

Abstract
Due to its potent anticancer activity, there is interest in repurposing of the FDA-approved anti-alcoholism drug, disulfiram (DSF). DSF forms potent complexes with copper (DSF/Cu) that induce apoptosis of many types of cancer cells. Here, we investigated the role of DSF/Cu in autophagy, a mechanism of cell death or survival, and its interplay with DSF/Cu induced apoptosis of human pancreatic and breast cancer cells. Methods: Levels of autophagy and apoptosis were assessed by Western blot, flow cytometry and immunofluorescence analysis. Cell viability was measured by MTT assays. Activation of inositol-requiring enzyme 1α (IRE1α)-mRNA X-box binding protein 1 (XBP1) pathway and spliced XBP1 (XBP1s) expression were analyzed by Western blot, Phos-tag gel assay, RT-PCR, qRT-PCR and flow cytometry. Results: The apoptosis induced by DSF/Cu in pancreatic and breast cancer cells is autophagy dependent. This is accomplished by activating IRE1α, the sensor of unfolded protein response (UPR) via promotion of phosphorylation of IRE1α and its downstream XBP1 splicing into active XBP1s. Conclusions: DSF/Cu induces ER-stress through activation of IRE1α-XBP1 pathway which is responsible, at least in part, for induction of autophagy-dependent apoptosis of cancer cells. Insight into the ER-stress inducing ability by DSF/Cu may open a new research area for rational design of innovative therapeutic strategies for pancreatic and breast cancers.
Keywords: Disulfiram, cancer, cytotoxic autophagy, apoptosis, ER stress, IRE1α pathway, XBP1s
Introduction
Drug repurposing has become an attractive area of cancer research for a variety of reasons. Increased knowledge of complexity and interrelationships of numerous signaling pathways involved in many human pathologies and the drugs developed for their treatment, sometimes even decades ago, are finding new applications in the field of cancer therapeutics. One such entity is DSF, a well-established agent for treatment of alcoholism based on its property of being a pan-inhibitor of aldehyde dehydrogenase (ALDH), a critical enzymatic activity in the metabolism of aldehydes, such as acetaldehyde, the initial metabolite of ethanol and genotoxic, unless converted to acetic acid. The interest in repurposing DSF for cancer therapy is based on its target, ALDH, which plays a key role in the resistance to the cytotoxicity in most standard chemo- and radio-therapies through metabolizing the genotoxic aldehydes they generate.
The anti-tumor activity of DSF, however, is not solely due to inhibition of ALDH enzymatic activities. DSF is a potent chelator of the trace element copper (Cu2+) and forms DSF-Cu2+ complexes (DSF/Cu), which effectively inhibit 26S proteasome activity [1]. Consequently, DSF/Cu interferes with the activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) [2-4], a master transcriptional factor. However, this activity alone does not define the anti-tumor effect of DSF/Cu. Proteasome inhibitors have emerged as a new class of endoplasmic reticulum (ER) stress agents [5,6]. Varied environmental, nutritional and pathological stresses to both normal and tumor cells can cause protein misfolding and accumulation in the ER and activation of the UPR. This is particularly pertinent to tumor cells, which due to their relatively higher proliferation and growth rates than normal cells are prone to ER stress. To ensure survival, the UPR plays a major role in blocking translation and promoting proper protein folding, while simultaneously enhancing the degradation of misfolded proteins and autophagy. If these activities do not restore proper cellular functions, the UPR leads ultimately to apoptosis. Consequently, the UPR interfaces survival as well as apoptotic pathways. ER stress induced autophagy activation could also induce a cytotoxic autophagic response depending on the intensity of the stimulus factors, the cell type (normal vs. cancer cells), and the cellular context (hypoxia, starvation, treatment with anti-tumor agents) [7,8]. Therefore, therapeutic exploitation of cytotoxic autophagy to drive cancer cell death is an emerging novel concept for novel cancer treatments. Therefore, the anti-tumor activity of DSF/Cu was investigated in vitro relative to its ability to promote cytotoxic autophagy of breast and pancreatic carcinoma cells via its impact on the ER stress and UPR pathways.
Materials and methods
Cell culture
The human breast carcinoma MDA-MB-231 cell line was acquired from the Duke Cancer Center Cell Culture Facility (Durham, NC, USA). The human breast cancer UACC-812 and pancreatic ductal adenocarcinoma (PDAC) PANC-1 cell lines were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). The pancreatic cancer PDAC6 cell line was established from the ascites of a patient with metastatic pancreatic ductal adenocarcinoma [4]. PANC-1, PDAC6 and MDA-MB-231 cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal calf serum (FCS; Atlanta Biologicals, Flowery Branch, GA, USA). UACC-812 was cultured in RPMI 1640 medium supplemented with 10% fetal calf serum. All cells were cultured at 37°C in a 5% CO2 atmosphere.
Chemical reagents and antibodies
Tetraethylthiuram disulfide (disulfiram, DSF), copper (II) D-gluconate (copper, Cu), autophagy inhibitor wortmannin, IRE1α inhibitors STF-083010 and 4µ8C, proteasome inhibitor bortezomib, and NF-κB inhibitor IMD-0354 were purchased from Sigma-Aldrich (St. Louis, MO, USA). The apoptosis inhibitor Z-VAD-FMK and the autophagy inhibitor chloroquine (CQ) were purchased from MedChem Express (Monmouth Junction, NJ, USA). The following rabbit monoclonal antibodies (mAb) used for Western blot analyses were purchased from Cell Signaling Technology (Danvers, MA, USA), and used at the following indicated dilutions: anti-LC3A/B (#4108) (1:1000), anti-human cleaved PARP (#9541) (1:1000), anti-human and anti-mouse ß-actin (#4970) (1:2000), anti-human eIF2α (#5324) (1:1000) and anti-peIF2α (#3398) (1:1000), anti-human XBP1s (D2C1F) (#12782) (1:1000) and goat anti-rabbit IgG, HRP-linked secondary antibody (#7074) (1:2000). The rabbit polyclonal anti-human phosphorylated IRE1α antibody (ab48187) (1:1000) was purchased from Abcam, Inc (Burlingame, CA, USA), and the anti-calnexin mouse mAb TO-5 was developed in our laboratory, as described previously [9]. All the primary antibodies were diluted in Phosphate Buffered Saline (PBS) containing 5% nonfat dry milk and secondary antibody was diluted in Tris Buffered Saline with 0.1% Tween® 20 (TBST). The following antibodies and the dilutions used for immunofluorescence and flow cytometry analysis were: Alexa Fluor® 488-conjugated rabbit anti-cleaved Caspase-3 (Asp175) (D3E9) mAb (#9603) (1:50), and Phycoerythrin (PE) conjugated-rabbit anti-LC3B (D11) XP® mAb (#8899) (1:50), and anti-LC3B (#2775) (1:200) were purchased from Cell Signaling Technology (Danvers, MA, USA), Mouse anti-human XBP1s mAb (MAB4257) (1:1000) was purchased from R&D Systems Inc (Minneapolis, MN, USA), and R-PE-conjugated AffiniPureF(ab’)2 Fragment Goat Anti-mouse IgG (115-116-071) (1:3000) and FITC- conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) (111-095-003) (1:2000) were purchased from Jackson ImmunoResearch Inc (West Grove, PA, USA). All antibodies were diluted in TBST containing 1% BSA. The restriction enzyme PstI (#R0140S) was purchased from New England Biolabs (Ipswich, MA, USA). Phos-tag™ SuperSep™ precast (50 μmol/l), 7.5% polyacrylamide gel was purchased from FUJIFILM Wako Chemicals U.S.A. Corporation (Richmond, VA, USA). The plasmid for retroviral transduction, pQCXI Puro DsRed-LC3-GFP (DsRed-LC3-GFP), was a gift from David Sabatini (Addgene plasmid #31182, Cambridge, MA, USA) [10].
