

Abstract
ErbB family members that contain EGFR, HER2, HER3 and HER4 play important roles in many cancer types, including head and neck; however, inhibition of these receptors by small molecule kinase inhibitors showed limited results due to compensatory up-regulation of some key survival signaling pathways. Here, we explore the effectiveness of Afatinib, an irreversible inhibitor of EGFR, HER2, and HER4, in combination with the MEK inhibitor PD0325901 to inhibit cisplatin-resistant head and neck squamous cell carcinoma (HNSCC). We treated two cisplatin-resistant HNSCC cell lines, UMSCC74B and O28, with Afatinib, PD0325901, or a combination, and measured signaling pathways, cell proliferation, and survival. We found that Afatinib blocked Akt/mTOR activity and phosphorylation of EGFR, HER2 and HER3, but up-regulated MEK/ERK signaling. Interestingly, MEK inhibitor PD0325901 blocked ERK phosphorylation, but elevated phosphorylation of Akt and mTOR pathways. Similarly, Afatinib and PD0325901 inhibited all these pathways and synergistically suppressed cell proliferation and survival. Our data demonstrate that Afatinib in combination with MEK inhibitors could provide a potential novel therapy for cisplatin-resistant head and neck squamous cell cancer.
Keywords: Head and neck squamous cell carcinoma, HNSCC, cisplatin-resistant HNSCC, EGFR, PI3K, Akt, mTOR, MEK, ERK, EGFR inhibitors, Afatinib, MEK inhibitor, PD0325901
Introduction
Head and neck cancers are cancers that occur in different organs in the head and neck regions, including the oral cavity, tongue, pharynx, paranasal sinuses, the nasal cavity, and salivary glands. Head and neck cancers account for approximately 4% of all cancers in the United States [1]. Pathologically, most head and neck cancers are squamous cell types (HNSCC). Although there have been many advances in basic research and clinical therapy, the 5-year overall survival rate of head and neck cancer patients has remained below 50% [1-3]. The poor prognosis of head and neck cancer is mainly due to cancer recurrence and metastasis in a large of potion of patients after classic therapies such as surgery, radiation, or combination. Cisplatin is the only treatment option available for patients with recurrent and metastatic HNSCC, but nearly all these patients eventually become resistant to this treatment and die within one year [4,5]. Therefore, finding effective regimens for cisplatin-resistant head and neck is the key to improving patient survival rate.
ErbB family members that contain EGFR, HER2, HER3, and HER4 play important roles in head and neck cancer [6-9]. Clinical results demonstrated that targeting EGFR using the EGFR antibody Cetuximab enhanced the effectiveness of radiation therapy for early-stage HNSCC patients and improved prognosis for late-stage patients taking cisplatin-based multiple therapy, which led to 2 to 3 months of prolonged life [10-12]. Since Cetuximab is extremely expansive, it would be ideal to use small molecule kinase inhibitors instead. Currently, the FDA-approved treatments that include ErbB family targets mainly contain the EGFR inhibitors, Gefitinib and Erlotinib, dual EGFR and HER2 inhibitor, Lapatinib, and Afatinib that targets EGFR, HER2, and HER4. These small inhibitors have already been included as a routine part of therapy in lung and breast cancers, but not for head and neck cancer to date [13]. Although clinical studies demonstrated that addition of Gefitinib, Erlotinib, or Afatinib into chemotherapies containing cisplatin had no improvement on patient survival rate, the potential roles of these inhibitors for clinical use in of head and neck cancer treatments have been an important focus of study [10,14,15]. One would expect to identify the crucial druggable targets that are up-regulated by these kinase inhibitors through compensatory mechanisms. Currently, Afatinib, as an irreversible inhibitor of EGFR, HER2 and HER4, seems to be the most attractive one of these inhibitors [10,15-19].
In this study, we explored the role of the MEK/ERK pathway to regulate the effectiveness of Afatinib to inhibit cisplatin-resistant HNSCC. We found that Afatinib inhibited phosphorylation of EGFR, HER2, and HER3, as well as the Akt/mTOR pathway activity, but enhanced phosphorylation of ERK. MEK inhibitor PD0325901 blocked basal and Afatinib-induced phosphorylation of ERK and synergized with Afatinib to suppress cell proliferation and survival of cisplatin-resistant HNSCC.
Materials and methods
Cell lines
HNSCC cell line UMSCC74B was the generous gift of Dr. Thomas E. Carey (University of Michigan, Ann Arbor, MI, USA). The O28 cell line was obtained from Dr. Zhongmin Guo and originally from Johns Hopkins University School of Medicine (Baltimore, MD) [20]. All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 100 U/mL penicillin and streptomycin (Gibco).
