV function. The presence and amount of viable ventricular myocardium have been shown to determine the progression or potential regression of the ongoing LV remodeling process 5, 6, 7, 8, 9.
Importantly, ICM is not just associated with remote remodeling in the ventricle, but it may also contribute to the development of a progressive atrial cardiomyopathy (10) characterized by impaired atrial emptying function and increased risk of atrial fibrillation (AF) 11, 12, 13. It remains unclear whether atrial cardiomyopathy in ICM is solely a consequence of LV systolic dysfunction or whether it may represent the atrial manifestation of the cardiac structural remodeling process remote from the ventricular infarct region, which correlates clinically with the presence and amount of viable remote LV myocardium 5, 6, 7, 8.
Cardiac ECM remodeling is characterized by changes in collagen composition, which contributes to interstitial fibrosis formation and impaired electrical and functional properties of the myocardium 14, 15, 16. The balance between ECM synthesis and degradation is of crucial relevance in maintaining cardiac structural integrity and is regulated by proteolysis as a key mechanism to control ECM function and turnover 16, 17. A set of proteolytic enzymes, including cathepsins, degrade ECM components and target a broad range of intracellular and extracellular proteins. Cysteine proteases such as cathepsin B, K, L, and S have been shown to play a pathophysiological role in myocardial infarction, congestive heart failure, and diabetes (17). The serine protease cathepsin A (CatA) is a multifunctional protein 18, 19, 20. Inside the lysosome, CatA exerts its catalytic function as a carboxypeptidase and protects beta-galactosidase and neuraminidase-1 from intralysosomal proteolysis by the formation of a lysosomal multienzyme complex (19). In addition, CatA is localized on the cell surface and is secreted into the extracellular space, where its proteolytic function has been suggested to be involved in ECM formation as well as degradation of different extracellular regulatory peptides 18, 19, 20. Recently, we showed that pharmacological inhibition of CatA activity prevented atrial fibrosis formation and reduces susceptibility to AF without significant effects on LV systolic function in an animal model of type 2 diabetes, suggesting a crucial role for CatA in ECM remodeling (21).
The cardiac regulation of CatA in ICM, its effect on viable LV myocardium after revascularization, and its role in the progression of ventricular and atrial cardiomyopathy as manifestations of remote remodeling remain unknown. Using a rat model for ICM induced by ventricular ischemia/reperfusion (I/R), we investigated the regulation of LV and left atrial (LA) CatA expression and the effect of pharmacological CatA inhibition on the induction of ICM and cardiac remodeling within and remote from the infarct area, including atrial cardiomyopathy. We compared the effect of pharmacological inhibition of CatA activity versus the effect of the angiotensin-converting enzyme inhibitor ramipril as an established drug for prevention of cardiac remodeling.
Methods
For a more detailed description of all methods used see the Supplemental Methods.
Human tissue
Human failing myocardium was obtained from patients with end-stage ICM who were scheduled for heart transplantation (n = 8). Eight donor hearts of patients with no signs of heart disease that could not be used for transplantation due to ABO-mismatch were used as nonfailing controls (n = 8). Information about clinical parameters and medication were not available. This investigation was reviewed by the regional ethics committee and conforms to the principles outlined in the Declaration of Helsinki.
Animal models
All animal studies were performed in accordance with the German law for the protection of animals. The investigation conforms to the guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (eighth edition; revised 2011). All procedures were in accordance with current Sanofi-Aventis Laboratory Animal Science and Welfare guidelines. The study was approved by the regional animal ethics commission in Darmstadt, Germany. Ninety male Wistar rats (12 weeks old) were purchased from Harlan Winkelmann GmbH (Borchen, Germany), housed 3 per cage under standardized conditions (room temperature 24°C, relative humidity 55%, 12 h dark/light cycle) with free access to a standardized diet (#1320, Altromin, Lage, Germany) and tap drinking water.
Permanent ligation of left anterior descending coronary artery in rats
Thirty male Wistar rats were anesthetized with 5% isoflurane in 95% oxygen followed by a deep anesthesia via intraperitoneal injection of ketamine hydrochloride (80 mg/kg body weight) and xylazine hydrochloride (6 mg/kg body weight). In 20 rats, the left anterior descending (LAD) coronary artery was permanently ligated; 10 sham-operated rats served as controls. Post-operative pain management was performed by using carprofen (5 mg/kg subcutaneously). After 8 weeks, rats were anesthetized (as discussed earlier), and the hearts were quickly removed. Additional details are provided in the Supplemental Methods.
Ventricular I/R was performed as previously described 22, 23. Briefly, 60 male Wistar rats were anesthetized with 5% isoflurane in 95% oxygen followed by a deep anesthesia via intraperitoneal injection of ketamine hydrochloride (80 mg/kg body weight) and xylazine hydrochloride (6 mg/kg body weight). In 51 rats, myocardial ischemia was induced by temporary occlusion of the left coronary artery for 30 min followed by reperfusion upon release of the ligation. Nine rats underwent sham operation. Eight I/R-rats died during the 24-h post-operative period. The day after surgery, animals were randomized into 4 groups: sham-operated rats (I/R-Sham, n = 9), I/R-rats given placebo (I/R-Placebo, n = 15), ramipril-treated I/R-rats (I/R-Ramipril, n = 14), and CatA inhibitor [(S)-3-{[1-(2-Fluoro-phenyl)-5-methoxy-1H-pyrazole-3-carbonyl]-amino}-3-o-tolyl-propionic-acid (SAR)]-treated I/R-rats (I/R-SAR, n = 14). SAR, a new orally active CatA inhibitor, was administered daily via oral gavage (30 mg/kg per day) (22), and ramipril (1 mg/kg per day) (23) was administered in chow in accordance with the literature.
After 9 weeks of treatment, rats were anesthetized for magnetic resonance imaging (MRI) of the heart with 1.5% to 2.5% isoflurane in an oxygen/nitrous oxide mixture (30%/70%) to determine LA emptying function at rest and global and regional LV function under basal and dobutamine stress conditions as previously described (21).
After 10 weeks of treatment, electrophysiological measurements were performed during general anesthesia by intraperitoneal injection of pentobarbital (100 mg/kg). Atrial electrophysiological measurements were performed in open-chest experiments to assess local conduction disturbances and susceptibility to AF by direct contact epicardial mapping (21). One I/R-Sham, four I/R-Placebo, and one I/R-SAR rat died during the open-chest experiments. Hearts were removed and quickly preserved for biochemical and histological analyses.
Additional details are provided in the Supplemental Methods.