Western blot analysis
Cells were plated in 6-well plates at a density of 3×105 cells/well (PANC-1), 4×105 cells/well (UACC-812, MDA-MB-231), or 5×105 cells/well (PDAC6) in 2 mL of the appropriate complete medium and grown overnight. All the cells were treated with DMSO, DSF (0.25 μM), Cu (1 µM) or DSF/Cu (0.25/1 µM) for 24 hours in the absence or presence of CQ (20 µM, 16 hours). When cells were treated at different times with DSF/Cu, the cells were plated in 6-well plates at a density of 4×105 cells/well (UACC-812) or 5×105 cells/well (PDAC6) in 2 mL of the appropriate complete medium and grown overnight. All the cells were then treated with DSF/Cu (0.25/1 µM) and collected at 0, 4, 8, 12, 16, 24, 36 and 48 hours. When treated with wortmannin and DSF/Cu, cells were plated in 6-well plates at a density of 5×105 cells/well in 2 mL of the appropriate complete medium and grown overnight. The cells were treated with wortmannin (1 µM) or not for 6 hours followed by DSF/Cu (0.25/1 µM) or not for another 24 hours. When treated with STF-083010 and DSF/Cu, cells were plated in 6-well plates at a density of 3×105 cells/well in 2 mL of the appropriate complete medium and grown overnight. The cells were treated with STF-083010 (1 µM) or not for 30 min, then co-treated with DSF/Cu (0.25/1 µM) or not for an additional 12 hours.
Treated cells were collected and lysed in lysis buffer (10 mM Tris-HCl [pH 8.2], 1% NP40, 1 mM EDTA, 0.1% BSA, 150 mM NaCl) containing 1/50 (vol./vol.) of a protease inhibitor cocktail (Calbiochem, San Diego, CA, USA). The supernatants of the lysed cells were analyzed by Western blot for expression of related proteins as described [4].
Phos-tag gel assay
Cells were plated in 6-well plates at a density of 4×105 cells/well in 2 mL of the appropriate complete medium and grown overnight. Cells were then treated with DSF/Cu (0.125/1 μM or 0.25/1 μM) for 8 or 24 hours. Treated cells were collected and lysed, and 10 μg of each cell lysate was loaded on Phos-tag™ SuperSep™ precast polyacrylamide gels. After electrophoresis, Phos-tag gels were soaked in a transfer buffer containing 5 mM EDTA for 10 min and repeated twice with gentle agitation to eliminate the manganese ions from the gels. Next, the gels were soaked in a transfer buffer without EDTA for 10 min with gentle agitation. The proteins were electrophoretically transferred to PVDF membranes in the transfer buffer without SDS/EDTA. The other steps were the same as regular Western Blotting experiments.
Flow cytometry
Cells were plated in 6-well plates at a density of 3×105 cells/well (PANC-1) or 4×105 cells/well (UACC-812, MDA-MB-231, PDAC6) in 2 mL of the appropriate culture medium and grown overnight. Cells were then treated with DMSO, Cu (1 μM), DSF (0.25 μM) or DSF/Cu (0.25/1 µM) for 24 hours. For detection of intracellular molecules, such as cleaved Caspase-3 and LC3II, cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% triton X-100 prior to being stained with Alexa Fluor® 488- or PE-conjugated antibodies for 1 hour. For detection of intracellular XBP1s, cells that were fixed and permeabilized as described above were stained with mouse anti-human XBP1s mAb for 1 hour and then PE-conjugated goat anti-mouse IgG for 30 min. Cells were analyzed using a BD Biosciences FACSAria II flow cytometer.
Immunofluorescence analysis (IFA)
MDA-MB-231 cells were plated in 6-well plates (with a cover slip at the bottom) at a density of 4×105 cells/well in 2 mL of the appropriate culture medium, cultured overnight and then treated with DSF/Cu (0.25/1 μM) for 24 hours. For the detection of autophagosome puncta in cytoplasm, cells were stained with anti-LC3B after they had been fixed with 4% paraformaldehyde and permeabilized with 0.1% triton X-100. Then the secondary FITC-conjugated Ab anti-Rabbit IgG was added and counterstained for 5 min with the nuclear stain DAPI at 1 μg/mL in PBS. The stained cells on the cover slip were sealed on a slide glass and observed by immunofluorescence microscope (10×100).
For the detection of autophagy and apoptosis in cytoplasm, fixed and permeabilized PANC-1 cells were treated with DSF/Cu (0.25/1 μM) for 24 hours and stained with Alexa Fluor® 488-conjugated anti-cleaved Caspase-3 and Phycoerythrin (PE) conjugated-rabbit anti-LC3B, and counterstained for 5 min with the nuclear stain DAPI at 1 μg/mL in PBS. The stained cells on the cover slip were sealed on a slide glass and observed by confocal laser scanning microscope (10×60).
Cell proliferation and MTT assay
Cells were plated in 96-well plates at a density of 5×103 cells/well in 100 µL of the appropriate complete medium and grown overnight. Cells were i) pretreated with STF-083010 (10 µM), or 4μ8C (10 μM), for 30 min, and then co-treated with DSF/Cu (0.25/1 µM) for another 24 hours or ii) co-treated with wortmannin (10 µM) or Z-VAD-FMK (20 µM), or CQ (20 μM) with DSF/Cu (0.25/1 µM) for 24 hours. Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Data was generated in triplicate and shown as mean ± SD (*P<0.05, **P<0.01, and ***P<0.001).
