

Abstract
Early metastasis and local recurrence are the major causes of mortality and poor prognosis of non-small cell lung cancer (NSCLC). However, the underlying mechanisms of these processes are poorly understood. In this study, we aimed to investigate the roles of the PTGES/PGE2 pathway in lung cancer progression. We found that prostaglandin E synthase (PTGES), a key enzyme for PGE2 synthesis in the arachidonic acid pathway, was highly dysregulated in NSCLC. Dysregulated PTGES was essential for the promotion of tumor migration and metastasis of NSCLC cells. Knockdown of PTGES in lung cancer cells resulted in suppressed cell migration, which was reversed by exogenous PGE2. Consistent with this, PTGES knockdown also reduced the expression of CSC markers, tumor sphere formation, colony forming activity, tumorigenicity, and lung metastasis in vivo. Dysregulated PTGES is mainly attributed to protein stabilization by USP9X, a deubiquitination enzyme. USP9X physically interacted with PTGES and prevented it from proteasome-directed degradation via deubiquitination. Consistent with this, USP9X expression was highly correlated with PTGES expression in NSCLC tumor tissues. Taken together, our results show that the upregulated USP9X-PTGES-PGE2 axis contributes significantly to the metastatic features of NSCLC.
Keywords: PTGES, PGE2, USP9X, metastasis, non-small cell lung cancer
Introduction
Lung cancer is the leading cause of cancer death worldwide, and non-small cell lung cancer (NSCLC) accounts for ~85% of all diagnosed lung cancers. Despite progress in early diagnosis and many areas of cancer therapeutics, the overall survival rate for patients with lung cancer remains low [1]. Tumor recurrence and metastasis are the major causes of treatment failure and death. More than half of patients with lung cancer have advanced or metastatic disease at the time of diagnosis. Metastasis is a complex process in which cells acquire multiple aggressive properties. Although much progress has been made in this area, the complexities of cancer metastasis are still not fully understood.
An inflammatory microenvironment plays an important role in the promotion of tumorigenesis, tumor progression, and metastasis of cancer, including lung cancer. Prostaglandins (PGs) are lipids with important physiological and pathological functions. Among them, prostaglandin E2 (PGE2) is a key mediator of inflammation, pain, and fever [2]. PGE2 is one of the most abundant prostaglandins synthesized from arachidonic acid (AA). Oxygenation of AA via cyclooxygenase-1 and 2 (COX-1/2) produces PGG2, which is subsequently reduced to PGH2. PGH2 is then converted by a variety of synthases into several prostanoids (e.g. PGF2a, PGD2, PGI2), including PGE2 and thromboxane A2 (TXA2). The terminal step in the production of PGE2 involves the action of PGE2 synthases (PTGES) for the conversion of PGH2 to PGE2 [3]. Three different PGES have been identified, namely cytosolic PGES (cPGES), microsomal PGES-1 (mPGES-1), and mPGES-2. Among them, mPGES-1 (PTGES) is highly upregulated in inflammatory tissues and overexpressed in tumors [4]. PTGES has been recognized as a key enzyme in inflammation and inflammatory diseases, including cancer.
PGE2 levels are upregulated in a variety of human tumors [5]. By its secretion into the tumor microenvironment, PGE2 is able to induce tumor growth and suppress immune functions. In addition, PTGES and COX-2 are overexpressed in NSCLC, but the upregulation of these two enzymes in individual tumors is not identical. Cellular transformation and cytokines have been shown to contribute to the upregulation of PTGES in NSCLC. However, targeted overexpression of PTGES and elevated PGE2 production in alveolar and airway epithelial cells alone is not sufficient for lung tumorigenesis in transgenic mice [6-8]. This suggests that PGE2 promotes tumor progression in a cellular context-dependent manner. Nevertheless, the mechanisms of PGE2-mediated tumor progression and dysregulation of PGE2 signaling remain unclear.
Ubiquitination is a major post-translational modification in protein activation, playing a central role in the degradation of proteins, both through proteasomal targeting and by direct sorting to the lysosome [9]. The reversal of ubiquitination is accomplished through deubiquitinases (DUBs). In humans, DUBs are subdivided into five families: ubiquitin-specific proteases (USPs), ovarian tumour proteases (OTUs), ubiquitin C-terminal hydrolases (UCHs), Josephins, and JAMMs. The ubiquitin-specific protease 9X (USP9X/FAM) is a substrate-specific DUB that is evolutionarily highly conserved [10]. The upregulation of USP9X has been implicated in prostate cancer, osteosarcoma, myeloma, esophageal squamous cell carcinoma, breast cancer, oral squamous cell carcinoma, and lung cancer [11]. Interestingly, USP9X has also been implicated both as an oncogene and tumor suppressor, depending on the type and stage of cancer. USP9X-mediated functions are dependent on its substrates. To date, USP9X is known to interact with 35 proteins, including 21 substrates (such as ASK1, β-catenin, ErbB2, ERG, SMAD4, and SURVIVIN) and 14 interacting proteins (such as FOXO3, p100, SOX2, and TANK) [10]. In prostate cancer cells, USP9X deubiquitinates and stabilizes ERG protein levels. USP9X deubiquitinates MCL and prevents its proteasomal degradation, thus promoting its anti-apoptotic functions in lymphomas, colon adenocarcinoma, and small cell lung carcinoma [9,12]. However, the role of USP9X in human lung cancer is not clear.
In this study, we aimed to investigate the role of the PTGES/PGE2 pathway in lung cancer progression. Our study provides insight into the contribution of the USP9X-PTGES-PGE2 axis to the metastatic features of NSCLC.
Materials and methods
Plasmids, reagents and antibodies
Antibodies for E-cadherin (#3195), N-cadherin (#13116), vimentin (#5741), SOX2 (#4900), CD44 (#3570), ABCG2 (#42078) and TWIST (#46702) were purchased from Cell Signaling Technology (Beverley, MA). Antibodies Anti-β-actin-HRP (#60008) and Flag (#20543-1-AP) were purchased from Proteintech (China). PTGES (#160140) were purchased from Cayman chemical. PGE2 (#p5640) were purchased from Sigma-Aldrich. GAPDH (#5174) and USP9X (#14898) were purchased from Santa Cruz (Santa Cruz, CA). GFP (#M048-3) were purchased from MBL (Nagoya, Japan). Antibody for HA (#CW0092M) were purchased from cwbio (China). PTGES (#PA5-33000) for IHC were purchased from Thermo fisher. The concentration of PGE2 levels in the supernatants was determined by ELISA kit (Cayman chemical). Oligonucleotides corresponding to these guide sequences (PAM motif in bold) were cloned into the BbsI site of pX330, a bicistronic expression vector encoding both Cas9 and the sgRNA.
