

Abstract
Neoadjuvant chemotherapy (NAC) may provide survival benefits for patients with advanced esophageal squamous cell carcinoma. However, tumor cells can display primary or secondary resistance to paclitaxel (PTX), a primary component of induction chemotherapy regimen. To identify genes capable of conveying PTX resistance, we performed a genome-wide CRISPR transcriptional activation library in human KYSE-180 cells. High throughput next generation sequencing was further applied to establish the phenotype-to-genotype relationship. Our highest-ranking hits are CDKN1A, TSPAN4, ELAVL2, JUNB and PAAF1. We generated evidence that esophageal tumors with high CDKN1A, ELAVL2 and TSPAN4 levels, quantified using qRT-PCR and Western blot assays, showed poorer chemotherapy response. Higher expression levels of TSPAN4 and ELAVL2 protein are independent risk factors for poor chemotherapy response in ESCC patients. We then found that overexpression of CDKN1A, ELAVL2 or TSPAN4 in ESCC cell lines significantly promoted the resistance to PTX by inhibiting cell apoptosis. Interestingly, ESCC cells overexpressed CDKN1A, ELAVL2 or TSPAN4 also acquired resistance to cisplatin (DDP). This phenomenon may be explained by cross-resistance of chemotherapy. We additionally found an association between ELAVL2 and CDKN1A, which may be regarded as the upstream and downstream factors that synergistically involved in the regulation of chemo-resistance in ESCC. Therefore, our study demonstrated that the genome-wide CRISPR activation library is a powerful strategy for the discovery of chemo-resistant genes critical for ESCC and we reported the first evidence that the ELAVL2-CDKN1A axis may be an important mechanism involved in chemo-resistance in ESCC.
Keywords: Esophageal squamous cell carcinoma, CRISPR library, neoadjuvant chemotherapy, drug-resistant genes, high throughput
Introduction
Esophageal cancer (EC) is the ninth most frequently invasive malignancy and ranks as the sixth cancer-related mortality in worldwide [1]. It mainly comprises of two different histopathological subtypes: esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC). Located in the so-called EC belt, China is one of the countries with high incidence of EC, and approximately 90% of cases occur are ESCC [2]. Currently, EC has become the third most common and the fourth most lethal cancer in China [3]. Despite the advances in contemporary treatment, the overall 5-year survival of EC remains dismal. Only 15%~25% of patients with EC survive for 5 years after diagnosis [4].
Surgery is still the most important single treatment for early staged tumors, but surgery combined with neoadjuvant therapy is recommended for the large majority of locally advanced EC [2,5-7]. Several randomized controlled trials showed that neoadjuvant chemotherapy (NAC) combined with surgery could significantly improve the R0 resection rate and prolong the overall survival for patients with advanced ESCC compared with surgery alone or surgery combined with adjuvant chemotherapy [8-10]. However, not all patients have good responses to NAC. The effective rate of NAC is less than 50% in ESCC, and the pathological complete response (pCR) rate of NAC is only 0~5% [7,11,12].
Drug resistance of tumor cells is one of the main causes of heterogeneity of clinical drug response [13-15]. Chemotherapy in a considerable proportion of ESCC patients often was hampered by drug-resistance and showed varying degrees of disease progression [16-18]. Drug-resistant phenotypes in tumor cells were often accompanied by changes in certain genotypes. Cisplatin (DDP) and paclitaxel (PTX) are two conventional drugs for NAC of EC. At present, many genes related to DDP resistance have been reported, while the mechanism of PTX resistance remains to be further studied. To identify the key genes and molecular mechanisms of PTX resistance in ESCC patients is of great clinical significance for helping to risk-stratify patients and promoting individualized treatment of ESCC [19,20].
Whole genome clustered regularly interspaced short palindromic repeats/CRISPR associated (CRISPR/Cas)-based lentiviral library is a powerful tool for genome-scale gain-of-function or loss-of-function screening [21,22]. This system has been proved to be highly effective in identifying drug resistant genes in vitro. Indeed, Shalem, Kurata and Joung have screened out essential genes for drug resistance in melanoma and AML using the CRISPR konckout (CRISPRi) or activation (CRISPRa) library [23-25]. Here, we attempted to combine the CRISPR library screening strategy with RNA sequencing technology to explore the critical genes and potential mechanism for chemo-resistance in ESCC.
Materials and methods
Cell culture, plasmids and reagents
Human ESCC cell lines KYSE-70, KYSE-150, KYSE-180, KYSE-450, and KYSE-510 were purchased from the Japanese Collection of Research Biosources cell bank (Osaka, Japan). Identities of the cell lines were confirmed by standard short tandem repeat (STR) analysis. Resulting STR profiles were matched with the American Tissue Culture Collection (ATCC) and Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ). All cells were passaged for less than 1 year before use and cultured in RPMI-1640 medium (Gibco, Life Technologies Inc., Australia) containing 10% heat-inactivated fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Gibco) at 37°C in a humidified atmosphere of 5% CO2.
Human CRISPR 2-plasmid activation pooled library (SAM) was a gift from Feng Zhang Lab (Addgene, #1000000078). Lentiviral infected ESCC cells were established in KYSE-180 by relevant lentiviral vectors transduction. Human TrueORFTM cDNA clones of CDKN1A, TSPAN4, ELAVL2 were purchased from OriGene Technologies (Beijing, China). Transient overexpressed ESCC cells were established in KYSE-180 and KYSE-150 by relevant plasmids transfection.
Cisplatin (DDP) was purchased from Sigma-Aldrich (P4394, St. Louis, MO) and prepared as a 1 μg/mL stock solution in saline, and further diluted with culture medium for treatment. Paclitaxel (PTX, T1912, Sigma-Aldrich) was dissolved in DMSO (final concentration 1 mM). All other chemicals and solvents were of the highest analytical grade available.
Determination of the chemosensitivity of ESCC cells
ESCC cells were seeded into 96-well plates at 5×103 cells/well overnight and then incubated with different concentrations of PTX (0, 0.5, 1, 2, 4, 8, 10, 30, 50 μM) for 24 h. At the end of the drug exposure, the effect of PTX on cell viability was measured by Cell Counting Kit-8 reagent (CCK-8, Dojindo Molecular Technologies Inc., Japan). Briefly, 10 μL of CCK-8 was added to each well and the samples were incubated for two hours at 37°C. The absorbance at 450 nm was measured using an iMark™ microplate absorbance reader (Bio-Rad Laboratories Inc., Hercules, CA, USA). Then, the half inhibitory concentration (IC50) values were calculated to evaluate the sensitivity of each cell line to PTX using GraphPad Prism 7.0 (La Jolla, CA, USA). Meanwhile, the IC50 of DDP in ESCC cells were also evaluated.