Retroviral transduction
PANC-1 cells were retrovirally transduced with DsRed-LC3-GFP to establish a stable cell line according to the manufacturer’s instructions. DsRed-LC3-GFP- transfected PANC-1 cells were seeded into a 6-well plate (3×105 cells/well) and grown overnight. The cells were subsequently treated with DSF/Cu (0.25/1 μM) for 24 hours. Treated cells were harvested, fixed for detection of LC3 positive cell population in red fluorescence by flow analysis.
Real-time quantitative reverse transcription PCR (qRT-PCR)
Cells were plated in 6-well plates at a density of 3×105 cells/well (PANC-1) or 4×105 cells/well (UACC-812, MDA-MB-231, PDAC6) in 2 mL of the appropriate complete medium and treated with DMSO, Cu (1 μM), DSF (0.25 μM) and DSF/Cu (0.25/1 μM) for 24 hours. On the other hand, the PANC-1 cells treated with different doses (0.125/1 μM or 0.25/1 μM) of DSF/Cu for 8 or 24 hours. Total RNA from all samples was isolated using TRIzol and 1 μg total RNA was reverse transcribed using M-MLV reverse transcriptase purchased from Life Technologies (Carlsbad, CA, USA), according to the manufacturer’s instructions. All samples within each experiment were reverse transcribed at the same time. The resulting cDNA was diluted to a concentration of 100 ng/μL in nuclease-free water. qRT-PCR using cDNA as a template with SYBR green detection was performed using a LightCycler96 system with FastStart SYBR Green Master Mix purchased from Roche (Penzburg, Germany). The following real-time PCR primers (forward and reverse) were used: spliced XBP1s, 5’-TGCTGAGTCCGCAGCAGGTG-3’ and 5’-GCTGGCAGGCTCTGGGGAAG-3’ [11]; ERdj4, 5’-TCTTAGGTGTGCCAAA ATCGG-3’ and 5’-TGTCAGGGTGGTACTTCATGG-3’; P58IPK, 5’-GGCTCGGTATTCCCCTTCCT-3’ and 5’-AGTAGCCCTCCGATAATAAGCAA-3’. Appropriate no-RT and non-template controls were included in each 96-well PCR reaction, and dissociation. Each gene expression level was normalized to mRNA levels of the housekeeping gene r18S. Fold changes of each gene’s expression were compared to that of the untreated sample using the 2-ΔΔCT method.
Semi-quantitative RT-PCR
The PANC-1 cells were treated with different doses (0.125/1 μM or 0.25/1 μM) of DSF/Cu for 8 or 24 hours. Tunicamycin (1 μg/mL) treated cells were used as a positive control. Total RNA was extracted with TRIZOL Reagent. Semi-quantitative RT-PCR was performed to amplify the spliced and unspliced XBP1 mRNA. Human XBP1 primers were used as described previously [12]: (Forward 5’-AAACAGAGTAGCAGCTCAGACTGC-3’, Reverse 5’-TCCTTCTGGGTAGACCTCTGGGAG-3’). Actin (Forward 5’-CATGTACGTTGCTATCCAGGC-3’, reverse 5’-CATGTACGTTGCTATCCAGGC-3’) was used as a loading control. PCR products were electrophoresed on a 2.5% agarose gel to distinguish the size difference, i.e., 26 nucleotides (nt) between the spliced (XBP1s) and the unspliced XBP1 (XBP1u). Furthermore, the amplified XBP1 cDNA was digested by restriction enzyme PstI, which has its specific recognition site located within the 26-nt region of XBP1 cDNA removed by IRE1α-mediated splicing, as previously described [12].
Results
DSF/Cu induces autophagy and apoptosis in cancer cells
The ability of DSF/Cu to induce autophagy and subsequent apoptosis was initially studied using a panel of breast and pancreatic carcinoma cell lines, which consisted of the breast cancer UACC-812 and MDA-MB-231 cell lines and the pancreatic cancer PDAC6 and PANC-1 cell lines. The cell lines were cultured in the presence of DSF/Cu in the presence or absence of CQ, an inhibitor of both fusion of autophagosome with lysosome and lysosomal protein degradation [13]. The conversion of the cytosolic form of the microtubule-associated protein 1A/1B light chain 3B (LC3I) to lipid bound LC3II is a critical step in autophagy, because LC3II is a central protein in the pathway, where it functions in autophagosome biogenesis. In the presence of CQ, accumulation of LC3II positive autophagosomes demonstrates efficient autophagic flux, while little to no detection of LC3II expression indicates a defect or delay earlier in the process, prior to degradation at the autolysosome. Thus, autophagic flux is considered a more reliable indicator of autophagic activity than measurement of autophagosome numbers. Accordingly, the difference in LC3II expression levels in cells treated in the presence and absence of CQ represents LC3II being delivered to lysosomes for degradation [14]. Consequently, Western blot analyses of cells treated with DSF/Cu and CQ were performed relative to the following two parameters: i) autophagic flux, i.e, LC3II expression levels in the presence or absence of CQ, and ii) the cleavage by Caspase-3 of poly (ADP-ribose) polymerase (PARP) to cleaved PARP, a marker of apoptosis. As expected, DSF/Cu (0.25/1 μM) induced the autophagy and apoptosis in all four cell lines tested as evidenced by expression levels of LC3II and cleaved PARP. However, DSF (0.25 μM) or Cu (1 μM) alone induced neither autophagy nor apoptosis. Although increased accumulation of LC3II was detected in all 4 cell lines in the presence of CQ, apoptosis was reduced in the presence of CQ compared to that in its absence in both breast carcinoma cell lines UACC-812 and MDA-MB-231, but remained at similar levels with or without CQ, as indicated by cleaved PARP, in pancreatic carcinoma cell lines PDAC6 and PANC-1 (Figure 1A-D). The latter may reflect that CQ is a late inhibitor of autophagy and reduced the viability of PDAC cells [15,16]. Concurrently, flow cytometry analysis confirmed autophagy and apoptosis were induced by DSF/Cu. Increased autophagy activity, indicated by DsRed-LC3 puncta in DsRed-LC3-GFP-transfected PANC-1 cells [10] was evident after 12 h and 24 h culture in the presence of DSF/Cu (Figure 1E, 1F). To confirm the sequence of DSF/Cu-induced autophagy and apoptotic events that define its anti-tumor activity, a kinetic analysis of treatment of the UACC-812 and PDAC6 cell lines over the course of a 48 h exposure to DSF/Cu was performed. Western blot analyses of the cell cultures indicated that conversion of LC3I to LC3II was detectable within 8 h of exposure, whereas cleaved PARP was detected at or around 12 h in both cell lines (Figure 1G, 1H). The falloff in the levels of LC3II and cleaved PARP after 24 h can be attributed to the decreased viability of the cell cultures. Gray intensity values for LC3II or cleaved PARP in the Western blot (Figure 1) were determined by Image J software with the mean gray value of the protein band areas method (Figure S1). In addition, IFA staining of PANC-1 cells for LC3II and cleaved Caspase-3 indicated that DSF/Cu induced autophagy and apoptosis simultaneously at a single cell level (Figure 1I), while the number of autophagosome puncta increased in DSF/Cu treated-MDA-MB-231 cells compared to untreated cells (Figure 2). Furthermore, cleaved Caspase-3 expression, another indicator of apoptosis, was detected by flow cytometry analysis in all four cell lines treated with DSF/Cu (Figure 3).