Biochemical and histological analyses
To visualize LA tissue fibrosis, cardiomyocyte diameter, and connexin 43 (Cx43), tissue preparation was performed as previously described (21), and 5-μm sections were either stained with Picro-Sirius Red (#13422.00500, Morphisto, Frankfurt am Main, Germany), hematoxylin and eosin (#2C-163, Waldeck, Münster, Germany), or anti-Cx43 (#MAB3068, Merck Millipore, Darmstadt, Germany). NIS-Elements BR 3.2 software (Nikon Instruments, Melville, New York) was used for the analysis. Gene expression analysis from human left ventricle, rat left ventricle, and rat LA tissue was performed by using TaqMan polymerase chain reaction. Western blot technique was used to determine protein expression. Additional details are provided in the Supplemental Methods.
Statistical analysis
All data are expressed as mean ± SEM. An unpaired Student’s t-test (2-tailed) was used for statistical analysis comparing 2 groups. For assessment of statistical significance between 4 groups, one-way analysis of variance followed by Tukey’s multiple comparison test was applied. Statistical analysis was conducted by using Prism version 6.01 software (GraphPad Software, La Jolla, California). The p values that reached a value <0.0001 were reported as p < 0.0001. Additional details are provided in the Supplemental Methods.
Results
Expression pattern of CatA in human ICM, in rats with permanent LAD ligation, and in rats with ventricular I/R
Human ICM
In LV samples of patients with heart failure and end-stage ICM, CatA gene and protein expression was enhanced compared with healthy nonfailing LV tissue (p = 0.0271 for messenger ribonucleic acid [mRNA] and p = 0.0333 for protein level) (Figures 1A and 1B). LV CatA up-regulation in ICM was associated with increased gene transcription of the heart failure marker atrial natriuretic peptide (p = 0.0296).
Figure 1.

Differential Expression Pattern of CatA in Human ICM and in Rats With Permanent Ligation and in Rats With Ventricular I/R
(A and B) Human ischemic cardiomyopathy (ICM). (A) Messenger ribonucleic acid (mRNA) expression of cathepsin A (CatA) and atrial natriuretic peptide (ANP) in left ventricular tissue from healthy nonfailing donor hearts (Ctr, n = 8) and from patients with end-stage ICM (n = 8). (B) Representative Western blot and quantification of CatA protein levels in the left ventricle of Ctr and ICM patients (n = 8 per group). (C and D) Rats with 30 min of ventricular ischemia followed by 10 weeks of reperfusion (ischemia/reperfusion [I/R]). (C) CatA and ANP mRNA content in the left ventricle (I/R-Sham, n = 9-8; I/R-Placebo, n = 14-13). (D) Left atrial tissue 10 weeks after I/R in sham-operated rats (I/R-Sham, n = 9) and rats receiving placebo (I/R-Placebo; n = 15-9). Values are mean ± SEM. GAPDH = glyceraldehyde 3-phosphate dehydrogenase; IOD = integrated optical density.
Rats with permanent LAD ligation
In a rat model with permanent LAD ligation (PL-rats), CatA protein abundance was significantly increased within the infarct area at 2 weeks (1.9-fold increase compared with sham control; p = 0.0009) and dropped back to control levels at 8 weeks. In contrast, CatA mRNA levels in the remote ventricular myocardium and in the LA myocardium were unchanged after 2 weeks but showed a significant increase at 8 weeks after PL (1.5-fold increase in left ventricle; p = 0.0342) and 1.8-fold increase in left atrium (compared with sham control; p = 0.0086), indicating a differential temporal and spatial pattern of CatA expression during PL (Supplemental Figure S1).
Rats with ventricular I/R
Given the late increase in CatA expression in the remote LV and LA myocardium at 8 weeks in PL-rats, we investigated the regulation of CatA expression in LV and LA myocardium in rats with 30 min of LAD ligation followed by 10 weeks of reperfusion (I/R); this animal model mimics the clinical situation of early revascularization after myocardial infarction and is suitable for investigating cardiac remote remodeling during ICM. In I/R-Placebo rats, mRNA levels of CatA were significantly increased in the remote left ventricle (1.7-fold; p = 0.0072) (Figure 1C) and left atrium (2.9-fold; p = 0.0042) (Figure 1D) compared with I/R-Sham rats; this finding was associated with increased gene transcription of atrial natriuretic peptide (vs. I/R-Sham, p = 0.0008).
LV structure and function in rats with ventricular I/R
I/R-Placebo rats exhibited an increased heart-weight-to-body-weight ratio compared with I/R-Sham rats (0.29 ± 0.02 g/100 mg body weight vs. 0.24 ± 0.01 g/100 mg body weight; p = 0.0360). Treatment of I/R-rats with the CatA inhibitor SAR or ramipril did not significantly reduce the heart-weight-to-body-weight ratio (I/R-SAR 0.26 ± 0.01 g/100 mg body weight vs. I/R-Ramipril 0.27 ± 0.01 g/100 mg body weight) but reduced plasma brain natriuretic peptide levels (I/R-SAR 81 ± 4 pg/ml vs. I/R-Placebo 172 ± 31 pg/ml [p = 0.0030]; I/R-Ramipril 105 ± 9 pg/ml vs. I/R-Placebo, p = 0.0455).
Infarct size, determined at the midventricular level, was comparable between I/R-Placebo rats (15.1 ± 1.6% fibrotic wall surface/total wall surface), I/R-SAR (14.4 ± 1.9% fibrotic wall surface/total wall surface), and I/R-Ramipril (15.6 ± 1.5% fibrotic wall surface/total wall surface) rats (Supplemental Figure S2).
Global LV function
I/R-Placebo rats developed a significant impairment in LV function, indicated by a reduced ejection fraction and increased end-diastolic and end-systolic volume at rest. EF reduction was prevented by SAR but not by ramipril (Table 1). LV systolic function and cardiac output in I/R-Placebo rats did not improve during dobutamine perfusion. During dobutamine stress testing, I/R-SAR, but not I/R-Ramipril, rats exhibited higher ejection fraction and cardiac output. However, ramipril, but not SAR, significantly reduced LV end-systolic and end-diastolic mass compared with findings in I/R-Placebo rats at rest and during dobutamine stress testing.
Table 1.