Lentiviral SAM library production and transduction
The lentiviral production, purification and stable cell lines establishment were performed according to our previous report [26]. The lentivirus titer of MS2-p65-HSF1v2 lentivirus (lenti-MPHv2, #89308, Addgene) was measured by p24 ELISA assay. KYSE-180 cells were transduced with lenti-MPHv2 at a multiplicity of infection (MOI) of 5 and supplemented 10 ug/ml polybrene (Sigma). Selecting with hygromycin (300 μg/mL) for 5 days to establish a MPHv2-expressing KYSE-180 stable cell line. As the efficiency of lentivirus transduction may vary per cell line, we precisely determine the actual MOI of purified SAM library lentivirus by small-scale transduction, antibiotic selection and transduction efficiency calculation in KYSE-180 cells according to a published protocol [25]. Then 7×107 MPHv2-expressing KYSE-180 cells were subjected to large-scale transducing with lentivirus particles containing the human sgRNA library (lentiSAMv2, #1000000078, Addgene) at MOI=0.4, expecting 2.8×107 transducted cells. Blasticidin (4 μg/mL) was added to the cells 24 h post transduction and maintained for 4 days. Cells were cultured for an additional 10 days which allowed enough time to achieve sufficient genome modification by CRISPR system. Finally, a pooled cell library of KYSE-180 stably that transduced with CRISPR-dCas9-sgRNA components was constructed (SAM-KYSE180s library). Cells in the pooled cell library were split into replicate flasks and cryopreserved at least one replicate (5×106 cells) for genomic DNA analysis as unselected control. The resting replicates were subjected for PTX-resistant screening.
Positive selection of PTX-resistant cells
We firstly determined the optimal PTX treatment concentration and duration that is required for 100% kill of KYSE-180 (IC100). PTX positive selection of chemo-resistant cells in the SAM library was performed according to the protocol described by Jong et al [25]. Briefly, the KYSE-180-SAM library was treated with PTX at the optimal screening condition to obtain resistant cells. Survived cells were subjected to genomic DNA (gDNA) extraction followed by PCR amplification of the sgRNA-coding regions and deep sequencing.
Amplification and purification of candidate sgRNAs
The gDNA was isolated using the Wizard® Genomic DNA Purification Kit (Promega, A1125). gRNA-coding regions integrated into the chromosomes were then PCR-amplified (Invitrogen™ Platinum™ SuperFi™ Green PCR Master Mix, MAN0014885) using sgRNA primer pairs listed in Table 1. PCR conditions were as follows: 5 μg of DNA template for each reaction, initial denaturation at 98°C for 3 min, followed by 25 cycles of denaturation at 98°C for 10 s, annealing at 63°C for 10 s and extension at 72°C for 25 s, with extension at 72°C for 2 min in the final cycle. PCR products of each replicate were pooled together and subjected to rapid purification from agarose gels using AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, AP-GX-250). Concentration and purity of the products were quantified by Nanodrop2000 ultraviolet spectrophotometer (Thermo Scientific) and Qubit2.0 fluorometer (Invitrogen, Carlsbad, CA, USA). The DNA fragments were further validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and only qualified samples (level B or above) can be used for library construction and deep sequencing.
Table 1.
Primer lists for PCR and qRT-PCR
| Gene | Primer Sequence (5’-3’) | Annealing temperature | Product Size |
|---|---|---|---|
| sgRNA | GTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACC | 63°C | 150 bp |
| ATTTTAACTTGCTAGGCCCTGCAGACATGGGTGATCCTCATGTTGGCCTAGCTCTAAAAC | |||
| GAPDH | GCCAAAAGGGTCATCATCTC | 58°C | 287 bp |
| GTAGAGGCAGGGATGATGTTC | |||
| CDKN1A | TGTCCGTCAGAACCCATGC | 58°C | 139 bp |
| AAAGTCGAAGTTCCATCGCTC | |||
| TSPAN4 | GTACCTGACTCCTGCTGCTT | 58°C | 116 bp |
| AGCAGGTTCTCCTGAAGC | |||
| ELAVL2 | TCTTCTGCCTCAATTCGC | 52°C | 139 bp |
| CAATGACCCAGAAGGAGTTG | |||
| JUNB | CAAGGTGAAGACGCTCAAGG | 58°C | 95 bp |
| TCATGACCTTCTGTTTGAGCTG | |||
| PAAF1 | GATGCCCAGCTGAAGATATG | 58°C | 120 bp |
| AGCAGACACCACATTCCTCC |
Sequencing of candidate sgRNAs
NGS library preparations were constructed following the manufacturer’s protocol (VAHTS Universal DNA Library Prep Kit for Illumina). For each sample, >50 ng purified PCR fragment was used for direct library preparation. The fragments were treated with End Prep Enzyme Mix for end repairing, 5’-phosphorylation and dA-tailing in one reaction, followed by a T-A ligation to add adaptors to both ends. Size selection of adaptor-ligated DNA was then performed using VAHTSTM DNA Clean Beads, each sample was then amplified by PCR for 8 cycles using P5 and P7 primers, with both primers carrying sequences which can anneal with flowcell to perform bridge PCR and P7 primer carrying a six-base index allowing for multiplexing. The PCR products were cleaned up using VAHTSTM DNA Clean Beads, validated using an Agilent2100 bioanalyzer, and quantified by Qubit2.0 fluorometer. Then libraries with different indexes were multiplexed and loaded on an Illumina HiSeq instrument according to manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequencing was carried out using a 2×150 paired-end (PE) configuration; image analysis and base calling were conducted by the HiSeq Control Software (HCS) + OLB + GAPipeline-1.6 (Illumina) on the HiSeq instrument. And then the sequencing results were processed and analyzed by GENEWIZ, Inc. (Suzhou, China).
Bioinformatic analysis of candidate resistant genes
Reads were aligned to the sgRNA sequences in SAM library and the counts of every unique sgRNA were subjected to differential abundance analysis using DESeq2. Individual sgRNA with a p value smaller than 0.01 were identified as enriched sgRNA (EN-sgRNA) between the PTX-treated group and the control group (DMSO). We then calculated an enrichment gene score (EG score) for each gene according to a method described in a previous report [27]. Briefly, sgRNA score (Ss) was firstly calculated for each EN-sgRNAs: Ss=log10 (Total types of EN-sgRNAs/Ranked position for each EN-sgRNAs based on abundance). Then, the enrichment gene score (EG score) for each gene was calculated as the sum of the Ss scores for any EN-sgRNAs targeting that gene. Finally, genes with higher Sg score were confirmed as enriched genes (EN-genes).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the top hits across the entire ranked gene list were implemented with the Database for Annotation, Visualization and Integrated Discovery 6.8 (DAVID 6.8), which uses a modified Fisher’s exact test followed by Benjamini-Hochberg multiple hypothesis testing correction.
Additionally, we calculated the log2-Flold Change between KYSE-150 and KYSE180 for each gene: Log2FC=log2TPMKYSE-150-log2TPMKYSE-180 using RNA-sequencing data in the Cancer Cell Line Encyclopedia (CCLE) project (GEO: GSE2454). Genes with a log2FC over 1 and under -1 were identified as differentially expressed genes (DE-genes) between KYSE-150 and KYSE-180 cell lines. We then integrated the EN-genes and DE-genes to identify candidate PTX resistance genes. Genes belonged to both EN-genes and DE-genes were selected as candidate genes that might mediates PTX resistance.
