

Abstract
OBJECTIVE
Elevated serum uric acid (SUA) is increasingly recognized as a risk factor for kidney disease in adults with diabetes, but data in youth are limited. We hypothesized that elevated SUA predicts development of elevated urinary albumin excretion (UAE) and hypertension over time in teens with type 2 diabetes (T2D).
RESEARCH DESIGN AND METHODS
Serum creatinine, cystatin C, SUA, and the urine albumin-to-creatinine ratio (UACR) were assessed in 539 obese youth, ages 12–17 years, with T2D duration <2 years at baseline in the Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) study. Estimated glomerular filtration rate (eGFR) was calculated using creatinine and cystatin C. Hypertension was defined as systolic or diastolic blood pressure ≥130/80 mmHg and elevated UAE as UACR ≥30 mg/g. Cox proportional hazards models evaluated the relationship between SUA and outcome variables longitudinally over an average follow-up of 5.7 years, adjusting for age, sex, race/ethnicity, BMI, HbA1c, eGFR, ACE inhibitor/angiotensin receptor blocker use, and TODAY treatment group assignment.
RESULTS
At baseline, hyperuricemia (≥6.8 mg/dL) was present in 25.6% of participants, hypertension in 18.7%, and elevated UAE in 6.1%. During follow-up of up to 7 years, hypertension developed in 37.4% and UAE in 18.0%. Higher baseline SUA increased the risk of incident hypertension (hazard ratio [HR] 1.19, 95% CI 1.03–1.38, per 1 mg/dL increase in SUA) and elevated UAE (HR 1.24, 95% CI 1.03–1.48) in adjusted models.
CONCLUSIONS
Hyperuricemia was common in youth with T2D. Higher baseline SUA independently increased the risk for onset of hypertension and elevated UAE. Research is needed to determine whether SUA-lowering therapies can impede development of diabetic kidney disease and hypertension in T2D youth.
Introduction
Diabetic kidney disease (DKD) and cardiovascular disease (CVD) are leading causes of morbidity and mortality in type 2 diabetes (T2D) (1) and develop at an alarming rate in adolescents with T2D (2–4). Previous analyses of the longitudinal data from the Treatment Options for type 2 Diabetes in Adolescents and Youth (TODAY) study demonstrated that the prevalence of elevated urinary albumin excretion (UAE) increased from 6.3% at baseline to 16.6%, while the prevalence of hypertension increased from 11.6% at baseline to 33.8% at an average follow-up of 3.9 years (3). Hypertension is closely related to DKD and serves as both a risk factor and clinical manifestation of DKD and CVD in T2D.
Once overt DKD and CVD manifest, progression can often be postponed, but rarely prevented, by current strategies for glycemic, blood pressure, and lipid control (5). Accordingly, alternative therapeutic targets are needed to supplement traditional treatments in preventing the development and progression of DKD and CVD in youth-onset T2D.
Elevated serum uric acid (SUA) is increasingly recognized as an important risk factor for hypertension and DKD in adults with type 1 diabetes (T1D) (6) or T2D and the general population (7). Mechanisms whereby elevated SUA confers increased risk for CVD and DKD are incompletely defined. Studies have suggested that inflammation (8), insulin resistance (9), intrarenal hemodynamic dysfunction (10), vascular, glomerular, and tubular injuries (9,11), and loss of nephron mass (12,13) could explain an etiologic relationship between elevated SUA and vascular disease in T2D and how elevated SUA may accelerate progression of DKD and CVD. To our knowledge, there are no longitudinal data on the relationship between SUA and hypertension or elevated UAE in adolescents with T2D.
Our aim was to investigate the longitudinal relationship between SUA and hypertension or elevated UAE during the 7 years of study. We hypothesized that higher SUA in youth with T2D would confer greater risk of incident hypertension and elevated UAE. Moreover, we sought to add to our previous work (3,14) by describing the prevalence of hyperuricemia (defined as SUA ≥6.8 mg/dL) in the TODAY study of youth-onset T2D at baseline and to extend the elevated UAE and hypertension data in TODAY participants to 7 years.
Research Design and Methods
TODAY Randomized Clinical Trial
The rationale, design, and methods of the TODAY trial have been reported in detail (15). Beginning in July 2004 and ending in February 2009, 699 participants were randomly assigned to metformin monotherapy, metformin plus rosiglitazone, or metformin plus an intensive lifestyle intervention program. Eligibility criteria included youth 10–17 years of age with T2D according to American Diabetes Association 2002 criteria with diabetes duration <2 years, overweight or obese (BMI ≥85th percentile), negative diabetes-associated autoantibodies, fasting C-peptide >0.6 ng/mL, and an adult caregiver willing to support study participation. Participants were excluded for refractory hypertension, defined as blood pressure ≥150/95 mmHg despite appropriate medical therapy, or a calculated Cockcroft and Gault creatinine clearance <70 mL/min. Eligible subjects entered a 2- to 6-month run-in period with goals of weaning from nonstudy diabetes medications, tolerating metformin up to a dose of 1,000 mg twice daily but no less than 500 mg twice daily, attaining glycemic control (HbA1c <8% for at least 2 months) on metformin alone, mastering standard diabetes education, and demonstrating adherence to study medication and visit attendance. Study medication arms were masked to investigators, study personnel, and participants.
The primary objective of the parent study was to compare the three treatment arms (metformin alone, metformin plus rosiglitazone, and metformin plus intensive lifestyle intervention) on time to treatment failure, classified as time to loss of glycemic control, defined as HbA1c ≥8% for 6 months or sustained metabolic decompensation requiring insulin. Half of the cohort reached the primary end point, and the results demonstrated that adding rosiglitazone to metformin was associated with more durable glycemic control after an average follow-up of 3.9 years (range 2–6.5). Insulin was initiated at the time of the primary outcome. Secondary aims included comparison of hypertension and microvascular complications. Participants received standardized treatment for confirmed hypertension and/or elevated UAE.