Cell lines and cell culture
Human embryonic kidney cells, HEK293T and NSCLC cells (A549 and HCC827) were tested and authenticated by DNA typing at Shanghai Jiao Tong University Analysis Core. Primary mouse lung cancer cells (SJT1601) were isolated from NNK-induced lung tumor of Gprc5a-ko mouse [13]. Cells were digested into single cells and seed the cells onto the palate over 3 days, then passing on the living cells. We constructed the stable cell line SJT-1601-ko-Ptges by CRISPR/Cas9 technology. Human NSCLC cells A549 and HCC827 stably transfected with nonspecific (sh-NS) or PTGES sh-RNA. Unless otherwise stated, all cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Gibco), penicillin and streptomycin, at 37°C in a humidified incubator in an atmosphere of 95% air and 5% CO2. HCC827 were cultured in RPMI-1640 essential medium with 10% fetal bovine serum.
Quantitative real-time (Q)-PCR and primers
Cells were firstly lysed using trizol. Then, total RNA was extracted with RNA Extract Kit (TIANGEN, Cat: #RK123) and cDNA were prepared from 1 μg total RNA using Fast Quant Kit. The Q-PCR analysis was performed on ABI 7300 real-time PCR machine. All Ct values were standardized by β-actin’s Ct value.
Following primers were used: hPTGES: F: 5’-TCC-TAA-CCC-TTT-TGT-CGC-CTG-3’ R: 5’-CGC-TTC-CCA-GAG-GAT-CTG-C-3’ β-actin: F: 5’-GCT-CTT-TTC-CAG-CCT-TCC-TT-3’ R: 5’-CTT-CTG-CAT-CCT-GTC-AGC-AA-3’; Caspase 9 to knock out SJT-1601 Ptges gene using primer: gRNA1: 1F: 5’-CAC-CGG-GAA-TGA-GTA-CAC-GAA-GCC-GAG-G-3’ 1R: 5’-AAA-CCC-TCG-GCT-TCG-TGT-ACT-CAT-TCC-C-3’ gRNA2: 2F: 5’-CAC-CGG-AAC-CCA-CGC-CTT-CGC-TCC-GGG-G-3’ 2R: 5’-AAA-CCC-CCG-GAG-CGA-AGG-CGT-GGG-TTC-C-3’.
Immunoblotting and immunoprecipitation
Experimental protocols for immunoblotting and immunoprecipitation follow those previously described [13]. Cells were lysed with RIPA buffer (150 mM NaCl, 50 mM Tris (PH 8.0), 25 mM NaF, 2 mM Na3VO4, 0.5% deoxycholate, 1% NP40, 0.1% SDS) and add protease inhibitor cocktail (biomark.cn). Then equal amounts of the protein samples (30 μg protein) were mixed with loading buffer and boiled for 5 minutes. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose (NC) membranes. Non-specific binding sites were blocked with 5% (w/v) non-fat dry milk in TBST, and the membranes were incubated overnight on a shaking platform at 4°C with the specific primary antibodies diluted in 5% BSA with 0.05% sodium azide. HRP-conjugated secondary antibody was incubated at room temperature for 1 h. Finally, the proteins were visualized by exposure with Immobilon Western Reagents. All antibodies were diluted for use according to manufacturers’ instructions.
Flow cytometry analysis
Flow cytometry was performed as described previously [14]. Trypsinized cells washed twice with PBS. Then the cells were incubated in 400 μL PBS containing 1% CD44-APC (BD Pharmingen) at 4°C for 30 minutes, however the control were incubated with IgG. After incubation, the cells were suspended in 400 μL PBS for Flow Cytometry analysis. The gates were established using the negative controls cells stained with IgG. Finally, the data was analyzed in the Flowjo 7.6.1 software.
Migration and wound healing assay
Experiments were performed as described previously [15]. For the migration assay, cells (2 × 104) were seeded onto the upper chamber in 200 μL of serum-free medium; the lower compartment was filled with 0.5 mL of DMEM media supplemented with 10% of FBS with and without 1 μM PGE2. After 24 h incubation, migrated cells on the lower surface of the filter were fixed and stained using 0.1% crystal violet; cells on the upper side were removed using a rubber scraper. Fluorescent images were obtained; reported data are counts of migrated cells with experiments performed in triplicate. Cells overspread the culture plate scratch the cells with micro pipette tips in 6-well plate. Then the picture captured as needed.
Sphere culture assay
The lung adenocarcinoma cells were resuspended in culture medium and were put into low-adhered dishes in order to simulate a 3D culture room. Every well of 96-well plate contains 5000 cells. The medium was changed every 3 days. Take care of the spheroids size to avoid necrosis in the center of the spheres. Recorded data are colony number obtained from 10 separate views using a microscope. Experiments were repeated three times with duplication in each experiment.
Immunohistochemical staining and clinical samples
A tissue microarray composed of tumor and adjacent normal tissue was stained to identify PTGES and USP9X proteins. The IHC protocol and score method were performed as previously described [16]. All antibodies were diluted for use according to manufacturers’ instructions. Human lung cancer tissue samples were obtained from Shanghai Chest Hospital, Shanghai Jiao Tong University (Shanghai, CHINA). Shanghai Chest Hospital approved the use of the tumor samples and animals in this study.
Soft agar colony-formation assays
Soft-agar assays were performed as described previously [13]. A 2% agarose solution was combined with DMEM-10% FBS (1:3 v/v; final concentration, 0.7%) and added to 24-well plates (0.5 mL per well), and allowed to solidify for 10 minutes at 4°C. SJT-1601 or NSCLC cells (500 cells in 50 μL medium) were added over the solidified agarose. A top 0.5 mL agarose layer was added over the cells, consisting of the 2% agarose solution combined with DMEM-10% FBS (1:6 v/v; final concentration, 0.35%) along with matrigel (1:30 v/v). The plates were incubated in 37°C, 5% CO2 incubator. After 2 weeks, colonies were counted using an inverted microscope at 40 × magnification and photographed. Reported data are the means and standard error from two independent experiments performed, in triplicate.
