

Abstract
Extracellular acidification is a very common cause of stress in tumor microenvironment and of Darwinian pressure. In acid areas of the tumor, most cancer cells are-albeit slowly proliferating-more resistant to cell death than those in well-perfused regions. Tumor acidosis can directly regulate the expression of pro-survival proteins since a low extracellular pH activates the caspase-dependent cell death machinery. This mechanism has never been explored in bone sarcomas. We cultured osteosarcoma and Ewing sarcoma cells under low pH (pH 6.5), and we performed deep-sequencing and protein analysis. Both in in vitro and in vivo models, acidification activity enhanced tumor cells survival. However, we did not observe any change in ERK1 phosphorylation. On the contrary, both at the mRNA and protein level, we found a significant induction of TRAF adaptor proteins and of cIAP proteins (BIRC2 and/or BIRC3). As a consequence, the downstream nuclear transcription factor kappa B (NF-κB) survival pathway was increased. Furthermore, the treatment with the cIAP inhibitor LCL161 reverted the protection from apoptosis under low pH. In vitro results were confirmed both in Ewing sarcoma xenograft and in osteosarcoma patients, since the analysis of tumor tissues demonstrated that the levels of expression of TRAF1 or NF-κB1 significantly correlate with the level of expression of the vacuolar ATPase (V-ATPase), the most important proton pump in eukaryotes. Moreover, in the tissue sections of xenograft model, the nuclear translocation of RelB, a key subunit of the NF-κB transcriptional complex, localized in the tumor region that also corresponded to the acid microenvironment associated with the highest levels of expression of LAMP2 and V-ATPase, in the internal area of the tumor, as revealed by immunohistochemistry. Our data confirm that tumor acid microenvironment activates a stress-regulated switch to promote cell survival of bone sarcoma, and support the hypothesis that this mechanism is mediated by the recruitment of TRAF/cIAP complexes. Altogether, these results suggest that TRAF/cIAP can be considered as a target for anti-cancer therapies.
Keywords: Bone sarcoma, acid tumor microenvironment, cell survival, BIRC/TRAF pathway
Introduction
Interstitial acidification, i.e. decreased extracellular pH (pHe), is now recognized as a hallmark of cancer, resulting from the combination of dysfunctional tissue perfusion and a high rate of cellular metabolism [1,2]. Indeed, solid malignancies are characterized by increased glycolysis that sustains uncontrolled proliferation, and that results in extracellular proton extrusion through the activity of ion/proton pumps and transporters which reduce the toxic accumulation of protons in the cytosol. Like hypoxia [3], due to Darwinian pressure [4], acidosis that is formed at the extracellular space, in turn, significantly influences cancer cell behavior and clinical outcome [1]. In primary malignant bone tumors, cancer cells have a high glycolytic and acidification activity that, with the help of several ion/proton pumps and transporters [5-7], gathers an extracellular pH within the range 6.4-7.4 [8] which, in turn, strongly promotes sarcoma aggressiveness [5,7,9]. Osteosarcoma is the most common malignant bone tumor in children and adolescents, with an incidence of 4.4 per million. The peak age incidence is in the second decade of life, with a smaller peak in older adults [10,11]. Ewing sarcoma is the second most common malignant bone tumor, with an incidence of 2.9 per million [12]. Despite their low incidence, osteosarcoma and Ewing sarcoma are both aggressive malignant tumors that, owing to their rapid and invasive growth, are a dismal cause of mortality and disability in children. Clarifying the mechanisms underlying the survival of tumor cells that cause chemoresistance is therefore essential to improve outcomes. To date, several mechanisms of drug resistance have been indentified, including altered drug uptake and transport [13,14] and apoptosis inhibition [15]. Our recent findings, indicate that in osteosarcoma a low pHe directly accounts for drug resistance through the inversion of pH gradient at the plasmalemma, resulting in a reduced drug uptake, or through the induction of lysosomal acidification and drug compartmentalization [7].
Apoptosis is a mechanisms of cell death that is used to maintain cellular homeostasis [16]. Tumor cells, and even more cancer stem cells, are commonly resistant to this process [17] as a defense mechanism. Additional indirect mechanisms might be involved, such as the activation of acid-induced anti-apoptotic intracellular signaling to counteract non-permissive microenvironmental conditions. This might allow long-term survival in a hostile environment and resistance to the cytotoxicity of anti-cancer drugs. Indeed, acidosis can directly regulate the expression of pro-survival and anti-apoptotic proteins like the BCL-2 family of proteins, frequently overexpressed in cancer and upregulated by the extracellular acid-sensing receptor GPR65 [18,19]. Extracellular acidosis has also been associated with ROS production in breast carcinoma [20], and ROS accumulation activates multiple pathways and exerts discrepant impacts on cancer cell survival in different tumor histotypes [21].
For the identification of effective anticancer strategies, an improved understanding of the molecular mechanisms of cancer cell survival is required. Most importantly, to obtain relevant information it is crucial to resemble, in in vitro preclinical models, the same stress conditions occurring in the tumor microenvironment. According to this aim and method, we intended to dissect the anti-apoptotic mechanisms and the downstream molecular pathways that are acutely and directly promoted by extracellular acidosis in osteosarcoma and in Ewing sarcoma.
Material and methods
Reagents
Iscove’s Modified Dulbecco’s Medium (IMDM) (Life Technologies, Carlsbad, California, USA); penicillin, streptomycin (Thermo Fisher Scientific, Waltham, Massachussetts, USA); fetal bovine serum (Euroclone, Milan, Italy); Matrigel (BD Bioscences, San Jose, California, USA); anti-human AKT, anti-human phospho-AKT (ser473), anti-human p44/42 MAP kinase, anti-human phospho-p44/42 Map kinase/Thr202/tyr204 (Cell Signalling Technology, Danvers, Massachusetts, USA); Restore Western Blot Stripping buffer, SuperSignal West Pico Chemiluminescent Substrate, TRIzolTM Reagent, BCA protein assay kit (Thermofisher Scientific, Waltham, Massachusetts, USA); bisBenzimide Hoechst 33342, rabbit anti-ATP6V1B2 antibody, rabbit anti-LAMP2 antibody, rabbit anti-RelB antibody (Sigma, Saint Louis, Missouri, USA); TruSeq RNA Sample Prep Kit v2 (Illumina); NucleoSpin RNA II (Macherey-Nagel, Duren, Germany); Advantage RT-for-PCR Kit, Trizol (Life Technologies, Monza, Italy); Universal ProbeLibrary system (Roche Applied Science, Monza, Italy); TRAF1 elisa kit: Human TNF receptor-associated factor 1 ELISA Kit, Human baculoviral IAP repeat-containing protein 2/3 (BIRC2/3) ELISA Kit (MyBiosource, San Diego, California, USA); Nuclear Extraction Kit (Cayman Chemical, Ann Arbor, Michigan, USA); ELISA-based TransAM NFκB Family kit (Active Motif, Carlsbad, CA, USA); Antra 40 mg (omeprazole) (AstraZeneca, Cambrige, UK); LCL161 10 mM (Clinisciences, Nanterre, France); siRNA TRAF1 (siRNA ID s14377) and silencer select negative control No. 2 (Ambion, Foster City, CA, USA).
Cell culture
HOS, Saos2, MG-63 (osteosarcoma), and A673 (Ewing sarcoma) human cell lines were purchased from the American Type Culture Collection, cultured in IMDM plus 20 unit/mL penicillin, 100 μg/mL streptomycin, and 10% FBS at pH 7.4, and incubated at 37°C in a humidified 5% CO2 atmosphere. In assays with different pH, cells were seeded in medium at pH 7.4, and after 24 h media were changed. New media were set at a specific pH by using different concentrations of sodium bicarbonate needed to preset pH in 5% CO2 atmosphere, according to the Henderson-Hasselbach equation. During different intermediate time points and at the end-point of the different assays, the maintenance of medium pH in the supernatant was always as certain by a digital pH-meter (6230N, Jenco, San Diego, CA, USA) (Figure S1).
In vivo model
Procedures involving animals and their care were performed after institutional approval and in accordance with national and international laws and policies (EEC Council Directive 86/609, OJ L 358, 1, Dec. 12, 1987; Italian Legislative Decree 116/92, Gazzetta Ufficiale della Repubblica Italiana n. 40, Feb. 18, 1992). Severe combined NOD/SCID mice were housed and maintained in a pathogen-free environment. A673 human cells (1 × 106) were subcutaneously injected with the reduced growth factor matrigel in the flank of 5 weeks old male mice (Charles River Laboratories International). Mice were randomly separated into groups and assigned to pharmacological treatments. Omeprazole (Antra) was reconstituted in saline solution and injected intraperitoneally (0, 2.5, and 10 mg/kg). Treatment was administered 3 times per week (every other day) for 4 weeks, starting the day after tumor cells inoculation. Weights were taken during treatment, and drug concentrations were recalculated to ensure that mice received a constant dose of Antra. Tumor size was estimated at every pharmacological treatment with a caliper and calculated by using the formula: tumor volume [mm3] = (length [mm] × width2 [mm2])/2 [22]. Total RNA extract were obtained using the acid guanidinium thiocyanate-phenol-chloroform method. For histological analysis, tumor xenografts were fixed in 10% buffered formalin and embedded in paraffin. Five μm sections were stained with hematoxylin/eosin (H&E). The area of necrosis was quantified by NIS element image analysis software in 10 different random fields for each sample (Nikon).