Patients selection and tissue samples collection
Tumor tissue samples of 31 ESCC patients who received NAC before surgical excision were collected at the Department of Thoracic Surgery I, Peking University Cancer Hospital (Beijing, China) between April 2018 and September 2018. Clinicopathological data of these patients including age, gender, smoking, drinking, family history, clinical stage, tumor location, tumor size, pathological differentiation, pathological primary tumor and lymph node stage, chemotherapy response and hematological markers were retrieved from our EC database. The clinical features of the tumor samples were defined according to the eighth edition of the American Joint Committee on Cancer/the Union for International Cancer Control (AJCC/UICC) Tumor Node Metastasis (TNM) classification. Our standard surgical procedures were a subtotal esophagectomy with mediastinal lymphadenectomy via right thoracotomy, upper abdominal lymphadenectomy, reconstruction with a gastric tube via the posterior mediastinum, and anastomosis in the cervical incision. NAC is a doublet regimen for 2~4 cycles, consisted of DDP 75 mg/m2 intravenous infusion over 2 h on day 1 followed by PTX 175 mg/m2 on day 1 or divided into day 1, day 8, and day 15. The clinical response to NAC was evaluated according to RECIST1.1 criteria. The included patients were divided into two groups (PR/CR group vs. PD/SD group) and the correlations between the chemotherapy response and other clinicopathological characteristics of the patients were analyzed. The study was approved by the ethics committee of the Peking University Cancer Hospital and signed consent was obtained from each participant.
Real-time quantitative reverse transcription PCR (qRT-PCR) analysis
Total RNA was extracted from 31 tumor samples using Trizol reagent (Invitrogen, Carlsbad, CA) and quantitated by NanoDropTM2000 spectrophotometer (Thermo Scientific). First-strand cDNA was synthesized with SuperScriptTM II Reverse Transcriptase (Invitrogen), according to the manufacturer’s instructions. Oligonucleotid sequences for CDKN1A, TSPAN4, ELAVL2, JUNB, PAAF1 and GAPDH from GenBank were used as a template for the construction of the primer pairs using SnapGene 2.3.2 software. The qRT-PCR primer set sequences were list in Table 1. qRT-PCR reactions were performed in triplicate in 20 μL volumes with SYBR Green I Master in an LightCycler® 480 Real-Time PCR System (Roche Diagnostics, Mannheim, Germany). Cycle parameters were as follows: 1 cycle with a pre-incubation step (98°C for 10 min) and 45 cycles with an amplification step (98°C for 10 s, 58°C for 10 s and 72°C for 30 s), followed by a melting step (95°C for 10 s, 55°C for 1 min and 95°C continuous) and a cooling step of 1 s at 40°C. Gene expression was normalized to GAPDH, and the fold change was calculated using 2-ΔCT.
Western blot analysis
Total protein from tumor tissues or plasmids overexpressed cell lines was extracted using RIPA lysis buffer. Protein concentrations were determined using the BCA method (Thermo Scientific). The protein samples were then separated on a NuPAGE® Novex® 10% Bis-Tris Gel (Life Technologies) with 10 μg protein per lane and transferred to nitrocellulose membranes. The membranes were blocked with Tris-buffered saline (pH 7.5) containing 0.2% Tween-20 and 5% nonfat milk at room temperature for 1 h. Primary antibodies used were as follows: rabbit anti-CDKN1A (Cell Signaling Technology, 1:1000 dilution), rabbit anti-TSPAN4 (Abcam, 1:5000 dilution), rabbit anti-ELAVL2 (Proteintech, 1:5000 dilution), rabbit anti-JUNB (Proteintech, 1:1000 dilution), rabbit anti-PAAF1 (Proteintech, 1:2000 dilution), rabbit anti-Bax (Proteintech, 1:1000 dilution), rabbit anti-p53 (Proteintech, 1:2000 dilution) and rabbit anti-GAPDH (Cell Signaling Technology, 1:5000 dilution). Goat anti-Rabbit HRP conjugated secondary antibodies were utilized at 1:10000 dilution (Santa Cruz Biotechnology). Antibodies binding were visualized following application of enhanced chemiluminescence (ECL, Millipore). Densitometric analysis was performed using ImageJ software and normalized to GAPDH.
CCK-8 assay
Plasmids overexpressed KYSE-180 and KYSE-150 cells were seeded in 96-well plates at a density of 5000 cells per well and then treated with gradient dilutions of PTX (0, 2, 4, 8, 16, 32, 64 and 100 μM) and DDP (0, 1, 2, 4, 8, 16, 32 and 64 μg/mL) for 24 hours. CCK-8 reagent was added to measure the absorbance at 450 nm. Finally, the IC50 values were calculated to evaluate the changes of chemosensitivity in each cell line.
Apoptosis assay
ESCC cells transiently transfected corresponding plasmids were further subjected to PTX and DDP exposure for 24 h at near IC50 concentration, respectively. After that, cells were collected and rinsed 3 times in PBS then resuspended in 1× Annexin V binding buffer at a concentration of 1×106 cells/mL, with 20 μL of Annexin V-FITC antibody and 5 μL of 7-AAD antibody (BD Biosciences). The samples were then incubated for 15 minutes in the dark at room temperature and analyzed by FACS Aria II (BD Biosciences) within 1 h.
Statistical analysis
All experiments were performed with three replicates. Values were expressed as means ± SD. SPSS software (version 24.0; IBM SPSS, Armonk, NY, USA) was used to perform the statistical analysis. Association between the efficacy of chemotherapy and other clinicopathological parameters and protein expression were assessed by the Chi-square test. The relationship between protein levels and gene expressions were evaluated using the Mann-Whitney test. Pearson’s correlation coefficients were calculated to determine correlations among the variables. Student’s t tests were performed to evaluate the significances of the differences between groups. All p-values were based on two-tailed statistical analysis and P<0.05 was considered statistically significant.
Results
Genome-scale CRISPR transcriptional activation screening in KYSE-180
To evaluate the chemo-sensitivity of ESCC cell lines, we determined the IC50 values of PTX and DDP in five ESCC cell lines by CCK-8 assay. KYSE-180 was regarded as the relatively PTX-sensitive cell line with the lowest IC50 (IC50PTX=1.37±0.93 μM), while KYSE-150 was considered to be the relatively PTX-resistant cell line with the highest IC50 (IC50PTX=31.11±1.22 μM) (Figure 1B). The IC50DDP of KYSE-180 (2.03±1.20 μg/mL) and KYSE-150 (13.61±1.14 μg/mL) were also calculated for subsequent apoptosis experiments (Figure 1B).
Figure 1.