Tumor growth and migration study in xenograft
A549 (sh-NS) or PTGES sh-RNA were injected subcutaneously (2.5 × 106 cell in 0.2 mL serum-free DMED medium supplemented with 30% matrigel) in nude mice. HCC827 (sh-NS) or PTGES sh-RNA were injected subcutaneously (1 × 106 cell in 0.2 mL serum-free RPMI-1640 medium supplemented with 30% matrigel) in nude mice. SJT-1601 and ko-Ptges were injected subcutaneously (2 × 105 cell in 0.2 mL serum-free DMED medium supplemented with 30% matrigel) in nude mice. After 3 weeks, the tumor volume was measured thrice a week. When the sh-NS tumor volume reached ~2000 mm3, the nude mice were sacrificed and the tumor volume (mm3) was calculated by the formula: (a × b2)/2, where “a” is the long diameter and “b” is the short diameter (mm). Then, xenografts consisting of 1 × 106 SJT-1601, ko-Ptges and ko-Ptges restore mice Ptges cells were injected tail vein (0.2 mL) in C57/BL mice. About 3 weeks, mice were subjected to killing when the following situations happened: displaying pain or distress, such as weight loss (> 10% of body weight), not eating or drinking, lethargy, lying down, or difficulty in breathing. Then, we measured the tumor and analyzed the data. Animal studies were conducted following the guidelines of the Experimental Animal Ethics Committee of Shanghai Jiao Tong University and were approved by the Experimental Animal Ethics Committee of Shanghai Jiao Tong University (Shanghai, China).
Statistical analysis
Statistical significance was determined using an unpaired two-tailed Student’s t test, one-way ANOVA, two-way ANOVA, Fisher’s exact test, log rank test, and the Pearson correlation coefficient as indicated. A p value < 0.05 was considered significant (*P < 0.05; **P < 0.01; ***P < 0.001).
Result
PTGES overexpression is associated with poor prognosis in NSCLC
To investigate the relationships between PTGES and human tumor samples, we analyzed PTGES mRNA expression levels in human tumor samples in TCGA using the GEPIA database (Figure 1A) and GEO database (GSE19804) (Figure 1C). PTGES expression levels were significantly elevated in human lung adenocarcinoma (LUAD) (n=483 tumor samples vs 347 normal samples) and lung squamous cell carcinoma (LUSC) (n=486 tumor samples vs 338 normal samples) compared to those in normal tissues (Figure 1B). A similar result was obtained by analysis of PTGES expression in NSCLC (n=60) from the GSE19804 data set (Figure 1C). Moreover, lung cancer patients with high PTGES expression exhibited shorter disease-free survival and overall survival than those with low PTGES expression (Figure 1D and 1E). Multiple databases with other data sources support the same conclusion (Figure S1A-D). Taken together, these results suggest that high expression of PTGES is strongly associated with poor clinical outcome in patients with NSCLC.
Figure 1.
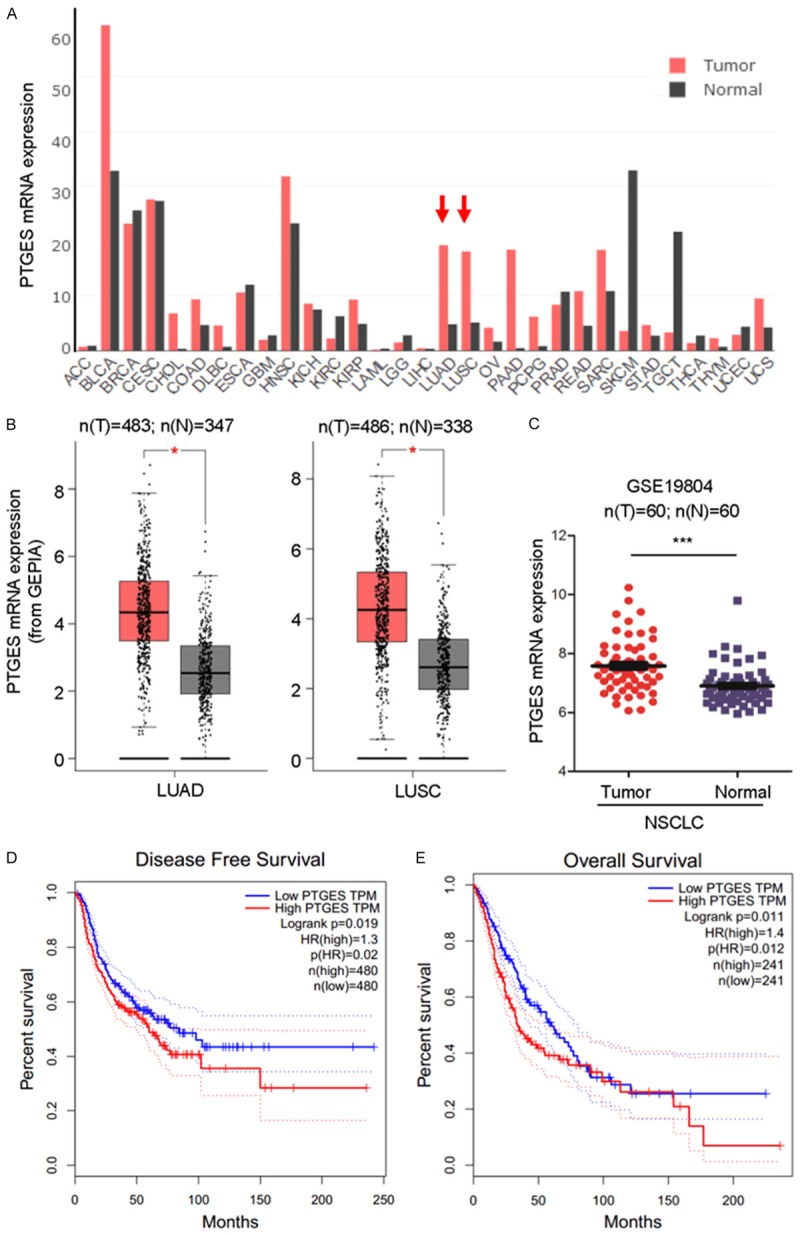
PTGES overexpression is associated with poor prognosis in non-small cell lung cancer (NSCLC). A. Comparison of mRNA expression levels of PTGES in cancer and adjacent normal tissues of different cancer types in TCGA database. B. PTGES mRNA expression levels in human NSCLC tumor samples compared with those in adjacent normal tissues in TCGA database (mean ± SD, P < 0.05). C. PTGES mRNA expression levels in human NSCLC tumor samples compared with those in adjacent normal tissues in the GEO database (GSE19804). D. PTGES expression was negatively correlated with patient disease-free survival. E. PTGES expression was negatively correlated with patient overall survival.