Global Left Ventricular Function Determined by Using Magnetic Resonance Imaging in Rats
| Left Ventricle at Rest |
Left Ventricle Under Dobutamine Stress |
|||||||
|---|---|---|---|---|---|---|---|---|
| I/R-Sham (n = 9) | I/R-Placebo (n = 15) | I/R-Ramipril (n = 14) | I/R-SAR (n = 14) | I/R-Sham (n = 9) | I/R-Placebo (n = 15) | I/R-Ramipril (n = 14) | I/R-SAR (n = 14) | |
| EF, % | 59.4 ± 1.6 | 39.2 ± 2.1∗ | 43.5 ± 3.2† | 49.1 ± 3.0‡ | 73.8 ± 2.6 | 43.6 ± 3.1∗ | 51.8 ± 4.8† | 58.5 ± 4.0‡ |
| CO, ml/min | 126.6 ± 5.2 | 121.0 ± 3.4 | 111.2 ± 4.6§ | 129.8 ± 3.5 | 159.5 ± 7.2 | 133.6 ± 6.3‖ | 132.2 ± 6.6‖¶ | 158.3 ± 3.6‡ |
| SV, μl | 327.7 ± 17.9 | 325.3 ± 8.6 | 296.7 ± 10.3 | 333.5 ± 7.9 | 372.2 ± 18.0 | 330.2 ± 12.4 | 312.8 ± 13.1‖¶ | 370.0 ± 10.0 |
| EDV, ml | 0.55 ± 0.03 | 0.86 ± 0.05† | 0.73 ± 0.06 | 0.70 ± 0.03 | 0.50 ± 0.02 | 0.80 ± 0.05† | 0.67 ± 0.06 | 0.66 ± 0.04 |
| ESV, ml | 0.22 ± 0.01 | 0.53 ± 0.05# | 0.43 ± 0.06‖ | 0.37 ± 0.04 | 0.13 ± 0.01 | 0.47 ± 0.05† | 0.36 ± 0.07 | 0.29 ± 0.04 |
| LVED mass, mg | 487.1 ± 13.9 | 534.3 ± 17.7 | 451.1 ± 16.2∗∗ | 503.6 ± 13.4 | 499.2 ± 8.8 | 528.3 ± 21.0 | 432.8 ± 17.5††¶ | 506.1 ± 13.9 |
| LVES mass, mg | 612.9 ± 14.7 | 646.9 ± 21.5 | 534.4 ± 20.4‖††§ | 622.3 ± 14.7 | 639.9 ± 12.7 | 642.2 ± 23.9 | 534.1 ± 25.4‖††§ | 646.5 ± 19.1 |
| Heart rate, beats/min | 389 ± 8 | 373 ± 7 | 375 ± 9 | 389 ± 3 | 427 ± 7 | 402 ± 8 | 421 ± 7 | 429 ± 5 |
Values are mean ± SEM. One-way analysis of variance followed by Tukey’s multiple comparisons test was used.
CO = cardiac output; EDV = end-diastolic volume; EF = ejection fraction; ESV = end-systolic volume; LVED = left ventricular end-diastolic; LVES = left ventricular end-systolic; SV = stroke volume.
p > 0.0001 vs. ischemia/reperfusion (I/R)-Sham.
p < 0.01 vs. I/R-Sham.
p < 0.05 vs. I/R-Placebo.
p < 0.01 vs. I/R–(S)-3-{[1-(2-Fluoro-phenyl)-5-methoxy-1H-pyrazole-3-carbonyl]-amino}-3-o-tolyl-propionic-acid (SAR).
p < 0.05 vs. I/R-Sham.
p < 0.05 vs. I/R-SAR.
p < 0.001 vs. I/R-Sham.
p < 0.01 vs. I/R-Placebo.
p < 0.001 vs. I/R-Placebo.
Regional LV function
In I/R-Placebo rats, MRI analysis of slices selected from the midventricular level of each heart revealed impaired wall motion and nearly akinesia in the infarcted area, which did not improve significantly during dobutamine stress testing, indicative of dysfunctional nonviable myocardium (Figures 2A and 2B, lateral and anterior, infarct area). Wall motion of the noninfarcted remote area was reduced at rest but still responsive to dobutamine (Figures 2A and 2B, septal and posterior, remote area), indicating dysfunctional but viable residual remote ventricular myocardium. Ramipril preserved better wall motion in the lateral infarcted area under basal conditions, but both ramipril and SAR improved dobutamine response in this area (I/R-Ramipril rats vs. I/R-Placebo rats; p = 0.0331; I/R-SAR rats vs. I/R-Placebo rats; p = 0.0289) (Figure 2B, under dobutamine). However, SAR, but not ramipril, was able to preserve better wall motion in the noninfarcted remote area at rest, which significantly further improved during dobutamine stress testing, suggesting more viable remote myocardium (under dobutamine: I/R-SAR vs. Ramipril; p = 0.0380; I/R-SAR vs. placebo; p = 0.0203).
Figure 2.

Regional Left Ventricular Function
Using magnetic resonance imaging, 16 short-axis cine imaging slices of ischemia/reperfusion (I/R)-Sham (n = 9), I/R-rats given placebo (n = 15), and I/R-rats treated with ramipril (n = 14) or (S)-3-{[1-(2-Fluoro-phenyl)-5-methoxy-1H-pyrazole-3-carbonyl]-amino}-3-o-tolyl-propionic-acid (SAR; n = 14) were performed; slices were divided into 97 cords. (A) Every cord was analyzed for left ventricular wall motion (WM) at rest and (B) during dobutamine stress testing. Analysis of left ventricular systolic wall thickness (SWTS) for every cord (C) at rest and (D) during dobutamine stress testing. Values are mean ± SEM. #I/R-SAR vs. I/R-Ramipril. ψI/R-SAR vs. I/R-Placebo. §I/R-Ramipril vs. I/R-Placebo.
I/R-Placebo rats displayed a dramatic thinning of the infarcted myocardial wall (anterolateral) in the infarct area and a moderate thinning in the noninfarcted myocardial wall (posteroseptal) in the remote area, determined by MRI-derived systolic wall thickness at rest and during dobutamine stress testing (Figures 2C and 2D). Thinning of the infarcted myocardial wall was partially prevented by ramipril and SAR, whereas treatment with SAR proved to be more effective in preventing thinning of the noninfarcted myocardial wall in the remote area compared with ramipril.