The TODAY study was extended for postintervention follow-up (TODAY2 phase 1, T2P1) that lasted 3 years and began immediately after the TODAY clinical trial was completed. One objective of the T2P1 longitudinal follow-up study was to continue observation of the TODAY cohort beyond the end of the TODAY randomized clinical trial after randomized treatment was discontinued. T2P1 provided medications for diabetes management (i.e., metformin and/or insulin) as well as for hypertension and elevated UAE. Treatment for hypertension included dietary intervention with a registered dietitian, initiation of an ACE inhibitor, and, if blood pressure remained elevated, additional antihypertensive medications at the discretion of the treating physician. Participants with elevated UAE with or without hypertension were treated with an ACE inhibitor, unless contraindicated. If elevated UAE persisted despite the maximum dose of the ACE inhibitor, additional medications could be added at the discretion of the treating physician or in consultation with a nephrologist. Results presented in this report are secondary analyses using observational data from the parent TODAY clinical trial in addition to T2P1. The protocol was approved by the institutional review boards of all participating institutions, and appropriate informed consent and assent was obtained.
Study Sample
The analysis included 539 of the 699 TODAY participants. Those excluded for lack of baseline SUA data did not differ significantly on any demographic or baseline characteristic (sex, race/ethnicity, age, BMI, or HbA1c) from the 539 included in this study. Analyses included all data available for the TODAY and T2P1 participants at each annual visit time points up to 84 months of follow-up (baseline and 12, 24, 36, 48, 60, 72, and 84 months). Participants were monitored for an average of 5.7 ± 1.7 years.
Study Measures and Laboratory Methods
Demographic data were collected at randomization. Weight, height, and calculated BMI were obtained at randomization and at every study visit thereafter. Blood samples were obtained at baseline and annually and processed immediately according to standardized procedures and shipped on dry ice for analysis at the TODAY central biochemical laboratory (15). SUA was measured by colorimetric method on Roche Modular P autoanalyzer assay (Roche Diagnostics, Indianapolis, IN). Hyperuricemia was defined as SUA ≥6.8 mg/dL, which represents the solubility of uric acid at normal physiological pH and temperature (16). As a sensitivity analysis, we also conducted analyses with sex-specific cutoffs for hyperuricemia (SUA: male ≥7 mg/dL; female ≥6 mg/dL), which provided similar findings. HbA1c (high-performance liquid chromatography) and insulin (double-antibody radioimmunoassay) assays were performed as previously described (3). Insulin sensitivity was calculated annually from 1/fasting insulin (mL/µU), which correlates strongly with hyperinsulinemic-euglycemic clamp–derived in vivo insulin sensitivity in obese youth with or without T2D (17).
Concentrations of creatinine in serum and urine were determined annually by using the Creatinine Plus enzymatic Roche reagent on a Modular P analyzer (Roche Diagnostics). The results of this procedure are traceable to the isotope dilution mass spectrometry reference method and allow for accurate estimated glomerular filtration rate (eGFR). The reportable range of creatinine in is 0.03–60.0 mg/dL serum/plasma samples and 0.03–1,200.0 mg/dL in urine samples. Concentration of cystatin C in serum was determined at baseline and annually by immunochemistry using Siemens reagents (Siemens Healthcare Diagnostics, Newark, DE) on a Siemens nephelometer autoanalyzer (BNII). This method is standardized against the International Federation of Clinical Chemistry and Laboratory Medicine/European Reference Material DA-471 Reference Material (RT Corp, Laramie, WY). Because of the expected normal-to-elevated GFRs for age, we calculated eGFR by the Zappitelli combined creatinine and cystatin C equation (eGFR = 25.38 * [1/serum cystatin C]0.331 * [1/serum creatinine]0.602 * [1.88height]), which has demonstrated strong agreement with measured GFRs at these ranges for adolescents (18,19). As a sensitivity analysis, we also calculated eGFR by the Full Age Spectrum (FAS) combined serum creatinine (SCr) and serum cystatin C (ScysC) equation, which has been newly validated in both pediatrics and adults and lends itself well to studies examining the transition from pediatric to early adulthood (20):
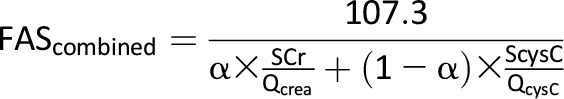 |
The FAS equation is based on normalized serum creatinine (SCr/Q), where Qcrea is the median SCr from healthy populations to account for age and sex, and QcysC is defined as 0.82 mg/L for ages <70 years. The coefficient α in the denominator is a weighting factor for the normalized renal biomarkers. We used α = 0.5, which means the denominator is equal to the average of both normalized biomarkers (20).
Blood pressure was measured using a CAS 740 monitor with standardized oscillometric cuff sizes at every visit, after a 5-min rest with the participant sitting. Three measurements were taken at 1-min intervals. The average of the second and third systolic and diastolic measures was calculated to obtain blood pressure at that visit. Hypertension was defined as an average systolic (SBP) or diastolic (DBP) blood pressure ≥130/80 mmHg or ≥95th percentile for age, sex, and height measured on at least two consecutive study visits and one interim visit. Mean arterial pressure was calculated as (2 × DBP + SBP)/3. The urine albumin-to-creatinine ratio (UACR) was measured at baseline and annually thereafter unless a result was abnormal. Spot urine samples were obtained after an overnight fast of 10 to 14 h. Elevated UAE (referred to as albuminuria in previous TODAY publications) was defined as a UACR of ≥30 mg/g on two of three urine samples collected during a 3-month minimal period (21). Macroalbuminuria was defined as a UACR ≥300 mg/g. Monitoring and treatment of confirmed elevated albumin excretion with ACE inhibitor therapy were conducted as previously described (21). Treatment was monitored by a safety oversight process using central data to enhance study site compliance and consistency with treatment protocols.
