

Abstract
One of the major goals of the Chromosome-Centric Human Proteome Project (C-HPP) is to fill the knowledge gaps between human genomic information and the corresponding proteomic information. These gaps are due to “missing” proteins (MPs)—predicted proteins with insufficient evidence from mass spectrometry (MS), biochemical, structural, or antibody analyses—that currently account for 2579 of the 19587 predicted human proteins (neXtProt, 2017–01). We address some of the lessons learned from the inconsistent annotations of missing proteins in databases (DB) and demonstrate a systematic proteogenomic approach designed to explore a potential new function of a known protein. To illustrate a cautious and strategic approach for characterization of novel function in vitro and in vivo, we present the case of Na(+)/H(+) exchange regulatory cofactor 1 (NHERF1/SLC9A3R1, located at chromosome 17q25.1; hereafter NHERF1), which was mistakenly labeled as an MP in one DB (Global Proteome Machine Database; GPMDB, 2011–09 release) but was well known in another public DB and in the literature. As a first step, NHERF1 was determined by MS and immunoblotting for its molecular identity. We next investigated the potential new function of NHERF1 by carrying out the quantitative MS profiling of placental trophoblasts (PXD004723) and functional study of cytotrophoblast JEG-3 cells. We found that NHERF1 was associated with trophoblast differentiation and motility. To validate this newly found cellular function of NHERF1, we used the Caenorhabditis elegans mutant of nrfl-1 (a nematode ortholog of NHERF1), which exhibits a protruding vulva (Pvl) and egg-laying-defective phenotype, and performed genetic complementation work. The nrfl-1 mutant was almost fully rescued by the transfection of the recombinant transgenic construct that contained human NHERF1. These results suggest that NHERF1 could have a previously unknown function in pregnancy and in the development of human embryos. Our study outlines a stepwise experimental platform to explore new functions of ambiguously denoted candidate proteins and scrutinizes the mandated DB search for the selection of MPs to study in the future.
Keywords: C-HPP, missing protein, proteogenomics, preeclampsia, NHERF1, SLC9A3R1
Graphical Abstract
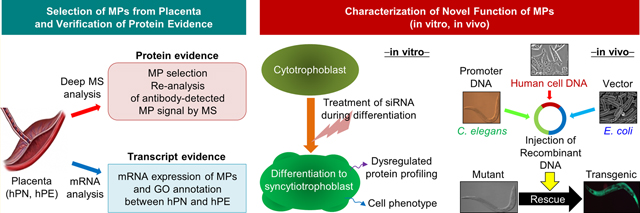
INTRODUCTION
Knowledge of the human genome and transcriptome (genes) is currently fragmented. The complete set of human proteome parts (proteins) encoded by chromosomes remains a work in progress because many proteins are not yet known or characterized. To address this challenge, the Chromosome-Centric Human Proteome Project (C-HPP), a consortium of 25 international teams from 20 countries, was established in 2012.1,2 The C-HPP aims to fill the gaps between the omics disciplines by mapping at least one representative protein per coding gene and by identifying and characterizing the alternative splicing variants and post-translational modifications of each protein. The C-HPP project has pioneered new methods of collaborative research by developing the ProteomeXchange database coupled to PRIDE (PRoteomics IDEntifications), making all data sets available to the proteome community.3 The protein mapping is also closely coordinated with the PeptideAtlas (www.peptideatlas.org), neXtProt (www.nextprot.org), the global proteome machine database (GPMDB; www.gpmdb.org), UniProtKB/Swiss-Prot (https://www.ebi.ac.uk/uniprot), and ProteinAtlas (www.proteinatlas.org).4,5
Despite the well-defined number of predicted human protein- coding genes, 19587 (neXtProt, 2017–01-23 release), there are notable barriers to the reliable generation of protein evidence or existence (PE) between the omics disciplines of genomics, transcriptomics, and proteomics.1,2 Currently, five PE levels indicate the quality or type of experimental evidence: PE1 for the protein level, PE2 for the transcript level, PE3 for the homology level, PE4 for the prediction level, and PE5 for dubious or uncertain gene.5 The most significant gap in knowledge across the omics disciplines is the set of ~2579 predicted proteins,4–8 known as “missing” proteins (MPs), that belong to PEs 2 through 4. These MPs are encoded by human chromosomes but lack conclusive mass spectrometric (MS) evidence or detection by other protein methods, according to neXtProt, and classifications based on transcript, species homology, or gene models.2 New guidelines have been adopted for MS-based claims of the identification of MPs.5 Because no systematic multiomics approach to the identification of MPs from initial discovery to functional validation has arisen, we attempted to explore potential pitfalls or mistakes for the detection of MP candidates.
The human placenta is a fetomaternal organ and plays an important role in gestation, fetal nutrients transport and production of several steroid hormones that regulate maternal physiology and fetal organ development (e.g. lung, liver, gastrointestinal tract and endocrine).9,10 In the placenta development process, the cytokine interaction between placental trophoblast and intracellular environment immune cells (e.g., natural killer (NK) cells and macrophage) seems very important in healthy pregnancy in keeping normal homeostasis of arterial intervillous vessel trophoblasts, which play a critical role in the structural developing bloodstream between maternal and fetal systems.11,12 Preeclampsia (hPE), which is a disorder of pregnancy (3–5% of all pregnancies), was characterized by high blood pressure (systolic blood pressure ≥140 mmHg or diastolic blood pressure ≥90 mmHg before pregnancy or before 20 weeks gestation) and abnormal amount of protein in the urine (≥300 mg of protein in 24 h), which can seriously affect both the mother and the fetus (e.g., premature birth).13 One of the known hPE etiologies is invasion and migration of placental trophoblast cells by inadequate trophoblast differentiation and acute phase interaction of immune cells into maternal spiral arteries that are too shallow, leading to abnormal blood pressure and hypoxia.14,15 Therefore, the function of trophoblast cells is vitally important in fetal development.
Here we propose and demonstrate a comprehensive proteogenomic platform that can be equally applied to functional characterizations of both MPs and a known protein, Na(+)/H(+) exchange regulatory cofactor 1 (NHERF1/SLC9A3R1; hereafter NHERF1). We address precautions and potential pitfalls based on the experience we gained. Despite the status of NHERF1 as a well-annotated protein, it was once labeled as an MP in the GPMDB (2011–09) when this study was initiated. We therefore demonstrate how a systematic approach to studying candidate proteins led us to uncover a new function of NHERF1 as a potential cellular regulator for a complicated human reproductive disorder, hPE. We also emphasize the importance of a highly scrutinized survey of all available public DBs when exploring candidate proteins.
EXPERIMENTAL PROCEDURES
Sample Preparation
Human placental tissues were obtained after cesarean delivery from women with and without hPE13 with informed consent from donors. The protocols for sample isolation and use for research purposes were approved by the Institutional Review Board of the CHA University College of Medicine, South Korea. hPE was defined as a blood pressure reading of140/90 mmHg or greater for the first time following midpregnancy and associated with abnormal protein excretion or multiorgan involvement (Table S1). The clinical characteristics of patients with and without diagnoses of hPE are outlined in Table S2. The systolic and diastolic blood pressures of the hPE patients were significantly higher than those of the control patients (hPN), and the infant birthweights of the hPE patients were significantly lower than those of the control patients (P < 0.05). The tissue samples were obtained from the placentas by peeling off the chorioamniotic membranes and removing the decidua regions. After the tissue samples were washed with phosphate-buffered saline solution (PBS), they were stored at –80 °C until use.