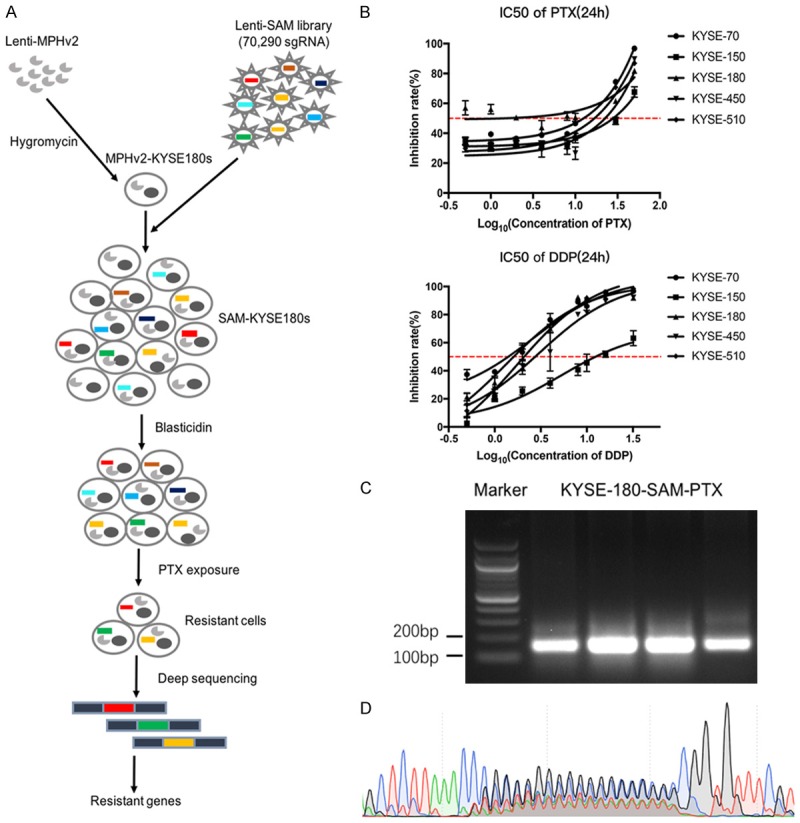
Genome-scale CRISPR activation screening in KYSE-180. A. Schema of SAM library screening. B. Identify the PTX and DDP-sensitivity of ESCC cell lines by CCK-8 assay. C. Amplification of sgRNAs through PCR reactions. D. Evaluation the diversity of sgRNAs by Sanger sequencing.
We firstly generated a pooled cell library of KYSE-180 that stably transduced with CRISPR-dCas9-sgRNA components (SAM-KYSE180s library) (Figure 1A). SAM-KYSE180s library contained 70,290 sgRNAs, targeting the promoter region of 23,430 protein-coding genes, in more than 2.8×107 lentiviral transducted cells for a coverage of >380 cells per sgRNA to guarantee a sufficient representation of each perturbation. SAM-KYSE180s hybrid cell library was selected in blasticidin for 4 days and expanded for additional 10 days before it was splitted into two equal cell libraries (more than 2.8×107 cells for each split). We subjected one split to lethal treatment with 2 μM PTX for 48 h to enrich the cell clones that resistant to PTX (experiment condition). The other split was treated DMSO as a control (control condition). We performed 4 independent screening experiments as biological replications.
Survival cell clones in the experiment conditions and cells in the control conditions were harvested for genomic DNA extraction. Integrated sgRNAs were amplified from genomic DNA and subjected to deep sequencing to evaluate their frequency distribution. The presence of sgRNA fragments was confirmed by PCR, with its size was approximately 150 bp and typical nested peaks representing the diversity of sgRNAs were appeared by sanger sequencing (Figure 1C, 1D). An average of 1×107 qualified reads were yield for each sample, achieving a coverage of >140 reads per sgRNA in the SAM library.
Top gene hits that might mediate PTX resistance
A total of 6300 sgRNA with a p-value <0.01 were identified as differentially presented sgRNA (EN-sgRNA) between the PTX-treated group and the control group. We then identified differentially presented genes (EN-genes) by calculated an enrichment gene score (EG score) for each gene. 194 genes with an EG score >1 were selected as EN-genes for subsequent validation (Figure 2A).
Figure 2.

Screening enrichment analysis. A. Bubble chart of EN-genes from SAM library screening, sorted on the x-axis by symbol. B. GO term enrichment analysis of EN-genes. C. KEGG enrichment analysis of EN-genes. D. Integrated analysis of enrichment gene score, determined by SMA library screening in KYSE-180, and Log2FC, determined by RNA-seq of KYSE-150 and KYSE-180 from CCLE dataset. E. Enrichment of sgRNAs that targeting top 5 gene hits.
Enrichment analysis of EN-genes were performed to identify biological processes and signaling pathways that might regulate PTX-resistance. GO term enrichment analysis on EN-genes showed that cell proliferation, cell apoptosis regulation, response to drug, protein complex biogenesis, amino acid transport, DNA replication are the main biological processes that may be closely related to the acquisition of PTX-resistance (Figure 2B). The KEGG pathway enrichment analysis showed that EN-genes were mostly involved in renal cell carcinoma, pathways in cancer, T cell receptor signaling pathway, focal adhesion, ubiquitin mediated proteolysis, as well as genes involved in ErbB and MAPK signaling pathway (Figure 2C).
In order to increase the chance of identifying essential genes involved in PTX resistance, an integrated analysis was performed to combine EN-genes in genome-scale CRISPRa screening and differentially expressed genes (DE-genes) between KYSE-150 and KYSE-180. Among the 991 overlapping hits between EN-genes and DE-genes, the top five hits were CDKN1A, TSPAN4, ELAVL2, JUNB and PAAF1, with EG scores >1.75 and log2FC >2, respectively, which suggest that activated expression of these five genes could lead to PTX resistance in KYSE-180 cells, and that all these genes were highly expressed in PTX-resistant KYSE-150 cells (Figure 2D).
Further tracing the enrichment of sgRNA targeting CDKN1A, TSPAN4, ELAVL2, JUNB and PAAF1 during the SAM library screening, it could be found that these sgRNAs were significantly enriched in the surviving cells after PTX exposure, with 8 to 21-fold increase in adjusted sgRNA count, respectively (Figure 2E).
Expression levels of CDKN1A, ELAVL2 and TSPAN4 associate with chemo-resistance in PTX-treated ESCC patients
A total of 31 cases of patients with ESCC who received surgery after NAC were included in this study, including 17 cases of PR/CR and 14 cases of PD/SD. None of clinical pathology parameters, including age, gender, smoking, drinking, family history, tumor location, tumor size, tumor differentiation, clinical stage, pathological stage, T stage, N stage, serum CEA and serum SCCA, are significantly correlated with cancer’s response to chemotherapy (Table 2).
Table 2.