Apoptosis assay
HOS, MG63 and A673 cells (5 × 103 cells) were seeded on glass coverslips in complete IMDM. After cell adhesion, medium was changed with complete IMDM at pH 6.5 or 7.4. After 24 h apoptosis was induced by incubation of cells on ice for 2 h, followed by rewarming at 37°C in an incubator with 5% CO2 (stress thermal shock). After 48 hr for HOS and MG63, and after 72 h for A673, cells were fixed with 3.7% paraformaldehyde, permeabilized with PBS containing 0.1% TritonX-100, and stained with 2.25 µg/mL of Hoechst 33258. Cells with apoptotic nuclear bodies were counted in twelve different fields by using 20X objective. Results were expressed as percentage of apoptotic cells over the field.
Western blot
For direct immunoblotting of total protein, cells were serum starved for 24 h and then cultured for 4, 8, 12 and 24 h with medium at pH 6.5 or 7.4. Protein lysates were obtained in RIPA buffer supplemented with protease inhibitors. Cell lysates containing 100 µg of total proteins were resolved by electrophoresis on a polyacrylamide gel, transferred to nitrocellulose membranes, and subjected to immunoblot analysis. Blots were probed with Phospho-44/42 MAP kinase or p44/42 MAP kinase or Phospho-Akt, or Akt polyclonal antibodies. Incubation with a horseradish peroxidase-conjugated anti-rabbit antibodies followed. To detect different antigens within the same blot, nitrocellulose membranes were stripped with Restore Western Blot Stripping buffer and reprobed. The reaction was revealed by a chemiluminescence substrate. Western blot were repeated thrice.
Illumina genome analyzer sequencing and data analysis
HOS, Saos2 and MG63 cells were cultured at pH 6.5 or 7.4 for 24 h. mRNA expression was evaluated by extracting total RNA using Trizol from cells cultured at pH 6.5 and pH 7.4 for 24 h. We quantified the RNA using a Bioanalyzer (Agilent). The RNA integrity numbers (RIN) and A260/A280 ratios were all equal to 10, and greater than 1.8, respectively. We converted the total RNA to a library of template molecules for high-throughput DNA sequencing using the TruSeq RNA Sample Prep Kit v2. We quantified the library using a Bioanalyzer. Library (7 pM) was subjected to cluster amplification to cluster generation on a Single Read Flow Cell v4 with a cluster generation instrument (Illumina). Sequencing was performed on a Genome Analyzer GAIIx for 76 cycles using Cycle Sequencing v4 regents. Image analysis and base calling were performed using Off-Line Basecaller Software 1.6 (Illumina). Reads were aligned using ELAND v2 of CASAVA Software 1.7 with the sequence data sets. Human genome build 19 (hg19) were downloaded from University of California, Santa Cruz genome browser (http://genome.ucsc.edu/) as the analytic reference. Transcript coverage for every gene locus was calculated from the total number passing filter reads that mapped, by ELAND-RNA, to exons. These analyses were performed using default parameters. The advanced analysis for quantification with Quantile normalization algorithm was performed using Avadis NGS software (version1.5, Strand Scientific Intelligence Inc., San Francisco, CA). The filtering was performed using default parameters. We registered the obtained data at DDBJ/EMBL/GenBank (DRA004087 and DRA004091).
siRNA transfection
Specific gene silencing was obtained by siRNA technology associated with pipette-type electroporation. A-673 cells were trypsinized at semi-confluence, and counted after erythrosine dye staining. 150 μL of cell suspension containing 1,500,000 cells and 2 nmol of specific siRNA TRAF1, or Silencer select negative control siRNA were transferred into a 1-mm cuvette (Neon® Transfection System, Invitrogen, Life Technologies). Electroporated cells were transferred into 30 mL of complete medium, and seeded in 12-well plates (100,000 cells/well) for RNA isolation or in 96-well plates for vitality assay (7,500 cells/well). After one day (T0), medium was changed with complete medium at pH 6.5 and apoptosis was induced by stress thermal shock (see above). At T0, cells were also used to isolate RNA, as previously described. After additional 24 h (T1) cell number was measured by an acid phosphatase assay, as previously described [7].
Subject and sample collection
Six patients with newly diagnosed high-grade OS were seen at our institution from February 2009 through July 2010, signed informed consent regarding the use of their biological materials for research studies, and entered the study. For clinical features of the patients included in the study see Table 1. Tumor tissues were collected after the institutional ethical committee approval (Ethics Committee of Istituto Ortopedico Rizzoli, No. of approval 0033626).
Table 1.
Clinical and pathological features of osteosarcoma patients
| Age | Sex | Site | Histological subtype | Stage | |
|---|---|---|---|---|---|
| Case 1 | 42 | F | Ankle | Chondroblastic | Local recurrence, no lung metastases |
| Case 2 | 23 | F | Sacrum | - | Primary, no lung metastases |
| Case 3 | 62 | F | Distal femur | Fibroblastic | Recurrence with lung metastases |
| Case 4 | 67 | F | Proximal femur | - | Local recurrence, no lung metastases |
| Case 5 | 19 | F | Proximal tibia | - | Primary, no lung metastases |
| Case 6 | 51 | M | Proximal humerus | Osteoblastic | Local recurrence, no lung metastases |
Real-time PCR
Total RNA of cells cultured at different pH was isolated after 24 h with the NucleoSpin RNA II. Total RNA extract from human or xenograft tumor tissues were obtained using Trizol. Total mRNA, obtained both from cell cultures and from human or xenograft tumor tissues, was reverse transcribed with the Advantage RT-for-PCR Kit. The expression of baculoviral IAP repeat containing 2 (BIRC2) (NM_001166.4), baculoviral IAP repeat containing 3 (BIRC3) (NM_001165.4), TNF receptor-associated factor 1 (TRAF1) (NM_001190945.1), TNF receptor-associated factor 2 (TRAF2) (NM_021138.3), nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 (NFKB1) (NM_003998.3) and v-rel reticuloendotheliosis viral oncogene homolog B (RELB) (NM_006509.3) was evaluated by using the Light Cycler instrument (Roche Diagnostics). One mg of cDNA was amplified and the Universal ProbeLibrary system used. Probes and primers were selected by using web-based assay design software ProbeFinder (https://www.roche-applied-science.com): BIRC2 fwd 5’-GACGTCATCGTGCGTCAG-3’, rev 5’-CGCCGACAAGGAGATACG-3’; BIRC3 fwd 5’-CAGCAAGGCCTGTCTGTTC-3’, 5’-AATATCCCCATCCCATACAGG-3’; TRAF1 fwd 5’-TTTTGGACCTGTCTCCAACAC-3’, rev 5’-CGATAAAAATCCCCTGGATG-3’; TRAF2 fwd 5’-GGAACACACCTGTCCCTCTT-3’, rev 5’-GGTCGAGCAGCATTAAGGTC-3’; NFKB1 fwd 5’-CCTGGAACCACGCCTCTA-3’, rev 5’-GGCTCATATGGTTTCCCATTTA-3’; RELB fwd 5’-GATTGTCGAGCCCGTGAC-3’, rev 5’-CCACGCCGTAGCTGTCAT-3’; ATPase V1B2 fwd 5’-TGGCCGAAGACTTCCTTG-3’, rev 5’-CCGAAATGCCAGTCTGAATC-3’, CA9 fwd 5’-TGCCTATGAGCAGTTGCTGT-3’, rev 5’-CCAGTCCTGGGACCTGAGT-3’; LAMP2 fwd 5’-AATGGCACAGTGAGCACAAA-3’, rev 5’-GAGATGGCACAGTGGTGTGT-3’. Results were expressed as ratio between the gene of interest and the reference gene rRNA18s (X03205.1) (rRNA18s fwd 5’-GCAATTATTCCCCATGAACG-3’, rev 5’-GGGACTTAATCAACGCAAGC-3’) according to the 2-ΔΔCT method.
Immunostaining analyses
Tumor sections from in vivo models were deparaffinized and rehydrated for immunostaining. Five μm sections were incubated in 3% hydrogen peroxide solution to block the endogenous peroxidase reaction. Nonspecific binding was blocked by incubation in 5% bovine serum albumin. The following primary antibodies were used: rabbit anti-ATP6V1B2 antibody, rabbit anti-LAMP2 antibody, and rabbit anti-RelB antibody. Sections were washed, incubated with a biotinylated secondary antibody, covered with DAB and counterstained with Mayer’s hematoxylin. Negative controls were also performed by omitting the primary antibody.