Isolation and Expansion of Primary Trophoblast Cells
Trophoblast cells were harvested from fresh placentas obtained after cesarean sections. After the chorioamniotic membrane and decidua regions were removed from the placentas, the tropho-blast area (~3 cm3, or 10 g) of the placental tissue was collected and washed thoroughly with 50 mL of sterilized cold PBS until the supernatant was nearly free of blood. The trophoblast tissues were then minced with scalpel blades. The minced tissues were transferred into a 50 mL centrifuge tube and incubated with a digestion solution containing collagenase IV in a 37 °C shaking incubator for 30 min. The harvested cells were suspended with 1 mL of Hank’s balanced salt solution, and the trophoblast cells were isolated from the mononuclear cells using a Ficoll density gradient separation. These cells were placed in an alpha-minimum essential medium (MEM-α) that was supplemented with 10% fetal bovine serum, 25 ng/mL of fibroblast growth factor 4, and 1 mg/mL of heparin and were cultured in a 37 °C incubator. After confluence, the cells were washed with PBS and collected.
RNA Sequencing
We carried out a comparative RNA sequence analysis for differentially expressed genes of individual three-paired trophoblasts isolated from the hPN and hPE tissues. The total RNA was extracted by the Trizol reagent (Invitrogen, USA) according to the manufacturer’s recommended protocol, and the RNA quality was assessed by analysis of the rRNA (rRNA) band integrity on an Agilent RNA 6000 Nano kit (Agilent Technologies, CA). Ahead of the complementary DNA (cDNA) library construction, 2 μg of total RNA and magnetic beads with Oligo (dT) were used to enrich its poly(A) mitochondrial RNA (mRNA) content. The purified mRNAs were then disrupted into short fragments, and double-stranded cDNAs were immediately synthesized. The cDNAs were subjected to end-repair and poly(A) addition and connected to sequencing adapters using the TruSeq RNA sample prep kit (Illumina, CA). Suitable fragments automatically purified by a BluePippin 2% agarose gel cassette (Sage Science, MA) were selected as templates for polymerase chain reaction (PCR) amplification. The final library sizes and qualities were evaluated via electrophoresis with an Agilent High Sensitivity DNA kit (Agilent Technologies), and the fragment lengths were found to be between 350 and 450 base pairs (bp). The library was then sequenced using an Illumina HiSeq2500 sequencer (Illumina, CA).
Transcriptome Data Analysis
Low-quality reads were filtered out according to the following criteria: reads with >10% of skipped bases (marked as “N”), reads with >40% bases with quality scores <20, and reads with an average quality score of <20. The filtering process was performed using in-house scripts. The filtered reads were mapped to the human reference genome (Ensemble release 72) using the aligner STAR v.2.3.0e. The gene expression levels were measured with Cufflinks v2.1.1 using the gene annotation database of Ensemble release 72. The noncoding gene regions were removed with the “-mask” option. To improve measurement accuracy, the multiread-correction and frag bias-correct options were applied. All other options were set to their default values. For the differential expression analysis, the gene level count data were generated using HTSeq-count v0.5.4p3 with the option “-m intersection-nonempty” and the “-r” option considering paired-end sequences. On the basis ofthe calculated read count data, the differentially expressed genes were identified using the R package TCC,16 which applies robust normalization strategies to compare tag count data. The normalization factors were calculated using the iterative DEGES/edgeR method. The q value was calculated based on the P value using the p.adjust function of the R package with the default parameter settings. Significant differentially expressed genes were identified based on a q -value threshold of <0.05.
Western Blot Analysis
Placental tissue, trophoblast, and cell line pellets were extracted using a modified radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 150-mM NaCl, 1% NP-40, pH 8.6) with protease inhibitor. The protein was loaded onto a 10% sodium dodecyl sulfate–polyacrylamide gel and transferred to a nitrocellulose membrane after electrophoresis. Blocking was carried out with PBS containing 5% skim milk and 0.2% Tween-20 for 1 h at room temperature. The membrane was incubated with primary antibodies overnight at 4 °C. After washing with PBS containing 0.2% Tween-20, the membrane was incubated with secondary antibodies for 1 h at room temperature. The band signal was developed by ECL plus reagents (GE Healthcare, Uppsala, Sweden) and scanned using a Typhoon 9400 scanner (GE Healthcare). The signal intensity was quantified via densitometry using ImageQuant TL v2005 software (GE Healthcare). The antibodies for Western blots were purchased from Santa-Cruz (polyclonal anti-PLC9, polyclonal anti-CBX7, polyclonal anti-PHF11, monoclonal anti-OAS2, polyclonal anti-OAS3, polyclonal anti-RNF121, monoclonal anti-NHERF1, monoclonal anti-KRT7, monoclonal anti-KRT18, monoclonal anti-COX5A, polyclonal anti-GAPDH, and monoclonal anti-β-actin antibody) and Abcam (polyclonal anti-INPPL1, monoclonal anti-eNOS, and polyclonal anti-IL-1β antibody).
Identification of Antibody-Captured Protein by LC–MS/MS
The target band of the preparative gel was sliced, destained, reduced with 20-mM dithiothreitol (DTT), alkylated with 25 mM iodoacetamide, and then digested with 50 ng trypsin per gel band containing 25-mM ammonium bicarbonate buffer. The peptide separation and identification were analyzed using liquid chromatography (LC) and MS with a LTQ-Orbitrap XL MS (Thermo Fisher Scientific, CA) coupled to an EASY-nLC 1000 system (Thermo Fisher Scientific). A C18 silica-packed nanobore column (ZORBAX 300SB-C18, 150 mm × 0.1 mm, 3 μm pore size, Agilent) was used for peptide separation. The settings used for the MS analyses for identification of antibody-captured proteins were: mass resolution setting for full MS scan: 15 000; mass range for MS1: 350 to 1200; isolation window for precursor ion selection: 2; number or precursors selected for tandem MS in each scan event: 9; intensity threshold for MS2: 100 000.0; charge-state screening parameters: 2 or more; relative collision energy: 35.0 in CID (collision-induced dissociation); mass analyzer for MS2: ion trap; dynamic exclusion settings: enable (repeat count: 1; repeat duration: 20.00, exclusion list size: 20.00, exclusion mass width relative to mass; exclusion list size: 20.00, exclusion mass width relative to low (ppm): 10.00; and exclusion mass width relative to high (ppm): 10.00). The MS data files were processed using Proteome Discoverer (version 1.4; Thermo Fisher Scientific), which managed database searching by Mascot (version 2.6.0, Matrix Science). The peptides were identified using UniProt (release date June 2012) with the following database search criteria: taxonomy, Homo sapiens (86 875 sequences; 36 599 288 residues); carboxyamidomethylated (C, cysteine residues) for fixed modifications; oxidized (M, methionine residues) for variable modifications; maximum missed cleavages: 2; 10 ppm MS tolerance; and a 0.8 Da CID. The reversed sequences were used for evaluation of the FDR < 1% on a spectrum match and at the peptide and protein levels.