Statistical Analysis
Analyses were performed using SAS 9.4 for Windows (SAS Institute, Cary, NC). F tests and χ2 tests were used to compare quantitative and categorical characteristics by sex at baseline. Variables with a skewed distribution (UACR, insulin sensitivity, triglycerides, and hs-CRP) were log-transformed as appropriate. We report the incidence rate per 100 person-years and the cumulative incidence of hypertension and elevated UAE over an average follow-up of 5.7 years. The longitudinal association of SBP, DBP, and UACR with baseline SUA and hyperuricemia was evaluated in linear mixed models to account for the serial correlations over time and included all observations collected in participants up to 84 months of follow-up. Measures of correlation over time are presented as β ± SE, percentage change, mean difference ± SE, or percentage difference in geometric means as appropriate, depending on whether the dependent variable was log-transformed (for models using UACR as the outcome) and/or the independent variable was categorical (for models using baseline hyperuricemia as a covariate).
Cox proportional hazards models were used to examine the relationships between baseline SUA or hyperuricemia and incident hypertension and elevated UAE. Participants with existing elevated UAE or hypertension were excluded from all time-to-event analyses. The proportional hazards assumption in the Cox proportional hazards model was assessed with graphical methods and with models including time-by-covariate interactions; all covariates tested met the assumption. All longitudinal and time-to-event models were adjusted for baseline eGFR, age, HbA1c, BMI, sex, race/ethnicity, randomized treatment group, and antihypertensive medication use. In sensitivity analyses, we further adjusted for insulin sensitivity to examine whether the relationship between SUA, elevated UAE, and hypertension was independent of insulin sensitivity. Analyses were also repeated, replacing eGFR via the Zappitelli equation with eGFR via the FAS equation in the models to see whether the change in equation had an impact. Similarly, analyses were repeated using a sex-specific cutoff for baseline hyperuricemia. In the time-to-event models, the interaction of sex and race/ethnicity with SUA was also evaluated to test whether the relationship between baseline SUA and the outcomes differed by sex or race/ethnicity. Where an interaction was present, the analysis was stratified and results within each subgroup were presented. All analyses were considered exploratory, and α < 0.05 was the cutoff used to determine statistical significance.
Results
Baseline Characteristics and Hyperuricemia
Table 1 reports participant characteristics at baseline for the 539 participants included in the analysis. The mean SUA at baseline was 5.8 ± 1.4 mg/dL, with 26% of participants found to have hyperuricemia. Compared with girls, boys had greater baseline SUA (5.4 ± 1.2 vs. 6.6 ± 1.4 mg/dL, P < 0.0001) and hyperuricemia prevalence (12 vs. 50%, P < 0.0001). In addition, non-Hispanic black and Hispanic participants had lower baseline SUA (5.6 ± 1.2 and 5.8 ± 1.4, respectively, vs. 6.3 ± 1.6 mg/dL, P < 0.0001 and P = 0.0004) and hyperuricemia prevalence (20% and 25%, respectively, vs. 37%, P = 0.0048) compared with non-Hispanic white participants. Baseline SUA concentrations and hyperuricemia prevalence stratified by both sex and race/ethnicity are reported in Supplementary Table 1.
Table 1.
Baseline characteristics of participants stratified by sex
| Overall (n = 539) | Female (n = 344) | Male (n = 195) | P value for difference by sex | |
|---|---|---|---|---|
| Female | 63.8 | – | – | – |
| Age (years) | 13.9 ± 2.0 | 13.6 ± 2.0 | 14.4 ± 1.9 | <0.0001 |
| Diabetes duration (months) | 7.9 ± 5.9 | 8.1 ± 6.0 | 7.5 ± 5.7 | NS |
| Race/ethnicity | ||||
| Black non-Hispanic | 33.6 | 36.0 | 29.2 | NS |
| Hispanic | 43.0 | 41.3 | 46.2 | |
| White non-Hispanic | 20.0 | 18.3 | 23.1 | |
| Other | 3.3 | 4.4 | 1.5 | |
| Tanner stage | ||||
| 4–5 | 89.8 | 90.4 | 88.7 | NS |
| <4 | 10.2 | 9.6 | 11.3 | |
| BMI (kg/m2) | 34.6 ± 7.8 | 34.3 ± 7.5 | 35.2 ± 8.3 | NS |
| Blood pressure (mmHg) | ||||
| Systolic | 112.7 ± 11.1 | 110.6 ± 10.4 | 116.5 ± 11.2 | <0.0001 |
| Diastolic | 66.4 ± 8.2 | 65.8 ± 8.3 | 67.4 ± 7.9 | 0.031 |
| Mean arterial pressure | 81.8 ± 8.4 | 80.7 ± 8.4 | 83.8 ± 8.0 | <0.0001 |
| ACE inhibitor/ARB use | 4.6 | 4.9 | 4.1 | NS |
| Lipid-lowering medication use | 0.9 | 1.2 | 0.5 | NS |
| UACR (mg/g)* | 30.5 ± 116.0, 7 (8) | 35.1 ± 128.9, 7 (10) | 22.6 ± 88.9, 6 (8) | 0.043 |
| Serum cystatin-C (mg/L) | 0.75 ± 0.12 | 0.72 ± 0.11 | 0.80 ± 0.11 | <0.0001 |
| Serum creatinine (mg/dL) | 0.61 ± 0.13 | 0.58 ± 0.11 | 0.66 ± 0.13 | <0.0001 |
| eGFR (mL/min/1.73 m2) | ||||
| Zappitelli combined equation | 109.7 ± 15.1 | 112.2 ± 15.2 | 105.3 ± 13.9 | <0.0001 |
| FAS equation | 116.7 ± 16.4 | 119.6 ± 16.4 | 111.6 ± 15.2 | <0.0001 |
| HbA1c (%) | 6.0 ± 0.7 | 6.0 ± 0.7 | 5.9 ± 0.7 | NS |
| Insulin sensitivity (1/IF) (mL/µU)* | 0.05 ± 0.05 | 0.05 ± 0.04 | 0.05 ± 0.04 | NS |
| HDL cholesterol (mg/dL) | 38.9 ± 8.8 | 39.7 ± 9.0 | 37.3 ± 8.1 | 0.0021 |
| Triglycerides (mg/dL)* | 113.6 ± 74.3 | 112.0 ± 69.6 | 116.6 ± 82.0 | NS |
| Triglycerides–to–HDL cholesterol ratio | 3.1 ± 2.3 | 3.1 ± 2.2 | 3.3 ± 2.5 | NS |
| hs-CRP (mg/dL)* | 0.44 ± 0.88 | 0.43 ± 0.64 | 0.45 ± 1.21 | NS |
| SUA (mg/dL) | 5.83 ± 1.41 | 5.36 ± 1.17 | 6.65 ± 1.43 | <0.0001 |
| SUA ≥6.8 mg/dL | 25.6 | 11.6 | 50.3 | <0.0001 |
Data are presented as mean ± SD or percentage, and for UACR, the median (interquartile) is also given. P values for differences by sex were calculated from F tests or χ2 tests. ARB, angiotensin-receptor blocker; IF, fasting insulin.