Figure 1.

DSF/Cu induces in vitro both autophagy and apoptosis in breast and pancreatic cancer cell lines. The cell lines UACC-812 (A), MDA-MB-231 (B), PDAC6 (C) and PANC-1 (D) were treated as indicated for 24 hours in the presence or absence of CQ (20 μM, 16 hours) for detection of autophagic flux (increased LC3II/LC3I ratio in the presence of CQ) and apoptosis (cleaved PARP). DsRed-LC3-GFP transfected PANC-1 cells treated with DSF/Cu for 12 hours (E) or 24 hours (F) were analyzed for red fluorescence LC3 positive cells by flow cytometry. UACC-812 (G) and PDAC6 (H) cells were treated with DSF/Cu at different indicated time points, lysed and analyzed by Western blot for expression of LC3II/I and cleaved PARP. PANC-1 cells treated with DSF/Cu were stained for autophagy and apoptosis at the same time and analyzed by IFA. A representative cell stained by immunofluorescence and viewed under confocal microscopy (600×) (OLYMPUSDeltaVision RT IX70) is shown (I): Blue- DAPI for nucleus (top left), Red-LC3 (bottom left) and Green-Cleaved Caspase-3 (top right) and merged (bottom right).
Figure 2.
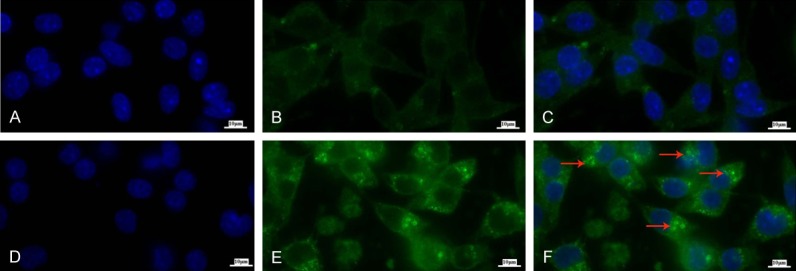
DSF/Cu increases levels of autophagosome puncta. The cell line MDA-MB-231 was treated in the absence (A-C) or presence (D-F) of DSF/Cu for 24 hours. Cells were harvested, fixed and permeabilized for intracellular LC3B staining. The autophagosomes in the cytoplasm were observed by an immunofluorescence microscope (10×100) (ZEISS AXIOScope. A1). Blue-DAPI for nucleus (A, D), Green-LC3B (B, E) and merged (C, F), autophagosomes as indicated by the red arrows in (F).
Figure 3.
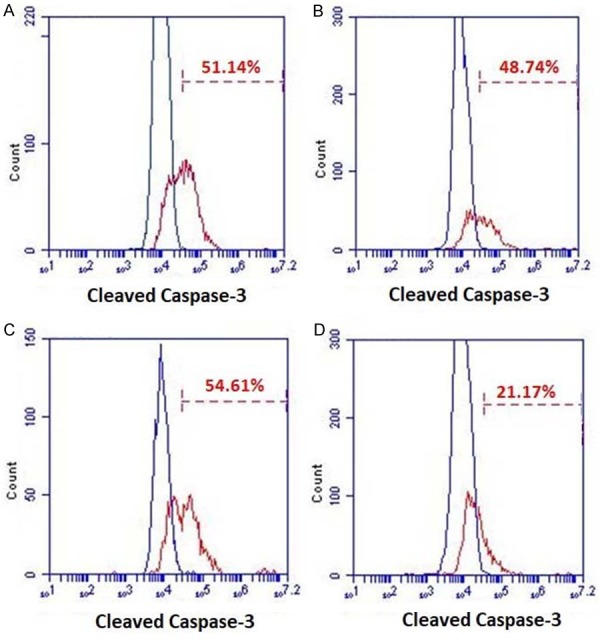
DSF/Cu induces apoptosis via Caspase-3 activation. The cell lines UACC-812 (A), MDA-MB-231 (B), PDAC6 (C) and PANC-1 (D) were treated in the absence or presence DSF/Cu for 24 hours. Treated cells were harvested, fixed, and permeabilized for intracellular staining of cleaved Caspase-3. Negative controls are superimposed on each graph. The percentage of cleaved Caspase-3 positive cells of each cell line is shown.
DSF/Cu induced apoptosis is autophagy dependent
Since the autophagy pathway balances cell survival with cell death relative the extent to which a cell can counteract various intensities of stress, the dependency of DSF/Cu induced apoptosis on autophagy was studied. Wortmannin, is an inhibitor of PI3K, which is required for autophagy [17]. Exposure of all four carcinoma cell lines to wortmannin resulted in abrogation of DSF/Cu-induced autophagy as well as apoptosis (Figure 4A-D), as indicated by the intensity values for LC3II and cleaved PARP in the Western blot analyses of the treated cells (Figures 4, S2). Relative to this point, cell viability analysis of the four carcinoma cell lines tested indicated that CQ (Figure 4E-H) or wortmannin (Figure 4I-L) significantly blocked the ability of DSF/Cu to induce cell death in the four human carcinoma cell lines tested. Finally, as expected, in the presence of Z-VAD-FMK, which inhibits Caspase-3, a required component of the apoptotic pathway, the ability of DSF/Cu to induce apoptosis of the four breast and pancreatic carcinoma cell lines was significantly reduced as indicated by cell viability assays (Figure 4I-L). In summary, the results of this set of experiments indicate that induction of apoptosis in tumor cells by DSF/Cu is dependent on the initiation of autophagy prior to the engagement of the apoptotic pathway.
Figure 4.

DSF/Cu-induced apoptosis is autophagy dependent. The cell lines UACC-812 (A), MDA-MB-231 (B), PDAC6 (C) and PANC-1 (D) were pretreated with or without wortmannin for 6 hours. The cells were further treated with or without DSF/Cu for 24 hours. Treated cells were lysed and analyzed by Western blot for autophagy and apoptosis. Cells were treated with DSF/Cu for 24 hours in the presence or absence of CQ (E - UACC-812, F - MDA-MB-231, G - PDAC6, H - PANC-1) or in the presence or absence of wortmannin and Z-VAD-FMK (I - UACC-812, J - MDA-MB-231, K - PDAC6, L - PANC-1). Cell viability was determined by the MTT assay. Data generated in triplicate were expressed relative to the mean of vehicle-treated cells in each experiment, for three independent experiments, and shown as mean ± SD (*P<0.05, **P<0.01, and ***P<0.001).