Cell Differentiation and Cell Migration Assay
The JEG-3 cell lines were purchased from the Korean Cell Line Bank and cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C with 5% CO2. The JEG-3 cells were seeded at 1 × 104 cells into 96-well plates, treated with 10 μM forskolin (differentiation inducer), and transfected with a pool of NHERF1 Stealth Select RNA interference (RNAi) small interfering (siRNA; three set; catalog numbers HSS145123, HSS145124, and HSS145125, Invitrogen) or a nonspecific scrambled siRNA negative control (Invitrogen) using Lipofectamine 2000 (Invitrogen). The proliferation of the JEG-3 cells was determined by a stable tetrazolium salt (WST-1) colorimetric assay kit (Sigma-Aldrich, St. Louis, MO). After continuous incubation for 48 h, 20 μL of WST-1 was treated, incubated for 2 h, and determined at 450 nm using an enzyme-linked immunosorbent assay reader. The secreted β-subunit of human chorionic gonadotropin (β-hCG) was analyzed using 100 μL of cultured media with an enzyme-linked immunosorbent assay kit (Abcam, Cambridge, U.K.). Cell migration was measured by a wound-healing assay in six-well plates. The confluent cells (1 × 106 cells) were scratched using a tip 1 mm wide, treated with forskolin, a pool of NHERF1 siRNAs and a scrambled siRNA negative control, and incubated for 48 h. Cell images were taken with an Olympus IX71 microscope (Tokyo, Japan).
Comparative Proteomic Analysis by Tandem Mass Tag Labeling
The JEG-3 cells were seeded at 2 × 106 cells into 100 mm dishes and treated with 50 nM of the NHERF1 siRNA and siRNA negative control under differentiation conditions. After 48 h, the cells were collected and extracted by a lysis buffer (6 M urea, 2 M thiourea, and 2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) with ultrasonication. Each 100 μg of the cell lysates containing 20 μL of 500-mM triethylammonium bicarbonate (TEAB) was reduced, alkylated, and digested with Pierce MS-grade trypsin protease (Thermo Fisher Scientific) and labeled with tandem mass tag (TMT) reagents (Thermo Fisher Scientific), as previously reported.17,18 To avoid technical errors, we split the samples into two groups, and each group was divided into three sets. We replicated the experiments three times under the following conditions: TMT chemicals 126,128, and 130 were labeled in samples that had been treated with the scrambled siRNA negative control with forskolin (FK-SC) and 127, 129, and 131 were labeled in samples that had been treated with NHERF1 siRNAs with forskolin (FK-siRNA). We excluded the proteins that showed a variation of ≥1 ± 0.2 among the FK-SC samples and selected the proteins within a ratio of triplicates ≤1 ± 0.2 for further analysis. The labeled peptides were identified and quantified with an LTQ-Orbitrap XL MS (Thermo Fisher Scientific).17,18 We used a Nano-LC system and a C18 column for peptide separation. The TMT-MS condition was set by the following: mass resolution setting for full MS scan: 15 000; mass range for MS1: 350 to 1200; isolation window for ion selection: 2; number or precursors selected for tandem MS in each scan event: 4; intensity threshold for MS2: 100 000.0; charge-state screening parameters: 2 or more; relative collision energy: 35.0 in CID and 70.0 in HCD (high-energy collision dissociation); mass analyzer for MS2: ion trap mass spectrometry (ITMS) for protein identification and Fourier transform MS (FTMS) for the case of TMT analysis; dynamic exclusion settings: enable (repeat count: 1; repeat duration: 20.00, exclusion list size: 20.00, exclusion mass width relative to mass; exclusion list size: 20.00, exclusion mass width relative to low (ppm): 10.00; and exclusion mass width relative to high (ppm): 10.00). Xcalibur (v2.1) and Proteome Discoverer (v1.4) software programs were used to generate peak lists and quantify unique peptides. Proteome Discoverer software was used only to quantify the unique peptides of target proteins. To identify peptide sequences, MASCOT (Matrix Science, London, U.K.; version 2.2.04) was used with the following database search criteria: taxonomy, Homo sapiens (86 875 sequences; 36 599 288 residues); TMT 6plexand carboxy-amidomethylated (C, cysteine residues) for fixed modifications; oxidized (M, methionine residues) and deamidated (N, Q; asparagine and glutamine residues) for variable modifications; maximum missed cleavages: 2; 10 ppm MS tolerance; and a 0.8 Da CID. The MS/MS tolerance was 50 ppm of HCD in the FTMS for TMT analysis. The reversed sequences were used for evaluation of the FDR < 1% on a spectrum match and at the peptide and protein levels. Quantification was performed by calculating the ratio between the peak areas of the TMT reporter groups. To eliminate the masking of changes in expression due to peptides that were shared between proteins, we calculated the protein ratio using only ratios from the spectra that were unique to each protein. All quantitative results were normalized using protein medians (minimum protein count: 20, default level), and in cases of one or more missing quant channels, the quant values were rejected. We estimated P values using a Student’s t test from the mean and standard deviation (SD) of each fold ratio for the 127/126, 129/128, and 131/130 TMT-labeling data.
Nematode Strains and Generation of Extra-Chromosomal Human Gene Rescue Transgenic C. elegans
Wild-type Bristol (N2) and VC320 (nrfl-1 (ok297), an ortholog of human NHERF1) mutants of Caenorhabditis elegans, were obtained from the Caenorhabditis Genetics Center and cultivated at 20 °C on Escherichia coli 0P50 strain-seeded nematode growth medium (NGM) plates.19–21 The transgenic worm YP0078, nrfl-1 (ok297);ykpEx018[Pnrfl-1(C. elegans)::NHERF1 (Human)::gfp, rol-6(su1006)], was created by microinjecting the construct Pnrfl-1::NHERF1::gfp into the N2 and transferring it into the nrfl-1(ok297) strain. The promoter nrfl-1 gene, like upstream fragments of 2.5 kb from C. elegans genomic DNA and the cDNA containing the full length of NHERF1 from the human HeLa cell line, were amplified by PCR and ligated into a green fluorescent protein (GFP) vector (pPD95.75) (Figure S1). The GFP vector was provided by the A. Fire laboratory. The transgenic constructs Pnrfl-1(C. elegans)::NHERF1 (Human)::gfp and rol-6(su1006), as phenotype markers, were coinjected at 50 and 10 ng/μL, respectively. The primers of the promoter cloning C. elegans nrfl-1 gene F1 were 5′-TGC CAT GAT TTT CGC CAC AA-3′ (sense) and 5′-TGC TAC TTT CGA CTT ACCA-3′ (antisense). The primers of F2 were 5′-TGG TAA GAA GTC GAA AGT AGC A-3′ (sense) and 5′-AGT CTT GGA GTC ACG TC-3′ (antisense). The primers of cDNA cloning human NHERF1 were 5′-CCC ATC GGA ACC CCA AGT C-3′ (sense) and 5’-GGG ATG TGG GTG CTG ATT TG-3′ (antisense). For brood size assays of the N2 wild type and nrfl-1 (ok297) mutant and transgenic worms, the number of eggs laid and hatched worms were counted every 24 h and transferred to fresh nematode growth media (NGM) plates to prevent mixing with progeny. The morphology of the worms and localization of the NHERF1::GFP signal were captured using a Nomarski microscope (Carl Zeiss Meditec, Jena, Germany) and LSM 880 confocal microscope (Carl Zeiss Meditec).