Reagents and antibodies
Protease and phosphatase inhibitors were purchased from Roche and CHAPS was obtained from Pierce. Afatinib and PD0325901 were purchased from Selleckchem. Antibodies against phospho-EGFR-Y1068 (CST-3733), phospho-HER2-Y1248 (CST-2247), HER2 (CST-4290), phospho-HER3-Y1289 (CST-2842), HER3 (CST-12708), phospho-Akt-S473 (CST-4508), Akt (CST-2938), phospho-S6 (CST-2211), S6 (CST-2317), phospho-ERK-T202/Y204 (CST-4370), ERK (CST-4348), cleaved caspase 3 (CST-9664), and GAPDH (CST-5174) were purchased from Cell Signaling. Anti-EGFR (SC-03) was purchased from Santa Cruz Biotechnology.
Cell lysis and western blot analysis
Cells were lysed and immunobotted as described previously [21].
Cell proliferation assays
Cells were split into in 96-well plates in triplicate at 3 × 103 cells/well and cultured in the presence or absence of single or double inhibitors with indicated concentrations. After three days, 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) compound (Promega, Cat#: G3580) was added and incubated at 37°C for 1 hour prior to colorimetric readouts at 490 nm on a Versamax Microplate Reader (Molecular Devices). The combination index values were determined according to the Chou-Talalay method using CalcuSyn software [22].
Measuring apoptosis by Annexin V/propidium iodide staining
Cells were trypsinized, washed once with PBS and Annexin V binding buffer, and re-suspended in 1 mL Annexin V binding buffer. Cells were then stained with 0.5 μL of Annexin V and 0.7 μL of propidium iodide (PI) for 15 minutes at room temperature. Cells stained were then analyzed by flow cytometry on the BD FACSCanto II™ Cell Analyzer (BD Biosciences). We used FCS Express 6 to analyze data. Experiments were performed twice in triplicate and statistical analysis was performed.
Statistical analysis
Data are presented as mean ± SD. Statistical analysis was performed using GraphPad Prism version 7.04 (GraphPad Software, Inc.). P values < 0.05 were considered as statistically significant.
Results
Afatinib induces the MEK/ERK pathway in cisplatin-resistant HNSCC cells
UMSCC74B cells were derived from a patient with recurrent tongue squamous cell carcinomas after cisplatin and radiation therapies [23]. We demonstrated that this cell line was resistant to cisplatin in vitro with an IC50 of 18 µmol/l (data not shown). We examined the effects of Afatinib treatment on the phosphorylation and total protein expression of EGFR, HER2, HER3, HER4, Akt, S6 (a downstream target of mTOR), and ERK (a downstream target of MEK kinase) in these cells. Cells were first treated with increasing doses for 24 hours before they were lysed. As shown in Figure 1A, the phosphorylation of EGFR, HER2, and HER3 decreased in a dose-dependent manner (Figure 1A, left panel). We found that phosphor-HER4 and total HER4 were undetectable in this cell line (data not shown). Consistent with the above inhibition results, Akt and S6 phosphorylation were also inhibited, while the total protein levels remained the same (Figure 1A, left panel). Interestingly, phosphorylation of ERK was elevated, while total ERK was not affected by Afatinib treatment (Figure 1A, left panel). Similarly, we found that Afatinib inhibited phosphorylation of EGFR, HER2, HER3, Akt, and S6, but induced ERK phosphorylation in cisplatin-resistant O28 cells (Figure 1A, right panel). Our data suggested that Afatinib inhibited PI3K/Akt/mTOR pathways but up-regulated MEK/ERK pathways in some cisplatin-resistant HNSCC cells (Figure 1B).
Figure 1.
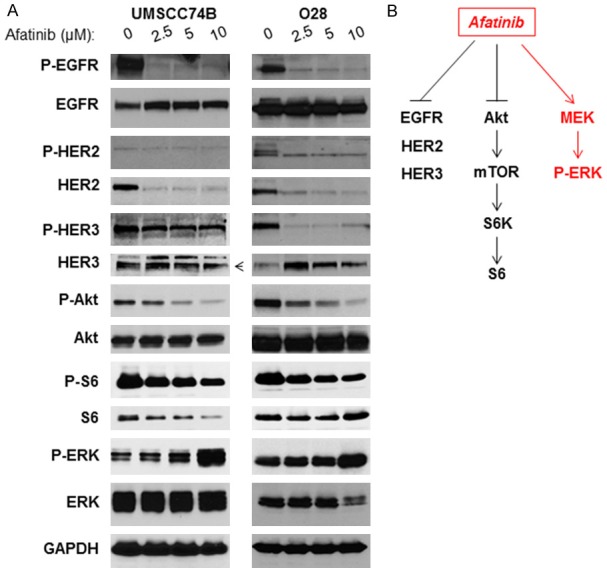
Afatinib inhibits EGFR, HER2, and HER3, as well as Akt/mTOR signaling, but induces MEK/ERK pathway in cisplatin-resistant HNSCC cells. A: Cell lysates were prepared from UMSCC74B (Left) and O28 (Right) treated with different doses of Afatinib for 24 hours and phosphorylation and total levels of EGFR, HER2, HER3, Akt, S6, and ERK, as well as GAPDH expression were detected by Western blot analysis. B: Inhibition of EGFR, HER2, HER3, Akt, S6, and ERK, but induction of ERK pathway by Afatinib.