DSF/Cu causes ER-stress by promoting IRE1α-mRNA splicing
The UPR is a stress response pathway triggered by sensors located at the ER membrane whose function is to reduce an excessive accumulation of misfolded protein in the ER. ER stress activates UPR to alleviate the stress and restore ER homeostasis. It promotes cell survival and adaptation by increasing transcription of ER-resident proteins that facilitate protein folding and degradation in the ER and attenuating translation of proteins targeted to the ER [6,18,19].
In order to further investigate the mechanistic pathways involved in DSF/Cu-induced autophagy, given that DSF/Cu is a functional proteasome inhibitor and can disrupt UPR, one can hypothesize that DSF/Cu inhibits UPR and causes increased and unresolved ER stress leading to autophagic cell death in cancer cells. Therefore, the impact of DSF/Cu on the three distinct pathways that constitute the UPR, namely, the IRE1α, ATF6α, and PERK pathways, was evaluated by analyzing the expression levels of phosphorylated IRE1α (pIRE1α) and XBP1s (IRE1α branch), elF2α/pelF2α (PERK branch) and calnexin (ATF6α branch), in DSF/Cu treated and untreated cells. Surprisingly, it was observed in Western blot analyses on all four cell lines that DSF/Cu activated, instead of inhibited the UPR IRE1α branch by upregulating phosphorylated IRE1α (pIRE1α) in a dose and time dependent manner (Figure 6A-D). Activated IRE1α, i.e., pIRE1α, possesses both kinase and RNase activity and triggers unconventional cytoplasmic splicing of XBP1 mRNA by splicing 26 nucleotides from the unspliced XBP1 (XBP1u) mRNA. The resulting spliced XBP1 mRNA is translated into the highly active transcription factor, XBP1s, that controls genes involved in ER membrane biosynthesis, protein import, chaperoning, ER-associated degradation (ERAD), and cell type-specific genetic programs [20,21]. Therefore, the impact of DSF/Cu on XBP1s expression was determined in several of the cancer cell lines used in this study. As expected, DSF/Cu increased the levels of spliced XBP1 at the mRNA level as determined by real-time quantitative PCR analysis (Figure 5A, 5B) and the protein levels in the breast carcinoma UACC-812 cell line as well as the pancreatic carcinoma PDAC6 line, as determined by flow cytometry analysis (Figure 5C, 5D). To visualize XBP1u and XBP1s by gel electrophoresis, we used XBP1 splicing assay and found that both bands were detected in PANC-1 cells treated by DSF/Cu and the XBP1s was increased in a dose and time dependent manner. Furthermore, unlike XBP1u, XBP1s does not contain a PstI restriction site, thus PstI could not digest the DNA of XBP1s. DNA gel electrophoresis of RT-PCR products digested with the restriction enzyme PstI from cells treated with DSF/Cu revealed an increased non-digested ~500 bp band (XBP1s), and digested ~200 and 300 bp bands (XBP1u) (Figure 5E). The classical XBP1s downstream genes ERdj4 and P58IPK as well as XBP1s were also elevated, quantified by qRT-PCR, in PANC-1 cells in responding to DSF/Cu treatment in a dose and time dependent manner (Figure 5F).
Figure 6.
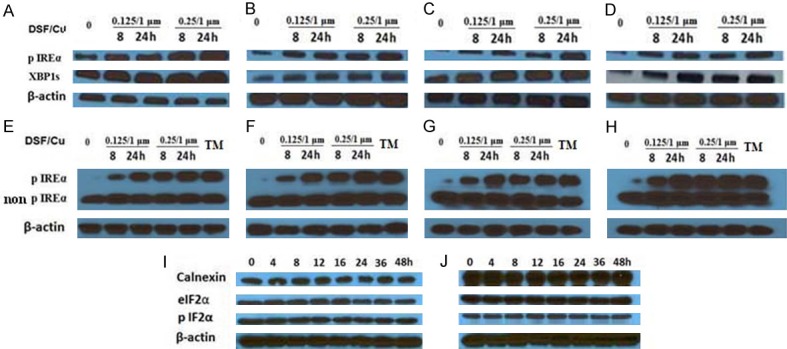
DSF/Cu activates the IRE1α-XBP1 axis. The cell lines were treated with the indicated doses of DSF/Cu for 8 and 24 hours. Treated cells were lysed and analyzed for expression of pIRE1α and XBP1s by Western blot (UACC-812 (A), MDA-MB-231 (B), PDAC6 (C) and PANC-1 (D)) or pIRE1α vs. non-phosphorylated IRE1α by Phos-tag gels (UACC-812 (E), MDA-MB-231 (F), PDAC6 (G) and PANC-1 (H)), cells treated for 8 hours with tunicamycin (TM) for inducing ER-stress were used as a positive control in Phos-tag gel assay. The UACC-812 (I) and PDAC6 (J) cell lines were treated with DSF/Cu (0.25/1 µM) for 0, 4, 8, 12, 16, 24, 36, and 48 hours. Treated cells were lysed and analyzed by Western blot for levels of calnexin and elF2α/pelF2α, downstream components of the other two UPR branches, ATF6α and PERK, respectively.
Figure 5.

DSF/Cu-induced spliced XBP1 is found at mRNA and protein levels. The UACC-812 (A) and PDAC6 (B) cell lines were treated as indicated for 16 hours. RNA was harvested from treated cells and the spliced XBP1 mRNA detected by qRT-PCR. Fold changes are shown as mean ± SD (*P<0.05, **P<0.01, and ***P<0.001). Treated cells were also harvested, fixed, and permeabilized for intracellular staining for XBP1s and analyzed by flow cytometry (UACC-812 - C, PDAC6 - D). The PANC-1 cells were treated with DSF/Cu at the indicated doses and time points. Then the total RNA was extracted from treated cells for XBP1 splicing assay using RT-PCR to amplify both XBP1u and XBP1s (E, top); RT-PCR products were also digested with PstI (E, middle) and actin was used as a loading control (E, bottom). Cells treated with tunicamycin (1 μg/mL) for 8 hours were used as a positive control (E). Furthermore, XBP1s and its downstream targets gene ERdj4, P58IPK were detected by qRT-PCR. Fold changes are shown as mean ± SD (*P<0.05, **P<0.01, and ***P<0.001) (F).