Bioinformatics and Statistical Analysis
The protein interaction network and functional classification of differentially expressed genes and protein lists was analyzed using the STRING database v10.0 (string-db.org) and the PANTHER (www.pantherdb.org) bioinformatics resource. As the SPRING network shows functional relationships by lining (no classification), gene ontology (GO) terms were classified according to their functional similarity based on the PANTHER database. Heat maps for protein expression and similarity were created using the gplots package in the R statistics software (version 3.2.3). If a protein expression value was higher in the FK-siRNA group, the heatmap was coded red; if a protein expression value was higher in the FK-SC group, then the heatmap was coded turquoise. The graph data were represented as means and SD. GraphPad Prime 5 for Windows (GraphPad Software, San Diego, CA) and Microsoft Excel 2010 were used for all statistical analyses, and the significance level of the Student’s t test was set at P < 0.05.
RESULTS
Stepwise Experimental Strategy for Exploring Protein Functions
To explore the possible functional roles (e.g., cellular regulator or biomarker) of the candidate proteins involved in the hPE clinical specimens,17 we first established the systematic proteogenomic experimental strategy outlined in Figure 1A. We performed a deep MS analysis (large-scale profiling, glycol-/phosphoprotein enrichment, fractionations) of normal (hPN) and diseased (hPE) human placental tissue (Step 1; Figure 1B). As described previously, we also obtained a list of candidate proteins (Step 2) in accordance with the GPMDB (2011–09 release; Table S3). The candidates of missing protein were selected in accordance with GPMDB guidelines, which state any proteins with either the low probability (log(e-2) or lower) or no previous report in GPMDB. The “log(e)” means the lowest (best) expectation value observed for these proteins, as described in the guidelines. The candidate proteins were further screened for molecular identification based on the availability of antibodies (Step 3) for Western blot assay using the placental tissues or corresponding cell lines. To ensure that the selected candidate proteins were fully supported by MS evidence, the protein bands detected by Western blotting were rigorously reanalyzed with high-resolution MS (e.g., LTQ-Orbitrap; Step 4). A comparative transcriptome analysis by RNA sequencing was performed (Step 5) that generated comprehensive information about differentially expressed mRNAs, including candidate proteins in the placental tissues. The hPE tissue was analyzed with GO terms for biological function, molecular pathways, and cell compartmentalization (Figure 1B, lower panel). When MS validation of the candidate proteins was completed (Figure 1B), we selected appropriate cell lines to further explore the target genes encoding candidate proteins by modulation through either siRNA treatment (suppression) or overexpression under well-established cell growth conditions (cell differentiation induced by forskolin; Step 6). This in vitro study included the knockdown of candidate protein genes, phenotype examination, and quantitative MS analysis in the presence or absence of modulators (forskolin, siRNA; Figure 1C). The function of each target gene was further investigated with in vivo phenotype observations of C. elegans mutants (Step 7) and by a rescue experiment involving the microinjection of the target human candidate protein gene into mutant C. elegans that lacked the target gene (Figure 1D). The cell lines and animal models were used interchangeably depending on the experimental purpose and their availability. Once the selected candidate proteins were functionally validated, we investigated their biomedical applications, such as whether they were cellular regulators for specific diseases or new biomarkers or drug targets.
Figure 1.
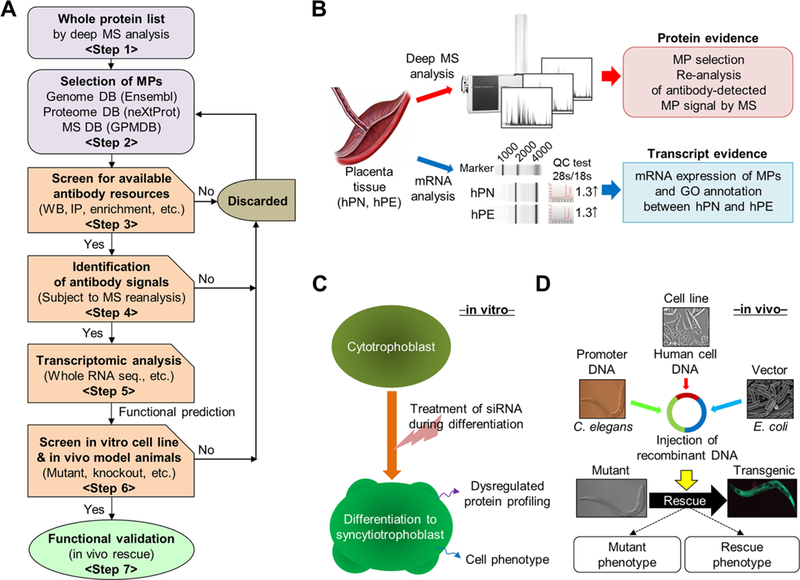
Workflow for the discovery and functional characterization of MPs. (A) Overall workflow. (B) MPs were first analyzed by high-sensitivity MS, annotated by bioinformatics databases (Ensembl, UniProt, GPMDB, 2011–09 release), then characterized by high-throughput RNA expression analysis and gene network databases using tissue from hPN and hPE. (C) For in vitro characterization of MPs identified as candidates for placental cellular function, cell phenotype analysis (cell growth, disease marker) was carried out using siRNAs under placental trophoblast-differentiation conditions. Differentially expressed proteins were further validated by Western blot or comparative MS analysis. (D) For in vivo animal model study, the mutant of C. elegans with an ortholog of a human target-gene was rescued by recombination of the human gene, and the phenotypes of the rescued mutant were analyzed in comparison with the wild-type control.
Validation for Protein Evidence of Candidate Proteins
Although we started this stepwise screening with 39 candidate proteins, only eight of them were tested for immunological validation by Western blot analysis with available commercial antibodies in placental tissue lysates. The antibody-captured signal bands were reverified by LC–MS/MS. Although five candidate proteins (PLAC9, CBX7, PHF11, OAS2, and OAS3) were not detected by immunoblot, the candidate proteins RNF121 and INPPL1 were detected (Figure S2A). Their expression levels were not different between the hPN and hPE tissues (RNF121, P = 0.0881; INPPL1, P = 0.1598; Figure S2B). Among the protein bands, only INPPL1 was identified, with a total score of 106 by LC-MS/MS (Table S4A). The MS/MS fragmentation of the representative unique peptides is presented in Figure S2C and Table S4B. RNF121 was detected very well by Western blot analysis, but the strong signal band was not identified by MS analysis. Although INPPL1 was validated by both methods, an in vivo study was not possible due to the absence of a mutant model organism (e.g., C. elegans). Thus we turned our focus on another candidate, NHERF1, which passed the threshold for MS validation of antibody-captured bands by immunoblot and the existence of a mutant animal model. This validating step for protein evidence is presented in Table S5. The expression of NHERF1 was examined using individual clinical samples (placental tissues and trophoblasts) and was found to be increased compared with hPN sample lysates (Figure 2A). Although there was essentially no significant difference in their expression levels between the hPN and hPE tissues (Figure 2B; tissue, P = 0.4530; trophoblast, P = 0.1496), we wanted to continue exploring its possible new functions. The target band in 1D gel was reverified as authentic NHERF1 by MS analysis with a total score of 417 (Table S6A). The MS/MS fragmentation of a unique peptide is presented in Figure 2C and Table S6B. Although RNF121 was detected as a strong signal by Western blot, but we failed to identify this protein by MS analysis. Most previous claims of MP discovery have been based only on primary MS profiling or antibody detection.22–25 Therefore, our result implies that the bands detected by antibody should also be scrutinized to confirm by MS analysis.