ELISA assay
BIRC2/3 and TRAF1 were measured in the cells lysates with Human baculoviral IAP repeat-containing protein 2/3, BIRC2/3 ELISA Kit and Human tumor necrosis factor-associated factor 1, TRAF1 ELISA KIT according to the manufacturer’s instructions. The absorbance was read by using Infinite1 200 PRO plate reader (Tecan). The amount of detected BIRC2/3 and TRAF1 was normalized based on the total protein content of the culture media, as evaluated by BCA method. To assess nuclear translocation of NF-κB component, nuclear protein extracts were obtained and quantified with a Nuclear Extraction Kit. Equal amounts of protein lysates were then used for NF-κB Transcription Factor Assay Kit quantification (TransAM) according to manufacturer’s instructions. The signal was quantified by using Infinite1 200 PRO plate reader (Tecan). The experiment was performed with four replicates.
Statistical analysis
Statistical analysis was performed with the StatViewTM 5.0.1 software (SAS Institute, Inc., Cary, Nord Carolina, USA). Due to the low number of observations, data were considered as not normally distributed, and nonparametric tests with exact p value were used. Mann-Whitney U test was used as unpaired comparison of two independent variables. Wilcoxon signed-rank test was used for paired analysis, and Spearman correlation test (one-tailed) for the analysis of correlation. Data were expressed as mean ± standard error (SE). Only p values < 0.05 were considered for statistical significance.
Results
Extracellular acidosis promotes cell survival in bone malignant tumors
As already demonstrated in a xenograft model of human osteosarcoma [7], the treatment with the V-ATPase inhibitor omeprazole significantly increased the tumor necrotic area in a xenograft model of Ewing sarcoma, and already at a lowest dosage (Figure 1A and 1C, 2.5 mg/kg P = 0.0189; 10 mg/kg P = 0.003), whereas tumor volume was unaffected (Figure 1B). In different model of primary malignant bone tumors, we found that extracellular acidosis significantly reduced the percentage of apoptotic cells treated with a stressing thermal shock (Figure 2A). However, the acute treatment with extracellular acidosis did not promote the phosphorylation of signal-regulated kinase (ERK) of MAP kinase (MAPK) family that is usually activated to induce the apoptotic process [23]. In more details, in HOS cells, after a few hours from the exposure to extracellular acid pH, we did observe a decrease of ERK phosphorylation that, however, returned to initial level already after 12 hours (Figure 2B), implying that the pro-apoptotic pathways mediated by MAPK were quickly counteracted by the activation of pro-survival signaling that prevailed already after 12-24 h (Figure 2B). The lack of induction or inhibition of ERK phosphorylation, after 24 h from the acid stress was confirmed in 3 different OS cell lines (Figure 2C).
Figure 1.
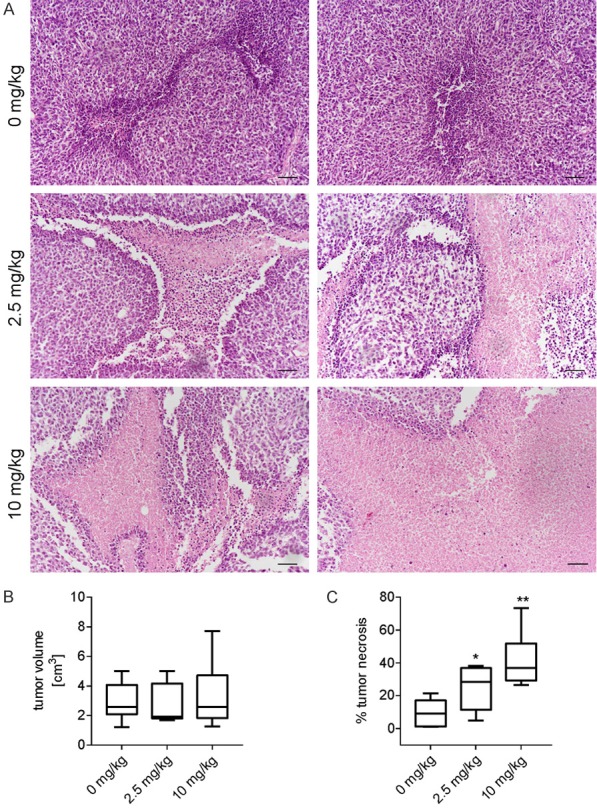
Pharmacological inhibition of the proton pump V-ATPase causes tumor necrosis in a Ewing’s sarcoma xenograft model. A. A673 cells were subcutaneously injected in NOD/SCID mice. Mice were then treated with different concentration of omeprazole, a V-ATPase inhibitor (0 mg/kg n = 14, 2.5 mg/kg n = 7, 10 mg/kg n = 14), to reduce intratumoral acidosis. H&E staining of tumor sections, representative images of the necrotic area, scale bar, 100 µm; B. Box plot of the tumor volume. Mean ± SE; C. Box plot of the tumor necrosis. Mean ± SE (*P < 0.05, **P < 0.01).
Figure 2.
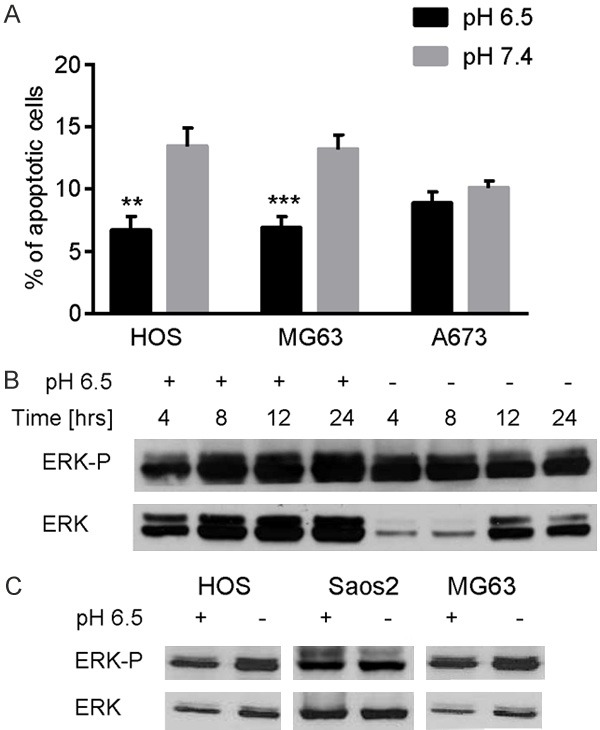
The anti-apoptotic effect of extracellular acidosis. A. Osteosarcoma and Ewing sarcoma cells were cultured under different pH (6.5 and 7.4) for 24 h and the induction of apoptosis was evaluated by the counting of apoptotic bodies after thermal shock. Mean ± SE (n = 12, **P < 0.01, ***P < 0.005); B. Western blot analysis of ERK phosphorylation after 24 h of acute acidosis (pH 6.5), representative images (where not specified, cells were cultured at standard condition, at pH 7.4); C. Western blot analysis of ERK phosphorylation over the time, up to 24 h after the culturing at low pH, in HOS cells, representative image (where not specified, cells were cultured at standard condition, at pH 7.4).
Extracellular acidosis activates the anti-apoptotic NF-κB pathway
To go deeper inside the molecular pathways involved in acid-induced sarcoma cell survival that might counteract MAPK pro-apoptotic signaling, we investigated whether osteosarcoma cells cultured at low pH showed an altered transcriptome related to the anti- or pro-apoptotic pathways. We found a trend of increase in the expression of anti-apoptotic genes, with particular regard to Baculoviral inhibitors of apoptosis repeat containing (BIRC) family, and a trend of decrease of the pro-apoptotic family of genes (Figure 3A). BIRC proteins are deeply involved in the activation of the NF-κB signaling and, according to this finding, previous reports showed that acid extracellular stimulus exerts NF-κB activation [24-27]. By mRNA and protein analysis, we confirmed the same trend in bone sarcoma cells: with the exception of a slight increase in the Ewing sarcoma cell line A673 (Figure 3B), extracellular acidosis did not induce any change in NF-κB1 gene expression, but it produced, in both osteosarcoma and Ewing sarcoma cells, a clear trend of increase of NF-κB proteins that are translocated into the nucleus, as for p50 and RelB (Figure 3C).
Figure 3.

Extracellular acidosis promotes cell survival in bone sarcoma via the NF-κB pathway. (A) Heat map representation of the fold increase of the expression of apoptosis and stress related-genes of osteosarcoma cells (MG63, HOS, Saos-2) after short-term acidosis. mRNA were analyzed by deep-sequencing after that cells were cultured at pH 6.5 compared to physiological medium (pH 7.4) for 24 h. Colors on the heat map indicate the log2 ratios of expression (representing normalized read counts). Red, upregulation; green, downregulation. (B) Cells were cultured under different pH (6.5 and 7.4) for 24 h as described in (A), and NF-κB mRNA expression and activation were quantified in cell lysates. (C) NF-κB1 (p50) and RelB nuclear protein concentration of osteosarcoma and Ewing sarcoma cells. Mean ± SE (n = 3 for HOS and MG63, and n = 5 for A673, *P < 0.05). (D) Signaling pathways of the tumor necrosis factor receptor (TNFR) family that mediates both survival and apoptotic pathways. The draw represents the pathways and adaptor molecules that are involved in the signaling that follows the acid stress: the NF-κB activation pathway via Akt (1), via the canonical pathway (2), via the non-canonical pathway (3), the activation of the AP1 complex transcriptional factor (4), and the TRAF-mediated death signaling with the activation of caspase cascade (5).