PTGES/PGE2 pathway promotes metastatic features of NSCLC cells in vitro
Metastatic activities are important features of tumor malignancy and tumor progression [17]. To determine the roles of PTGES in the metastatic activity of lung cancer cells, we silenced PTGES via shRNA in the A549 and HCC827 human NSCLC cell lines, which express relatively high levels of PTGES (Figure 2A and 2B). In addition, we used the CRISPR/Cas9 system to knock out Ptges in the mouse lung cancer cell line SJT-1601. Next, we compared the expression of molecular biomarkers of epithelial to mesenchymal transition (EMT) in these cell lines. The results showed that PTGES knockdown in HCC827 cells resulted in reduced levels of N-cadherin, Vimentin and Twist in HCC827-shPTGES cells compared to those in HCC827-NS cells (Figure 2A). Similarly, Twist and Vimentin were also reduced in A549-shPTGES cells compared to those in A549 cells. Interestingly, E-cadherin and Snail were not changed (Figure 2B). Since E-cadherin is mainly regulated by transcription repressor Snail [18], this observation suggests that PTGES promotes EMT-like features in lung cancer cells, mainly through the Twist-Vimentin pathway, but not the Snail-E-cadherin pathway. In mouse lung tumor SJT-1601 cells, the effect of Ptges knockout was more dramatic than those in human NSCLC cells. Ptges knockout resulted in dramatic suppression of mesenchymal markers, including vimentin, N-cadherin, and twist, while E-cadherin was slightly reduced (Figure 2C). Consistent with this, the PTGES product PGE2 was significantly reduced in SJT-1601-ko-Ptges cells compared to levels in SJT-1601-NS cells (Figure 2D). To determine the biological effects of PTGES knockdown, we examined the migration of these cells via Transwell and migration assays in vitro. The results showed that PTGES knockdown or Ptges knockout significantly reduced the migration of human and mouse lung cancer cells compared to that of the parental cancer cells (Figure 2E-I), suggesting that PTGES expression is required for migration of these lung cancer cells. Importantly, the addition of exogenous PGE2, the product of PTGES, largely restored the migration activity lost in the shPTGES cell lines, suggesting that PGE2 is the effector molecule for PTGES-mediated function. Taken together, these results suggest that the PTGES/PGE2 pathway promotes metastatic features of lung cancer cells in vitro.
Figure 2.

The PTGES/PGE2 pathway promotes migration of NSCLC cells in vitro. A. Western blotting analysis of E-cadherin, N-cadherin, vimentin, twist, snail and PTGES protein levels in HCC827 and HCC827-KD-PTGES cell lines. B. Western blotting analysis of E-cadherin, N-cadherin, vimentin, twist, snail and PTGES protein levels in A549 and A549-KD-PTGES cell lines. C. Western blotting analysis of E-cadherin, N-cadherin, vimentin, twist, snail and PTGES protein levels in SJT-1601 cells with and without knockout of Ptges via CRISPR/Cas9 technology. D. ELISA kit analysis of the secretion of PGE2 from SJT-1601 and SJT-1601-ko-Ptges cell lines, HCC827 and HCC827-KD-PTGES cell lines, and A549 and A549-KD-PTGES cell lines. E. Migration of HCC827, A549, and SJT-1601 cells with or without PTGES silencing analyzed by Transwell migration assay. Scale bar=100 μm. F. Graph demonstrates the mean ± SD percentage of migrated cells in HCC827, A549, and SJT-1601 cells with or without PTGES silencing from 3 separate experiments. G-I. Wound healing and migration analysis of HCC827, A549, and SJT-1601 cells with or without PTGES silencing and with or without treatment with PGE2 (1 μM) at 24 hours. Scale bar=100 μm.
Upregulated PTGES is essential for malignant features and stemness of lung cancer cells
Metastatic features have been linked to stemness-like characteristics in cancer cells [19]. To determine if the dysregulation of PTGES is also involved in enhancing the stemness-like features of lung cancer cells, we next examined the related characteristics in vitro. We found that parental mouse lung cancer SJT-1601 cells were capable of forming colonies in both clonogenic and soft agarose assays, whereas SJT-1601-ko-Ptges cells exhibit considerable suppression of this activity (Figure 3A-E). Consistent with this, stem cell biomarkers SOX2, CD44, and ABCG2, which are expressed in control A549, HCC827, and SJT-1601 cells, were greatly suppressed in PTGES knockdown and Ptges knockout cells (Figure 3F-H). Next, we examined numbers of CD44+ cells, a stem cell-enriched population, in these cell lines by FACS analysis. The results showed that the CD44+ population was significantly reduced in HCC827-shPTGES cells compared to that in HCC827-shNS cells (Figure 3I). Moreover, the CD44+ population was also dramatically reduced in SJT-1601-ko-Ptges cells compared to that in SJT-1601 cells (Figure 3J), suggesting that PTGES is essential for maintaining stemness in these lung cancer cells. In addition, we also examined the side population (SP), a stem cell-enriched population, in these lung cancer cells. The results showed that PTGES knockdown reduced the SP from 0.4% to 0.22% in A549 cells, while Ptges knockout reduced the SP from 7.77% to 1.96% in mouse lung cancer SJT-1601 cells (Figure 3K, 3L). Taken together, these findings demonstrate that the PTGES/PGE2 pathway is essential for the upregulation of stemness and oncogenic activities in NSCLC and mouse lung cancer cells.
Figure 3.

Dysregulation of PTGES is essential for maintaining the increased proliferation and stemness of NSCLC cells. (A) Morphology changes associated with metastasis in SJT-1601 and SJT-1601-ko-Ptges cells shown in phase contrast images. Scale bar = 200 μm. (B) Numbers of SJT-1601 and SJT-1601-ko-Ptges cells grown in a culture dish for 5 days with the same number of starting cells. (C) Measurement of 3D tumorsphere formation (mean ± SD from 3 separate experiments) in SJT-1601 and SJT-1601-ko-Ptges cells. (D) Colony formation assay of SJT-1601 control and SJT-1601-ko-Ptges cells. Colonies were measured after 1 week of cultivation and stained with crystal violet. (E) Measurement of tumorsphere formation (mean ± SD from 3 separate experiments) in SJT-1601 and SJT-1601-ko-Ptges cells. Graph shows the mean ± SD percentage; scale bar = 100 μm. Western blotting analysis of SOX2, CD44, ABCG2, and PTGES protein levels in HCC827 (F), A549 (G), and SJT-1601 cells (H) with or without PTGES silencing. (I) Flow cytometry analysis of CD44+ in HCC827 (red) and HCC827-shPTGES (blue) cells. (J) Flow cytometry analysis of CD44+ in SJT-1601 (red) and SJT-1601-ko-Ptges (blue) cells. (K) SP sorting in A549 and A549-shPTGES cells using Hoechst 33342. A polygonal live gate was created to exclude debris and dead cells. At least 50,000 cells were acquired to analyze the SP phenotype in each sample (top panel). The percentage of SP cells decreased when A549 cells were pre-incubated with verapamil to block the ATP transporter (bottom panel). (L) SP sorting in SJT-1601 and SJT-1601-ko-Ptges cells.