Inhibition of MEK/ERK pathway elevates PI3K/Akt/mTOR cascade
We next determined the effects of MEK/ERK inhibitor treatment on the phosphorylation of ERK and PI3K/Akt/mTOR pathways. PD0325901 is a novel MEK inhibitor that has been heavily studied in vitro and in vivo and is currently under clinical trials for treatment of multiple cancer types [24-27]. UMSCC74B cells were treated with increasing doses of PD0325901 for 24 hours. As shown in Figure 2A, the phosphorylation of ERK was reduced in a dose-dependent manner, whereas the total level of ERK remained unchanged (Figure 2A, left panel). In addition, the phosphorylation of Akt and S6 (downstream of mTOR) was elevated while the total levels of these proteins remained the same (Figure 2A, left panel). Similar results were found in O28 cells (Figure 2A, right panel). Our data demonstrated that the PI3K/Akt/mTOR cascade was elevated in some cisplatin-resistant HNSCC, while MEK/ERK pathways were inhibited (Figure 2B).
Figure 2.
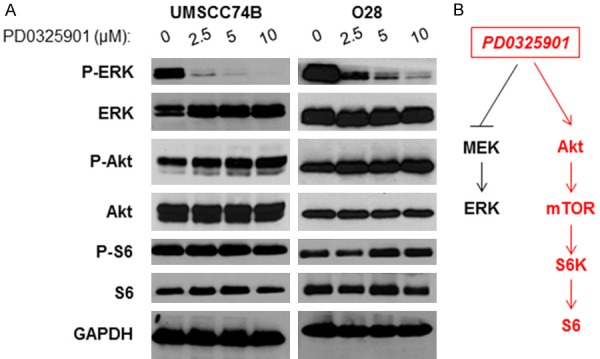
MEK inhibitor PD0325901 inhibits ERK, but induces Akt/mTOR signaling pathway in cisplatin-resistant HNSCC cells. A: Cell lysates were prepared from UMSCC74B (Left) and O28 (Right) treated with different doses of PD0325901 for 24 hours and phosphorylation and total levels of ERK, Akt, and S6, as well as GAPDH expression were detected by Western blot analysis. B: Inhibition of ERK, but induction of Akt/mTOR pathways by PD0325901.
Inhibition of both PI3K/Akt/mTOR and MEK/ERK pathways by combination of Afatinib and PD0325901
We next determined whether a combination of Afatinib and PD0325901 could block PI3K/Akt/mTOR and MEK/ERK pathways. UMSCC74B cells were treated with either DMSO, Afatinib, PD0325901 or a combination for 48 hours and Western blot analysis was performed. Similar to Figure 1, Afatinib blocked Akt and S6 phosphorylation, but induced ERK phosphorylation (Figure 3A, left panel, lanes 1 and 2). Conversely, PD0325901 blocked ERK phosphorylation, but induced Akt and S6K phosphorylation (Figure 3A, left panel, lanes 1 and 3). The combination, however, was able to block phosphorylation of ERK, Akt, and S6 (Figure 3A, left panel, lane 4). Similar results were found when UMSCC74B cells were treated with DMSO, Afatinib, PD0325901, or a combination for 72 hours (Figure 3A, right panel). Furthermore, the combination treatment also blocked phosphorylation of ERK, Akt, and S6 in O28 cells (Figure 3B, left and right panels). Our data demonstrated that both PI3K/Akt/mTOR and MEK/ERK cascades were blocked by Afatinib and PD0325901 combination treatment (Figure 3C).
Figure 3.