Histological Picro-Sirius Red staining of the left ventricle confirmed thinning of the infarcted wall in I/R-Placebo rats (I/R-Sham 2.28 ± 0.11 mm vs. I/R-Placebo 1.78 ± 0.17 mm; p = 0.1348) (Supplemental Figures S3A and S3B). LV wall thinning was significantly attenuated in hearts from I/R-SAR rats (2.33 ± 0.13 mm) compared with I/R-Placebo rats (p = 0.0407) and considerably improved by ramipril (2.15 ± 0.15 mm vs. I/R-Placebo; p = 0.2604). LV septal wall thickness was unchanged in all groups (Supplemental Figure S3C). LV fibrosis formation was significantly increased upon I/R in rats receiving placebo (I/R-Sham 2.59 ± 0.46% vs. I/R-Placebo 23.62 ± 2.83%; p < 0.0001) and was not prevented by treatment with either ramipril (I/R-Ramipril 16.41 ± 3.34% vs. I/R-Sham; p = 0.0092; I/R-Ramipril vs. I/R-Placebo; p = 0.2195) or SAR (I/R-SAR 18.19 ± 2.29% vs. I/R-Sham; p = 0.0027; vs. I/R-Placebo; p = 0.4613) (Supplemental Figure S3D).
Atrial emptying function
In I/R-Placebo rats, LA diameter was significantly increased and LA fractional shortening was reduced compared with I/R-Sham rats (Table 2). LA emptying function as assessed by total percent emptying and active percent emptying was diminished in I/R-Placebo rats. Pharmacological inhibition of CatA activity by SAR did not affect LA dimensions in I/R-rats, and LA fractional shortening was significantly higher compared with I/R-Placebo rats. SAR preserved the LA emptying function in I/R-rats. Ramipril treatment reduced LA diameter in I/R-rats, but similar dimensions in I/R-Sham rats were not achieved. In I/R-Ramipril rats, LA fractional shortening and total percent emptying were only mildly depressed compared with I/R-Sham rats.
Table 2.
Atrial Emptying Function Determined by Using Magnetic Resonance Imaging in Rats
| I/R-Sham (n = 9) | I/R-Placebo (n = 15) | I/R-Ramipril (n = 14) | I/R-SAR (n = 14) | |
|---|---|---|---|---|
| LA diameter, cm | 0.47 ± 0.01∗ | 0.57 ± 0.02 | 0.52 ± 0.02∗ | 0.54 ± 0.01 |
| LA volume, maximum, ml | 0.34 ± 0.04 | 0.48 ± 0.04 | 0.42 ± 0.03 | 0.41 ± 0.03 |
| LA fractional shortening, % | 20 ± 2† | 16 ± 1 | 18 ± 2 | 22 ± 1† |
| Total percent emptying | 50 ± 2† | 37 ± 3 | 46 ± 3† | 47 ± 2† |
| Active percent emptying | 37 ± 4† | 26 ± 3 | 33 ± 3† | 34 ± 2† |
| Passive percent emptying | 35 ± 6 | 24 ± 3 | 27 ± 1 | 28 ± 3 |
Values are mean ± SEM.
LA = left atrial; other abbreviations as in Table 1.
p < 0.05.
p < 0.01 vs. I/R-Placebo.
Atrial electrophysiology
AF could be induced in 6 of 8 sham-operated rats and in all I/R-Placebo (11 of 11) rats by atrial burst stimulation (Figure 3A). Inducible AF duration was markedly increased in I/R-Placebo rats compared with I/R-Sham rats (I/R-Placebo 45.9 ± 19.1 s vs. I/R-Sham 1.4 ± 0.4 s; p = 0.0196). The percentage of areas of slow conduction during rapid pacing was significantly increased in I/R-Placebo rats compared with I/R-Sham rats (p < 0.0001) (Figure 3B). Total atrial activation time was significantly longer in I/R-Placebo rats compared with I/R-Sham rats (I/R-Placebo 15.2 ± 0.5 ms vs. I/R-Sham 11.5 ± 0.5 ms; p = 0.0006) (Figure 3C). The atrial effective refractory period did not differ between the groups (Figure 3D). In I/R-rats, SAR and ramipril reduced AF inducibility (I/R-SAR: 10 of 13; I/R-Ramipril: 11 of 14) and inducible AF duration (I/R-SAR: 3.7 ± 1.6 s; p = 0.0108; I/R-Ramipril: 3.8 ± 1.8 s; p = 0.0092) compared with I/R-Placebo rats. SAR and ramipril attenuated changes in total atrial activation time (I/R-SAR 12.9 ± 0.34 ms; p = 0.0302 vs. I/R-Placebo; I/R-Ramipril 11.9 ± 0.6 ms; p = 0.0012 vs. I/R-Placebo). Both ramipril and SAR in particular significantly reduced the percentage of areas of slow conduction.
Figure 3.
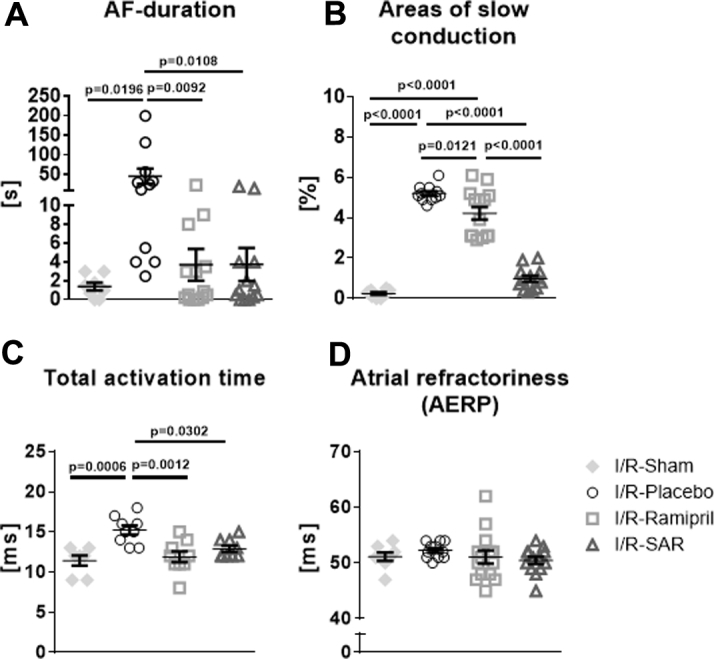
Atrial Electrophysiological Measurements in I/R-Rats
(A) Atrial fibrillation (AF) inducibility and duration induced by burst-stimulation in I/R-Sham (n = 8), I/R-rats given placebo (n = 11), and I/R-rats treated with ramipril (n = 14) or SAR (n = 13). (B) Areas of slow conduction. (C) Total atrial activation time (I/R-Sham, n = 7; I/R-Placebo, n = 9; I/R-Ramipril, n = 9; I/R-SAR, n = 9). (D) Left atrial effective refractory period (AERP) at a stimulation frequency of 400/min (basic cycle lengths: 150 ms). Values are mean ± SEM. Abbreviations as in Figures 1 and 2.