Dysregulated PTGES promotes tumorigenicity and metastasis of lung tumor cells in vivo
To determine the biological function of PTGES in vivo, we examined the tumorigenicity of lung tumor cells in mouse models. First, A549-NS vs A549-shPTGES, HCC827-NS vs HCC827-shPTGES, and SJT-1601 vs SJT-1601-ko-Ptges cells were s.c. injected in nude mice, and the tumor sizes in these groups were measured. The results showed that the tumorigenicity of A549-shPTGES and HCC827-shPTGES cells was significantly reduced compared to that of A549-NS and HCC827-NCS cells, respectively (Figure 4A, 4B, 4D). Additionally, the tumorigenicity of SJT-1601-ko-Ptges cells was significantly reduced compared to that of the parental SJT-1601 cells (Figure 4C). These results indicate that PTGES expression is essential for the tumorigenicity of lung cancer cells in vivo.
Figure 4.
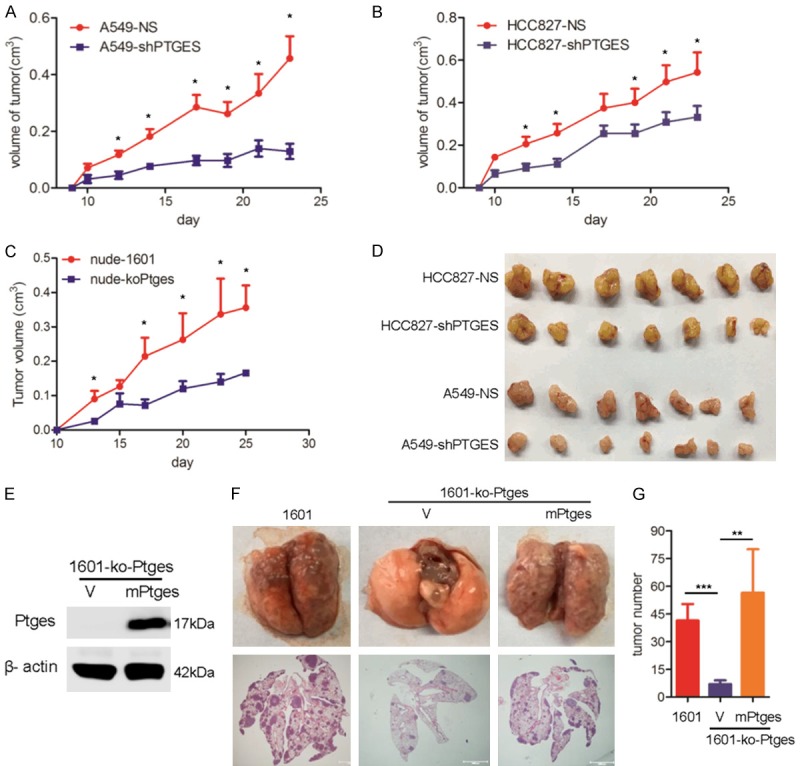
Dysregulation of PTGES promotes tumorigenicity of lung tumor cells in vivo. Lung tumor cells A549-NS and A549-shPTGES. A. HCC827-NS and HCC827-shPTGES. B. Were s.c. injected into nude mice. About 3 weeks later, tumor size was measured as indicated. C. Mouse lung tumor SJT-1601 and SJT-1601-ko-Ptges cells were s.c. injected into nude mice, and tumor size was measured as indicated. D. Tumors from indicated groups. E. Western blotting analysis of PTGES in SJT-1601 cells and restoration in SJT-1601-ko-Ptges cells after infection with Ptges-specific RNA lentivirus. F. C57/BL mice were infected through the tail vein with 1 × 106 SJT-1601 or SJT-1601-ko-Ptges cells, and Ptges was restored in SJT-1601-ko-Ptges cells. Lungs were removed from the mice at 3 weeks after inoculation. The experimental scheme is presented. Representative images of lung tissues stained with H&E are presented (bottom). Black arrow, tumor; scale bar, 1000 μm. G. Number of metastatic lesions in the lungs (n=5). ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To determine the role of PTGES expression in metastasis, we examined lung metastasis in an experimental metastasis model. The results showed that SJT-1601-ko-Ptges cells resulted in a dramatic suppression of lung metastasis compared to that of parental SJT-1601 cells (Figure 4F). To further confirm the role of PTGES, we re-expressed mouse Ptges (mPtges) in SJT-1601-ko-Ptges cells (Figure 4E) and examined their resulting metastatic potential. Re-expression of mPtges in SJT-1601-ko-Ptges cells restored lung metastasis to levels comparable to that of SJT-1601-ko-Ptges parent cells (Figure 4F and 4G), suggesting that Ptges is indeed required for the metastasis of lung cancer cells in vivo. Taken together, these results demonstrate that dysregulated PTGES is essential for the tumorigenicity and metastasis of lung cancer cells in vivo.