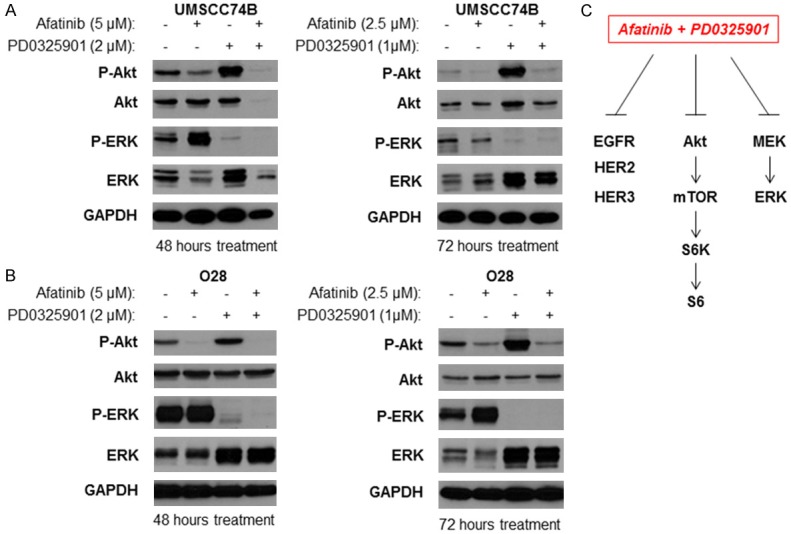
Inhibition of ErbB family, Akt/mTOR, and MEK/ERK by a combination of Afatinib and PD0325901 in cisplatin-resistant HNSCC. A: Cell lysates were prepared from UMSCC74B cells treated with different doses of PD0325901 for 48 (Left) or 72 (Right) hours and phosphorylation and total levels of Akt and ERK, as well as GAPDH expression were detected by Western blot analysis. B: Cell lysates were prepared from O28 cells treated with different doses of PD0325901 for 48 (Left) or 72 (Right) hours and phosphorylation and total levels of Akt and ERK, as well as GAPDH expression were detected by Western blot analysis. C: Inhibition of Akt and ERK following combination treatment with Afatinib and PD0325901.
Inhibition of MEK/ERK enhances Afatinib-induced caspase cleavage and apoptosis
We next determined whether a combination of Afatinib and PD0325901 could induce additional apoptosis compared to separate Afatinib and PD0325901 treatments in UMSCC74B cells. Afatinib alone induced cleaved caspase-3 whereas PD0325901 induced no cleaved caspase-3, however, combination of Afatinib and PD0325901 caused more cleaved caspase-3. These results indicated that Afatinib induced caspase cleavage more effectively in combination with MEK inhibitor (Figure 4A). To further confirm the above results, flow cytometry experiments were performed in UMSCC74B cells treated with either DMSO, Afatinib, PD0325901 or a combination of both. The results showed that Afatinib or PD0325901 alone caused apoptosis compared to vehicle control treatment, while the combination caused more significant apoptosis and cell death compared to either single treatment (Figure 4B and 4C).
Figure 4.

Afatinib and PD0325901 synergistically induces apoptosis in UMSCC74B cells. (A) Cleaved caspase-3 in cell lysates from Figure 3A was detected by Western blot. (B) Cells were treated with vehicle control, Afatinib, PD0325901, or a combination for 48 hours. Cell apoptosis was measured by Annexin V. (C) Experiments in (B) were performed in triplicate, early and late stage apoptosis counted, and statistical analysis performed. P values less than 0.05 were considered to be statistically significant.
We further tested whether a combination of Afatinib and PD0325901 could induce increased levels of apoptosis in comparison to single Afatinib or PD0325901 treatments in O28 cells. Treatment with Afatinib or PD0325901 alone slightly induced cleaved caspase-3, whereas combination of Afatinib and PD0325901 induced more cleaved caspase-3 cleavage (Figure 5A). Consistently, flow cytometry experiments showed that a combination of Afatinib and PD0325901 led to more significant apoptosis and cell death when compared to each single treatment (Figure 5B and 5C). Our data indicated that a combination of PD0325901 and Afatinib more dramatically induced apoptosis compared to either treatment alone.
Figure 5.

Afatinib and PD0325901 cooperate to induce apoptosis in O28 cells. (A) O28 cells were treated with vehicle control, Afatinib, PD0325901, or a combination for 48 hours before cells were lysed; cleaved caspase-3 in cell lysates was detected by Western blot. (B) Cells were treated with vehicle control, Afatinib, PD0325901, or a combination for 48 hours. Cell apoptosis and death were measured by Annexin V. (C) Experiments in (B) were performed in triplicate, early and late stage apoptosis counted, and statistical analysis performed. P values less than 0.05 were considered to be statistically significant.