Moreover, we detected increased protein levels of pIRE1α and XBP1s in all four cell lines treated by DSF/Cu in a dose and time dependent manner (Figure 6A-D). The detection of IRE1α phosphorylation on Phos-tag gels as a dose and time dependent response in all 4 cell lines to treatment with DSF/Cu further confirmed the results that DSF/Cu activated IRE1α-XBP1 axis (Figure 6E-H). In contrast, the Western blot analysis of breast carcinoma UACC-812 cells and pancreatic carcinoma PDAC6 cells indicated little to no effect of DSF/Cu on either calnexin or elF2α/pelF2α levels during a 48 h kinetic study, respectively, ruling out a significant participating role of the ATF6α and PERK pathways of the UPR (Figure 6I, 6J). This finding would indicate, therefore, that DSF/Cu induced-UPR activation was via the IRE1α-XBP1 signaling pathway. Additional confirmation that the IRE1α pathway is responsible for DSF/Cu induced autophagy was obtained by an analysis of the impact of STF-083010 and 4µ8C, inhibitors of IRE1α endonuclease activity involved in the formation of XBP1s mRNA. Following treatment of the four breast carcinoma and pancreatic carcinoma cell lines with DSF/Cu in the presence or absence of STF-083010, Western blot analysis indicated that the inhibitor partially blocked DSF/Cu-induced conversion of LC3I to LC3II and cleaved PARP in the four cell lines (Figure 7A-D). Gray intensity values in the Western blot (Figure 7) were determined by Image J software (Figure S3). An additional analysis of the cell viability on the four carcinoma cell lines treated with DSF/Cu in the presence or absence of STF-083010 or 4µ8C was also performed. The results reflected the involvement of IRE1α pathway in DSF/Cu induced autophagic apoptosis by the fact that culture in the presence of STF-083010 or 4µ8C significantly increased the cell viability of cells treated with DSF/Cu (Figure 7E-H).
Figure 7.

IRE1α inhibitors reduce DSF/Cu-induced autophagy and apoptosis. The UACC-812 (A), MDA-MB-231 (B), PDAC6 (C) and PANC-1 (D) cell lines were pretreated with or without STF-083010 for 30 minutes and further treated with DSF/Cu for 24 hours. Treated cells were lysed and analyzed by Western blot for levels of LC3II/I and cleaved PARP. In addition, the UACC-812 (E), MDA-MB-231 (F), PDAC6 (G) and PANC-1 (H) cell lines were pretreated with STF-083010 or 4μ8C for 30 min, and then co-treated with DSF/Cu for 24 hours. Cell viability was determined by the MTT assay. Data generated in triplicate were expressed relative to the mean of vehicle-treated cells in each experiment, for three independent experiments, and shown as mean ± SD (*P<0.05, **P<0.01, and ***P<0.001).
DSF/Cu acting as a proteasome inhibitor appears to induce autophagy
DSF/Cu is known to inhibit proteasome 26S and as a consequence, inhibit NF-κB activity [1,2]. To investigate whether this DSF/Cu functional activity is responsible, at least in part for induction of autophagic apoptosis in cancer cells, the effects of inhibitors of proteasome and NF-κB were directly compared to those of DSF/Cu on cancer cells. Bortezomib is a specific inhibitor of the 26S proteasome and clinically approved for treatment of myeloma [22]. It inhibits the activation of NF-κB and promotes ER stress and UPR [23,24]. Bortezomib has been shown to induce apoptosis of a wide range of human carcinoma cell lines, including breast and pancreatic carcinoma cell lines [25,26]. To this end, the human breast UACC-812 and pancreatic carcinoma PDAC6 cell lines were cultured in the presence of bortezomib, NF-κBi, or DSF/Cu and then analyzed by Western blot for levels of autophagic and apoptotic events, namely, conversion of LC3I to LC3II and cleavage of PARP. The results indicated that bortezomib and NF-κBi mimicked the effects of DSF/Cu on autophagy. Interestingly, however, only bortezomib mimicked DSF/Cu and induced both autophagy and apoptosis, whereas NF-κBi (at 1 and 10 µM) only induced autophagy of both human carcinoma cell lines tested (Figure 8).
Figure 8.
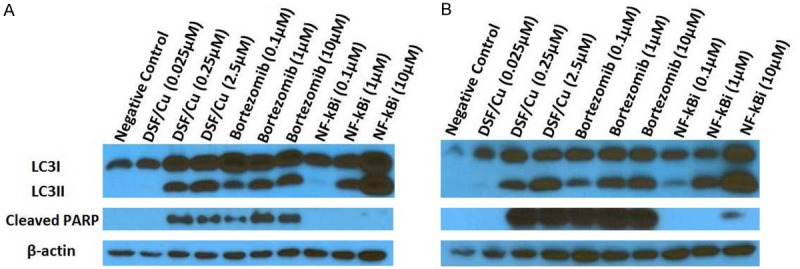
DSF/Cu and proteasome inhibitor bortezomib induce autophagy and apoptosis of cancer cells. The UACC-812 (A) and PDAC6 (B) cell lines were treated with either DSF/Cu, bortezomib, or NF-κBi, at indicated doses for 24 hours. Treated cells were lysed and analyzed by Western blot for levels of LC3II/I and cleaved PARP.
Discussion
Tumor cells, with their more rapid growth and proliferative rates than normal cells, have a greater potential of being manipulated by therapeutics to undergo autophagy and become apoptotic. In this study, it is evident that DSF/Cu, perhaps independent of its ability to inhibit ALDH activity, promotes ER stress, UPR and apoptosis, making it as an attractive candidate for drug repurposing in the area of cancer therapeutics.
DSF/Cu impacts various biochemical pathways related to cell survival and cell death. Earlier research, including the data from our laboratory, indicated that the anti-tumor activity of DSF/Cu was initiated by inhibition of IκBα, which blocks the activation of NF-κB, a master transcriptional factor controlling many pathways involved in the survival, death and stemness pathways. Consequently, the anti-tumor activity of DSF/Cu appears to be similar to that of bortezomib, a famous inhibitor of 26S proteasome and, in fact, DSF/Cu has also been shown to be an inhibitor of the 20S proteasome and 26S proteasome [1].