Upregulated PTGES in NSCLC cells is mainly attributable to protein stabilization via USP9X-mediated deubiquitination
The upregulation of PTGES may occur at either the mRNA or protein level. By analyzing data from TCGA, we surprisingly found that the gene amplification and upregulation of PTGES and USP9X, encoding an ubiquitin family protein, are mutually exclusive (Figure 5A). This suggests that the gene amplification and upregulation of these two genes are functionally redundant, meaning that the upregulation of USP9X functions similarly to the gene amplification of PTGES. USP9X is a deubiquitinase, and the ubiquitination pathway is important in the regulation of protein stability [20,21]. To investigate whether the ubiquitination pathway is involved in the regulation of PTGES, we sought to determine whether PTGES is a target of ubiquitin family proteins. By searching several proteomic databases, (https://www.ncbi.nlm.nih.gov/gene/9536#gene-expression, https://www.ncbi.nlm.nih.gov/gene/8239, and https://www.ncbi.nlm.nih.gov/gene/1080), we surprisingly found that PTGES is among the interacting proteins of USP9X. This suggests that USP9X and PTGES may form a complex. To determine the relationship between PTGES and USP9X, we examined the expression of the two proteins in NSCLC cell lines. The results showed that most NSCLC cells express both PTGES and USP9X (Figure 5B). Interestingly, USP9X knockdown in A549 and HCC827 cells resulted in a reduction in PTGES, which could be partially restored by treatment with the proteasome inhibitor MG132 (Figure 5C and 5D). Similar results were obtained in the mouse lung tumor SJT-1601 cell line (Figure 5E). Interestingly, USP9X knockdown-induced repression of PTGES does not appear to occur at the mRNA level, since shUSP9X did not reduce PTGES mRNA levels (Figure 5F). These observations suggest that USP9X-mediated upregulation of PTGES may occur at the protein level. Next, we aimed to determine whether USP9X physically interacts with PTGES. Upon co-immunoprecipitation with Flag-tagged PTGES, we detected endogenous USP9X via IP-western blot analysis (Figure 5G). Moreover, co-transfection of HEK293T cells with expression plasmids indicated that USP9X overexpression inhibits polyubiquitylated PTGES (Figure 5H). These results indicate that USP9X physically interacts with PTGES and mediates the deubiquitination or stabilization of PTGES. Thus, overexpression of USP9X may contribute to the upregulation of PTGES in NSCLC. To determine whether USP9X knockdown also inhibits PTGES-mediated biological functions, we examined the migration of A549 and HCC827 cells upon shUSP9X transfection. The results showed that USP9X knockdown reduced the migration of these cells. Moreover, addition of exogenous PGE2 largely restored the migration activity of these cells (Figure 5I and 5J). This suggests that USP9X knockdown destabilizes PTGES, leading to a reduction in PGE2 and consequently suppressing migration. Taken together, these results suggest that USP9X interacts with and stabilizes PTGES via deubiquitination, which enhances PGE2 signaling.
Figure 5.

USP9X interacts with and stabilizes PTGES via deubiquitination. A. Comparison of mRNA expression levels of PTGES and USP9X in human non-small cell lung cancer (NSCLC) tumor samples in TCGA database. B. PTGES and USP9X protein expression in a variety of NSCLC cell lines. C-E. A549, HCC827, and SJT-1601 cells with or without USP9X silencing were treated with or without CQ or MG-132, and the indicated protein levels were examined. F. A549 and HCC827 cells with or without USP9X silencing were subjected to qRT-PCR to examine the indicated mRNA levels. The PTGES mRNA level relative to that of β-actin was quantified (mean ± SD (n=6). G. HEK293T cells were lysed, and IP was performed with indicated antibodies. Immunocomplexes were subjected to western blotting. H. HEK293T cells were transfected with GFP-PTGES, FLAG-USP9X, and HA-Ub as indicated. The polyubiquitylated PTGES protein was detected by anti-HA antibody. I, J. Wound healing and migration analysis of HCC827 and A549 cells with or without USP9X silencing and with or without treatment with PGE2 (1 μM) at 24 hours. Scale bar=100 μm.
USP9X is highly correlated with PTGES in NSCLC tissues
To determine the correlation between USP9X and PTGES in tumor development, we next examined the expression of USP9X and PTGES in NSCLC tissue samples. Immunoblotting analysis showed that PTGES levels were dramatically increased in most NSCLC tissues compared to those in adjacent normal tissues (Figure 6A). Moreover, USP9X expression was highly correlated with PTGES expression in most NSCLC samples (Figure 6A and 6B). To extend the analysis further, we examined USP9X and PTGES levels via immunohistochemical (IHC) staining in NSCLC tissue microarrays. The results showed that both USP9X and PTGES were highly upregulated in NSCLC samples compared with levels in normal lung tissues, with PTGES and USP9X expression patterns being highly correlated (Figure 6C-F). Taken together, these results strongly support the theory that USP9X plays an important role in the upregulation of PTGES in NSCLC.
Figure 6.
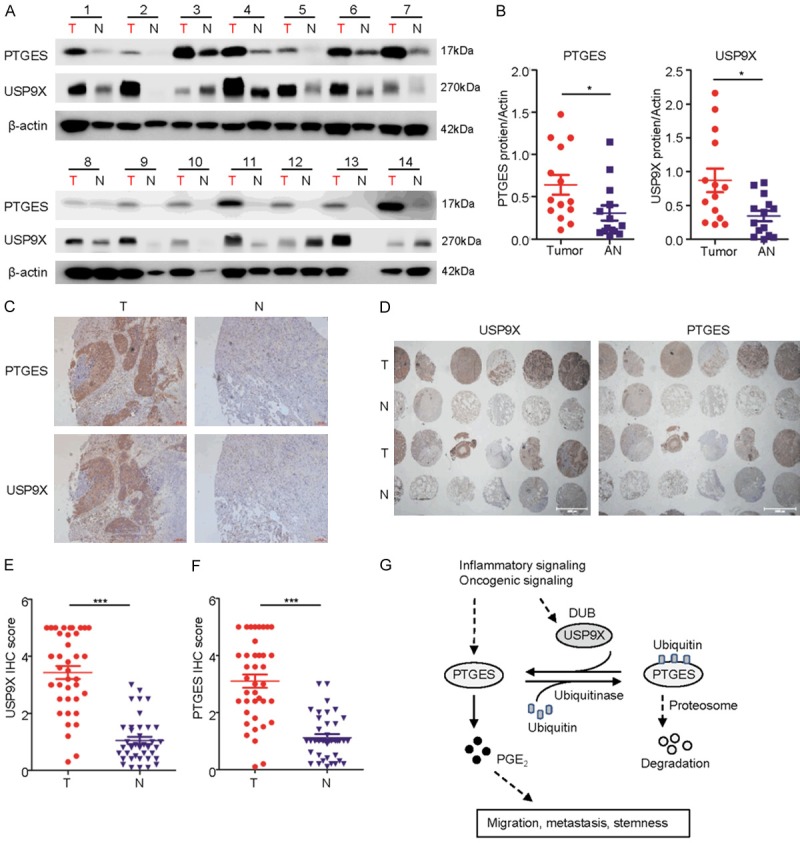
USP9X expression is significantly correlated with PTGES in lung cancer samples. A. A subset of lung tumor and normal tissues were subjected to western blotting to examine the PTGES and USP9X protein levels (n=14 pairs). B. Western blotting scores of PTGES and USP9X from the tissues as indicated. C. Representative staining of USP9X and PTGES in lung carcinoma and peritumoral lung tissues of patients from TCGA dataset (mean ± SD, P < 0.05). T, tumor; N, normal adjacent. D. Expression of USP9X and PTGES by IHC staining of a human lung tissue chip including tumor and adjacent normal tissues (n=76 pairs). E, F. IHC scores of USP9X and PTGES from the tissue chip as indicated. G. Experimental model of the mechanism by which USP9X regulates PTGES.