*Variables were log-transformed before testing.
Prevalence, Incidence Rate, and Cumulative Incidence of Hypertension and Elevated UAE
At baseline, 18.7% of the participants had hypertension, 6.1% had UACR ≥30 mg/g, and 1.5% had UACR ≥300 mg/g. The incidence rates over 7 years were 8.5 per 100 person-years for hypertension, 3.5 per 100 person-years for UACR ≥30 mg/g, and 0.7 per 100 person-years for UACR ≥300 mg/g. The cumulative incidence was 37.4% for hypertension, 18% for UACR ≥30 mg/g, and 4% for UACR ≥300 mg/g, when excluding baseline prevalence (Supplementary Fig. 1).
Associations Between SUA, SBP, DBP, and UACR
Baseline SUA correlated with an increase in SBP (β ± SE 0.81 ± 0.25, P = 0.001), DBP (0.61 ± 0.21, P = 0.004), and UACR (percent change 10.2%, P = 0.003) over time in multivariable models. Similar results were obtained using baseline hyperuricemia in the models. Indeed, baseline hyperuricemia associated with increase in SBP (β ± SE 2.59 ± 0.78, P = 0.0009), DBP (1.91 ± 0.65, P = 0.004), and UACR (percent change 26.3%, P = 0.01). Further adjustments by log insulin sensitivity did not attenuate the relationships between baseline SUA, SBP, DBP, and UACR (Table 2).
Table 2.
Longitudinal associations/correlations of SBP, DBP, and UACR with baseline SUA and hyperuricemia in multivariable models*
| SBP (mmHg) | DBP (mmHg) | UACR (mg/g) | |
|---|---|---|---|
| Baseline SUA (per 1 mg/dL) | |||
| Model 1—without adjustment for insulin sensitivity | |||
| β ± SE or % change | 0.81 ± 0.25 | 0.61 ± 0.21 | 10.2 |
| P value | 0.0013 | 0.0041 | 0.0030 |
| Model 2—with adjustment for insulin sensitivity | |||
| β ± SE or % change | 0.77 ± 0.25 | 0.60 ± 0.21 | 10.3 |
| P value | 0.0024 | 0.0046 | 0.0028 |
| Baseline hyperuricemia (≥6.8 mg/dL vs. not) | |||
| Model 1—without adjustment for insulin sensitivity | |||
| Mean difference ± SE or % difference in means | 2.59 ± 0.78 | 1.91 ± 0.65 | 26.3 |
| P value | 0.0009 | 0.0037 | 0.0133 |
| Model 2—with adjustment for insulin sensitivity | |||
| Mean difference ± SE or % difference in means | 2.50 ± 0.78 | 1.92 ± 0.66 | 27.4 |
| P value | 0.0014 | 0.0036 | 0.0098 |
*Measures of correlation over time are presented from linear mixed models adjusted for baseline eGFR, age, HbA1c, BMI, sex, race/ethnicity, randomized treatment group, and antihypertensive medication use before and after adjustment for insulin sensitivity. For the models relating baseline SUA to SBP and DBP, data are β estimate ± SE; for models relating baseline SUA to UACR (log-transformed before testing), data are percentage change in UACR per unit change in baseline SUA; for those relating baseline hyperuricemia to the outcomes, data are the difference in means in SBP or DBP or the percentage difference in geometric mean in UACR between the two SUA groups (≥6.8 mg/dL vs. not). P values between outcome and exposure are also shown.
Associations Between SUA, Incident Hypertension, and Elevated UAE
Higher baseline SUA was independently associated with an increased risk of hypertension over 7 years (hazard ratio [HR] 1.2, 95% CI 1.0–1.4, P = 0.01, per 1 mg/dL of SUA) in adjusted models. Male sex, HbA1c, BMI, and antihypertensive medication use also were associated with an increased risk of incident hypertension (Table 3). Baseline hyperuricemia conferred almost a twofold increased risk of hypertension over 7 years (Fig. 1A). Higher baseline SUA was also independently associated with an increased risk of elevated UAE over 7 years (HR 1.2, 95% CI 1.0–1.5, P = 0.004, per 1 mg/dL of SUA) in multivariable models. Higher baseline HbA1c also predicted a greater risk of elevated UAE (Table 3). Baseline hyperuricemia did not confer a greater risk of elevated UAE in fully adjusted models (Fig. 1B). Replacing baseline eGFR by the Zappitelli equation with baseline eGFR by the FAS equation in the models did not affect the results. Results from univariate Cox regression analyses for SUA and for each covariate evaluated are presented in Supplementary Table 2.