Recent findings also indicate that the complex of diethydithiocarbamate (a metabolite of DSF) and copper, CuET (bis(Diethyldithiocarbamate)-copper) [27], targets the p97 segregase adaptor NPL4, a component in the p97/NPL4/UFD1 pathway, which influences UPR and ER stress [28,29]. Inhibition of p97, which has ATPase activity, by CB-5083, a selective and orally bioavailable inhibitor, was shown to downregulate the degradation of post-ubiquitinated proteins by proteasomes, which is required for homeostasis [30]. As a result, CB-5083 greatly enhanced ER stress and activation of the 3 branches of UPR, including upregulation of XBP1s protein expression, and as expected, induction of autophagy and apoptosis [31]. Similarly, CuET promotes aggregation of p97/NPL4 preventing formation of p97/NPL4/UFD1 complexes and degradation of proteins marked for degradation, which leads to lethal ER stress [28]. It is not clear if CuET is fully responsible for the cytotoxic activity of DSF/Cu in vitro and in vivo as the interplay between the activation of the UPR IRE1α-XBP1 branch pathway and inhibition of p97 through targeting its adaptor NPL4 induced by DSF/Cu needs to be resolved. Moreover, the relationship between DSF/Cu induced-activation of IRE1α-XBP1 or inhibition of NPL4 and proteasome, i.e., as a cause or result of either one or both, needs to be elucidated in future studies. In vitro and in vivo-based data also demonstrate that DSF/Cu antagonizes antioxidant levels [32] and reduces the cellular antioxidant capacity to induce ROS and activate redox signaling. Consequently, DSF/Cu may contribute to ER stress through redox modulation, as well.
Presently there are seven active clinical cancer trials, including one for metastatic breast cancer, involving administration of DSF and Cu (https://clinicaltrials.gov). Unfortunately, they may not succeed because it is known that many tumors including breast tumors have elevated Cu levels compared to normal cells. While this provides an opportunity for selective DSF targeting tumor over normal tissues, Cu is tumor promoting [33,34]. In this regard, Cu depleting therapy using the Cu chelator, tetrathiomolybdate, increased relapse-free survival in patients at high risk for breast cancer recurrence [35]. In contrast, a new epidemiological study in Nature revealed the exciting result that administration of DSF (250 mg/daily) without exogenous copper significantly reduced risk of death from cancer, including breast cancer, in patients with either localized or metastatic disease [28]. Moreover, a DSF-related, post-adjuvant phase II trial involving breast cancer was completed involving 64 patients at high risk for metastasis. They received adjuvant chemotherapy with or without the non-FDA approved, DSF metabolite, DEDTC (600 mg/weekly), in the absence of exogenous copper. Interestingly, the trial clinicians were unaware of DEDTC being a DSF metabolite [36]. Overall survival at 6 years was 81% in the group given DEDTC and chemotherapy versus 55% for chemotherapy alone [37]. These findings strongly argue in favor of using DSF without exogenous Cu and relying on endogenous tumor Cu levels to form DSF/Cu complexes.
In summary, using well-established inhibitors of various parameters associated with autophagy, such as LC3I/II conversion and PARP cleavage, the results of this study indicate that DSF/Cu induced autophagy-dependent apoptosis in all 4 tested human breast and pancreatic carcinoma cell lines (Figures 1, 2, 4). Furthermore, among the three pathways associated with ER stress and UPR, the anti-tumor activity of DSF/Cu occurs selectively through the IRE1α pathway with upregulation of the pIRE1α-XBP1 pathway leading to autophagic apoptosis (Figures 5, 6 and 7). The results of this study clearly indicate the efficacy of DSF/Cu in promoting elevated ER-stress in cancer cells, and the potential mechanism that DSF/Cu activates directly or indirectly the UPR IRE1α axis, resulting in cytotoxic autophagy of cancer cells (Figure 9). This newly elucidated mechanism of the anti-tumor activity of DSF/Cu warrants further studies and highlights the utility of repurposing this established, safe and low-cost pharmaceutical in treating cancers.
Figure 9.
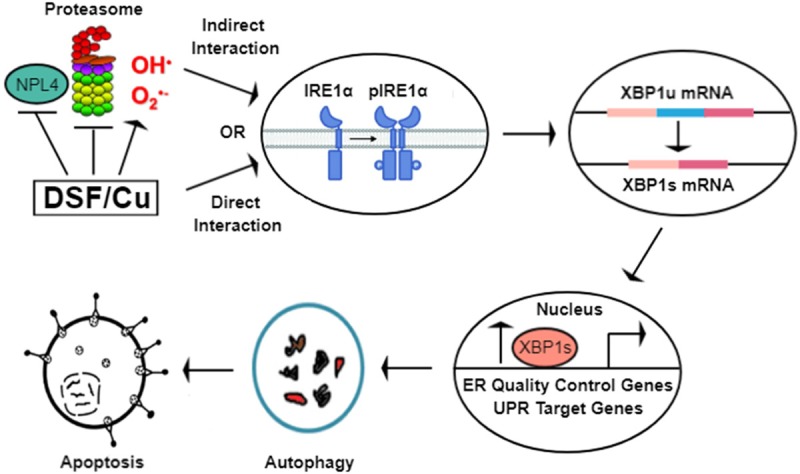
Schematic diagram demonstrating the potential mechanism of DSF/Cu in induction of ER-stress and cytotoxic autophagy. DSF/Cu may directly (via interaction with IRE1α) or indirectly (via inhibition of NPL4, proteasome, generation of reactive oxygen species, or a combination of these mechanisms) activate the UPR IRE1α-XBP1 pathway, resulting in cytotoxic autophagy-dependent apoptosis in cancer cells.