LA-ECM remodeling
LA fibrosis
In Picro-Sirius Red–stained LA sections, the degree of atrial total collagen content was highly increased in I/R-Placebo rats compared with I/R-Sham rats (Figures 4A to 4D). Increased fibrosis formation in I/R-Placebo rats was associated with significantly enhanced mRNA expression of collagens 1a1, 1a2, and 3a1 and a distinct increase in collagen 4a1. Inhibition of CatA by SAR treatment significantly prevented accumulation of tissue fibrosis and significantly dampened collagen 1a1 mRNA expression (vs. I/R-Placebo; p = 0.0334). In contrast, ramipril did not significantly affect fibrosis formation or collagen mRNA levels (Figures 4C and 4E). Polarized light microscopy of Picro-Sirius Red–stained LA sections (Figure 4B) revealed an increase in the ratio of collagen type I (red-yellow fibers) to collagen type III (green fibers) in I/R-Placebo rats (vs. I/R-Sham; p = 0.0023). The collagen type I/type III ratio was normalized by both ramipril (vs. I/R-Placebo; p = 0.0034) and even more pronounced by SAR (vs. I/R-Placebo; p < 0.0001) (Figure 4D).
Figure 4.

Atrial Extracellular Matrix Remodeling
Representative histological images of (A) conventional total collagen (red fibers) and (B) polarized light microscopy (collagen type I [red-yellow fibers] and collagen type III [green fibers]) of Picro-Sirius Red–stained left atrial preparations in I/R-Sham (n = 9), I/R-rats given placebo (n = 13), and I/R-rats treated with ramipril (n = 12) or SAR (n = 11). (C) Quantification of left atrial (LA) fibrosis amount. (D) Collagen type I/type III ratio and (E) mRNA expression of collagen 1a (Col1a1), Col1a2, Col3a1, and Col4a1 normalized to expression of GAPDH in I/R-Sham (n = 9-7), I/R-Placebo (n = 14), I/R-Ramipril (n = 11-10), and I/R-SAR (n = 14-12). Values are mean ± SEM. Abbreviations as in Figures 1 and 2.
LA hypertrophy
LA weight-to-body-weight ratio and LA myocyte diameter are shown in Figures 5A to 5C. The LA weight-to-body-weight-ratio was significantly enhanced in I/R-rats given placebo (Figure 5B). Although ramipril had only a slight effect, SAR significantly lowered the LA weight-to-body-weight-ratio to the I/R-Sham level. Myocyte diameters were unchanged with placebo, SAR, and ramipril (Figure 5C). The LA mRNA level of hypertrophy marker brain natriuretic peptide was significantly increased in I/R-Placebo rats (Figure 5D) and significantly lowered only by ramipril but not by SAR. In I/R-rats, neither treatment with SAR nor ramipril blunted the increased LA CatA gene expression (I/R-SAR 2.25 ± 0.43 relative gene expression/glyceraldehyde 3-phosphate dehydrogenase vs. I/R-Sham; p = 0.0427; I/R-Ramipril 4.33 ± 1.58 relative gene expression/glyceraldehyde 3-phosphate dehydrogenase vs. I/R-Sham; p = 0.0438).
Figure 5.

Atrial Hypertrophy
Representative histological images of (A) hematoxylin and eosin–stained LA tissue preparations in I/R-Sham rats, I/R-rats given placebo, and I/R-rats treated with ramipril or SAR. (B) Ratio of LA weight per 100 g body weight of I/R-Sham (n = 9), I/R-Placebo (n = 12), I/R-Ramipril (n = 12), and I/R-SAR (n = 13) rats. (C) Quantification of LA myocyte diameter and (D) brain natriuretic peptide (BNP) mRNA level in the left atrium in I/R-Sham (n = 9), I/R-rats given placebo (n = 11-12) and I/R-rats treated with ramipril (n = 11-12) or SAR (n = 11-14). Values are mean ± SEM. Abbreviations as in Figures 1, 2, and 4.
LA spatial distribution of Cx43
In I/R-Sham rats, we found the typical intracellular distribution of Cx43, with a clear accentuation of Cx43 at the cell poles, and only sparse localization of Cx43 at the lateral sides of the cells (Figure 6). In I/R-Placebo rats, there was a significantly enhanced lateral and polar Cx43 immunostaining. Lateralization of Cx43 was significantly prevented by SAR (vs. I/R-Placebo; p = 0.0004), whereas polar Cx43 expression remained enhanced compared with control (vs. I/R-Sham; p = 0.0039). Ramipril did not significantly prevent Cx43 lateralization (vs. I/R-Sham; p = 0.0018), and polar Cx43 localization remained unchanged compared with I/R-Sham rats (p = 0.0697).
Figure 6.
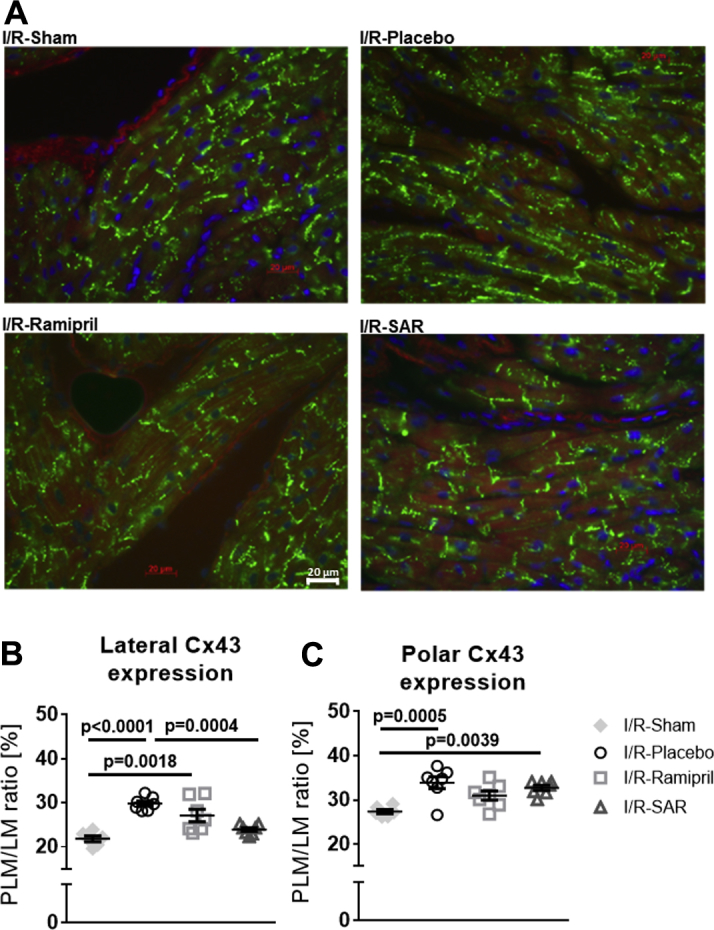
Atrial Cx43 Distribution
(A) Representative immunohistochemical staining for connexin 43 (Cx43) in left atrial tissue preparations in I/R-Sham (n = 6), I/R-rats given placebo (n = 7), and I/R-rats treated with ramipril (n = 7) or SAR (n = 7). (B) Quantification of Cx43-positively stained lateral and (C) polar cardiomyocyte membrane. Values are mean ± SEM. Other abbreviations as in Figure 1.