MEK inhibitor synergizes with Afatinib to inhibits cell proliferation
We next determined whether PD0325901 and Afatinib could cooperate to inhibit cell proliferation. UMSCC74B cells were treated with DMSO, Afatinib, PD0325901, or a combination for 48 hours, followed by microscopic observation of cell conditions. The cell density in samples treated with Afatinib or PD0325901 were slightly lower than that in dishes treated with the DMSO control, whereas the density in samples treated with the combination were significantly lower than that in either single treatment (Figure 6). Next, UMSCC74B cells were treated with DMSO, increasing doses of either Afatinib or MEK inhibitor, or a combination for 72 hours. Cell proliferation was measured by MST assay and cell viability was normalized to the DMSO control. The results showed that Afatinib or PD0325901 caused dose-dependent inhibition of cell proliferation, but a combination increased inhibition of cell proliferation compared to either single treatment (Figure 6B). In addition, we utilized the CalcuSyn 2.0 software to calculate the combination index values (CI) according to the Chou-Talalay method [22]. The CI values in most combination treatments shown in Figure 6B were less than 1 (Figure 6B).
Figure 6.
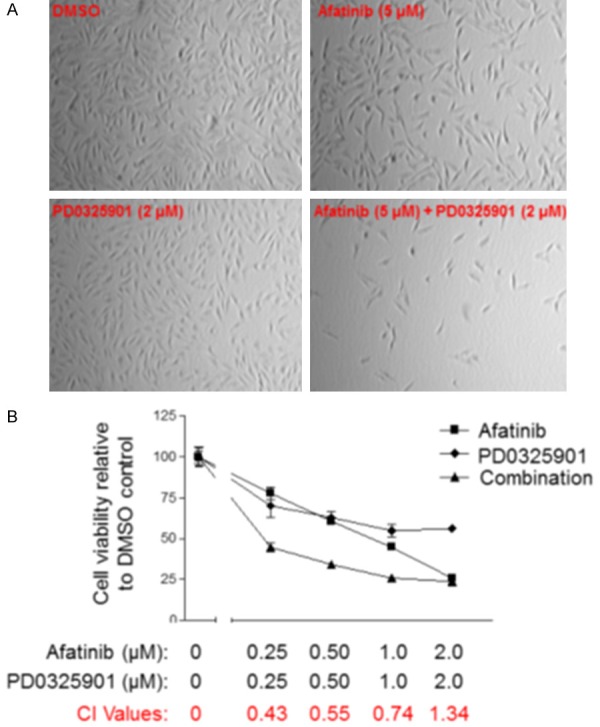
Afatinib and PD0325901 synergistically cooperate to suppress UMSCC74B cell proliferation. A: Synergistic suppression of cell growth by Afatinib and PD0325901. UMSCC74B cells were treated with vehicle control, Afatinib, PD0325901, or a combination for 48 hours and cell density was observed microscopically. B: Afatinib and PD0325901 synergistically inhibit cell proliferation. UMSCC74B cells were treated with DMSO or different doses of Afatinib, PD0325901, or a combination for 72 hours and cell proliferation was determined by MTS assay. The experiments were performed in triplicate, and the combination index values (CI values) were determined using CalcuSyn software.
We also determined whether Afatinib and PD0325901 cooperated to suppress cell proliferation in O28 cells. Consistent with the data in Figure 6A, the cell density in dishes treated with Afatinib or PD0325901 was lower than that in dishes treated with the control, whereas the density in cells treated with the combination were significantly decreased in comparison to either single treatment (Figure 7A). Furthermore, we found that treatment with Afatinib or PD0325901 alone inhibited cell proliferation; however, treatment with a combination of Afatinib and PD0325901 led to synergistic inhibition of cell proliferation (Figure 7B). Taken together, Afatinib and MEK inhibitors synergistically inhibit cell proliferation.
Figure 7.
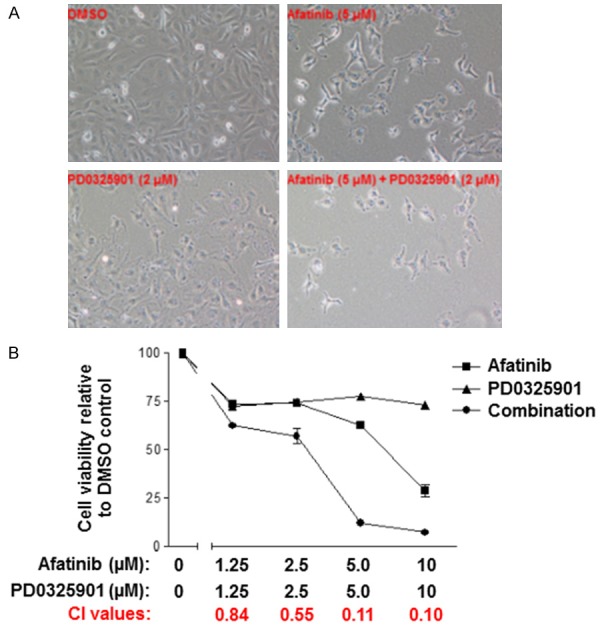
Afatinib and PD0325901 cooperate to suppress O28 cell proliferation. A: O28 cells were treated with vehicle control, Afatinib, PD0325901 or a combination for 72 hours and cell density was observed microscopically. B: O28 cells were treated with DMSO or different doses of Afatinib, PD0325901, or a combination for 72 hours and cell proliferation was determined by MTS assay. The experiments were conducted twice in triplicate and the combination index values (CI values) were determined using CalcuSyn software.