Table 3.
Multivariable Cox proportional hazards models predicting hypertension and elevated UAE (defined as UACR ≥30 mg/g)
| Hypertension |
UACR ≥30 mg/g |
|||
|---|---|---|---|---|
| Characteristics (reference group or unit change)* | HR (95% CI) | P value | HR (95% CI) | P value |
| Baseline SUA (per 1 mg/dL) | 1.19 (1.03–1.38) | 0.015 | 1.24 (1.03–1.48) | 0.022 |
| Baseline eGFR (per 1 mL/min/1.73 m2) | 1.01 (0.99–1.02) | 0.14 | 1.01 (0.99–1.03) | 0.12 |
| Age at baseline (per 1 year) | 1.04 (0.95–1.14) | 0.38 | 0.99 (0.88–1.12) | 0.93 |
| Male | 1.78 (1.20–2.65) | 0.0044 | 0.94 (0.57–1.55) | 0.82 |
| Race/ethnicity (reference group: non-Hispanic white) | ||||
| Non-Hispanic black | 0.98 (0.61–1.57) | 0.93 | 1.06 (0.56–2.04) | 0.85 |
| Hispanic | 0.70 (0.44–1.10) | 0.12 | 1.32 (0.72–2.41) | 0.37 |
| HbA1c (per 1%) | 1.32 (1.03–1.69) | 0.027 | 1.39 (1.04–1.86) | 0.026 |
| BMI (per 1 kg/m2) | 1.05 (1.02–1.08) | 0.0002 | 1.00 (0.97–1.03) | 0.97 |
| Antihypertensive medication use at baseline | 4.64 (1.85–11.7) | 0.0011 | 2.20 (0.94–5.15) | 0.07 |
| Treatment group (reference group: metformin) | ||||
| Metformin + rosiglitazone | 0.74 (0.49–1.14) | 0.96 | 0.75 (0.44–1.30) | 0.72 |
| Metformin + intensive lifestyle intervention | 0.99 (0.66–1.49) | 0.17 | 1.10 (0.67–1.81) | 0.31 |
*Reference groups are specified for categorical covariates and unit changes are given for continuous ones. HR (along with 95% CI and P values) is for specified unit change (for continuous covariates) or respective to reference group (for categorical covariates), where <1 indicates less risk and >1 indicates more risk.
Figure 1.

Forest plot portraying the HR, 95% CI, and P value of the association between baseline SUA or hyperuricemia and incident hypertension (A) or elevated UAE (UACR ≥30 mg/g) (B) in adjusted models over time in TODAY. All models were adjusted for baseline eGFR, age, HbA1c, BMI, antihypertensive medication use, sex, race/ethnicity, and randomized treatment group assignment.
There were sex (P = 0.03) and race/ethnicity (P = 0.04) interactions between baseline SUA and elevated UAE. Therefore, we also stratified our analyses by sex and race/ethnicity. In boys, higher baseline SUA independently conferred greater risk of UAE (P = 0.004), but the risk conferred by baseline hyperuricemia did not reach statistical significance (P = 0.28) (Fig. 2A). In girls, neither higher baseline SUA nor baseline hyperuricemia was associated with greater risk of UAE (Fig. 2A). In non-Hispanic white youth, both higher baseline SUA (HR 1.4, 95% CI 1.1–1.9; P = 0.02) and baseline hyperuricemia (HR 4.2, 95% CI 1.1–15.3, P = 0.03) conferred greater risk of elevated UAE. On the contrary, in non-Hispanic black and Hispanic youth, neither higher baseline SUA nor baseline hyperuricemia was associated with a greater risk of elevated UAE (Fig. 2B). There were no sex (P = 0.07) or race/ethnicity (P = 0.39) interactions between baseline SUA and hypertension, and therefore, these analyses were not stratified by sex or race/ethnicity.
Figure 2.

Forest plot portraying the HR, 95% CI, and P value of the association between baseline SUA or hyperuricemia and incident elevated UAE (UACR ≥30 mg/g) stratified by sex (A) and race/ethnicity (B) in adjusted models over time in TODAY. All models were adjusted for baseline eGFR, age, HbA1c, BMI, antihypertensive medication use, sex (for model B), and race/ethnicity (for model A).
Conclusions
Our study demonstrates for the first time that hyperuricemia is common in adolescents with T2D and that a higher baseline SUA independently increased risk for incident hypertension and elevated UAE over 7 years. The relationships between SUA, blood pressure, and UACR were independent of insulin sensitivity. We also found important sex and race/ethnicity interactions between SUA and elevated UAE, with the strongest relationships found between SUA and elevated UAE in boys and non-Hispanic white youth.
T2D in youth is increasing in prevalence in parallel with the rise in obesity (4). In the U.S., almost half of patients with renal failure have DKD, and ≥80% of DKD is due to T2D (5). Moreover, DKD causes early mortality and contributes to increased CVD risk (22). Compared with adult-onset T2D, youth-onset T2D has a more aggressive phenotype, with greater insulin resistance, more rapid β-cell decline, and a higher prevalence of DKD, arguing for dedicated studies in youth (2–4). While adolescents with T2D are at greater risk of developing CVD and DKD than youth with T1D or adults with adult-onset T2D (2–4), there are limited longitudinal studies of potential contributors to DKD in youth-onset T2D. Accordingly, TODAY provides the opportunity to examine the relationships between risk factors and the development of renal and vascular complications.