Correlation between chemotherapy response and clinical characteristics in 31 ESCC patients
| Clinicalopathological parameters | Patients (%) | Chemotherapy response (n=31) | P | |
|---|---|---|---|---|
|
| ||||
| PR/CR (n=17) | PD/SD (n=14) | |||
| Age (year) | 0.371 | |||
| ≤60 | 13 (41.9) | 5 | 8 | |
| >60 | 18 (58.1) | 12 | 6 | |
| Gender | 0.476 | |||
| Male | 24 (77.4) | 14 | 10 | |
| Female | 7 (22.6) | 3 | 4 | |
| Smoking | 0.444 | |||
| Yes | 20 (64.5) | 12 | 8 | |
| No | 11 (35.5) | 5 | 6 | |
| Drinking | 0.249 | |||
| Yes | 19 (61.3) | 12 | 7 | |
| No | 12 (38.7) | 5 | 7 | |
| Family history | 0.250 | |||
| Yes | 10 (32.3) | 7 | 3 | |
| No | 21 (67.7) | 10 | 11 | |
| Tumor location | 0.171 | |||
| Upper | 4 (12.9) | 4 | 0 | |
| Middle | 14 (45.2) | 7 | 7 | |
| Lower | 13 (41.9) | 6 | 7 | |
| Tumor size (cm3) | 0.451 | |||
| ≤15.5 | 24 (77.4) | 15 | 9 | |
| >15.5 | 7 (22.3) | 2 | 5 | |
| Tumor differentiation differentiation | 0.555 | |||
| High | 2 (6.5) | 1 | 1 | |
| Middle | 20 (64.5) | 12 | 8 | |
| Low | 9 (29.0) | 4 | 5 | |
| Clinical stage | 0.891 | |||
| II | 7 (22.6) | 4 | 3 | |
| III | 24 (77.4) | 13 | 11 | |
| Pathologic stage | 0.098 | |||
| 0/I/II | 14 (45.2) | 10 | 4 | |
| III/IV | 17 (54.8) | 7 | 10 | |
| T stage | 0.960 | |||
| T1-2 | 11 (35.5) | 6 | 4 | |
| T3-4 | 20 (64.5) | 11 | 10 | |
| N stage | 0.144 | |||
| N0 | 12 (38.7) | 9 | 3 | |
| N1-3 | 19 (61.3) | 8 | 11 | |
| Serum CEA | ||||
| Positive | 2 (6.5) | 1 | 1 | 0.889 |
| Negative | 29 (93.5) | 16 | 13 | |
| Serum SCC | ||||
| Positive | 1 (3.2) | 0 | 1 | 0.270 |
| Negative | 30 (96.8) | 17 | 13 | |
| Protein CDKN1A | 0.024* | |||
| Low | 18 (58.1) | 13 | 5 | |
| High | 13 (41.9) | 4 | 9 | |
| Protein TSPAN4 | 0.002** | |||
| Low | 14 (45.2) | 12 | 2 | |
| High | 17 (54.8) | 5 | 12 | |
| Protein ELAVL2 | 0.005** | |||
| Low | 18 (58.1) | 14 | 4 | |
| High | 13 (41.9) | 3 | 10 | |
| Protein JUNB | 0.473 | |||
| Low | 20 (64.5) | 10 | 10 | |
| High | 11 (35.5) | 7 | 4 | |
| Protein PAAF1 | 0.926 | |||
| Low | 18 (58.1) | 10 | 8 | |
| High | 13 (41.9) | 7 | 6 | |
Note: CEA: Carcinoembryonic antigen; SCCA: Squamous cell carcinoma antigen;
P<0.05;
P<0.01.
We performed qRT-PCR to determine the mRNA level of five candidate genes in the primary tumor issue of ESCC patients. The results showed that the mRNA expression levels of CDKN1A, TSPAN4 and ELAVL2 were significantly higher in the tumor tissue of PD/SD group compared to the PR/CR group. There was no significant difference in the mRNA level of PAAF1 between two groups. To our surprise, the mRNA transcription level of JUNB was much higher in PR/CR group than in PD/SD group, which is the opposite of the observation in KYSE-150 and KYSE-180 (Figure 3A).
Figure 3.

Validation of top gene hits in ESCC patients. A. qRT-PCR analysis of the mRNA expression of CDKN1A, TSPAN4, ELAVL2, JUNB and PAAF1 in ESCC tumor tissues. B. Western blot of the protein expression of CDKN1A, TSPAN4, ELAVL2, JUNB and PAAF1 in ESCC tumor tissues. C. Correlation analysis of mRNA and protein levels of CDKN1A and ELAVL2 in ESCC tumor tissues. *P<0.05, **P<0.01.
Western blot results also showed that the CDKN1A, TSPAN4 and ELAVL2 were significantly highly expressed in the PD/SD group compared with PR/CR group. But, there was no significant difference in the protein level of JUNB and PAAF1 (Figure 3B). Correlation analysis of protein and mRNA levels of target genes was further performed. A close relationship was observed between mRNA and protein expression level for CDKN1A (r2=0.7137 in PR/CR group, r2=0.7777 in PD/SD group) and ELAVL2 (r2=0.7043 in PD/SD group) (Figure 3C).
Univariate analysis showed that the protein levels of CDKN1A, TSPAN4 and ELAVL2 were risk factors associated to resistance to NAC in ESCC, while other parameters showed no impact on the chemo-resistance (Table 2). Multivariate Logistic regression analysis further showed that higher expression levels of TSPAN4 and ELAVL2 protein are independent risk factors for poor chemotherapy response in patients with ESCC, OR were 0.056 (P=0.021, 95% CI: 0.005-0.649) and 0.07 (P=0.033, 95% CI: 0.006-0.811), respectively (Table 3).
Table 3.
Multivariate Logistic regression analysis
| Variables | OR | 95% CI | P | |
|---|---|---|---|---|
|
| ||||
| Lower limits | Upper limits | |||
| CDKN1A | 0.727 | 0.088 | 5.986 | 0.767 |
| TSPAN4 | 0.056 | 0.005 | 0.649 | 0.021* |
| ELAVL2 | 0.07 | 0.006 | 0.811 | 0.033* |
Note: CI: confidence interval;
P<0.05.
CDKN1A, ELAVL2 and TSPAN4 promote resistant to chemotherapies in ESCC cell lines through regulating cell apoptosis
We evaluated the potential of CDKN1A, ELAVL2 and TSPAN4 to promote chemo-resistance in ESCC cells. Our results showed that overexpression of CDKN1A, ELAVL2 or TSPAN4 could significantly increase the resistance to PTX in both KYSE-180 and KYSE-150 cells. Moreover, overexpressed CDKN1A, ELAVL2 or TSPAN4 could also contribute to DDP-resistance in KYSE-150, while a strong trend but not a statistically significant IC50 of DDP increase was observed in KYSE-180, implying a cellular context-dependent effect in this process (Figure 4A).
Figure 4.

Validation of top gene hits in ESCC cell lines. A. Cell proliferation assay of ESCC cells overexpressed CDKN1A, ELAVL2 or TSPAN4 under the treatment of PTX and DDP. B. Annexin V staining of ESCC cells overexpressed CDKN1A, ELAVL2 or TSPAN4 under the treatment of PTX and DDP. C. Western blot analysis of CDKN1A, ELAVL2, TSPAN4, Bax, P53 and GAPDH in ESCC cells overexpressed CDKN1A or ELAVL2. *P<0.05, **P<0.01, ***P<0.001.
To clarify whether the increased chemo-resistance was due to the reduction of cell apoptosis, Annexin V staining assay was further performed. PTX and DDP induced apoptosis was both significantly suppressed when CDKN1A, ELAVL2 or TSPAN4 were overexpressed in KYSE-180 and KYSE-150 cells (Figure 4B).
Western blot results further confirmed that the overexpression of ELAVL2 could up-regulate the expression of CDKN1A, while the reverse was not true, suggesting that CDKN1A might serve as a downstream factor of ELAVL2 to participate in the chemo-resistance of ESCC (Figure 4C). We also monitored the expression level of P53 and Bax protein after overexpression of CDKN1A and ELAVL2. We found that there was no obvious change in the protein level of P53 and Bax.
Discussion
The comprehensive treatment strategy of NAC followed by surgery improves overall survival for locally advanced ESCC compared with surgery alone. It has become standard treatment modality that recommended in the NCCN guidelines for EC management. However, the responses of NAC were heterogeneous and the NAC failed in almost half of patients. Therefore, identifying the underlying biomarkers of NAC would be beneficial to understand the mechanism of chemo-resistance and develop new therapeutic targets for ESCC patients.