Discussion
The current standard treatment regimen for recurrent and distant metastatic HNSCC is chemotherapy that mainly contains cisplatin and 5-Fluorouracil (5-FU), as well as the EGFR antibody Cetuximab [28,29]. In addition, combing Cetuximab only modestly improves overall survival [10,30]. Clinical trials demonstrated that adding EGFR tyrosine kinase inhibitors, such as Gefitinib and Erlotinib, to therapies that contain cisplatin did not show any improvement in patient survival [10,14]. Multiple factors contribute to the effectiveness and resistance of EGFR inhibitors, such as up-regulation of some survival pathways and other ErbB family members, like HER2 and HER3. Since Afatinib inhibited more ErbB family members compared to EGFR inhibitor (Gefitinib and Erlotinib) treatment, it would be interesting to see if clinical trials increase in efficacy with the addition of Afatinib to cisplatin-containing therapies [10,16].
In the current study, we explored the therapeutic potential in the combination of the ErbB family inhibitor Afatinib with MEK inhibitor PD0325901 to inhibit cisplatin-resistant head and neck squamous cell cancer (HNSCC). We showed that the combination was effective in induction of apoptosis and inhibition of cell proliferation compared to single treatments with either Afatinib or PD0325901 alone. Afatinib inhibited EGFR, HER2, and HER3 phosphorylation and subsequently blocked Akt/mTOR activity, but it also induced the MEK/ERK pathway. In addition, MEK/ERK inhibition by PD0325901 led to up-regulation of the PI3K/Akt/mTOR pathway. Taken together, we have shown that the combination of these two inhibitors abolished all above signaling pathways. These data provided the rationale to use a combination of Afatinib and MEK inhibitors to treat cisplatin-resistant HNSCC in future clinical studies.
Combination of cisplatin-based chemotherapy with other chemotherapy drugs or target inhibitors greatly improves the effectiveness of HNSCC treatment. A recent study demonstrated that co-treatment of head and neck cancer with a combination of Afatinib and cisplatin more effectively inhibited cell proliferation [31]. However, for patients with cisplatin-resistant cancer, continual use of cisplatin for treatment might not benefit overall survival and could even be toxic to the patient. It is therefore important to find new combination target therapies that do not include cisplatin. Afatinib, in combination with other kinase inhibitors, such as MER/ERK inhibitors, could be a potential candidate for a portion of patients with cisplatin-resistant HNSCC.
Mechanisms by which Afatinib up-regulates the MEK/ERK pathway remain unclear. Given the fact that MER/ERK is a downstream target of RAS, which is thus controlled by multiple tyrosine kinases, it is important to identify which membrane receptors are elevated to enhance RAS/MEK/ERK pathways that confer Afatinib resistance, but can still inhibit EGFR, HER2, and HER4.
Our current data remain consistent with previous studies that show MEK/ERK inhibition leads to up-regulation of PI3K/Akt/mTOR pathway in multiple cancer cell lines, including HNSCC cells. These data suggest that the combination of MEK and PI3K inhibitors display synergistic inhibition of cell proliferation in vitro and in vivo [27,32-34]. Our data also demonstrate that Afatinib can inhibit both basal and MEK inhibitor-induced activation of the PI3K/Akt pathway. It is possible that the combination of Afatinib and MEK inhibitors will thus more effectively suppress cell proliferation.
It should be noted that Afatinib induction of MEK/ERK pathway could be cell-type specific. We found that Afatinib treatment led to inhibition of both PI3K/Akt/mTOR and MEK/ERK pathways in several cisplatin-resistant HNSCC cell lines (data not shown). Therefore, inhibition of cisplatin-resistant HNSCC proliferation by a combination of Afatinib and MEK inhibitors may be effective only in cells in which Afatinib induces MER/ERK activity.
We should also mention that very recently, Yee, PS et al., published a manuscript which described that Afatinib and Trametinib (another MEK inhibitor) synergistically inhibit HNSCC growth in a pre-clinical model of oral squamous cell carcinoma [35]. These findings demonstrate that a combination of Afatinib and MEK inhibitors would also effectively suppress cisplatin-sensitive HNSCC. In fact, we also found that the combination of Afatinib and Trametinib more effectively kills non-small cell lung cancer (NSCLC) cells when compared to single treatment in multiple NSCLC cell lines (data not shown). Taken together, we conclude that co-targeting ErbB family and MER/ERK pathways could be effective to inhibit cancer proliferation and survival in multiple cancers.