Extracellular acidosis induces the formation of TRAF and cIAP complexes
Although a great deal of evidence has been amassed on the ligands and receptors that mostly activate NF-κB pathway, to date it is unclear which cytoplasmic adapter molecules transduced the signals from receptors to nucleus when acid stress occurs. BIRC genes encode cellular inhibitors of apoptosis (IAPs) that bind the BIR domain (BIRC proteins) of TNF receptor family, with cIAP1 encoded by BIRC2 and cIAP2 encoded by BIRC3. These are essential regulators of apoptosis, preventing caspase activation or interfering with pro-apoptotic signaling intermediates [28], including MAP kinase pro-apoptotic signaling [29]. A common feature of the pro-survival NF-κB signaling is to recruit during ligation different TRAF members at the receptor complex, at TRAF-binding motif, together with stable cIAP proteins, thereby avoiding cell death cascade [30] (Figure 3D). By RT-PCR analysis, we confirmed the trend observed by deep-sequencing data showing a significant increase of BIRC2 expression in MG63 osteosarcoma cells, and of TRAF1 and TRAF2 in Ewing’s sarcoma A673 cells, and a trend of increased expression for BIRC3 for the other cell lines included in this study, with the exception of TRAF2 for HOS and MG63 cells (Figure 4A). Results on TRAF and BIRC were also validated by protein analysis (Figure 4B), suggesting the formation of a TRAF/cIAP protein complex at the BIR and TRAF domain to mediate anti-apoptotic signals via NF-κB activation. According to the literature, this might be achieved by multiple mechanisms, including both canonical and non-canonical NF-κB [31]. Also signaling via AKT is a possible mechanism for the activation of pro-survival NF-κB pathway (Figure 3D), especially after ROS production [20], and notably, we also found that acute acidosis induced AKT phosphorylation in both the osteosarcoma cell lines HOS and MG63 (Figure 4C). To strengthen the role of acidosis in promoting cell survival via TRAF and BIRC proteins, we used different strategies. For the targeting of cIAP proteins and TRAF1, we used the specific inhibitor LCL161, a small molecule that binds with high affinity to the BIR3 domains of cIAP1, disengages IAPs from caspases and induces proteasomal degradation of cIAP-1 and -2, and that has been included in different clinical trials [32]. For the inhibition of TRAF1, we used specific siRNA. Treatment of A673 cells with both LCL161 cIAP inhibitor and TRAF1 siRNA caused a significant inhibition of cell viability (Figure 5A-D). For LCL161 we also demonstrated that the inhibition of cell viability was significantly higher at low pH in respect to pH 7.4 (Figure 5A), and that the same drug concentration promoted an increase of apoptotic bodies in cells cultured at acid condition (Figure 5B), For silencing experiments, although also the unspecific not targeted siRNA was also toxic for Ewing Sarcoma cells, the specific TRAF1 silencing was more effective in inhibiting the cell viability (Figure 5D).
Figure 4.
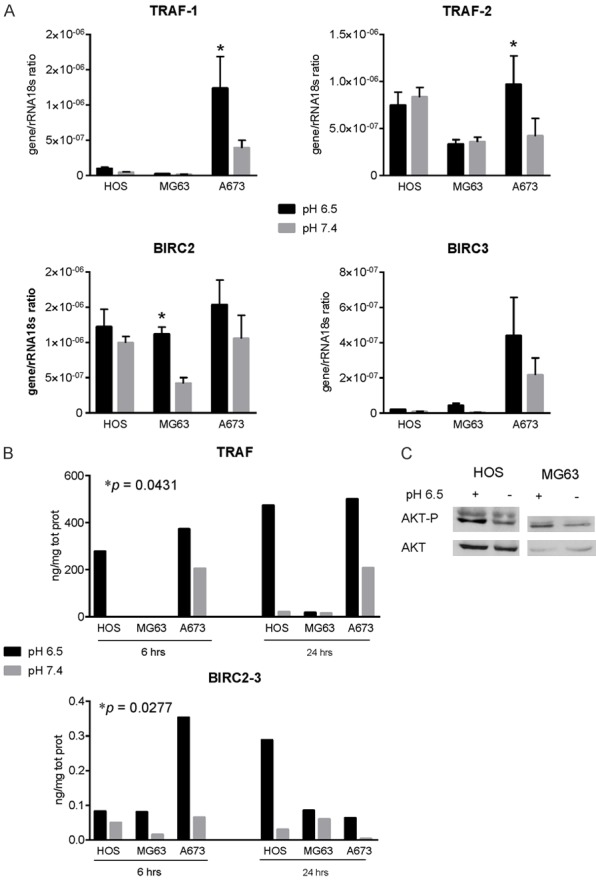
Extracellular acidosis promotes the induction of BIRC and TRAF expression. A. RT-qPCR analysis of the TRAF1 and 2 and of the BIRC2 and 3 expression in cells treated at the same condition of Figure 3A. Mean ± SE (n = 5, *P < 0.05); B. Protein quantification of TRAF and BIRC family of proteins in cells treated at the same condition of Figure 3A (statistical paired analysis, *P < 0.05), for 6 and 24 h. Mean ± SE (n = 3, P was obtained by Wilcoxon Rank paired analysis considering each osteosarcoma cell line as a single value); C. Western blot of AKT and the phosphorylated form in cells treated with acid medium (pH 6.5) for 24 h.
Figure 5.
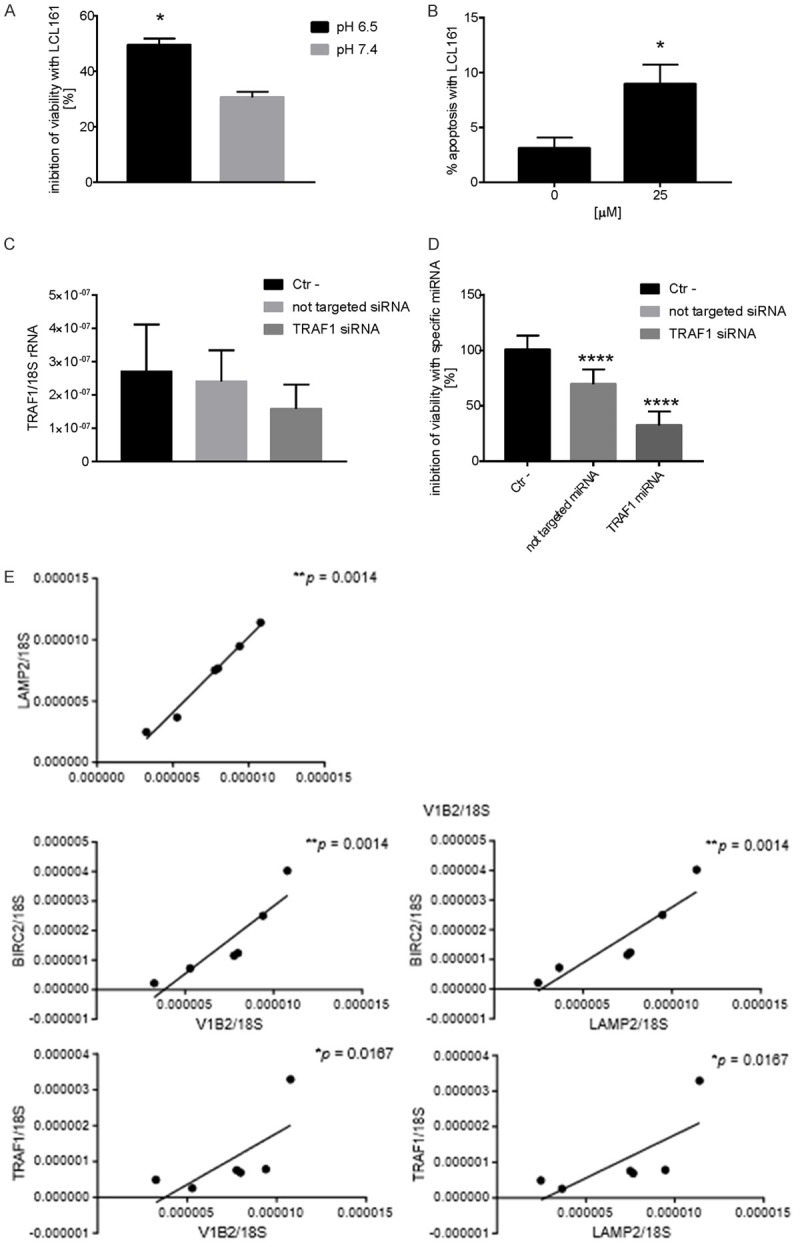
Targeting of cIAP protein or of TRAF1 inhibit cell viability of A673 cells when cultured in acid condition. A. Inhibition of cell viability as verified by cell counting after the treatment with LCL161 (25 mM). The inhibitor was more effective at pH 6.5 than at pH 7.4. Mean ± SE (n = 3, *P < 0.05); B. Induction of apoptosis as verified by the counting of apoptotic bodies after the treatment with LCL161 (25 mM) of cells cultured at low pH (6.5). Mean ± SE (n = 3, *P < 0.05); C. mRNA analysis of TRAF1 expression by Q-RT-PCR after the treatment with specific siRNA at low pH (pH 6.5); D. Inhibition of cell viability as verified by acid phosphatase indirect assay after the treatment with specific siRNA at low pH (pH 6.5). Mean ± SE (n = 19, ****P < 0.001); E. Correlation of mRNA level between different proteins of the TRAF-cIAP destruction complex in Ewing sarcoma xenografts with acid-related markers V-ATPase V1B2 or LAMP2 (n = 6, *P < 0.05, **P < 0.01).