The relationship between SUA and hypertension was first noted in the 1870s (23). SUA was initially thought to be a surrogate marker and by-product of decreased GFR that conferred greater risk of hypertension and CVD rather than a risk factor. Since then, SUA has been shown to be associated with the development of both DKD and CVD in adults with T2D (24). Several studies have further demonstrated an independent relationship between SUA and incident hypertension in adults (7,25) and also in adolescents (26). SUA as a unified risk factor for the development of both DKD and CVD does not necessarily imply causation, but increasing evidence implicates SUA in the pathogenesis of vascular complications in T2D. To our knowledge, there are limited—if any—longitudinal studies examining the effect of elevated SUA in adolescents with T2D and its relationship with the development of DKD and CVD.
Studies have suggested that inflammation (8), insulin resistance (9), intrarenal hemodynamic dysfunction (10,27), vascular, glomerular, and tubular injuries (9,11), and loss of nephron mass (12,13) could explain an etiologic relationship between elevated SUA and vascular disease in T2D and how elevated SUA may accelerate progression of DKD and CVD. Brenner et al. (28) proposed in a recent study that hypertension may be a function of nephron mass loss. In fact, Denic et al. (12) recently reported that higher SUA was associated with lower number of nephrons in 1,388 living kidney donors. The mechanism linking low nephron number to hypertension has been proposed to be renal arteriolopathy, interstitial inflammation, endothelial dysfunction, and renin expression (13,29). That hyperuricemia can result in hypertension independently of nephron numbers is also plausible, as suggested by experimental models (30). The pathogenesis of hypertension in DKD is complex and may also involve inappropriate sodium reabsorption, increased activity of the sympathetic nervous system and renin-angiotensin-aldosterone system (RAAS), endothelial cell dysfunction, and increased oxidative stress and inflammation. SUA has been implicated in several of these mechanisms in experimental models, including activating RAAS, the sympathetic nervous system, and mediating endothelial dysfunction, but the data are inconsistent and the relationships incompletely delineated (31,32). Further, there is increasing evidence that SUA may have a causal role in obesity and insulin resistance (33). It is therefore interesting that the relationships we observed between SUA, incident hypertension, and elevated albumin excretion were independent of estimated insulin sensitivity.
We demonstrated a sex interaction between SUA and elevated UAE that was not evident with hypertension. Whereas the pathogenesis underlying this interaction is unclear, potential mechanisms may relate to sex hormones, including increased renal clearance of urate due to estrogen in premenopausal women (34,35). The increased renal clearance may provide protection against development of elevated UAE but not incident hypertension, although this remains speculative. In addition, a race/ethnicity interaction was observed between SUA and elevated UAE but not with hypertension. The stronger relationship between SUA and elevated UAE in non-Hispanic white participants is unlikely to relate to race/ethnicity differences in SUA concentrations because they were similar across the race/ethnicity groups in our study. Further, the sex distribution across the race/ethnicity groups was not statistically different, and our sample size was not large enough to allow stratification for both race-ethnicity and sex. Different expressions and polymorphisms of urate transporters, including urate transporter 1 (URAT1), GLUT9b, and organic anion transporters OAT4 and OAT10, may explain race/ethnicity-related differences in renal handling of urate, but this continues to be incompletely understood and should be considered hypothesis generating (36).
Therapies to lower SUA, including allopurinol, have attenuated elevated albumin excretion and slowed eGFR decline in adults with T2D (37,38). Furthermore, Feig et al. (39) demonstrated a reduction of blood pressure in a randomized, double-blind, placebo-controlled, crossover trial in adolescents with newly diagnosed hypertension. To our knowledge, however, there are no such trials yet in youth-onset T2D. In adults with T1D, SUA lowering with febuxostat modestly lowered blood pressure without impacting the RAAS, suggesting that elevated SUA may augment other hemodynamic or inflammatory mechanisms (40). Results from the Preventing Early Renal Function Loss (PERL) allopurinol study, an ongoing multicenter randomized clinical trial in adults with T1D, are pending (41). It is also important to emphasize that in contrast to youth and adults with T2D, people with T1D tend to have lower SUA concentrations due to the uricosuric effect (6,9). Therefore, findings in T1D may not be generalizable to T2D and vice versa.
Our study does have important strengths and limitations. Limitations include the use of eGFR and insulin sensitivity rather than direct measurements. Repeated gold standard assessments of GFR and insulin sensitivity would have been difficult in such a large, long-term longitudinal study. Moreover, our data were limited to random UACR collections rather than timed urine collections, and information on use of hypouricemic drugs was not collected during the study. The lack of association between hyperuricemia and elevated UAE may relate to an inadequate follow-up period, treatment of hypertension, and a relatively limited number of observations with the dependent (e.g., elevated UAE) and independent (e.g., hyperuricemia) variables both being categorical. The exact timing of T2D onset remains difficult to ascertain, and therefore, our diabetes duration is likely subject to inaccuracy. Yet, we can probably better pinpoint T2D onset in adolescents compared with adults due to the rapid deterioration and onset of diabetes in youth.
The strengths of our study include up to 7 years of longitudinal data from the largest multicenter study of youth-onset T2D subjects, who were extensively phenotyped. The relatively large number of participants with available data at several time points for UACR, SBP, DBP, and also other important covariates (e.g., HbA1c, BMI) also allowed us to examine the independent relationships between SUA, elevated UAE, and hypertension. Finally, there was less than 10% missing data in TODAY over the 5.7 years of follow-up.
In summary, we demonstrate for the first time that hyperuricemia is present in more than one-quarter of youth with T2D and that greater SUA strongly predicts greater risk for hypertension and elevated albumin excretion. Future directions include examining the relationships between SUA, retinopathy, neuropathy, and other CVD markers, including echocardiographic and arterial stiffness measures.