Here, we conducted a genome-scale CRISPRa screening in human ESCC cell line KYSE-180 using SAM pooled library. Using positive screening strategy, we successfully identified a total of 6300 sgRNA and 194 genes that have been obviously enriched in the survival cells after PTX treatment in KYSE-180. All of these 194 EN-genes containing at least 2 independent EN-sgRNAs that were enriched during screening, which suggested that our CRISPR library screening was properly performed.
We further performed GO and KEGG enrichment analysis of EN-genes. GO term enrichment analysis on EN-genes showed that cell proliferation, cell apoptosis regulation, response to drug, and DNA replication are the main biological processes that may be closely related to the acquisition of PTX-resistance. The KEGG pathway enrichment analysis showed that EN-genes were mostly involved in renal cell carcinoma, pathways in cancer, ubiquitin mediated proteolysis, as well as genes involved in ErbB and MAPK signaling pathway. Most of these GO terms or KEGG pathways linked to the control of cell proliferation and cell fate control, processes that closely involved in the cellular adaptation after chemotherapy, which again support that our screening results are convincing.
Five candidate genes, CDKN1A, TSPAN4, ELAVL2, JUNB and PAAF1, were further selected as essential top gene hits that could mediate PTX-resistance. sgRNAs targeting these genes were significantly enriched in survived cells after PTX treatment in KYSE-180 cells. Meanwhile, all these genes were highly expressed in chemo-resistant KYSE-150 cells.
In order to verify whether the top gene hits screened above were actually involved in the chemo-resistance of ESCC, we did relevant experiments at the histological and cytological levels. To our surprise, we found that three out five top gene hits, CDKN1A, ELAVL2 and TSPAN4, were highly expressed in the tumor tissue of chemo-resistant ESCC patients and the elevated protein level of these three genes could serve as risk factors of chemo-resistance. Higher expression levels of TSPAN4 and ELAVL2 protein are even independent risk factors for poor chemotherapy response in patients with ESCC. We also proved that overexpression of CDKN1A, ELAVL2 and TSPAN4 in ESCC cell lines significantly promoted the resistance to both PTX and DDP by inhibiting cell apoptosis. Such a high validation rate of the biological role of candidate chemo-resistant genes, which were screened out in both clinical samples and cell lines suggested that the combination of CRISPR screening and RNAseq is a powerful strategy in the identification of essential genes.
CDKN1A is currently known as a cell cycle suppressor protein with extensive kinase activity. As one of the important downstream genes of oncogene p53, CDKN1A is involved in mediating cell cycle arrest after DNA damage [28]. CDKN1A has long been considered as a tumor suppressor gene, which plays an anticancer role in tumors [29]. Until recent years, CDKN1A has been reported to promote tumorigenesis in more and more solid tumors [30]. Therefore, we believe that CDKN1A has dual characteristics of oncogene and tumor suppressor gene. A recent literature reported the mechanism of chemo-resistance in triple-negative breast cancer, and the results showed that the up-regulated expression of PTN/PTPRZ1 induced by chemotherapy promoted tumor cells proliferation and anti-apoptosis, while CDKN1A was the key factor to regulate the expression of PTN/PTPRZ1. The up-regulated expression of CDKN1A/PTN/PTPRZ1 was related to the activation of NF-κB pathway, which revealed a new mechanism of chemo-resistance in breast cancer [31].
ELAVL2, also known as HuB, belongs to the family of ELAV-like RNA binding protein (RNAbps), which is mainly involved in regulating the functions of neurons and plays a crucial role in the normal cognition and behavior of the brain [32]. Studies have shown that 60% of small-cell lung cancers are associated with high ELAVL2 expression [33]. Recently, it has been reported that ELAVL1 (HuR), which is homologous with ELAVL2, was highly expressed in colorectal cancer and could regulate the proliferation and migration of tumor cells and the cancer-promoting effect of ELAVL1 may be realized through the JUN-miR-22-HuR axis [34]. ELAVL1 is also related to the prognosis of pancreatic ductal adenocarcinoma and can be used as an indicator of the efficacy of gemcitabine-based chemotherapy [35]. Further study is necessary to identify whether ELAVL2 has the same or similar function of ELAVL1.
TSPAN4 is one of the members of the transmembrane protein 4 superfamily, which usually forms complexes with interferon and other cell surface proteins, and is widely expressed in a variety of tissues and cell types, such as fibroblasts, endothelial cells, dendritic cells and mesenchymal cells [36]. TSPAN4 has been reported to be related to the occurrence of squamous cell carcinoma of oral cavity and gastric cancer [37,38].
Further validation is needed. But, at least, based on our present experimental results, we reported the first evidence that ELAVL2-CDKN1A axis contributed to the PTX-resistance in ESCC. We showed that overexpression of ELAVL2 obviously upregulated the protein level of CDKN1A in both KYSE-180 and KYSE-150 cell lines. We also revealed that overexpression of either one of ELAVL2, CDKN1A or TSPAN4 could distinctly inhibit PTX and DDP induced apoptosis in ESCC cell lines. And, this inhibition effect was not mediated through the regulation of p53-Bax pathway. Future studies need be performed to reveal the detailed mechanisms behind ELAVL2-CDKN1A axis and TSPAN4 gene.
Pooled library screenings based on CRISPR/Cas9 system, which functions as an adaptive immune defense in bacteria, is a powerful approach for genome-scale functional screening and revealing mechanisms of biological processes and diseases [25]. This strategy has already assist us in systematically validating key genes and mechanisms behind a series of processes, including drug resistance [24,39-41], virus infection [42], tumor growth and metastasis [43]. Andrew has recently reported that MSH2 plays vital role in DDP-mediated cell death in muscle-invasive bladder cancer also using CRISPR library screening strategy [41]. All these successful application, including this study, suggested that CRISPR library screening would dramatically promote the fast development of our understanding of essential biology processes. In summary, we revealed that ESCC patients with high expression of ELAVL2, CDKN1A and TSPAN4 had poorer response to PTX-based NAC. Consistent with our clinical analysis, we generated in vitro evidence that the ELAVL2-CDKN1A axis may be an underlying mechanism for PTX-resistance, which can be regarded as a novel therapy-overarching resistant biomarker for tailoring individual treatment of ESCC in the future.
Acknowledgements
This work was supported financially by the National Key Sci-Tech Special Project of China (No. 2018ZX10302207, No. 2017ZX10203202), the National High Technology Research and Development Programs of China (2015AA020403, 2017YFC0907504), the Special Fund of Beijing Municipal Administration of Hospitals of Gastroenterology Collaborative Development Center (XXT18), the Science Fund of Beijing Municipal Science & Technology Commission (D141100000214002), the Beijing Municipal Administration of Hospitals Young Scholars Program (QML20171106), the Beijing Municipal Excellent Talents Incubating Program (2016000021469G185), the Beijing Municipal Administration of Hospitals Incubating Program (PX2018044), the Hospital Fund of Peking University Cancer Hospital and the Latitudinal Project of Minzu University of China.
Disclosure of conflict of interest
None.