Discussion
CatA expression is significantly increased in human failing ICM ventricular myocardium and in ventricular and atrial myocardium from rats with experimental ICM induced by permanent LAD ligation or ventricular I/R in rats. Herein, increased ventricular and atrial expression of CatA was accompanied by LV global systolic dysfunction and the development of remote atrial cardiomyopathy characterized by atrial ECM remodeling, impaired LA emptying function, conduction disturbances, and longer inducible AF duration. Pharmacological inhibition of CatA initiated at the time point of ventricular reperfusion did not significantly improve LV global systolic function at rest but preserved more viable LV myocardium and prevented the progression of atrial cardiomyopathy.
Ventricular remote remodeling and atrial arrhythmogenic substrate in I/R-rats
Ventricular and atrial remodeling in rats with permanent LAD ligation has been previously described 12, 13. To the best of our knowledge, the present analysis is the first detailed characterization of the development of ICM-induced ECM remodeling and arrhythmogenic and functional atrial cardiomyopathy in a heart failure rat model with ventricular I/R 22, 23. Thirty minutes of myocardial ischemia induced by LAD ligation followed by 10 weeks of reperfusion led to global LV systolic dysfunction and impaired LV wall motion, with dysfunctional and nonviable myocardium in the infarct area and dysfunctional but partially viable myocardium in the LV remote noninfarcted myocardium. LV and LA CatA expression remote from the infarct area was increased at 10 weeks. This scenario was associated with the development of an atrial cardiomyopathy characterized by impaired atrial emptying function and increased AF susceptibility. The documented structural atrial changes, together with the disorganization of Cx43, resulted in the disruption of side-to-side electrical connections between muscle bundles, thereby contributing to the conduction abnormalities and increased susceptibility to AF observed in I/R-rats 24, 25. Epicardial mapping of these structurally remodeled atria revealed longer total atrial activation times, larger areas of slower local atrial conduction, and an unchanged atrial effective refractory period. Overall, I/R-rats display characteristics consistent with changes observed in patients with ICM and various animal models for ICM-associated AF 26, 27, 28, 29.
Role of CatA for ventricular remote remodeling and atrial cardiomyopathy
In our rat model for ICM, pharmacological inhibition of CatA activity did not significantly improve LV global systolic function at rest; however, it did prevent wall thinning and preserve more viable myocardium in the LV infarct area and the LV noninfarcted remote myocardium without reducing LV fibrosis. In each of these parameters, SAR was more efficacious than ramipril.
We showed that this antiremodeling effect occurred not only in the ventricular infarct area and noninfarcted remote myocardium but also in the atrium. Inhibition of CatA activity hampered LA fibrosis formation and reduced gene expression of fibrillary collagen types I and III, which are known to be dysregulated in cardiac disease 14, 30, 31. In the heart, the predominant fibrillary collagens are the rigid type I (80%) and the elastic type III (11%), providing structure and elasticity to the ECM 14, 31. The extent of this remodeling process is controlled by the balance between ECM synthesis and degradation, which is tightly regulated by proteolytic enzymes (e.g., cathepsins) as a key mechanism to control ECM function and turnover 16, 17. Increased fibrosis formation directly influences architecture of the interstitial matrix, cell-to-cell coupling, cardiomyocyte excitability, and Cx43 distribution, impairing proper electrical conduction of the myocardium 15, 32. In our rat model, I/R-Placebo rats exhibited increased atrial fibrosis formation and a higher collagen type I to type III ratio, providing the substrate for stiffer, less compliant atria. Attenuation of increased atrial fibrosis formation and normalization of the collagen type I to type III ratio by pharmacological CatA inhibition was associated with less lateralization of Cx43, preserving physiological coupling between the atrial bundles network and contributing to preserved atrial function and reduced AF susceptibility by fewer conduction disturbances. Importantly, the effect of SAR-mediated CatA inhibition on the development of atrial cardiomyopathy and atrial fibrosis in I/R-rats was more pronounced compared with ramipril, and it occurred independent of significant improvements in LV global systolic function or total LV fibrosis, suggesting prevention of atrial ECM remodeling beyond a simple consequence of improved LV function.
These findings support the concept that remote remodeling in ICM is not restricted to the noninfarcted ventricular myocardium 3, 4, 5, 6 but also manifests in the atrium and is critically mediated by ECM proteinases such as CatA. This theory is in line with our previous studies, showing that cardiomyocyte-specific overexpression of CatA in mice mediated AF susceptibility and initiates the development of structural remodeling in the atrium, even in the absence of ventricular dysfunction of other stressors (21). A previous proteomic analysis performed in mice with permanent LAD ligation found that pharmacological inhibition of CatA activity partially restored the infarction-induced alterations of proteins associated with ECM remodeling (33). Our study extended these findings by showing the cardiac functional and structural changes of CatA inhibition in rats with I/R. These changes translate the biochemical beneficial effects of CatA inhibition into a common clinical scenario of early reperfusion following myocardial infarction by using clinically relevant functional imaging techniques and in vivo electrophysiological measures of LV and LA function.
SAR exhibits high cell permeability (20). Pharmacological inhibition of CatA by SAR may therefore display its beneficial effects by inhibition of both intracellular and extracellular CatA. Due to the design of this in vivo study, which mainly focused on the effect of SAR on clinically relevant measures of ventricular and atrial remodeling and function, the mechanism by which SAR prevented cardiac remodeling could not be identified. Further biochemical studies are warranted to identify potential intracellular and extracellular target proteins of CatA to explain the mechanisms underlying the benefit of pharmacological CatA inhibition on ECM remodeling in the heart.