In conclusion, our data demonstrated that acute acidosis activates pro-survival signaling via NF-κB pathway in bone sarcoma cells through different mechanisms.
The expression of BIRC/IAP directly correlates with the proton pump V-ATPase in bone sarcoma tissues
To strengthen our hypothesis of a correlation between acidosis and the expression of BIRC and TRAF proteins, and the downstream activation of NF-κB pathway, we further analyzed in xenografts of Ewing Sarcoma (control group, the same animal models used in Figure 1A) the expression of BIRC2 and TRAF1 and its correlation with the expression of the indirect marker of acidosis vacuolar proton pump V-ATPase V1B2 and LAMP-2, by Q-RT-PCR (Figure 5E). As clearly demonstrated by previous authors, chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2, especially at the plasma membrane [33]. To validate V1B2 and LAMP2 level of expression as both indirect markers of extracellular acidosis, we found a significant correlation between the expression of the two genes (Figure 5E). Notably, we found a significant correlation also between the expression of BIRC2 and VI1B2 or LAMP2, and between TRAF1 and V1B2 or LAMP2 (Figure 5E). We further confirmed by immunohistochemistry of consecutive tissue sections of the same in vivo models that RelB activation (nuclear translocation) localized at the same region of the tumor where extracellular acidosis localized, as revealed by the staining of both V1B2 in the intracellular vesicles of tumors cells (cytoplasmic punctate patterns), and by the staining of LAMP2 (both in the intracellular vesicles and at the cell membrane of tumor cells) (Figure 6).
Figure 6.
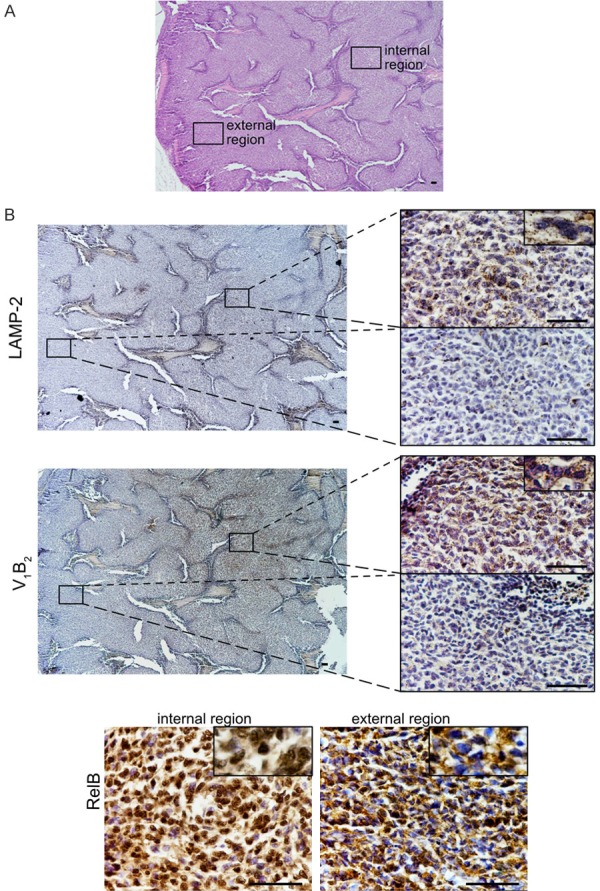
Immunoistaining of Ewing sarcoma xenografts showing the same localization for the indirect markers of acidosis V1B2, LAMP2, and the nuclear localization of RelB, a subunit of the NF-κb transcription complex that can be activated by the TRAF-cIAP destruction complex. (A) H&E staining of a section a representative xenograft of Ewing’s sarcoma model of the control group shown in Figure 1 (scale bar, 200 µm, 4 × objective). We focused our investigation on an external and an internal region of the tumor. (B) Consecutive tissue sections of the section showed in (A), and stained for different antigens. For LAMP2 and V1B2 in the left panel, lower magnification (scale bar 200 µm, 4 × objective), and in the right panels, higher magnification of the specified area unlighted at low magnification by the rectangle (scale bar 20 µm, objective 60 ×). Note in the rectangle showing an enlarged detail, for V-ATPase, the staining is punctuated suggesting a intracellular vesicular localization, for LAMP2 is both punctuated and at the cytoplasmic membrane. For RelB, only high magnification of the two different regions, external and internal are shown (scale bar 20 µm, objective 60 ×). Note in the rectangle showing an enlarged detail that cytoplasm was always positive, whereas nuclei in the internal region were almost stained suggesting the nuclear translocation of the protein, whereas in the external region of the tumor only a small fraction of nuclei were stained.
Finally, to confirm in clinical setting the data obtained in preclinical models, we analyzed a small number of samples of tumor tissues obtained from patients with osteosarcoma and we looked for the correlation between the mRNA expression of different proteins of the NF-κB pathway. As a confirmation of the formation of the TRAF-cIAP complex, although we did not find a correlation between BIRC3 expression and TRAF (data not shown), we found a significant correlation between TRAF1 and TRAF2 expression, and a trend of correlation between TRAF1 and BIRC2 (Figure 7A). Notably, as we already found in Ewing’s sarcoma xenografts, TRAF1 expression also significantly correlated with the expression of the V1B2 subunit of the proton pump V-ATPase, meaning that TRAF1 is the major TRAF protein recruited at the TRAF motif after the induction of the acid stressing stimulus (Figure 7B). The level of expression of TRAF1 also significantly correlated with those of both NF-κB1 (p50) and RelB, whereas TRAF2 level correlated only with RelB (Figure 7C). In turn, as the final effector of the acid-stress-mediated pro-survival signaling, p50 significantly correlated with the V-ATPase V1B2 (Figure 7D). Interestingly, we also found a trend of correlation between carbonic anhydrase 9 (CA9) and RelB (Figure 7D). CA9 is enzymes that catalyze the interconversion between carbon dioxide and water and the dissociated ions of carbonic acid with the net results of increasing proton concentration, and is responsible for intratumoral acidosis in several types of cancer, including sarcomas [34-36]. However, CA9 expression did not correlate with none of the other players of the non-canonical NF-κB pathway in human osteosarcoma tissue samples, implying that CA9 might activate NF-κB signaling via different pathways.
Figure 7.