References
- 1.Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, Wolfe C, Hamadeh RR, Moore A, Werdecker A, Gessner BD, Te Ao B, McMahon B, Karimkhani C, Yu C, Cooke GS, Schwebel DC, Carpenter DO, Pereira DM, Nash D, Kazi DS, De Leo D, Plass D, Ukwaja KN, Thurston GD, Yun Jin K, Simard EP, Mills E, Park EK, Catala-Lopez F, deVeber G, Gotay C, Khan G, Hosgood HD 3rd, Santos IS, Leasher JL, Singh J, Leigh J, Jonas JB, Sanabria J, Beardsley J, Jacobsen KH, Takahashi K, Franklin RC, Ronfani L, Montico M, Naldi L, Tonelli M, Geleijnse J, Petzold M, Shrime MG, Younis M, Yonemoto N, Breitborde N, Yip P, Pourmalek F, Lotufo PA, Esteghamati A, Hankey GJ, Ali R, Lunevicius R, Malekzadeh R, Dellavalle R, Weintraub R, Lucas R, Hay R, Rojas-Rueda D, Westerman R, Sepanlou SG, Nolte S, Patten S, Weichenthal S, Abera SF, Fereshtehnejad SM, Shiue I, Driscoll T, Vasankari T, Alsharif U, Rahimi-Movaghar V, Vlassov VV, Marcenes WS, Mekonnen W, Melaku YA, Yano Y, Artaman A, Campos I, MacLachlan J, Mueller U, Kim D, Trillini M, Eshrati B, Williams HC, Shibuya K, Dandona R, Murthy K, Cowie B, Amare AT, Antonio CA, Castaneda-Orjuela C, van Gool CH, Violante F, Oh IH, Deribe K, Soreide K, Knibbs L, Kereselidze M, Green M, Cardenas R, Roy N, Tillmann T, Li Y, Krueger H, Monasta L, Dey S, Sheikhbahaei S, Hafezi-Nejad N, Kumar GA, Sreeramareddy CT, Dandona L, Wang H, Vollset SE, Mokdad A, Salomon JA, Lozano R, Vos T, Forouzanfar M, Lopez A, Murray C, Naghavi M. The global burden of cancer 2013. JAMA Oncol. 2015;1:505–527. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lagergren J, Smyth E, Cunningham D, Lagergren P. Oesophageal cancer. Lancet. 2017;390:2383–2396. doi: 10.1016/S0140-6736(17)31462-9. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 4.Boonstra JJ, Kok TC, Wijnhoven BP, van Heijl M, van Berge Henegouwen MI, Ten Kate FJ, Siersema PD, Dinjens WN, van Lanschot JJ, Tilanus HW, van der Gaast A. Chemotherapy followed by surgery versus surgery alone in patients with resectable oesophageal squamous cell carcinoma: long-term results of a randomized controlled trial. BMC Cancer. 2011;11:181. doi: 10.1186/1471-2407-11-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jang R, Darling G, Wong RK. Multimodality approaches for the curative treatment of esophageal cancer. J Natl Compr Canc Netw. 2015;13:229–238. doi: 10.6004/jnccn.2015.0029. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro J, van Lanschot JJB, Hulshof MCCM, van Hagen P, van Berge Henegouwen MI, Wijnhoven BPL, van Laarhoven HWM, Nieuwenhuijzen GAP, Hospers GAP, Bonenkamp JJ, Cuesta MA, Blaisse RJB, Busch ORC, Ten Kate FJW, Creemers GM, Punt CJA, Plukker JTM, Verheul HMW, Bilgen EJS, van Dekken H, van der Sangen MJC, Rozema T, Biermann K, Beukema JC, Piet AHM, van Rij CM, Reinders JG, Tilanus HW, Steyerberg EW, van der Gaast A CROSS study group. Neoadjuvant chemoradiotherapy plus surgery versus surgery alone for oesophageal or junctional cancer (CROSS): long-term results of a randomised controlled trial. Lancet Oncol. 2015;16:1090–1098. doi: 10.1016/S1470-2045(15)00040-6. [DOI] [PubMed] [Google Scholar]
- 7.Sjoquist KM, Burmeister BH, Smithers BM, Zalcberg JR, Simes RJ, Barbour A, Gebski V Australasian Gastro-Intestinal Trials Group. Survival after neoadjuvant chemotherapy or chemoradiotherapy for resectable oesophageal carcinoma: an updated meta-analysis. Lancet Oncol. 2011;12:681–692. doi: 10.1016/S1470-2045(11)70142-5. [DOI] [PubMed] [Google Scholar]
- 8.Medical Research Council Oesophageal Cancer Working Group. Surgical resection with or without preoperative chemotherapy in oesophageal cancer: a randomised controlled trial. Lancet. 2002;359:1727–1733. doi: 10.1016/S0140-6736(02)08651-8. [DOI] [PubMed] [Google Scholar]
- 9.Allum WH, Stenning SP, Bancewicz J, Clark PI, Langley RE. Long-term results of a randomized trial of surgery with or without preoperative chemotherapy in esophageal cancer. J. Clin. Oncol. 2009;27:5062–5067. doi: 10.1200/JCO.2009.22.2083. [DOI] [PubMed] [Google Scholar]
- 10.Ando N, Kato H, Igaki H, Shinoda M, Ozawa S, Shimizu H, Nakamura T, Yabusaki H, Aoyama N, Kurita A, Ikeda K, Kanda T, Tsujinaka T, Nakamura K, Fukuda H. A randomized trial comparing postoperative adjuvant chemotherapy with cisplatin and 5-fluorouracil versus preoperative chemotherapy for localized advanced squamous cell carcinoma of the thoracic esophagus (JCOG9907) Ann Surg Oncol. 2012;19:68–74. doi: 10.1245/s10434-011-2049-9. [DOI] [PubMed] [Google Scholar]
- 11.Urschel JD, Vasan H. A meta-analysis of randomized controlled trials that compared neoadjuvant chemoradiation and surgery to surgery alone for resectable esophageal cancer. Am J Surg. 2003;185:538–543. doi: 10.1016/s0002-9610(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 12.Pasquali S, Yim G, Vohra RS, Mocellin S, Nyanhongo D, Marriott P, Geh JI, Griffiths EA. Survival after neoadjuvant and adjuvant treatments compared to surgery alone for resectable esophageal carcinoma: a network meta-analysis. Ann Surg. 2017;265:481–491. doi: 10.1097/SLA.0000000000001905. [DOI] [PubMed] [Google Scholar]
- 13.Wilson TR, Longley DB, Johnston PG. Chemoresistance in solid tumours. Ann Oncol. 2006;17(Suppl 10):x315–324. doi: 10.1093/annonc/mdl280. [DOI] [PubMed] [Google Scholar]
- 14.Saunders NA, Simpson F, Thompson EW, Hill MM, Endo-Munoz L, Leggatt G, Minchin RF, Guminski A. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol Med. 2012;4:675–684. doi: 10.1002/emmm.201101131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu D, Wang DC, Cheng Y, Qian M, Zhang M, Shen Q, Wang X. Roles of tumor heterogeneity in the development of drug resistance: a call for precision therapy. Semin Cancer Biol. 2017;42:13–19. doi: 10.1016/j.semcancer.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Jiang L, Wang W, Li G, Sun C, Ren Z, Sheng H, Gao H, Wang C, Yu H. High TUG1 expression is associated with chemotherapy resistance and poor prognosis in esophageal squamous cell carcinoma. Cancer Chemother Pharmacol. 2016;78:333–339. doi: 10.1007/s00280-016-3066-y. [DOI] [PubMed] [Google Scholar]