Acknowledgements
We also thank the Translation Core Lab of the University of Maryland Greenebaum Comprehensive Cancer Center for their help in carrying out several experiments. This research was supported, in part, by the NIH National Cancer Institute (NCI) to H.D. (R00CA149178 and R01CA212094), and the University of Maryland Marlene and Stewart Greenebaum Cancer Center.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Santuray RT, Johnson DE, Grandis JR. New therapies in head and neck cancer. Trends Cancer. 2018;4:385–96. doi: 10.1016/j.trecan.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammerman PS, Hayes DN, Grandis JR. Therapeutic insights from genomic studies of head and neck squamous cell carcinomas. Cancer Discov. 2015;5:239–44. doi: 10.1158/2159-8290.CD-14-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart DJ. Mechanisms of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol. 2007;63:12–31. doi: 10.1016/j.critrevonc.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Xu MJ, Johnson DE, Grandis JR. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017;36:463–73. doi: 10.1007/s10555-017-9687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du Y, Peyser ND, Grandis JR. Integration of molecular targeted therapy with radiation in head and neck cancer. Pharmacol Ther. 2014;142:88–98. doi: 10.1016/j.pharmthera.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Cohen RB. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC) Cancer Treat Rev. 2014;40:567–77. doi: 10.1016/j.ctrv.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Roskoski R Jr. ErbB/HER protein-tyrosine kinases. Structures and small molecule inhibitors. Pharmacol Res. 2014;87:42–59. doi: 10.1016/j.phrs.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Zibelman M, Mehra R. Overview of current treatment options and investigational targeted therapies for locally advanced squamous cell carcinoma of the head and neck. Am J Clin Oncol. 2016;39:396–406. doi: 10.1097/COC.0000000000000283. [DOI] [PubMed] [Google Scholar]
- 11.Sacco AG, Cohen EE. Current treatment options for recurrent or metastatic head and neck squamous cell carcinoma. J. Clin. Oncol. 2015;33:3305–13. doi: 10.1200/JCO.2015.62.0963. [DOI] [PubMed] [Google Scholar]
- 12.Tian X, Zhou JG, Zeng Z, Shuai T, Yi LJ, Ma L, Wang Y, Cao H, Song GM. Cetuximab in patients with esophageal cancer: a systematic review and meta-analysis of randomized controlled trials. Med Oncol. 2015;32:127. doi: 10.1007/s12032-015-0521-2. [DOI] [PubMed] [Google Scholar]
- 13.Herbst RS, Bunn PA Jr. Targeting the epidermal growth factor receptor in non-small cell lung cancer. Clin Cancer Res. 2003;9:5813–24. [PubMed] [Google Scholar]
- 14.Rabinowits G, Haddad RI. Overcoming resistance to EGFR inhibitor in head and neck cancer: a review of the literature. Oral Oncol. 2012;48:1085–9. doi: 10.1016/j.oraloncology.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 15.Mok TSK, Kim SW, Wu YL, Nakagawa K, Yang JJ, Ahn MJ, Wang J, Yang JC, Lu Y, Atagi S, Ponce S, Shi X, Rukazenkov Y, Haddad V, Thress KS, Soria JC. Gefitinib plus chemotherapy versus chemotherapy in epidermal growth factor receptor mutation-positive non-small-cell lung cancer resistant to first-line gefitinib (IMPRESS): overall survival and biomarker analyses. J. Clin. Oncol. 2017;35:4027–34. doi: 10.1200/JCO.2017.73.9250. [DOI] [PubMed] [Google Scholar]
- 16.Modjtahedi H, Cho BC, Michel MC, Solca F. A comprehensive review of the preclinical efficacy profile of the ErbB family blocker afatinib in cancer. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:505–21. doi: 10.1007/s00210-014-0967-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machiels JP, Haddad RI, Fayette J, Licitra LF, Tahara M, Vermorken JB, Clement PM, Gauler T, Cupissol D, Grau JJ, Guigay J, Caponigro F, de Castro G Jr, de Souza Viana L, Keilholz U, Del Campo JM, Cong XJ, Ehrnrooth E, Cohen EE. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): an open-label, randomised phase 3 trial. Lancet Oncol. 2015;16:583–94. doi: 10.1016/S1470-2045(15)70124-5. [DOI] [PubMed] [Google Scholar]