Discussion
In this study, we showed that dysregulated PTGES leads to activated PGE2 signaling, which promotes the metastatic features of lung cancer cells. Importantly, for the first time, we demonstrated that USP9X, a deubiquitinase, can physically interact with and stabilize PTGES via deubiquitination. Thus, the USP9X-PTGES-PGE2 axis plays an important role in lung cancer progression.
Dysregulation of the PGE2 pathway plays an important role in mediating the inflammatory response. Depending on the types of tumor cells and models used, PGE2 also exhibits many tumor-promoting activities, including the promotion of proliferation [22], anti-apoptosis [23], angiogenesis [24], invasion, and metastasis [25], and it also inhibits host immune function [26]. The upregulation of PGE2 is usually attributed to the dysregulation of COX-2. However, the targeting of COX-2, either by non-steroidal anti-inflammatory drugs (NSAIDs) or selective COX-2 inhibitors such as celecoxib, produces side effects since its inhibition also reduces other prostanoids, including PGF2a, TXA2, PGE2, PGI2, and PGD2 [27]. For example, COX-2 inhibitors increase the risk of cardiovascular problems. In this study, we showed that dysregulated PTGES plays a critical role in the upregulation of PGE2 signaling in NSCLC. PTGES-mediated migration is mainly attributed to PGE2, since exogenous PGE2 restored this activity in PTGES knockdown NSCLC cells. Thus, the targeting of PTGES should be a more specific method for inhibiting PGE2-mediated metastatic features while reducing the side effects generated by affecting other prostanoids.
EMT is an important molecular process in tumor metastasis and recurrence [28]. In this study, we showed that PTGES knockdown significantly reduced the expression of EMT biomarkers such as vimentin, twist, and N-cadherin. Via wound healing and migration assays, we found that exogenous PGE2 restored the expression of these markers. EMT is also involved in regulating stem-like properties in epithelial cells [29]. CSCs represent a subpopulation of cells with self-renewal ability that are thought to confer chemical and radiotherapy resistance and be responsible for tumor maintenance and metastasis [30]. In fact, many studies have shown that EMT confers stem cell-like characteristics on tumor cells [31,32]. Nevertheless, our study showed that the dysregulation of the PTGES/PGE2 pathway is essential for both induction of EMT-like features and maintenance of stem cell-like properties. Thus, targeting the PTGES/PGE2 axis may represent a novel approach for preventing metastatic NSCLC.
Deubiquitylating enzymes, by removing ubiquitin chains from substrates, regulate multiple cellular processes, such as cell survival, differentiation, chromosome remodeling, and apoptosis [10]. USP9X is a deubiquitinase that is known to have 21 substrates and 14 interacting proteins [10]. In this study, we found that USP9X can stabilize PTGES via deubiquitination. High expression of USP9X has been observed in several types of human cancer, such as esophageal squamous cell carcinoma [33], follicular lymphoma [34], colon adenocarcinoma [35], and NSCLC [36]. In particular, elevated expression of USP9X correlates with poor prognosis in human NSCLC [36]. However, the mechanism and biological significance of its upregulation were previously unclear. Here, for the first time, we revealed a connection between USP9X and the PTGES pathway in NSCLC. Moreover, we showed that the upregulation of USP9X is correlated with dysregulated PTGES in lung cancer. Thus, we propose that the USP9X-PTGES-PGE2 axis is crucial for the promotion of metastatic features in lung cancer. Targeting this pathway may provide an effective approach for the treatment of NSCLC.
Acknowledgements
This work was supported by the following funding agencies: National Natural Science Foundation of China (81620108022, 91129303, 91729302 and 81572759 to JD); The Fundamental Research Funds for the Central Universities (to HYS) and Youth Research Program of Shanghai Municipal Commission of Health and Family Planning (20164Y0261 to HYS).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 2.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 3.Ke Y, Oskolkova OV, Sarich N, Tian Y, Sitikov A, Tulapurkar ME, Son S, Birukova AA, Birukov KG. Effects of prostaglandin lipid mediators on agonist-induced lung endothelial permeability and inflammation. Am J Physiol Lung Cell Mol Physiol. 2017;313:L710–L721. doi: 10.1152/ajplung.00519.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isono M, Suzuki T, Hosono K, Hayashi I, Sakagami H, Uematsu S, Akira S, DeClerck YA, Okamoto H, Majima M. Microsomal prostaglandin E synthase-1 enhances bone cancer growth and bone cancer-related pain behaviors in mice. Life Sci. 2011;88:693–700. doi: 10.1016/j.lfs.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi R, Amano H, Satoh T, Tabata K, Ikeda M, Kitasato H, Akira S, Iwamura M, Majima M. Roles of microsomal prostaglandin E synthase-1 in lung metastasis formation in prostate cancer RM9 cells. Biomed Pharmacother. 2014;68:71–77. doi: 10.1016/j.biopha.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Avis I, Martínez A, Tauler J, Zudaire E, Mayburd A, Abu-Ghazaleh R, Ondrey F, Mulshine JL. Inhibitors of the arachidonic acid pathway and peroxisome proliferator-activated receptor ligands have superadditive effects on lung cancer growth inhibition. Cancer Res. 2005;65:4181–4190. doi: 10.1158/0008-5472.CAN-04-3441. [DOI] [PubMed] [Google Scholar]
- 7.Jones VC, Birrell MA, Maher SA, Griffiths M, Grace M, O’Donnell VB, Clark SR, Belvisi MG. Role of EP2 and EP4 receptors in airway microvascular leak induced by prostaglandin E2. Br J Pharmacol. 2016;173:992–1004. doi: 10.1111/bph.13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sastre B, del Pozo V. Role of PGE2 in asthma and nonasthmatic eosinophilic bronchitis. Mediators Inflamm. 2012;2012:645383. doi: 10.1155/2012/645383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Liu T, Li Y, Wu C, Luo K, Yin Y, Chen Y, Nowsheen S, Wu J, Lou Z, Yuan J. The deubiquitinase USP9X promotes tumor cell survival and confers chemoresistance through YAP1 stabilization. Oncogene. 2018;37:2422–2431. doi: 10.1038/s41388-018-0134-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murtaza M, Jolly LA, Gecz J, Wood SA. La FAM fatale: USP9X in development and disease. Cell Mol Life Sci. 2015;72:2075–2089. doi: 10.1007/s00018-015-1851-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Yu C, Gao J, Zhu H, Cui B, Zhang T, Zhou Y, Liu Q, He H, Xiao R, Huang R, Xie H, Gao D, Zhou H. A novel USP9X substrate TTK contributes to tumorigenesis in non-small-cell lung cancer. Theranostics. 2018;8:2348–2360. doi: 10.7150/thno.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DC, Dixit VM. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 13.Song H, Sun B, Liao Y, Xu D, Guo W, Wang T, Jing B, Hu M, Li K, Yao F, Deng J. GPRC5A deficiency leads to dysregulated MDM2 via activated EGFR signaling for lung tumor development. Int J Cancer. 2019;144:777–787. doi: 10.1002/ijc.31726. [DOI] [PubMed] [Google Scholar]