Figure 2.
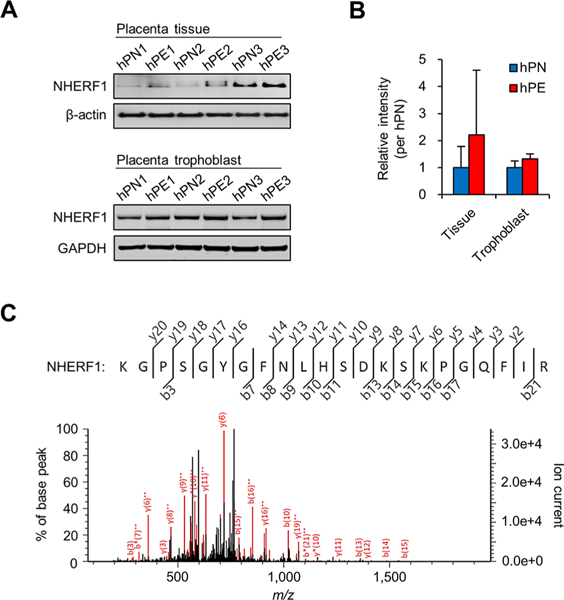
Validation of the protein evidence for NHERF1. (A) NHERF1 was analyzed by Western blotting using placenta tissue and trophoblast isolated from individual donors. (B) Relative band intensity was estimated based on the average levels obtained from three hPN samples. (C) Antibody-captured signal band was identified as bona fide NHERF1 by LC-MS/MS. The unique peptide of NHERF1 is illustrated.
The transcriptomic data for 18 157 genes in the trophoblasts from hPN and hPE revealed that 111 genes were differentially expressed (q < 0.05, Figure 3A and Table S7). Although 32 cases of the 39 candidate proteins were not significant, their transcript evidence is presented at the fragments per kilobase million (FPKM) level. Seven candidate proteins were not detected (Table S8). The GO classifications of the 111 genes were divided into mainly cellular (19.0%), metabolic (16.5%), and developmental processes (11.0%; Figure 3B). Among those 111 genes, 23 showed a functional relationship with cell migration/invasion, differentiation, and cytokine production (Figure 3C). Upon closer examination, the molecular expression levels of the genes in the categories of both cell migration/invasion and cell differentiation were significantly higher in the trophoblasts of hPE than hPN (upper panel and left side of lower panel in Figure 3D), whereas those involved in cytokine production were significantly decreased (right side of lower panel in Figure 3D). This result suggests that the causes of hPE might include (but are not limited to) the abnormal expression of certain genes involved in cell migration/invasion, differentiation, and cytokine induction in placental trophoblasts.
Figure 3.
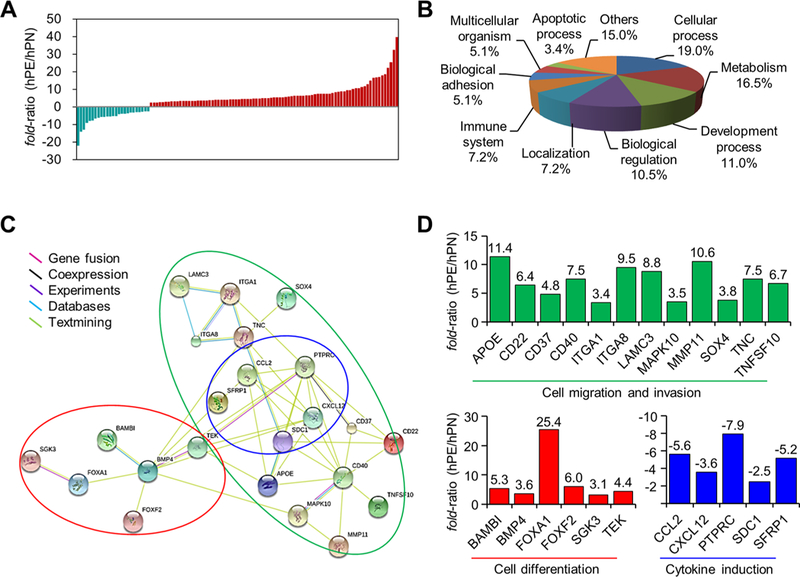
Gene network analysis of hPE from RNA sequencing. (A) Significant differentially expressed transcripts of trophoblasts isolated from placenta tissue of three hPN and hPE patients totaled111by RNA sequencing (q < 0.05, n = 3). Turquoise and red bars indicate down-regulated and up-regulated genes, respectively. (B) GO classifications of biological function were obtained from the PANTHER database. (C) Twenty-three genes were classified by interaction type using the STRING network, PANTHER, and the literature. The original data were modified to group the genes according to molecular and biological function. The green line indicates cell migration/invasion factors, the red line indicates cell differentiation factors, and the blue line indicates cytokine induction factors. (D) The fold ratio of each gene indicated by the colored lines in panel C.
Potential Regulatory Function of NHERF1
Although the mRNA and protein levels of NHERF1 in the hPN and hPE trophoblasts were not significantly different (see Table S8 and Figure 2B), we determined whether NHERF1 played a role in the differentiation of the human placental trophoblast cell line (JEG-3), which has been widely studied in the context of human reproduction.26 We performed a cell proliferation WST-1 assay to identify any cellular regulatory function of NHERF1. In this experiment, placenta-specific secreted β-hCG was used as a cell differentiation marker.27,28 The expression of NHERF1 was decreased in the differentiated condition by forskolin (FK-SC, treated with forskolin and 50-nM scrambled siRNA) compared with the untreated control (treated with DMSO only), and the expression level was also significantly decreased in FK-siRNA (treated with forskolin and 50-nM NHERF1 siRNA) compared with FK-SC (P < 0.05, Figure 4A). The levels of cell proliferation and secreted β-hCG were increased under the FK-SC condition compared with the untreated control but were significantly decreased in the FK-siRNA condition (Figure 4B; P < 0.05). These results suggest that NHERF1 played a potential regulatory role in trophoblast differentiation. We next screened those proteins that had been differentially expressed under the FK-siRNA condition compared with FK-SC under the same conditions using TMT-labeled MS/MS analysis. Among the 1001 identified proteins, 155 were differentially expressed after filtering by the following criteria: peptide count ≥2, peptide sequence number ≥9, FDR <1%,5,7 and detection in at least two TMT-labeled sets (Figure 4C and Table S9). Because the proteins were identified at a FDR < 1%, we could not exclude the possibility of false-positive results. Among the 39 proteins with significantly differential expressions (Table S9, P < 0.05), 21 were classified by functional network and GO classification using the STRING and PANTHER resources. These 21 proteins yielded major functional classifications—protein folding, RNA metabolic processes, and cell motility—that could indicate a potential regulatory role of NHERF1 (Figure 4D).
Figure 4.