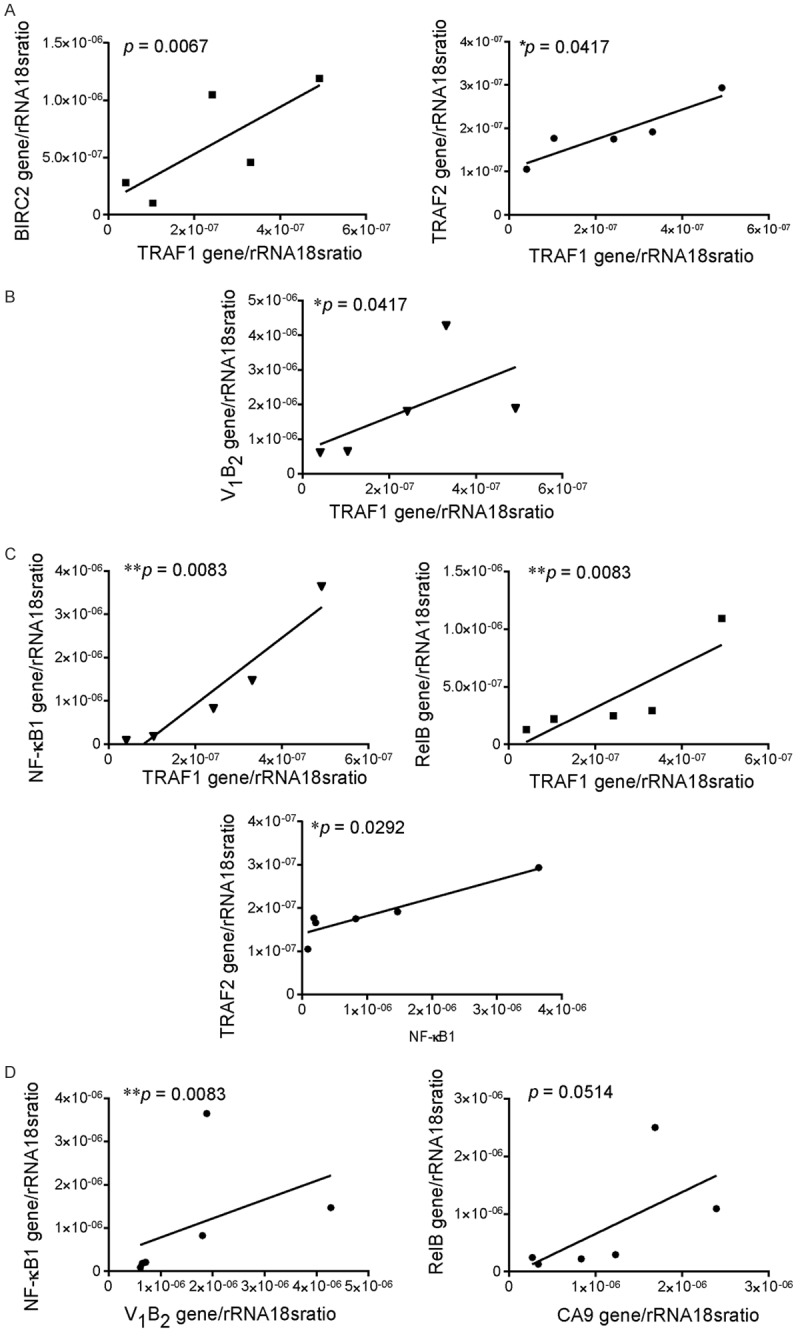
The expression of BIRC/IAP mRNA directly correlate with the expression of the acidifying proton pump V-ATPase in tumor tissues from osteosarcoma patients. A. Correlation of mRNA level between different proteins of the TRAF-cIAP destruction complex in human osteosarcoma tissue samples; B. Correlation of the V-ATPase V1b2 with TRAF1 expression; C. Correlation of the mRNA expression of the different proteins of the TRAF-cIAP destruction complex with the mRNA expression of the proteins of the NF-κB complex; D. Correlation of proton pump and transporters with the mRNA expression of the proteins of the NF-κB complex. For all the analysis, Mean ± SE (n = 5, *P < 0.05, Spearman correlation test).
Discussion
Due to the high expression of the most important proton pump V-ATPase [37,38], tumor cells from osteosarcoma and other types of sarcoma strongly acidify the extracellular space [5,7,35]. Proton pump inhibitors like omeprazole can block the V-ATPase activity [39] in different sarcoma cell types [35], and the combined treatment of doxorubicin and omeprazole in osteosarcoma xenografts significantly reduced tumor volume and increased the tumor necrosis [7]. Also in a pilot study with osteosarcoma patients, the use of omeprazole in combination with doxorubicin and other conventional chemotherapeutics increased the necrotic index [6]. We hypothesized that, following omeprazole treatment and following the blocking of proton pumping, alkalinisation of the extracellular space paralleled the cytosolic acidification, ultimately leading to a reversal of the pH gradient at the plasma membrane of tumor cells. This is a permissive condition for the intracellular uptake and enhanced cytotoxicity of doxorubicin [7]. Indeed, under physiological conditions, pH homeostasis inside/outside the cell is crucially balanced and has to be strictly maintained to keep a selective permeability to different agents at the plasmalemma. Furthermore, in the early nineties, several authors have reported that pH homeostasis most importantly accounts for the life/death balance of the single cell as intracellular acidification is concomitant of cell death [40-43]. Falling in line with this concept, here we speculated that, in a symmetrical way, acidification of the extracellular environment, followed by intracellular alkalinisation in cancer cells [44], is a pro-survival signal, and that the increased cytotoxicity of doxorubicin in osteosarcoma, when combined with omeprazole, was due both to a high drug uptake and to the blockage of pH-mediated pro-survival signaling that follows omeprazole-induced intracellular acidification. As a matter of fact, in a xenograft model, omeprazole produced a trend of increase of the necrotic tumor area also as a single treatment [7]. Likewise, in this study, in a Ewing sarcoma xenograft, omeprazole significantly enhanced tumor necrosis in a dose-dependent manner, and acute exposure to a low pHe promoted in vitro a reduction of the number of apoptotic cells after a thermal shock in both Ewing and osteosarcoma cells. We thus went deeper inside into the apoptosis-related molecular machinery that is regulated by pH homeostasis.
Multiple stress-inducible molecules, including c-Jun N-terminal kinase (JNK), mitogen-activated protein kinase (MAPK)/extracellular signal-regulated protein kinase (ERK), NF-κB or ceramide have been involved in transmitting the apoptotic signal [45]. Among these, the MAPK are key regulators and inducers of cell apoptosis [46]. In our model, acid stress produced an inhibition of ERK1 (p44 MAPK) phosphorylation. However, this inhibition was fully replaced at 12 hours, suggesting that pro-survival pathways might prevail. As a confirmation, by wide-screening analysis, after acute incubation with acidosis, the molecular signature confirmed a clear trend of induction of pro-survival mechanisms rather than the activation of pro-apoptotic signaling in bone sarcoma cells.
In diverse cell types, Rel/NF-κB transcription factors have a role in regulating the apoptotic program, either as essential for the induction of apoptosis or, perhaps more commonly, as blockers of apoptosis [47]. In particular, NF-κB pathway plays a central role in the cellular response to stress and is also known as a redox-sensitive transcription factor activated by reactive oxygen species (ROS). Notably, acidosis induces the generation of ROS which can be suppressed by ROS scavengers [20], and our previous works have demonstrated in MSC, sharing with sarcoma cells a mesenchymal origin, a strong induction of the expression of members of the NF-κB pathway, like RelB and p50 or p52, after the acid stress stimulus [24,25]. Likewise, in this study we observed NF-κB activation in acid-induced bone sarcoma cells, with an increase in NF-κB1 (p50) and RelB protein expression.
The NF-κB transcription factor family consists of 5 subunits-p65 (RelA), p105/p50, c-Rel, RelB, and p100/p52-acting as dimers in two distinct pathways-the classical (or canonical) pathway and alternative (or non-canonical) pathway [30]. Regarding the non canonical pathway, NF-κB-inducing kinase (NIK) activates the downstream kinase IκB kinase-α (IKKα) that triggers p100 phosphorylation and processing to produce p52 and a RelB-specific inhibitor [30] (point 3 in Figure 3D). The activity of NIK is strongly determined by its degradation that, in turn, is modulated by the TRAF-IAP complex [31]: the continuous degradation of NIK induced by TRAF3 prevents NIK accumulation and NF-κB pathway activation; adversely, when ligation of specific TNFR superfamily members by their ligands occurs, TRAF2, TRAF3, cIAP1 and cIAP2 are recruited to the receptor complex and TRAF3 ubiquitilated and degraded, thereby resulting in stabilization and accumulation of NIK and the induction of p100 processing, followed by the translocation of NF-κB p52-RelB heterodimers into the nucleus [48]. Among the most expressed genes stimulated by acute acidosis, we found an up-regulation of the TRAF1 and TRAF2 and of cIAP1 (BIRC2) and cIAP2 (BIRC3). These observations suggest that, under acidosis, TRAF/cIAP protein complexes form at the receptor complex and, simultaneously NIK leads to the formation of active RelB and p52 NF-κB pro-survival transcriptional complex. Furthermore, previous data showing that TRAF-mediated NF-kappaB activation suppresses the TNF-induced ROS accumulation that subsequently induces prolonged MAPK activation and necrotic cell death [49] apparently justify the temporary ERK1 inhibition that we observed in sarcoma cells cultured in acid medium.