Study limitations
Although we found strong indications for the development of an arrhythmogenic substrate in the atrium, we observed no spontaneous, nonsustained episodes of AF in these study rats, probably due to the small size of the atrium. The determination of atrial activation times by using conventional contact mapping procedures in small animals has several technical limitations, including spatial and time resolution disparities. Due to the tissue size of the small rat atria, we had to focus on detailed histological and biochemical analyses but could not identify the underlying cellular mechanisms by which pharmacological CatA inhibition attenuates the development of an arrhythmogenic substrate. We did not investigate whether a combination of ramipril and SAR would show synergistic effects and whether the agents tested are able to reverse pre-existing structural and functional atrial remodeling. These findings would be of clinical relevance, because upstream therapy to prevent development of the AF substrate cannot be started early enough. We performed LAD ligation distal to possible atrial branches. Therefore, ischemic effects on atrial myocardium, which have been described following ligation of the right coronary artery or the left circumflex artery, should be limited 34, 35.
Conclusions
Cardiac CatA gene expression was significantly increased in human ICM and experimental ICM in various rat models. Ventricular I/R in rats was associated with reduced LV global systolic function and the development of atrial cardiomyopathy. Pharmacological CatA inhibition initiated at the time point of reperfusion preserved more viable LV myocardium and prevented ECM remodeling and the development of an arrhythmogenic and functional substrate for AF, independent of preserved LV function at rest. Whether preservation of LV viable myocardium, in the absence of improved LV systolic function, helps prevent atrial cardiomyopathy in the setting of ICM and whether LV viable myocardium represents a more suitable metric than LV systolic function and LV fibrosis for the assessment of remote remodeling to guide AF upstream therapy warrant further study (36).
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Herein we report that a pharmacological intervention, which preserves more viable ventricular myocardium in ICM with no significant improvements in LV global systolic function, goes hand in hand with a prevention of atrial cardiomyopathy. This observation suggests that atrial cardiomyopathy most likely represents the manifestation of global remote ECM remodeling in ICM and is not simply the result of ICM-related reduction in LV systolic function. Ventricular adverse remodeling and the amount of viable myocardium are powerful predictors for the development of chronic heart failure after myocardial infarction and therefore remain an important pharmacological treatment target, even independent from crude global parameters such as LV systolic function in heart failure.
TRANSLATIONAL OUTLOOK: Further clinical studies are warranted to test whether preservation of LV viable myocardium, even without improvements in LV global systolic function, translates into prevention of atrial cardiomyopathy in the setting of ICM and whether LV viable myocardium represents a better metric than LV global systolic function to guide AF upstream therapy. In this context, the CatA inhibitor SAR has already shown a favorable safety profile in early Phase I studies in healthy young and elderly human subjects alike.
Footnotes
This research was supported by the German Research Foundation (DFG SFB/TRR219-M02/-S02). Ms. Lang received a scholarship by the “Stiftung Begabtenförderung berufliche Bildung (SBB) GmbH im Auftrag und mit Mitteln des Bundesministeriums für Bildung und Forschung.” Drs. Sadowski, Hübschle, and Ruf are employees of Sanofi-Aventis Deutschland GmbH. Dr. Sanders is an advisory board member for Biosense-Webster, Medtronic, St. Jude Medical, Boston Scientific, and CathRx; has received lecture and/or consulting fees from Biosense-Webster, Medtronic, St. Jude Medical, and Boston Scientific; and is supported by a Practitioner Fellowship from the National Health and Medical Research Council of Australia and by the National Heart Foundation of Australia. Dr. Böhm has received funding from Amgen, Bayer, Servier, Medtronic, Boehringer Ingelheim, Vifor, and Bristol-Myers Squibb. Dr. Schotten has received funding from EP and YourRhythmics BV. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and U.S. Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
References
- 1.Køber L., Torp-Pedersen C., Carlsen J.E. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Trandolapril Cardiac Evaluation (TRACE) Study Group. N Engl J Med. 1995;333:1670–1676. doi: 10.1056/NEJM199512213332503. [DOI] [PubMed] [Google Scholar]
- 2.Neumann F.J., Sousa-Uva M., Ahlsson A. 2018 ESC/EACTS guidelines on myocardial revascularization. Eur Heart J. 2019;40:87–165. doi: 10.1093/eurheartj/ehy855. [DOI] [PubMed] [Google Scholar]
- 3.Carberry J., Carrick D., Haig C. Remote zone extracellular volume and left ventricular remodeling in survivors of ST-elevation myocardial infarction. Hypertension. 2016;68:385–391. doi: 10.1161/HYPERTENSIONAHA.116.07222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson E.M., Moainie S.L., Baskin J.M. Region- and type-specific induction of matrix metalloproteinases in post-myocardial infarction remodeling. Circulation. 2003;107:2857–2863. doi: 10.1161/01.CIR.0000068375.40887.FA. [DOI] [PubMed] [Google Scholar]
- 5.Wijns W., Vatner S.F., Camici P.G. Hibernating myocardium. N Engl J Med. 1998;339:173–181. doi: 10.1056/NEJM199807163390307. [DOI] [PubMed] [Google Scholar]
- 6.Bax J.J., Poldermans D., Elhendy A. Improvement of left ventricular ejection fraction, heart failure symptoms and prognosis after revascularization in patients with chronic coronary artery disease and viable myocardium detected by dobutamine stress echocardiography. J Am Coll Cardiol. 1999;34:163–169. doi: 10.1016/s0735-1097(99)00157-6. [DOI] [PubMed] [Google Scholar]
- 7.Meluzín J., Cerný J., Frélich M. Prognostic value of the amount of dysfunctional but viable myocardium in revascularized patients with coronary artery disease and left ventricular dysfunction. Investigators of this Multicenter Study. J Am Coll Cardiol. 1998;32:912–920. doi: 10.1016/s0735-1097(98)00324-6. [DOI] [PubMed] [Google Scholar]