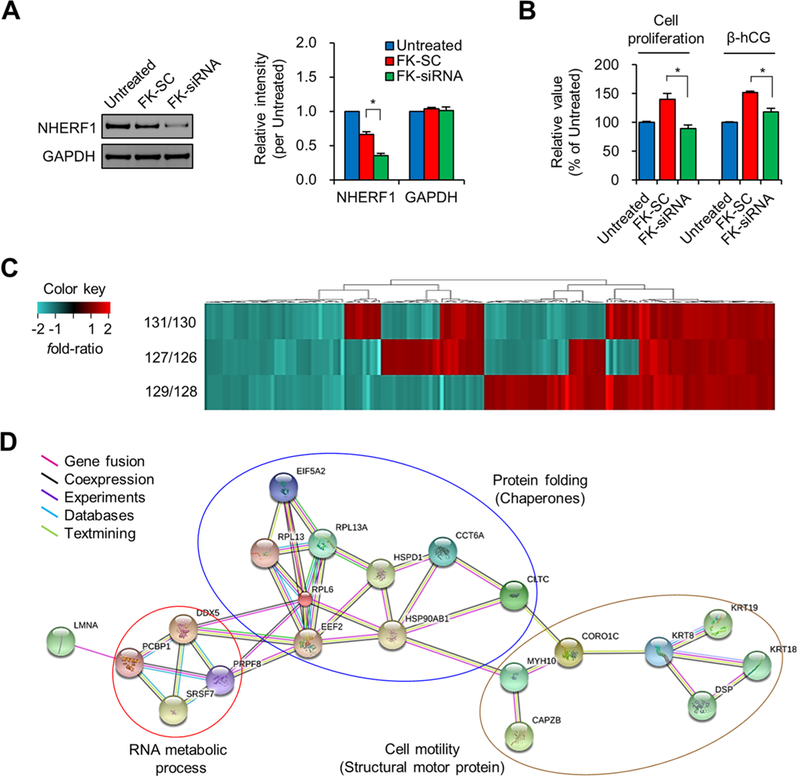
Effect of NHERF1 knockdown in cytotrophoblast JEG-3 cells. (A) NHERF1 expression in untreated, FK-SC, and FK-siRNA cells was confirmed by Western blotting, and their relative band intensity was estimated based on that of the untreated DMSO control. (B) Cell proliferation was determined by WST-1 assay and the secreted β-hCG level was determined by the corresponding enzyme-linked immunosorbent assay. Asterisk (*P < 0.05) represents a significant difference by Student’s t test (n = 3). (C) Each isobaric-labeled sample of FK-SC (126-, 128-, 130-TMT label) and FK-siRNA (127-, 129-, 131-TMT label) was comparatively analyzed by LTQ-Orbitrap XL MS, and the differentially expressed proteins (totaling 155 from duplicate running) were classified by heatmapping. (D) Protein–protein interaction network for the differentially expressed proteins (P < 0.05) is presented according to STRING where those proteins were classified into three major functional groups using colored lines by the PANTHER database.
Effect of NHERF1 on Cell Migration
Because NHERF1 exhibited a potential regulatory role in cell motility (Figure 4D), we performed a monolayer wound-healing assay. As anticipated, the cell movement after 48 h was found to be suppressed in the FK-siRNA condition compared with FK-SC under differentiation conditions (Figure 5A). We next examined the changes in the expression levels of trophoblast motility factor (IL-1β, eNOS, and KRT7), which regulates migration and invasion of trophoblasts,14,15,29,30 and TMT-MS candidates (KRT18 and COX5A) under the same conditions. The levels of three proteins, NHERF1, KRT7, and KRT18, were significantly decreased in the FK-siRNA condition compared with FK-SC (P < 0.05), whereas IL-1β, eNOS, and COX5A were not significantly different (IL-1β P = 0.1951; eNOS, P = 0.9041; COX5A, P = 0.4141; Figure 5B). Like the KRT7 protein, KRT18 is also known as a trophoblast motility factor,30 and it was classified as a structural motor protein in proteome DBs (Figure 4D). We concluded that NHERF1 regulated the cytoskeletal motor proteins with trophoblast motility function. Because the proteins we analyzed by TMT-MS accounted for ~5% of the total proteins (1001 of 19587), we thought that eNOS and IL-1β might be present at levels too low for detection by MS analysis (i.e., in the pg to ng range) and would only be detectable with an antibody assay.
Figure 5.
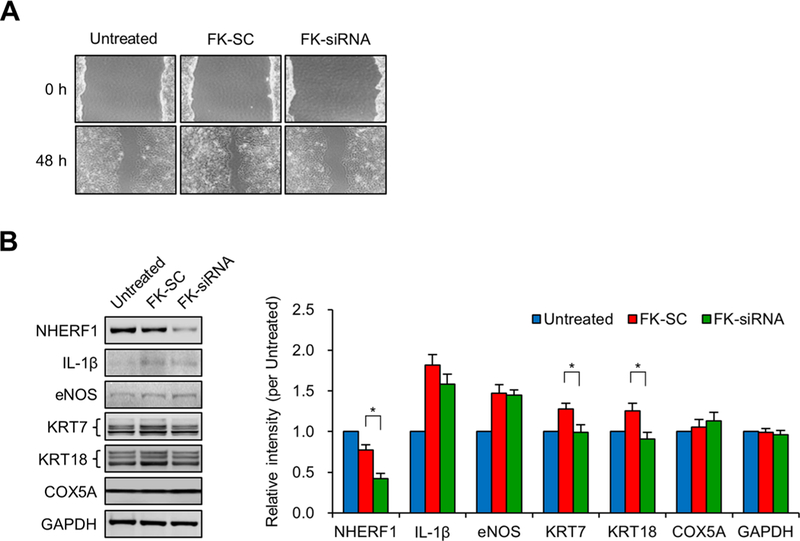
Effect of NHERF1 knockdown on migration of JEG-3 cells. (A) After confluence in six-well plates,JEG-3 cells were wounded by a tip of 1 mm width, treated with scrambled siRNA and a pool of NHERF1 siRNAs at 50-nM, and then incubated for 48 h. Morphological differences were captured using an Olympus IX71 microscope. (B) Expression of NHERF1, IL-1β, eNOS, KRT7, KRT18, COX5A, and GAPDH was analyzed by Western blotting, and their relative band intensity was estimated based on that of the untreated DMSO control (three independent experiments). The asterisk (*P < 0.05) represents a significant difference found by Student’s t test (n = 3).
C. elegans nrfl-1 Mutant, a Novel Model Organism To Study Human Reproductive Disease
Our in vitro results from human trophoblast cells indicated that NHERF1 could have an in vivo function relevant to hPE, a human reproductive disease. We therefore performed an in vivo rescue experiment using the C. elegans mutant nrfl-1 (ok297), an ortholog of human NHERF1. This mutant strain, nrfl-1 (ok297), contains an indel mutation within the nrfl-1 gene (724 bp insertion/16 bp deletion), which is located between exon 3 and exon 4 (Figure S3). Unlike the N2 wild-type animals, this mutant frequently ruptured under the pressure of the cover glass, probably due to its protruding vulva (Pvl) phenotype (N2 wildtype, Figure 6A; mutant, Figure 6B). In rescue experiments in which recombinant human NHERF1 cDNA was transfected into N2 by microinjection and transferred to nrfl-1 (ok297) mutants, the phenotype of this transgenic worm (YP0078) reverted to that of the N2 wild type (Figure 6C). The NHERF1::GFP was clearly expressed in the transgenic worm (Figure 6D), and its expression was detected in the head (pharynx, excretory canal [H form]), body (intestine), and cytosolic tail part (Figure 6E). Although Hagiwara’s group reported that C. elegans nrfl-1 was expressed in the intestine and the excretory system,31 in contrast with our data, our findings agreed with the information on nrfl-1 localization in the WormBase (http://www.wormbase.org/). This discrepancy could be due to differences in the promoter lengths used by the two groups because this usually controls the expression loci of genes. To analyze reproductive function, brood size was counted as the number of eggs laid. Although the egg-laying ability of nrfl-1(ok297) mutants was substantially lower than the N2 wild type 7 days after the L4 stage (P < 0.0001), the egg-laying ability of the transgenic YP0078 worms increased to 90.8% of the N2 wild type (P = 0.008, n = 10; Figure 6F, left panel). We observed this change only on the first day because on the second day the nrfl-1(ok297) mutant did not lay eggs (Figure 6F, right panel) and the eggs remained inside the body. The rate of unhatched eggs in the nrfl-1(ok297) mutant was 91.1% (n = 30), and it thus exhibited a “matricidal” phenotype. This phenotype was not detected in the N2 or transgenic worms (Figure 6G). Together, these results suggest that C. elegans NRFL-1, the nematode ortholog of human NHERF1, could play an important role in the morphogenesis of female genital organs and in fertilization. These findings mark the first reported potential regulatory function of NHERF1 in reproductive disorders.