In summary, by this study we demonstrated that in osteosarcoma and Ewing sarcoma cell lines the acid-promoted expression of TRAF proteins parallels with the increased expression of cIAP proteins, suggesting that TRAF activity is mainly devoted to the activation of the pro-survival signaling mediated by NF-κB pathway. We also confirmed our hypothesis by using cIAP specific inhibitor and TRAF1 silencing approaches. With the exception of TRAF6 expression in osteosarcoma that correlated with a high Enneking score [50], and of BIRC5, that has been considered as a poor prognostic marker in Ewing sarcoma [51], the role of TRAF proteins and cIAP have never been evaluated in sarcoma. As reflected by their name, cIAP were initially characterized as endogenous inhibitors of caspases. In particular, XIAP block caspase activation by binding to caspases -3, -7, and -9 via the BIR domains and negatively regulate both the intrinsic and extrinsic apoptosis pathways [52]. In addition to the non-canonical pathway via ubiquitination events of TRAF3 protein (point 3 in Figure 3D), cIAP cause the nondegradative ubiquitination of RIP1 and the following activation of inhibitor of NF-κB kinase (IKK, IkB kinase b) in the canonical NF-κB pathway (point 2 in Figure 3D). This leads to the phosphorylation and proteasomal degradation of IkBa followed by nuclear translocation of NF-κB subunits (p50). In addition to regulate cell survival and inflammation, both cIAP and TRAF proteins, with a lesser extent for cIAP, can also mediate the activation of MAPKK, resulting in the induction of the transcription of cytokines and regulators of cell differentiation [52,53] (point 4 in Figure 3D), whereas TRAF alone can activate the FADD/caspase death signaling (point 5 in Figure 3D). Finally, according to our data, in addition to the signaling pathway mediated by the formation of the complex between the adaptor protein TRAF and the IAP proteins, acidosis might also activate a pro-survival signaling after TNFR activation via the NF-κB inflammatory pathway through the phosphorylation of Akt (point 1, Figure 3D) [54,55]. This is not the first time that the Akt signaling is observed after acid stress, as it was already detected in breast cancer [20]. The involvement of the NF-κB complex downstream the acid-induced cIAP/TRAF activities was also demonstrated by both mRNA analysis and immunohistochemistry of Ewing’s sarcoma xenograft since the level of expression of the indirect marker of acidosis, the most important acidifying protein in eukariotic cells V-ATPase, and LAMP2, lysosome-associated membrane protein, significantly correlated with both BIRC2 and TRAF1. Moreover, nuclear staining of RelB in the tumor cells was found in the same internal region of the tumor that was also positive for V-ATPase in the lysosomal compartment, and for LAMP2, both at the plasma membrane level and in the lysosomes. Indeed, as reported by Damaghi et al., LAMP2 is located at the plasma membrane in clinical samples of breast cancer and this redistribution is acid-induced in in vitro model of the same type of cancer [33]. The internal regions of the tumor are most likely also the more hypoxic region and, thus, the most acid.
Finally, similarly to what we observed in animal models, by the analysis of tumor samples from osteosarcoma patients, as a clear demonstration of our hypothesis, we showed a strong correlation between the TRAF and cIAP mRNA contents, and downstream between the concentration of NF-κB1, with the acidification activity, which was indirectly quantified by the analysis of the level of the expression V-ATPase. Indeed, even by analyzing a small number of samples, we found not only a significant correlation between BIRC2 and TRAF1 or between TRAF1 and NF-κB, as expected, but also a significant correlation between V1B2 and TRAF1 or between V1B2 and NF-κB1 (p50). During evolution, the eukaryotic V-ATPases have acquired a unique regulatory mechanism of regulation of proton-pumping activity. Moreover, the eukaryotic V-ATPases have also acquired alternative roles, including as a receptor to sense and transmit transmembrane signaling as well as to directly and indirectly modulate the trafficking and signaling of other cellular receptors [56]. In particular, we have previously demonstrated that the level of expression of the V1B2 isoform of the B subunit of the V1 domain in several Ewing sarcoma cells directly correlates with the extent of the acidification activity and of tumor aggressiveness [5].
High levels of IAPs are detected in many tumors, and this is often associated with chemoresistance, metastasis, and poor prognosis [57]. IAPs are thus attractive pharmacologic targets for ameliorating therapeutic resistance in cancers. Several small molecule IAP inhibitors and antagonists, also known as second mitochondria-derived activator of caspases (SMAC) mimetic after the endogenous inhibitor, have been developed, some of which have been tested in phase I and II clinical trials [58]. According to our data, the use of these inhibitors might thus be considered for the identification of novel and more effective therapies in bone sarcomas.
In conclusion, by using both in vitro and in vivo preclinical models, and by the analysis of tumor tissues derived from bone sarcoma patients, we indirectly demonstrated that intratumoral acidosis is a crucial feature of bone sarcoma since it is an important trigger of pro-survival signaling via cIAP/TRAF/NF-κB pathway. These key adaptor and effector proteins might be thus considered as important markers of resistance to the therapy and considered as effective therapeutic targets.
Acknowledgements
The work was supported by the Italian Association for Cancer Research (n. 15608 to N.B.), and by the financial support for Scientific Research “5 per mille 2015” (to N.B.).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kolosenko I, Avnet S, Baldini N, Viklund J, De Milito A. Therapeutic implications of tumor interstitial acidification. Semin Cancer Biol. 2017;43:119–133. doi: 10.1016/j.semcancer.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Spugnini EP, Sonveaux P, Stock C, Perez-Sayans M, De Milito A, Avnet S, Garcia AG, Harguindey S, Fais S. Proton channels and exchangers in cancer. Biochim Biophys Acta. 2015;1848:2715–2726. doi: 10.1016/j.bbamem.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 3.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 4.Lloyd MC, Cunningham JJ, Bui MM, Gillies RJ, Brown JS, Gatenby RA. Darwinian dynamics of intratumoral heterogeneity: not solely random mutations but also variable environmental selection forces. Cancer Res. 2016;76:3136–3144. doi: 10.1158/0008-5472.CAN-15-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avnet S, Di Pompo G, Lemma S, Salerno M, Perut F, Bonuccelli G, Granchi D, Zini N, Baldini N. V-ATPase is a candidate therapeutic target for Ewing sarcoma. Biochim Biophys Acta. 2013;1832:1105–1116. doi: 10.1016/j.bbadis.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Ferrari S, Perut F, Fagioli F, Brach Del Prever A, Meazza C, Parafioriti A, Picci P, Gambarotti M, Avnet S, Baldini N, Fais S. Proton pump inhibitor chemosensitization in human osteosarcoma: from the bench to the patients’ bed. J Transl Med. 2013;11:268. doi: 10.1186/1479-5876-11-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avnet S, Lemma S, Cortini M, Pellegrini P, Perut F, Zini N, Kusuzaki K, Chano T, Grisendi G, Dominici M, De Milito A, Baldini N. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget. 2016;7:63408–63423. doi: 10.18632/oncotarget.11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engin K, Leeper DB, Cater JR, Thistlethwaite AJ, Tupchong L, McFarlane JD. Extracellular pH distribution in human tumours. Int J Hyperthermia. 1995;11:211–216. doi: 10.3109/02656739509022457. [DOI] [PubMed] [Google Scholar]
- 9.Salerno M, Avnet S, Bonuccelli G, Hosogi S, Granchi D, Baldini N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PLoS One. 2014;9:e110340. doi: 10.1371/journal.pone.0110340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bacci G, Ferrari S, Bertoni F, Ruggieri P, Picci P, Longhi A, Casadei R, Fabbri N, Forni C, Versari M, Campanacci M. Long-term outcome for patients with nonmetastatic osteosarcoma of the extremity treated at the istituto ortopedico rizzoli according to the istituto ortopedico rizzoli/osteosarcoma-2 protocol: an updated report. J. Clin. Oncol. 2000;18:4016–4027. doi: 10.1200/JCO.2000.18.24.4016. [DOI] [PubMed] [Google Scholar]
- 11.Longhi A, Errani C, De Paolis M, Mercuri M, Bacci G. Primary bone osteosarcoma in the pediatric age: state of the art. Cancer Treat Rev. 2006;32:423–436. doi: 10.1016/j.ctrv.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Arndt CA, Rose PS, Folpe AL, Laack NN. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin Proc. 2012;87:475–487. doi: 10.1016/j.mayocp.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldini N. Multidrug resistance--a multiplex phenomenon. Nat Med. 1997;3:378–380. doi: 10.1038/nm0497-378. [DOI] [PubMed] [Google Scholar]