- 8.Pagley P.R., Beller G.A., Watson D.D., Gimple L.W., Ragosta M. Improved outcome after coronary bypass surgery in patients with ischemic cardiomyopathy and residual myocardial viability. Circulation. 1997;96:793–800. doi: 10.1161/01.cir.96.3.793. [DOI] [PubMed] [Google Scholar]
- 9.Rizzello V., Poldermans D., Boersma E. Opposite patterns of left ventricular remodeling after coronary revascularization in patients with ischemic cardiomyopathy: role of myocardial viability. Circulation. 2004;110:2383–2388. doi: 10.1161/01.CIR.0000145115.29952.14. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen O.D., Søndergaard P., Nielsen T. Atrial fibrillation, ischaemic heart disease, and the risk of death in patients with heart failure. Eur Heart J. 2006;27:2866–2870. doi: 10.1093/eurheartj/ehl359. [DOI] [PubMed] [Google Scholar]
- 11.Lubitz S.A., Benjamin E.J., Ellinor P.T. Atrial fibrillation in congestive heart failure. Heart Fail Clin. 2010;6:187–200. doi: 10.1016/j.hfc.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardin S., Guasch E., Luo X. Role for MicroRNA-21 in atrial profibrillatory fibrotic remodeling associated with experimental postinfarction heart failure. Circ Arrhythm Electrophysiol. 2012;5:1027–1035. doi: 10.1161/CIRCEP.112.973214. [DOI] [PubMed] [Google Scholar]
- 13.Jumeau C., Rupin A., Chieng-Yane P. Direct thrombin inhibitors prevent left atria remodeling associated with heart failure in rats. J Am Coll Cardiol Basic Trans Science. 2016;1:328–339. doi: 10.1016/j.jacbts.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber K.T. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- 15.De Jong S., van Veen T.A., van Rijen H.V., de Bakker J.M. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011;57:630–638. doi: 10.1097/FJC.0b013e318207a35f. [DOI] [PubMed] [Google Scholar]
- 16.Müller A.L., Dhalla N.S. Role of various proteases in cardiac remodeling and progression of heart failure. Heart Fail Rev. 2012;17:395–409. doi: 10.1007/s10741-011-9269-8. [DOI] [PubMed] [Google Scholar]
- 17.Cheng X.W., Shi G.P., Kuzuya M., Sasaki T., Okumura K., Murohara T. Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation. 2012;125:1551–1562. doi: 10.1161/CIRCULATIONAHA.111.066712. [DOI] [PubMed] [Google Scholar]
- 18.Ruf S., Buning C., Schreuder H. Inhibition of CatA: an emerging strategy for the treatment of heart failure. Future Med Chem. 2013;5:399–409. doi: 10.4155/fmc.13.24. [DOI] [PubMed] [Google Scholar]
- 19.Hiraiwa M. Cathepsin A/protective protein: an unusual lysosomal multifunctional protein. Cell Mol Life Sci. 1999;56:894–907. doi: 10.1007/s000180050482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seyrantepe V., Hinek A., Peng J. Enzymatic activity of lysosomal carboxypeptidase (cathepsin) A is required for proper elastic fiber formation and inactivation of endothelin-1. Circulation. 2008;117:1973–1981. doi: 10.1161/CIRCULATIONAHA.107.733212. [DOI] [PubMed] [Google Scholar]
- 21.Linz D., Hohl M., Dhein S. Cathepsin A mediates susceptibility to atrial tachyarrhythmia and impairment of atrial emptying function in Zucker diabetic fatty rats. Cardiovasc Res. 2016;110:371–380. doi: 10.1093/cvr/cvw071. [DOI] [PubMed] [Google Scholar]
- 22.Ruf S., Buning C., Schreuder H. Novel β-amino acid derivatives as inhibitors of cathepsin A. J Med Chem. 2012;55:7636–7649. doi: 10.1021/jm300663n. [DOI] [PubMed] [Google Scholar]
- 23.Wohlfart P., Linz W., Hübschle T. Cardioprotective effects of lixisenatide in rat myocardial ischemia-reperfusion injury studies. J Transl Med. 2013;11:84. doi: 10.1186/1479-5876-11-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wakili R., Voigt N., Kääb S., Dobrev D., Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011;121:2955–2968. doi: 10.1172/JCI46315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schotten U., Verheule S., Kirchhof P., Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y., Ducharme A., Li D., Gaspo R., Nattel S., Tardif J.C. Remodeling of atrial dimensions and emptying function in canine models of atrial fibrillation. Cardiovasc Res. 2001;52:217–225. doi: 10.1016/s0008-6363(01)00377-7. [DOI] [PubMed] [Google Scholar]
- 27.Li D., Fareh S., Leung T.K., Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95. doi: 10.1161/01.cir.100.1.87. [DOI] [PubMed] [Google Scholar]
- 28.Lau D.H., Psaltis P.J., Mackenzie L. Atrial remodeling in an ovine model of anthracycline-induced nonischemic cardiomyopathy: remodeling of the same sort. J Cardiovasc Electrophysiol. 2011;22:175–182. doi: 10.1111/j.1540-8167.2010.01851.x. [DOI] [PubMed] [Google Scholar]
- 29.Sanders P., Morton J.B., Davidson N.C. Electrical remodeling of the atria in congestive heart failure: electrophysiological and electroanatomic mapping in humans. Circulation. 2003;108:1461–1468. doi: 10.1161/01.CIR.0000090688.49283.67. [DOI] [PubMed] [Google Scholar]
- 30.Boixel C., Fontaine V., Rucker-Martin C. Fibrosis of the left atria during progression of heart failure is associated with increased matrix metalloproteinases in the rat. J Am Coll Cardiol. 2003;42:336–344. doi: 10.1016/s0735-1097(03)00578-3. [DOI] [PubMed] [Google Scholar]
- 31.Li L., Zhao Q., Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018;68–69:490–506. doi: 10.1016/j.matbio.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 32.Jansen J.A., van Veen T.A., de Jong S. Reduced Cx43 expression triggers increased fibrosis due to enhanced fibroblast activity. Circ Arrhythm Electrophysiol. 2012;5:380–390. doi: 10.1161/CIRCEP.111.966580. [DOI] [PubMed] [Google Scholar]
- 33.Petrera A., Gassenhuber J., Ruf S. Cathepsin A inhibition attenuates myocardial infarction-induced heart failure on the functional and proteomic levels. J Transl Med. 2016;14:153. doi: 10.1186/s12967-016-0907-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishida K., Qi X.Y., Wakili R. Mechanisms of atrial tachyarrhythmias associated with coronary artery occlusion in a chronic canine model. Circulation. 2011;123:137–146. doi: 10.1161/CIRCULATIONAHA.110.972778. [DOI] [PubMed] [Google Scholar]
- 35.Alasady M., Shipp N.J., Brooks A.G. Myocardial infarction and atrial fibrillation: importance of atrial ischemia. Circ Arrhythm Electrophysiol. 2013;6:738–745. doi: 10.1161/CIRCEP.113.000163. [DOI] [PubMed] [Google Scholar]
- 36.Tillner J., Lehmann A., Paehler T. Tolerability, safety, and pharmacokinetics of the novel cathepsin A inhibitor SAR164653 in healthy subjects. Clin Pharmacol Drug Dev. 2016;5:57–68. doi: 10.1002/cpdd.201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.