- 17.Chang ZW, Jia YX, Zhang WJ, Song LJ, Gao M, Li MJ, Zhao RH, Li J, Zhong YL, Sun QZ, Qin YR. LncRNA-TUSC7/miR-224 affected chemotherapy resistance of esophageal squamous cell carcinoma by competitively regulating DESC1. J Exp Clin Cancer Res. 2018;37:56. doi: 10.1186/s13046-018-0724-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hara T, Makino T, Yamasaki M, Tanaka K, Miyazaki Y, Takahashi T, Kurokawa Y, Nakajima K, Matsuura N, Mori M, Doki Y. Effect of c-Met and CD44v6 expression in resistance to chemotherapy in esophageal squamous cell carcinoma. Ann Surg Oncol. 2019;26:899–906. doi: 10.1245/s10434-018-07126-5. [DOI] [PubMed] [Google Scholar]
- 19.Shekhar MP. Drug resistance: challenges to effective therapy. Curr Cancer Drug Targets. 2011;11:613–623. doi: 10.2174/156800911795655921. [DOI] [PubMed] [Google Scholar]
- 20.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- 22.Peng J, Zhou Y, Zhu S, Wei W. High-throughput screens in mammalian cells using the CRISPR-Cas9 system. FEBS J. 2015;282:2089–2096. doi: 10.1111/febs.13251. [DOI] [PubMed] [Google Scholar]
- 23.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurata M, Rathe SK, Bailey NJ, Aumann NK, Jones JM, Veldhuijzen GW, Moriarity BS, Largaespada DA. Using genome-wide CRISPR library screening with library resistant DCK to find new sources of Ara-C drug resistance in AML. Sci Rep. 2016;6:36199. doi: 10.1038/srep36199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie X, Wang X, Liao W, Fei R, Wu N, Cong X, Chen Q, Wei L, Wang Y, Chen H. PPPDE1 promotes hepatocellular carcinoma development by negatively regulate p53 and apoptosis. Apoptosis. 2019;24:135–144. doi: 10.1007/s10495-018-1491-6. [DOI] [PubMed] [Google Scholar]
- 27.Heaton BE, Kennedy EM, Dumm RE, Harding AT, Sacco MT, Sachs D, Heaton NS. A CRISPR activation screen identifies a pan-avian influenza virus inhibitory host factor. Cell Rep. 2017;20:1503–1512. doi: 10.1016/j.celrep.2017.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gartel AL. P21(WAF1/CIP1) may be a tumor suppressor after all. Cancer Biol Ther. 2007;6:1171–1172. doi: 10.4161/cbt.6.8.4712. [DOI] [PubMed] [Google Scholar]
- 29.Gou Q, Gao L, Nie X, Pu W, Zhu J, Wang Y, Liu X, Tan S, Zhou JK, Gong Y, He J, Wu K, Xie Y, Zhao W, Dai L, Liu L, Xiang R, Wei YQ, Zhang L, Peng Y. Long noncoding RNA AB074169 inhibits cell proliferation via modulation of KHSRP-mediated CDKN1a expression in papillary thyroid carcinoma. Cancer Res. 2018;78:4163–4174. doi: 10.1158/0008-5472.CAN-17-3766. [DOI] [PubMed] [Google Scholar]
- 30.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang P, Ouyang DJ, Chang S, Li MY, Li L, Li QY, Zeng R, Zou QY, Su J, Zhao P, Pei L, Yi WJ. Chemotherapy-driven increases in the CDKN1A/PTN/PTPRZ1 axis promote chemoresistance by activating the NF-kappaB pathway in breast cancer cells. Cell Commun Signal. 2018;16:92. doi: 10.1186/s12964-018-0304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zybura-Broda K, Wolder-Gontarek M, Ambrozek-Latecka M, Choros A, Bogusz A, Wilemska-Dziaduszycka J, Rylski M. HuR (Elavl1) and HuB (Elavl2) stabilize matrix metalloproteinase-9 mRNA during seizure-induced mmp-9 expression in neurons. Front Neurosci. 2018;12:224. doi: 10.3389/fnins.2018.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Alessandro V, Muscarella LA, Copetti M, Zelante L, Carella M, Vendemiale G. Molecular detection of neuron-specific ELAV-like-positive cells in the peripheral blood of patients with small-cell lung cancer. Cell Oncol. 2008;30:291–297. doi: 10.3233/CLO-2008-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Chen X, Cheng R, Yang F, Yu M, Wang C, Cui S, Hong Y, Liang H, Liu M, Zhao C, Ding M, Sun W, Liu Z, Sun F, Zhang C, Zhou Z, Jiang X, Chen X. The Jun/miR-22/HuR regulatory axis contributes to tumourigenesis in colorectal cancer. Mol Cancer. 2018;17:11. doi: 10.1186/s12943-017-0751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richards NG, Rittenhouse DW, Freydin B, Cozzitorto JA, Grenda D, Rui H, Gonye G, Kennedy EP, Yeo CJ, Brody JR, Witkiewicz AK. HuR status is a powerful marker for prognosis and response to gemcitabine-based chemotherapy for resected pancreatic ductal adenocarcinoma patients. Ann Surg. 2010;252:499–505. doi: 10.1097/SLA.0b013e3181f1fd44. discussion 505-496. [DOI] [PubMed] [Google Scholar]
- 36.Tachibana I, Bodorova J, Berditchevski F, Zutter MM, Hemler ME. NAG-2, a novel transmembrane-4 superfamily (TM4SF) protein that complexes with integrins and other TM4SF proteins. J Biol Chem. 1997;272:29181–29189. doi: 10.1074/jbc.272.46.29181. [DOI] [PubMed] [Google Scholar]
- 37.Hirano C, Nagata M, Noman AA, Kitamura N, Ohnishi M, Ohyama T, Kobayashi T, Suzuki K, Yoshizawa M, Izumi N, Fujita H, Takagi R. Tetraspanin gene expression levels as potential biomarkers for malignancy of gingival squamous cell carcinoma. Int J Cancer. 2009;124:2911–2916. doi: 10.1002/ijc.24297. [DOI] [PubMed] [Google Scholar]
- 38.Qi W, Sun L, Liu N, Zhao S, Lv J, Qiu W. Tetraspanin family identified as the central genes detected in gastric cancer using bioinformatics analysis. Mol Med Rep. 2018;18:3599–3610. doi: 10.3892/mmr.2018.9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou P, Wu C, Wang Y, Qi R, Bhavanasi D, Zuo Z, Dos Santos C, Chen S, Chen Y, Zheng H, Wang H, Perl A, Guo D, Huang J. A genome-wide CRISPR screen identifies genes critical for resistance to FLT3 inhibitor AC220. Cancer Research. 2017;77:4402–4413. doi: 10.1158/0008-5472.CAN-16-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodspeed A, Jean A, Costello JC. A whole-genome CRISPR screen identifies a role of MSH2 in cisplatin-mediated cell death in muscle-invasive bladder cancer. Eur Urol. 2019;75:242–250. doi: 10.1016/j.eururo.2018.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, Carette JE. Genetic dissection of flaviviridae host factors through genome-scale CRISPR screens. Nature. 2016;535:159–163. doi: 10.1038/nature18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, Lee H, Zhang F, Sharp PA. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]