Supplementary Material
Article Information
Acknowledgments. The TODAY Study Group thanks the following companies for donations in support of the study’s efforts: Becton, Dickinson and Company, Bristol-Myers Squibb, Eli Lilly and Company, GlaxoSmithKline, LifeScan, Pfizer, and Sanofi-Aventis. The authors also gratefully acknowledge the participation and guidance of the American Indian partners associated with the clinical center located at the University of Oklahoma Health Sciences Center, including members of the Absentee Shawnee Tribe, Cherokee Nation, Chickasaw Nation, Choctaw Nation of Oklahoma, and Oklahoma City Area Indian Health Service.
The opinions expressed in this paper are those of the authors and do not necessarily reflect the views of the respective Tribes and the Indian Health Service.
Funding. This work was completed with funding from National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health grant numbers T32-DK-063687, K23-DK-116720, U01-DK-61212, U01-DK-61230, U01-DK-61239, U01-DK-61242, and U01-DK-61254; from National Center for Research Resources General Clinical Research Centers Program grant numbers M01-RR-00036 (Washington University in St. Louis School of Medicine), M01-RR-00043-45 (Children’s Hospital Los Angeles), M01-RR-00069 (University of Colorado Denver), M01-RR-00084 (Children’s Hospital of Pittsburgh), M01-RR-01066 (Massachusetts General Hospital), M01-RR-00125 (Yale University), and M01-RR-14467 (University of Oklahoma Health Sciences Center); and from National Center for Research Resources Clinical and Translational Science Awards grant numbers UL1-RR-024134 (Children’s Hospital of Philadelphia), UL1-RR-024139 (Yale University), UL1-RR-024153 (Children’s Hospital of Pittsburgh), UL1-RR-024989 (Case Western Reserve University), UL1-RR-024992 (Washington University in St. Louis), UL1-RR-025758 (Massachusetts General Hospital), and UL1-RR-025780 (University of Colorado Denver).
The National Institute of Diabetes and Digestive and Kidney Diseases had no role in study design, collection, analysis, interpretation of data, or writing the report.
Duality of Interest. P.B. has acted as a consultant for Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, Sanofi, and Horizon Pharma and serves on the advisory board of XORTX. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. P.B. conceived the study design, contributed to data collection, data analysis, and interpretation, and wrote the manuscript. L.L., J.L., and K.J.N. conceived the study design, contributed to the data analysis, interpretation, and discussion, and reviewed and edited the manuscript. L.E.g. analyzed the data, contributed to the data interpretation, and reviewed and edited the manuscript. R.S.W. and S.E.T. contributed to the data analysis, interpretation, and discussion, and reviewed and edited the manuscript. L.E.g. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Data Availability. Materials developed and used for the TODAY standard diabetes education program and the intensive lifestyle intervention program are available to the public at https://today.bsc.gwu.edu/.
Prior Presentation. Parts of this study were presented in abstract form at the 78th Scientific Sessions of American Diabetes Association, Orlando, FL, 22–26 June 2018.
Footnotes
Clinical trial reg. no. NCT00081328, clinicaltrials.gov
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc18-2147/-/DC1.
A complete list of participants in the TODAY Study Group can be found in the online Supplementary Data.
References
- 1.Collins AJ, Foley RN, Herzog C, et al. . US Renal Data System 2010 Annual Data Report. Am J Kidney Dis 2011;57(Suppl. 1):A8, e1-526. [DOI] [PubMed] [Google Scholar]
- 2.Dabelea D, Stafford JM, Mayer-Davis EJ, et al.; SEARCH for Diabetes in Youth Research Group . Association of type 1 diabetes vs type 2 diabetes diagnosed during childhood and adolescence with complications during teenage years and young adulthood. JAMA 2017;317:825–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.TODAY Study Group Rapid rise in hypertension and nephropathy in youth with type 2 diabetes: the TODAY clinical trial [published correction appears in Diabetes Care 2013;36:2448]. Diabetes Care 2013;36:1735–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Saeed AH, Constantino MI, Molyneaux L, et al. . An inverse relationship between age of type 2 diabetes onset and complication risk and mortality: the impact of youth-onset type 2 diabetes. Diabetes Care 2016;39:823–829 [DOI] [PubMed] [Google Scholar]
- 5.Saran R, Li Y, Robinson B, et al. . US Renal Data System 2015 Annual Data Report: epidemiology of kidney disease in the United States. Am J Kidney Dis 2016;67(Suppl. 1):Svii, S1–S305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bjornstad P, Maahs DM, Rivard CJ, et al. . Serum uric acid predicts vascular complications in adults with type 1 diabetes: the coronary artery calcification in type 1 diabetes study. Acta Diabetol 2014;51:783–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuwabara M, Hisatome I, Niwa K, et al. . Uric acid is a strong risk marker for developing hypertension from prehypertension: a 5-year Japanese Cohort Study. Hypertension 2018;71:78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turak O, Ozcan F, Tok D, et al. . Serum uric acid, inflammation, and nondipping circadian pattern in essential hypertension. J Clin Hypertens (Greenwich) 2013;15:7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bjornstad P, Lanaspa MA, Ishimoto T, et al. . Fructose and uric acid in diabetic nephropathy. Diabetologia 2015;58:1993–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uedono H, Tsuda A, Ishimura E, et al. . U-shaped relationship between serum uric acid levels and intrarenal hemodynamic parameters in healthy subjects. Am J Physiol Renal Physiol 2017;312:F992–F997 [DOI] [PubMed] [Google Scholar]