- 14.Baldini N, Scotlandi K, Barbanti-Brodano G, Manara MC, Maurici D, Bacci G, Bertoni F, Picci P, Sottili S, Campanacci M, et al. Expression of P-glycoprotein in high-grade osteosarcomas in relation to clinical outcome. N Engl J Med. 1995;333:1380–1385. doi: 10.1056/NEJM199511233332103. [DOI] [PubMed] [Google Scholar]
- 15.Li S, Sun W, Wang H, Zuo D, Hua Y, Cai Z. Research progress on the multidrug resistance mechanisms of osteosarcoma chemotherapy and reversal. Tumour Biol. 2015;36:1329–1338. doi: 10.1007/s13277-015-3181-0. [DOI] [PubMed] [Google Scholar]
- 16.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryder C, McColl K, Zhong F, Distelhorst CW. Acidosis promotes Bcl-2 family-mediated evasion of apoptosis: involvement of acid-sensing G protein-coupled receptor Gpr65 signaling to Mek/Erk. J Biol Chem. 2012;287:27863–27875. doi: 10.1074/jbc.M112.384685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosko AE, McColl KS, Zhong F, Ryder CB, Chang MJ, Sattar A, Caimi PF, Hill BT, Al-Harbi S, Almasan A, Distelhorst CW. Acidosis sensing receptor GPR65 correlates with anti-apoptotic Bcl-2 family member expression in CLL cells: potential implications for the CLL microenvironment. J Leuk (Los Angel) 2014;2 doi: 10.4172/2329-6917.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta SC, Singh R, Pochampally R, Watabe K, Mo YY. Acidosis promotes invasiveness of breast cancer cells through ROS-AKT-NF-kappaB pathway. Oncotarget. 2014;5:12070–12082. doi: 10.18632/oncotarget.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marengo B, Nitti M, Furfaro AL, Colla R, Ciucis CD, Marinari UM, Pronzato MA, Traverso N, Domenicotti C. Redox homeostasis and cellular antioxidant systems: crucial players in cancer growth and therapy. Oxid Med Cell Longev. 2016;2016:6235641. doi: 10.1155/2016/6235641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geran RI, Greenberg NH, Macdonald MM, Abbott BJ. Modified protocol for the testing of new synthetics in the L1210 lymphoid leukemia murine model in the DR&D program, DCT, NCI. Natl Cancer Inst Monogr. 1977:151–3. [PubMed] [Google Scholar]
- 23.Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98:2603–2614. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- 24.Avnet S, Di Pompo G, Chano T, Errani C, Ibrahim-Hashim A, Gillies RJ, Donati DM, Baldini N. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-kappaB activation. Int J Cancer. 2017;140:1331–1345. doi: 10.1002/ijc.30540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Pompo G, Lemma S, Canti L, Rucci N, Ponzetti M, Errani C, Donati DM, Russell S, Gillies R, Chano T, Baldini N, Avnet S. Intratumoral acidosis fosters cancer-induced bone pain through the activation of the mesenchymal tumor-associated stroma in bone metastasis from breast carcinoma. Oncotarget. 2017;8:54478–54496. doi: 10.18632/oncotarget.17091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakanishi M, Morita Y, Hata K, Muragaki Y. Acidic microenvironments induce lymphangiogenesis and IL-8 production via TRPV1 activation in human lymphatic endothelial cells. Exp Cell Res. 2016;345:180–189. doi: 10.1016/j.yexcr.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Peppicelli S, Bianchini F, Contena C, Tombaccini D, Calorini L. Acidic pH via NF-kappaB favours VEGF-C expression in human melanoma cells. Clin Exp Metastasis. 2013;30:957–967. doi: 10.1007/s10585-013-9595-4. [DOI] [PubMed] [Google Scholar]
- 28.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 29.Varfolomeev E, Goncharov T, Maecker H, Zobel K, Komuves LG, Deshayes K, Vucic D. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5:ra22. doi: 10.1126/scisignal.2001878. [DOI] [PubMed] [Google Scholar]
- 30.Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mace PD, Smits C, Vaux DL, Silke J, Day CL. Asymmetric recruitment of cIAPs by TRAF2. J Mol Biol. 2010;400:8–15. doi: 10.1016/j.jmb.2010.04.055. [DOI] [PubMed] [Google Scholar]
- 32.West AC, Martin BP, Andrews DA, Hogg SJ, Banerjee A, Grigoriadis G, Johnstone RW, Shortt J. The SMAC mimetic, LCL-161, reduces survival in aggressive MYC-driven lymphoma while promoting susceptibility to endotoxic shock. Oncogenesis. 2016;5:e216. doi: 10.1038/oncsis.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Damaghi M, Tafreshi NK, Lloyd MC, Sprung R, Estrella V, Wojtkowiak JW, Morse DL, Koomen JM, Bui MM, Gatenby RA, Gillies RJ. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nat Commun. 2015;6:8752. doi: 10.1038/ncomms9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perut F, Carta F, Bonuccelli G, Grisendi G, Di Pompo G, Avnet S, Sbrana FV, Hosogi S, Dominici M, Kusuzaki K, Supuran CT, Baldini N. Carbonic anhydrase IX inhibition is an effective strategy for osteosarcoma treatment. Expert Opin Ther Targets. 2015;19:1593–1605. doi: 10.1517/14728222.2016.1086339. [DOI] [PubMed] [Google Scholar]
- 35.Perut F, Avnet S, Fotia C, Baglio SR, Salerno M, Hosogi S, Kusuzaki K, Baldini N. V-ATPase as an effective therapeutic target for sarcomas. Exp Cell Res. 2014;320:21–32. [PubMed] [Google Scholar]
- 36.Pastorek J, Pastorekova S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: from biology to clinical use. Semin Cancer Biol. 2015;31:52–64. doi: 10.1016/j.semcancer.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Martinez-Zaguilan R, Lynch RM, Martinez GM, Gillies RJ. Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Am J Physiol. 1993;265:C1015–1029. doi: 10.1152/ajpcell.1993.265.4.C1015. [DOI] [PubMed] [Google Scholar]
- 38.Sennoune SR, Martinez-Zaguilan R. Vacuolar H(+)-ATPase signaling pathway in cancer. Curr Protein Pept Sci. 2012;13:152–163. doi: 10.2174/138920312800493197. [DOI] [PubMed] [Google Scholar]
- 39.Moriyama Y, Patel V, Ueda I, Futai M. Evidence for a common binding site for omeprazole and N-ethylmaleimide in subunit A of chromaffin granule vacuolar-type H(+)-ATPase. Biochem Biophys Res Commun. 1993;196:699–706. doi: 10.1006/bbrc.1993.2306. [DOI] [PubMed] [Google Scholar]
- 40.Gottlieb RA, Nordberg J, Skowronski E, Babior BM. Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidification. Proc Natl Acad Sci U S A. 1996;93:654–658. doi: 10.1073/pnas.93.2.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barry MA, Eastman A. Endonuclease activation during apoptosis: the role of cytosolic Ca2+ and pH. Biochem Biophys Res Commun. 1992;186:782–789. doi: 10.1016/0006-291x(92)90814-2. [DOI] [PubMed] [Google Scholar]
- 42.Barry MA, Reynolds JE, Eastman A. Etoposide-induced apoptosis in human HL-60 cells is associated with intracellular acidification. Cancer Res. 1993;53:2349–2357. [PubMed] [Google Scholar]
- 43.Gottlieb RA. Cell acidification in apoptosis. Apoptosis. 1996;1:40–48. [Google Scholar]
- 44.Harguindey S, Arranz JL, Wahl ML, Orive G, Reshkin SJ. Proton transport inhibitors as potentially selective anticancer drugs. Anticancer Res. 2009;29:2127–2136. [PubMed] [Google Scholar]
- 45.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 46.Mandal R, Raab M, Matthess Y, Becker S, Knecht R, Strebhardt K. pERK 1/2 inhibit Caspase-8 induced apoptosis in cancer cells by phosphorylating it in a cell cycle specific manner. Mol Oncol. 2014;8:232–249. doi: 10.1016/j.molonc.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- 48.Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17:545–558. doi: 10.1038/nri.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meng Q, Zheng M, Liu H, Song C, Zhang W, Yan J, Qin L, Liu X. TRAF6 regulates proliferation, apoptosis, and invasion of osteosarcoma cell. Mol Cell Biochem. 2012;371:177–186. doi: 10.1007/s11010-012-1434-4. [DOI] [PubMed] [Google Scholar]
- 51.Hingorani P, Dickman P, Garcia-Filion P, White-Collins A, Kolb EA, Azorsa DO. BIRC5 expression is a poor prognostic marker in Ewing sarcoma. Pediatr Blood Cancer. 2013;60:35–40. doi: 10.1002/pbc.24290. [DOI] [PubMed] [Google Scholar]
- 52.Fulda S. Molecular pathways: targeting inhibitor of apoptosis proteins in cancer--from molecular mechanism to therapeutic application. Clin Cancer Res. 2014;20:289–295. doi: 10.1158/1078-0432.CCR-13-0227. [DOI] [PubMed] [Google Scholar]
- 53.Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors (TRAFs) Oncogene. 2001;20:6482–6491. doi: 10.1038/sj.onc.1204788. [DOI] [PubMed] [Google Scholar]
- 54.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 55.So T, Croft M. Regulation of PI-3-kinase and akt signaling in T lymphocytes and other cells by TNFR family molecules. Front Immunol. 2013;4:139. doi: 10.3389/fimmu.2013.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marshansky V, Rubinstein JL, Gruber G. Eukaryotic V-ATPase: novel structural findings and functional insights. Biochim Biophys Acta. 2014;1837:857–879. doi: 10.1016/j.bbabio.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 57.Falkenhorst J, Grunewald S, Muhlenberg T, Marino-Enriquez A, Reis AC, Corless C, Heinrich M, Treckmann J, Podleska LE, Schuler M, Fletcher JA, Bauer S. Inhibitor of apoptosis proteins (IAPs) are commonly dysregulated in GIST and can be pharmacologically targeted to enhance the pro-apoptotic activity of imatinib. Oncotarget. 2016;7:41390–41403. doi: 10.18632/oncotarget.9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27:6252–6275. doi: 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.