Figure 6.
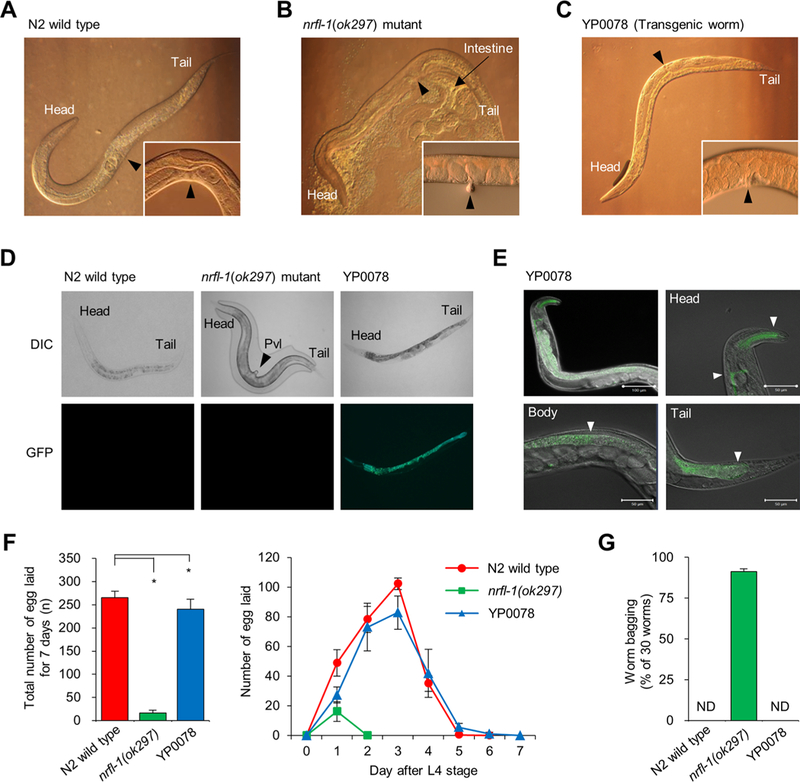
Rescue effect of C. elegans nrfl-1(ok297) by microinjection of human NHERF1 and cellular localization of nrfl-1 expression in C. elegans. The vulvar positions of N2 wild type (A), nrfl-1(ok297) mutant (B), and YP0078 transgenic worm (C) are shown by black arrows. (D) The NHERF1::GFP was detected by fluorescence microscopy. The morphology of nrfl-1(ok297) mutant was examined without a cover-glass to prevent protruding vulva (Pvl, black arrow). DIC, differential interference contrast. (E) To prevent interference from nonspecific autofluorescent E. coli, the YP0078 transgenic worm was starved for 30 min on nonfed media; then, NHERF1::GFP was visualized by confocal fluorescent microscopy. The GFP localization is shown by white arrows (pharynx and excretory canal in head, intestine in body, and cytosolic tail). Scale bars represent 100 μm in the top-left panel and 50 μm in head (top-right), intestine (bottom-left), and tail (bottom-right) part. (F) Total number of progeny (eggs laid) (left panel) and daily number of progeny for 7 days (right panel) were scored after the L4 stage (n = 10). (G) Reversibly laid eggs; the number of internally hatched worms (F0) was counted for 5 days after the L4 stage (n = 30, triplicate). “ND” means “not detected”. The asterisk (*P < 0.05) represents a significant difference detected by Student’s t test.
DISCUSSION
We present a proteogenomics-based experimental platform that offers a systematic approach to study NHERF1 from molecular reanalysis to functional validation. We anticipate that this integrated strategy will be directly applicable to study the remaining 2579 MPs and many known proteins that have less characterized functions.
There are three important points of this work. First, we clarified the history of the study of this protein and addressed the potential problems with databases on which we are dependent. Our report not only deals with the potential pitfalls of the identification of MPs but also outlines a discovery of their novel functions, if any.
The second important point is the notable inconsistency in the status of NHERF1 among the public DBs of the GPMDB, PeptideAtlas, and neXtProt. This inconsistency might also have been associated with the time between an initial identification of the candidate proteins and their functional characterization. For example, NHERF1 was selected as one of the 39 candidate proteins for functional characterization study from the Global Proteome Machine Database (GPMDB; 2011–09 release) and PeptideAtlas (2011). At that time, we did not use neXtProt as a standard reference DB for defining the status of MPs until after 2012 HUPO Congress. A gap in time exists between the identification and annotation of candidate proteins in public DBs. There were 39 candidate proteins listed at one point in time in the GPMDB (2011–9 release), but some, including NHERF1, later turned out to have been well known (PE1) for years before they had been classified as MP in some public DBs (e.g., SwissProt). This misclassification is why NHERF1 was included as one of the 39 candidate proteins in our previous publication on placenta protein profiling in January 2013.17 Thus this report conveys the potential problems associated with initial identification of candidate proteins in addition to the functional aspects of NHERF1. We believe that our learning experience could be useful for other investigators who might encounter the same situation when studying MPs in the future.
The third important point of our study is that already well-known proteins (e.g., regulator of G-protein signaling 13 (RGS13), NX 014921, 1q31.2), like NHERF1, seemed to have an inconsistent status of “MPs” (neXtProt; 2017–04-12 release). For example, RGS13 is currently classified as a PE2 protein and no post-translational modification (PTM) site is listed. Xie et al. (2010)32 reported that it has a Thr41 phosphorylated site in their cellular study, and there are multiple reports on the functional studies of this PE2 protein (e.g., protein, mRNA evidence, cell line, disease study, in vivo animal study; see refs 33–36). Information is available about the RGS13 protein that covers not only the level of transcription data but also the protein existence data (antibody detection, cellular functional characterization). Like NHERF1, RGS13 has likely been misannotated because of the time gap between functional characterization and annotation in the public DBs. Many more such cases may await exploration in the future.