- 11.Feig DI, Madero M, Jalal DI, Sanchez-Lozada LG, Johnson RJ. Uric acid and the origins of hypertension. J Pediatr 2013;162:896–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denic A, Mathew J, Lerman LO, et al. . Single-nephron glomerular filtration rate in healthy adults. N Engl J Med 2017;376:2349–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feig DI, Nakagawa T, Karumanchi SA, et al. . Hypothesis: uric acid, nephron number, and the pathogenesis of essential hypertension. Kidney Int 2004;66:281–287 [DOI] [PubMed] [Google Scholar]
- 14.Bjornstad P, Nehus E, El Ghormli L, et al.; TODAY Study Group . Insulin sensitivity and diabetic kidney disease in children and adolescents with type 2 diabetes: an observational analysis of data from the TODAY Clinical Trial. Am J Kidney Dis 2018;71:65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeitler P, Epstein L, Grey M, et al.; TODAY Study Group . Treatment options for type 2 diabetes in adolescents and youth: a study of the comparative efficacy of metformin alone or in combination with rosiglitazone or lifestyle intervention in adolescents with type 2 diabetes. Pediatr Diabetes 2007;8:74–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richette P, Bardin T. Gout. Lancet 2010;375:318–328 [DOI] [PubMed] [Google Scholar]
- 17.George L, Bacha F, Lee S, Tfayli H, Andreatta E, Arslanian S. Surrogate estimates of insulin sensitivity in obese youth along the spectrum of glucose tolerance from normal to prediabetes to diabetes. J Clin Endocrinol Metab 2011;96:2136–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacchetta J, Cochat P, Rognant N, Ranchin B, Hadj-Aissa A, Dubourg L. Which creatinine and cystatin C equations can be reliably used in children? Clin J Am Soc Nephrol 2011;6:552–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fadrowski JJ, Neu AM, Schwartz GJ, Furth SL. Pediatric GFR estimating equations applied to adolescents in the general population. Clin J Am Soc Nephrol 2011;6:1427–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pottel H, Hoste L, Dubourg L, et al. . An estimated glomerular filtration rate equation for the full age spectrum. Nephrol Dial Transplant 2016;31:798–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacha F, Pyle L, Nadeau K, et al.; TODAY Study Group . Determinants of glycemic control in youth with type 2 diabetes at randomization in the TODAY study. Pediatr Diabetes 2012;13:376–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pálsson R, Patel UD. Cardiovascular complications of diabetic kidney disease. Adv Chronic Kidney Dis 2014;21:273–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swales JD. Manual of Hypertension Oxford, U.K., Blackwell Science, 1995 [Google Scholar]
- 24.Tseng CH. Correlation of uric acid and urinary albumin excretion rate in patients with type 2 diabetes mellitus in Taiwan. Kidney Int 2005;68:796–801 [DOI] [PubMed] [Google Scholar]
- 25.Perlstein TS, Gumieniak O, Williams GH, et al. . Uric acid and the development of hypertension: the Normative Aging Study. Hypertension 2006;48:1031–1036 [DOI] [PubMed] [Google Scholar]
- 26.Loeffler LF, Navas-Acien A, Brady TM, Miller ER 3rd, Fadrowski JJ. Uric acid level and elevated blood pressure in US adolescents: National Health and Nutrition Examination Survey, 1999-2006. Hypertension 2012;59:811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uedono H, Tsuda A, Ishimura E, et al. . Relationship between serum uric acid levels and intrarenal hemodynamic parameters. Kidney Blood Press Res 2015;40:315–322 [DOI] [PubMed] [Google Scholar]
- 28.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens 1988;1:335–347 [DOI] [PubMed] [Google Scholar]
- 29.Perlstein TS, Gumieniak O, Hopkins PN, et al. . Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney Int 2004;66:1465–1470 [DOI] [PubMed] [Google Scholar]
- 30.Mazzali M, Kanellis J, Han L, et al. . Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol 2002;282:F991–F997 [DOI] [PubMed] [Google Scholar]
- 31.Mulé G, Castiglia A, Morreale M, et al. . Serum uric acid is not independently associated with plasma renin activity and plasma aldosterone in hypertensive adults. Nutr Metab Cardiovasc Dis 2017;27:350–359 [DOI] [PubMed] [Google Scholar]
- 32.Lambert EA, Hachem M, Hemmes R, et al. . Serum uric acid and the relationship with subclinical organ damage in adults. J Hypertens 2017;35:745–752 [DOI] [PubMed] [Google Scholar]
- 33.Kodama S, Saito K, Yachi Y, et al. . Association between serum uric acid and development of type 2 diabetes. Diabetes Care 2009;32:1737–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viazzi F, Parodi D, Leoncini G, et al. . Serum uric acid and target organ damage in primary hypertension. Hypertension 2005;45:991–996 [DOI] [PubMed] [Google Scholar]
- 35.Yoshitomi R, Fukui A, Nakayama M, et al. . Sex differences in the association between serum uric acid levels and cardiac hypertrophy in patients with chronic kidney disease. Hypertens Res 2014;37:246–252 [DOI] [PubMed] [Google Scholar]
- 36.Tasic V, Hynes AM, Kitamura K, et al. . Clinical and functional characterization of URAT1 variants. PLoS One 2011;6:e28641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Momeni A, Shahidi S, Seirafian S, Taheri S, Kheiri S. Effect of allopurinol in decreasing proteinuria in type 2 diabetic patients. Iran J Kidney Dis 2010;4:128–132 [PubMed] [Google Scholar]
- 38.Miao Y, Ottenbros SA, Laverman GD, et al. . Effect of a reduction in uric acid on renal outcomes during losartan treatment: a post hoc analysis of the reduction of endpoints in non-insulin-dependent diabetes mellitus with the Angiotensin II Antagonist Losartan Trial. Hypertension 2011;58:2–7 [DOI] [PubMed] [Google Scholar]
- 39.Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 2008;300:924–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lytvyn Y, Har R, Locke A, et al. . Renal and vascular effects of uric acid lowering in normouricemic patients with uncomplicated type 1 diabetes. Diabetes 2017;66:1939–1949 [DOI] [PubMed] [Google Scholar]
- 41.Maahs DM, Caramori L, Cherney DZ, et al.; PERL Consortium . Uric acid lowering to prevent kidney function loss in diabetes: the Preventing Early Renal Function Loss (PERL) allopurinol study. Curr Diab Rep 2013;13:550–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.