- 18.Clement PM, Gauler T, Machiels JP, Haddad RI, Fayette J, Licitra LF, Tahara M, Cohen EE, Cupissol D, Grau JJ, Guigay J, Caponigro F, de Castro G Jr, de Souza Viana L, Keilholz U, Del Campo JM, Cong XJ, Ehrnrooth E, Vermorken JB. Afatinib versus methotrexate in older patients with second-line recurrent and/or metastatic head and neck squamous cell carcinoma: subgroup analysis of the LUX-Head & Neck 1 trial. Ann Oncol. 2016;27:1585–93. doi: 10.1093/annonc/mdw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saada-Bouzid E, Le Tourneau C. Beyond EGFR targeting in SCCHN: angiogenesis, PI3K, and other molecular targets. Front Oncol. 2019;9:74. doi: 10.3389/fonc.2019.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen B, Chen J, House MG, Cullen KJ, Nephew KP, Guo Z. Role of neurofilament light polypeptide in head and neck cancer chemoresistance. Mol Cancer Res. 2012;10:305–15. doi: 10.1158/1541-7786.MCR-11-0300. [DOI] [PubMed] [Google Scholar]
- 21.Li Z, Yang Z, Passaniti A, Lapidus RG, Liu X, Cullen KJ, Dan HC. A positive feedback loop involving EGFR/Akt/mTORC1 and IKK/NF-kB regulates head and neck squamous cell carcinoma proliferation. Oncotarget. 2016;7:31892–906. doi: 10.18632/oncotarget.7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 23.Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, Bradford CR, Carey TE. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32:417–26. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown AP, Carlson TC, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59:671–9. doi: 10.1007/s00280-006-0323-5. [DOI] [PubMed] [Google Scholar]
- 25.Wang JY, Wilcoxen KM, Nomoto K, Wu S. Recent advances of MEK inhibitors and their clinical progress. Curr Top Med Chem. 2007;7:1364–78. doi: 10.2174/156802607781696837. [DOI] [PubMed] [Google Scholar]
- 26.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–64. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohan S, Vander Broek R, Shah S, Eytan DF, Pierce ML, Carlson SG, Coupar JF, Zhang J, Cheng H, Chen Z, Van Waes C. MEK inhibitor PD-0325901 overcomes resistance to PI3K/mTOR inhibitor PF-5212384 and potentiates antitumor effects in human head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21:3946–56. doi: 10.1158/1078-0432.CCR-14-3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Specenier PM, Vermorken JB. Recurrent head and neck cancer: current treatment and future prospects. Expert Rev Anticancer Ther. 2008;8:375–91. doi: 10.1586/14737140.8.3.375. [DOI] [PubMed] [Google Scholar]
- 29.Specenier PM, Vermorken JB. Current concepts for the management of head and neck cancer: chemotherapy. Oral Oncol. 2009;45:409–15. doi: 10.1016/j.oraloncology.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Sacco AG, Worden FP. Molecularly targeted therapy for the treatment of head and neck cancer: a review of the ErbB family inhibitors. Onco Targets Ther. 2016;9:1927–43. doi: 10.2147/OTT.S93720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brands RC, Muller-Richter UD, De Donno F, Seher A, Mutzbauer G, Linz C, Kubler AC, Hartmann S. Co-treatment of wild-type EGFR head and neck cancer cell lines with afatinib and cisplatin. Mol Med Rep. 2016;13:2338–44. doi: 10.3892/mmr.2016.4786. [DOI] [PubMed] [Google Scholar]
- 32.Yun MR, Choi HM, Kang HN, Lee Y, Joo HS, Kim DH, Kim HR, Hong MH, Yoon SO, Cho BC. ERK-dependent IL-6 autocrine signaling mediates adaptive resistance to pan-PI3K inhibitor BKM120 in head and neck squamous cell carcinoma. Oncogene. 2018;37:377–88. doi: 10.1038/onc.2017.339. [DOI] [PubMed] [Google Scholar]
- 33.Luangdilok S, Box C, Harrington K, Rhys-Evans P, Eccles S. MAPK and PI3K signalling differentially regulate angiogenic and lymphangiogenic cytokine secretion in squamous cell carcinoma of the head and neck. Eur J Cancer. 2011;47:520–9. doi: 10.1016/j.ejca.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 34.ElMokh O, Ruffieux-Daidie D, Roelli MA, Stooss A, Phillips WA, Gertsch J, Dettmer MS, Charles RP. Combined MEK and Pi3’-kinase inhibition reveals synergy in targeting thyroid cancer in vitro and in vivo. Oncotarget. 2017;8:24604–20. doi: 10.18632/oncotarget.15599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yee PS, Zainal NS, Gan CP, Lee BKB, Mun KS, Abraham MT, Ismail SM, Abdul Rahman ZA, Patel V, Cheong SC. Synergistic growth inhibition by afatinib and trametinib in preclinical oral squamous cell carcinoma models. Target Oncol. 2019;14:223–235. doi: 10.1007/s11523-019-00626-8. [DOI] [PubMed] [Google Scholar]