- 14.Kawano Y, Iwama E, Tsuchihashi K, Shibahara D, Harada T, Tanaka K, Nagano O, Saya H, Nakanishi Y, Okamoto I. CD44 variant-dependent regulation of redox balance in EGFR mutation-positive non-small cell lung cancer: a target for treatment. Lung Cancer. 2017;113:72–78. doi: 10.1016/j.lungcan.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Ye D, Guo W, Yu W, He Y, Hu J, Wang Y, Zhang L, Liao Y, Song H, Zhong S, Xu D, Yin H, Sun B, Wang X, Liu J, Wu Y, Zhou BP, Zhang Z, Deng J. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget. 2015;6:6887–6901. doi: 10.18632/oncotarget.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong S, Yin H, Liao Y, Yao F, Li Q, Zhang J, Jiao H, Zhao Y, Xu D, Liu S, Song H, Gao Y, Liu J, Ma L, Pang Z, Yang R, Ding C, Sun B, Lin X, Ye X, Guo W, Han B, Zhou BP, Chin YE, Deng J. Lung tumor suppressor GPRC5A binds egfr and restrains its effector signaling. Cancer Res. 2015;75:1801–1814. doi: 10.1158/0008-5472.CAN-14-2005. [DOI] [PubMed] [Google Scholar]
- 17.Zhuang X, Zhang H, Li X, Li X, Cong M, Peng F, Yu J, Zhang X, Yang Q, Hu G. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat Cell Biol. 2017;19:1274–1285. doi: 10.1038/ncb3613. [DOI] [PubMed] [Google Scholar]
- 18.Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP. G9a interacts with Snail and is critical for snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122:1469–1486. doi: 10.1172/JCI57349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Xu X, Jiang M, Bi Y, Xu J, Han M. Lipopolysaccharide induces inflammation and facilitates lung metastasis in a breast cancer model via the prostaglandin E2-EP2 pathway. Mol Med Rep. 2015;11:4454–4462. doi: 10.3892/mmr.2015.3258. [DOI] [PubMed] [Google Scholar]
- 20.Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L, Inui M, Moro S, Modena N, Argenton F, Newfeld SJ, Piccolo S. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–135. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 21.Opferman JT, Green DR. DUB-le trouble for cell survival. Cancer Cell. 2010;17:117–119. doi: 10.1016/j.ccr.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Howe LR, Subbaramaiah K, Kent CV, Zhou XK, Chang SH, Hla T, Jakobsson PJ, Hudis CA, Dannenberg AJ. Genetic deletion of microsomal prostaglandin E synthase-1 suppresses mouse mammary tumor growth and angiogenesis. Prostaglandins Other Lipid Mediat. 2013;106:99–105. doi: 10.1016/j.prostaglandins.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aghourian MN, Lemarié CA, Bertin FR, Blostein MD. Prostaglandin E synthase is upregulated by Gas6 during cancer-induced venous thrombosis. Blood. 2016;127:769–777. doi: 10.1182/blood-2015-02-628867. [DOI] [PubMed] [Google Scholar]
- 24.Cheng SY, Zhang H, Zhang M, Xia SK, Bai XM, Zhang L, Ma J, Rong R, Wang YP, Du MZ, Wang J, Chen M, Shi F, Yang QY, Leng J. Prostaglandin E(2) receptor EP2 mediates snail expression in hepatocellular carcinoma cells. Oncol Rep. 2014;31:2099–2106. doi: 10.3892/or.2014.3074. [DOI] [PubMed] [Google Scholar]
- 25.Che D, Zhang S, Jing Z, Shang L, Jin S, Liu F, Shen J, Li Y, Hu J, Meng Q, Yu Y. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/beta-catenin signalling pathway. Mol Immunol. 2017;90:197–210. doi: 10.1016/j.molimm.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 26.Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, Sahai E, Reis e Sousa C. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162:1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 28.Neil JR, Johnson KM, Nemenoff RA, Schiemann WP. Cox-2 inactivates smad signaling and enhances EMT stimulated by TGF-beta through a PGE2-dependent mechanisms. Carcinogenesis. 2008;29:2227–2235. doi: 10.1093/carcin/bgn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Visvader JE, Lindeman GL. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 31.Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, Bapat SA. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009;27:2059–2068. doi: 10.1002/stem.154. [DOI] [PubMed] [Google Scholar]
- 32.Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, Radisky DC, Ferrone S, Knutson KL. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69:2887–2895. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng J, Hu Q, Liu W, He X, Cui L, Chen X, Yang M, Liu H, Wei W, Liu S, Wang H. USP9X expression correlates with tumor progression and poor prognosis in esophageal squamous cell carcinoma. Diagn Pathol. 2013;8:177. doi: 10.1186/1746-1596-8-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson LF, Sun H, Liu Y, Potu H, Kandarpa M, Ermann M, Courtney SM, Young M, Showalter HD, Sun D, Jakubowiak A, Malek SN, Talpaz M, Donato NJ. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood. 2015;125:3588–3597. doi: 10.1182/blood-2014-10-605584. [DOI] [PubMed] [Google Scholar]
- 35.Kloosterman WP, Coebergh van den Braak RRJ, Pieterse M, van Roosmalen MJ, Sieuwerts AM, Stangl C, Brunekreef R, Lalmahomed ZS, Ooft S, van Galen A, Smid M, Lefebvre A, Zwartkruis F, Martens JWM, Foekens JA, Biermann K, Koudijs MJ, Ijzermans JNM, Voest EE. A systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 2017;77:3814–3822. doi: 10.1158/0008-5472.CAN-16-3563. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Liu Y, Yang B, Cao H, Yang CX, Ouyang W, Zhang SM, Yang GF, Zhou FX, Zhou YF, Xie CH. Elevated expression of USP9X correlates with poor prognosis in human non-small cell lung cancer. J Thorac Dis. 2015;7:672–679. doi: 10.3978/j.issn.2072-1439.2015.04.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.