Acknowledgements
This work was supported by grants R21 CA181851 (X.W.), Massachusetts General Hospital ECOR Formulaic Bridge Funding (X.W.), 1R01CA226981-01A1 (X.W.), the National Natural Science Foundation of China (81872426, X.Z.) and Natural Science Foundation of Jiangsu Province, China (BK20181372, X.Z.).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66:10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- 2.Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL, Xu B, Cassidy J, Darling JL, Wang W. Disulfiram modulated ROS-MAPK and NFkappaB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011;104:1564–1574. doi: 10.1038/bjc.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Li W, Patel SS, Cong J, Zhang N, Sabbatino F, Liu X, Qi Y, Huang P, Lee H, Taghian A, Li JJ, DeLeo AB, Ferrone S, Epperly MW, Ferrone CR, Ly A, Brachtel EF, Wang X. Blocking the formation of radiation-induced breast cancer stem cells. Oncotarget. 2014;5:3743–3755. doi: 10.18632/oncotarget.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cong J, Wang Y, Zhang X, Zhang N, Liu L, Soukup K, Michelakos T, Hong T, DeLeo A, Cai L, Sabbatino F, Ferrone S, Lee H, Levina V, Fuchs B, Tanabe K, Lillemoe K, Ferrone C, Wang X. A novel chemoradiation targeting stem and nonstem pancreatic cancer cells by repurposing disulfiram. Cancer Lett. 2017;409:9–19. doi: 10.1016/j.canlet.2017.08.028. [DOI] [PubMed] [Google Scholar]
- 5.Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol. 2010;2010:930509. doi: 10.1155/2010/930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–97. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 7.Blais J, Bell JC. Novel therapeutic target: the PERKs of inhibiting the integrated stress response. Cell Cycle. 2006;5:2874–2877. doi: 10.4161/cc.5.24.3597. [DOI] [PubMed] [Google Scholar]
- 8.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 9.Ogino T, Wang X, Kato S, Miyokawa N, Harabuchi Y, Ferrone S. Endoplasmic reticulum chaperone-specific monoclonal antibodies for flow cytometry and immunohistochemical staining. Tissue Antigens. 2003;62:385–393. doi: 10.1034/j.1399-0039.2003.00114.x. [DOI] [PubMed] [Google Scholar]
- 10.Sheen JH, Zoncu R, Kim D, Sabatini DM. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell. 2011;19:613–628. doi: 10.1016/j.ccr.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Schadewijk A, van’t Wout EF, Stolk J, Hiemstra PS. A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell Stress Chaperones. 2012;17:275–9. doi: 10.1007/s12192-011-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang J, Donsante A, Desai V, Patronas N, Kaler SG. Clinical outcomes in Menkes disease patients with a copper-responsive ATP7A mutation, G727R. Mol Genet Metab. 2008;95:174–181. doi: 10.1016/j.ymgme.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 15.Frieboes HB, Huang JS, Yin WC, McNally LR. Chloroquine-mediated cell death in metastatic pancreatic adenocarcinoma through inhibition of autophagy. JOP. 2014;15:189–197. doi: 10.6092/1590-8577/1900. [DOI] [PubMed] [Google Scholar]
- 16.Boone BA, Zeh HJ 3rd, Bahary N. Autophagy inhibition in pancreatic adenocarcinoma. Clin Colorectal Cancer. 2018;17:25–31. doi: 10.1016/j.clcc.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 18.Szokalska A, Makowski M, Nowis D, Wilczynski GM, Kujawa M, Wojcik C, Mlynarczuk-Bialy I, Salwa P, Bil J, Janowska S, Agostinis P, Verfaillie T, Bugajski M, Gietka J, Issat T, Glodkowska E, Mrowka P, Stoklosa T, Hamblin MR, Mroz P, Jakobisiak M, Golab J. Proteasome inhibition potentiates antitumor effects of photodynamic therapy in mice through induction of endoplasmic reticulum stress and unfolded protein response. Cancer Res. 2009;69:4235–4243. doi: 10.1158/0008-5472.CAN-08-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trusina A, Papa FR, Tang C. Rationalizing translation attenuation in the network architecture of the unfolded protein response. Proc Natl Acad Sci U S A. 2008;105:20280–20285. doi: 10.1073/pnas.0803476105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11:239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juvekar A, Manna S, Ramaswami S, Chang TP, Vu HY, Ghosh CC, Celiker MY, Vancurova I. Bortezomib induces nuclear translocation of IkappaBalpha resulting in gene-specific suppression of NF-kappaB--dependent transcription and induction of apoptosis in CTCL. Mol Cancer Res. 2011;9:183–194. doi: 10.1158/1541-7786.MCR-10-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kretowski R, Borzym-Kluczyk M, Cechowska-Pasko M. Efficient induction of apoptosis by proteasome inhibitor: bortezomib in the human breast cancer cell line MDA-MB-231. Mol Cell Biochem. 2014;389:177–185. doi: 10.1007/s11010-013-1939-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nawrocki ST, Carew JS, Dunner K Jr, Boise LH, Chiao PJ, Huang P, Abbruzzese JL, McConkey DJ. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki Y, Fujii S, Tominaga T, Yoshimoto T, Yoshimura T, Kamada H. The origin of an EPR signal observed in dithiocarbamate-loaded tissues. Copper(II)-dithiocarbamate complexes account for the narrow hyperfine lines. Biochim Biophys Acta. 1997;1335:242–245. doi: 10.1016/s0304-4165(97)00027-5. [DOI] [PubMed] [Google Scholar]
- 28.Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J, Ozdian T, Bartkova J, Turi Z, Moudry P, Kraus M, Michalova M, Vaclavkova J, Dzubak P, Vrobel I, Pouckova P, Sedlacek J, Miklovicova A, Kutt A, Li J, Mattova J, Driessen C, Dou QP, Olsen J, Hajduch M, Cvek B, Deshaies RJ, Bartek J. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. 2017;552:194–199. doi: 10.1038/nature25016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li JM, Wu H, Zhang W, Blackburn MR, Jin J. The p97-UFD1L-NPL4 protein complex mediates cytokine-induced IkappaBalpha proteolysis. Mol Cell Biol. 2014;34:335–347. doi: 10.1128/MCB.01190-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou HJ, Wang J, Yao B, Wong S, Djakovic S, Kumar B, Rice J, Valle E, Soriano F, Menon MK, Madriaga A, Kiss von Soly S, Kumar A, Parlati F, Yakes FM, Shawver L, Le Moigne R, Anderson DJ, Rolfe M, Wustrow D. Discovery of a first-inclass, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083) J Med Chem. 2015;58:9480–9497. doi: 10.1021/acs.jmedchem.5b01346. [DOI] [PubMed] [Google Scholar]
- 31.Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, Madriaga A, Soriano F, Menon MK, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou HJ, Wustrow D, Rolfe M. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell. 2015;28:653–665. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allensworth JL, Evans MK, Bertucci F, Aldrich AJ, Festa RA, Finetti P, Ueno NT, Safi R, McDonnell DP, Thiele DJ, Van Laere S, Devi GR. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Mol Oncol. 2015;9:1155–1168. doi: 10.1016/j.molonc.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F, Jiao P, Qi M, Frezza M, Dou QP, Yan B. Turning tumor-promoting copper into an anti-cancer weapon via high-throughput chemistry. Curr Med Chem. 2010;17:2685–2698. doi: 10.2174/092986710791859315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishida S, Andreux P, Poitry-Yamate C, Auwerx J, Hanahan D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc Natl Acad Sci U S A. 2013;110:19507–19512. doi: 10.1073/pnas.1318431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain S, Cohen J, Ward MM, Kornhauser N, Chuang E, Cigler T, Moore A, Donovan D, Lam C, Cobham MV, Schneider S, Hurtado Rua SM, Benkert S, Mathijsen Greenwood C, Zelkowitz R, Warren JD, Lane ME, Mittal V, Rafii S, Vahdat LT. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann Oncol. 2013;24:1491–1498. doi: 10.1093/annonc/mds654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cvek B. Comment on ‘cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells’. Br J Cancer. 2013;108:993. doi: 10.1038/bjc.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dufour P, Lang JM, Giron C, Duclos B, Haehnel P, Jaeck D, Jung JM, Oberling F. Sodium dithiocarb as adjuvant immunotherapy for high risk breast cancer: a randomized study. Biotherapy. 1993;6:9–12. doi: 10.1007/BF01877380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.