One of our goals was to increase awareness of discrepancies in the time between an initial recognition of an MP and the actual annotation or vice versa. We also note that close attention should be paid to this fact, and the literature and public DBs should be extensively searched at the beginning of a study on an MP. However, it could also be inevitable that the status of one of the 2579 MPs will be updated in the public DBs during a study, as occurred during our study of NHERF1. Although we discovered that NHERF1 was indeed listed as PE1 in neXtProt, we decided to continue our effort to explore the function of this protein through proteogenomic studies of placenta samples.17 It was worth trying to explore the previously undiscovered function, if any, of this protein in human reproduction in addition to its already well-known functions.37–40 From a technical point of view, our results suggest the importance of refining protein evidence by rigorous MS analysis combined with antibody-capture assays. Given the extremely doubtful specificity of commercial antibodies (mAb and pAb) that cross-bind to nonspecific proteins, we are also cautious about overreliance on Western blotting and emphasize the importance of subsequent MS reanalysis of antibody-captured bands. From a biological point of view, our strategy stresses complementary in vitro and in vivo investigations when seeking or validating a novel function of a protein. The full characterization of any candidate protein also requires cellular functional assays (in vitro) and molecular phenotype analysis (in vivo) regarding the disease or regulatory aspects of these proteins. Trophoblast differentiation has been proven to be extremely important in placental development through spiral artery remodeling by trophoblast migration and invasion.10,14 In our comparative transcript analysis of the trophoblasts of hPN and hPE women, the changes in gene expression within the functional network of a few key regulatory proteins proved to be closely related to the differentiation of trophoblasts and cell migration. The results of our in vitro study also support the possibility that NHERF1 regulates key proteins for trophoblast differentiation and motility factors such as β-hCG hormone, KRT7 and KRT18 (Figures 4B and 5B and Table S9). Together, our integrative analysis indicates that NHERF1 appears to regulate trophoblast differentiation by influencing placental hormone levels and cell migration factors.
Like other global projects, C-HPP emphasizes biomedical utility in the characterization of potential novel proteins known as transcript level in human disease research. In this respect, C. elegans has many advantages for the functional characterization of novel proteins over other vertebrate animals. These advantages include a short life cycle (3.5 days at 20 °C), ease of maintenance, and production of 300 eggs by self-fertilization.41 C. elegans mutants also mimic many morphological or functional defects that are encountered in aspects of human disease. Using rapid developments of mutants, the functional analysis of human orthologous genes can be analyzed by the rescue-transformation of mutant animals by microinjection.42 Like the mutant phenotypes of C. elegans nrfl-1, which is a human ortholog of NHERF1, most Pvl mutants have an egg-laying defective (Egl) phenotype.43 In mammalian studies, the NHERF1-null female mice yielded only two to four pups (they normally would have 8 to 10 pups) and showed defects of the intestinal villi and apical cyto-skeleton.44,45 Together, these data imply a close relationship between the phenotype exhibited by nrfl-1 (ok297) mutants of C. elegans (vulva abnormality and egg-laying defects) and that of its female mammalian counterpart. We confirmed that the Pvl mutant phenotypes were rescued under normal conditions by a recombinant human NHERF1 gene. Our findings on the potential roles of NHERF1 in reproductive processes (egg-laying and vulva development) suggest that the C. elegans nrfl-1(ok297) mutant strain serves as an experimental model organism for studying issues related to human embryo development and pregnancy (e.g., hPE).
CONCLUSIONS
As a first step toward the functional validation of candidate proteins, the antibody-captured signal bands corresponding to the target MP must be rigorously verified by MS analysis in accordance with HPP data guidelines5 (www.thehpp.org/guidelines) and followed by an extensive in vitro cellular study and in vivo genetic complementary workup (Figure 7). Because the functional study of MPs usually takes months to years and can result in significant gaps between the initiation or publication of a study and the time when a DB is released, the case of NHERF1 could be just one example of the functional characterization of the remaining 2579 MPs in the future.
Figure 7.
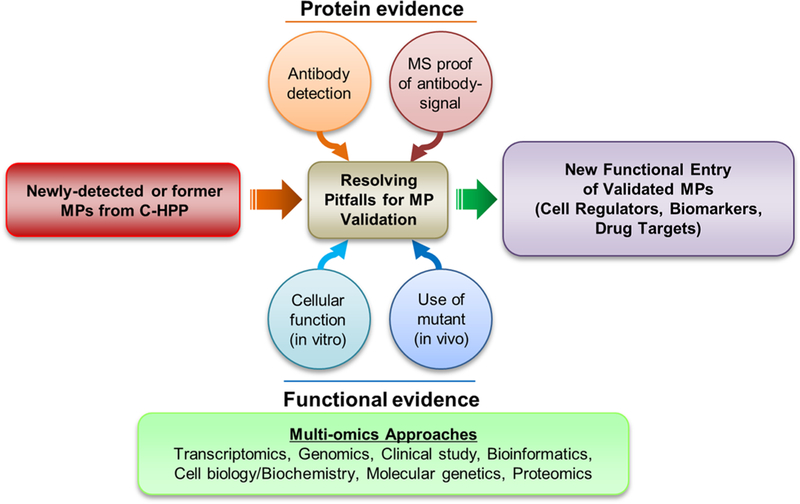
Potential solutions to pitfalls encountered during the physical and functional validation of MPs.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Korean Ministry of Health and Welfare (HI13C2098 and HI16C0257 to Y.-K.P.).
ABBREVIATIONS
- β-hCG
β-subunit of human chorionic gonadotropin
- CBX7
chromobox homologue 7
- C-HPP
Chromosome-Centric Human Proteome Project
- COX5A
cytochrome c oxidase subunit 5A
- DMSO
dimethyl sulfoxide
- eNOS
endothelial nitric oxide synthase
- FDR
false discovery rate
- FK
forskolin
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GO
gene ontology
- hPE
human preeclampsia
- hPN
human placenta normal
- IL-1β
interleukin-1β
- INPPL1
inositol polyphosphate-5 phosphatase-like 1
- KRT7
cytokeratin 7
- KRT18
cytokeratin 18
- MP
missing protein
- NHERF1/SLC9A3R1
Na+/H+ exchanger regulatory factor 1
- OAS2
2′-5′-oligoadenylate synthetase 2
- OAS3
2′-5′-oligoadenylate synthetase 3
- PE
protein evidence or existence
- PHF11
PHD finger protein 11
- PLAC9
placenta specific 9
- RNF121
ring finger protein 121
- TMT
tandem mass tags
- WST-1
water-soluble tetrazolium salts
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.7b00146.
The authors declare no competing financial interest.
REFERENCES
- (1).Paik YK; Jeong SK; Omenn GS; Uhlen M; Hanash S; Cho SY; Lee HJ; Na K; Choi EY; Yan F; Zhang F; Zhang Y; Snyder M; Cheng Y; Chen R; Marko-Varga G; Deutsch EW; Kim H; Kwon JY; Aebersold R; Bairoch A; Taylor AD; Kim KY; Lee EY; Hochstrasser D; Legrain P; Hancock WS The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat. Biotechnol 2013, 30, 221–223. [DOI] [PubMed] [Google Scholar]
- (2).Paik YK; Omenn GS; Overall CM; Deutsch EW; Hancock WS Recent advances in the Chromosome-Centric Human Proteome Project: missing proteins in the spot light. J. Proteome Res. 2015, 14, 3409–3414. [DOI] [PubMed] [Google Scholar]
- (3).Vizcaino JA; Deutsch EW; Wang R; Csordas A; Reisinger F; Rios D; Dianes JA; Sun Z; Farrah T; Bandeira N; Binz PA; Xenarios I; Eisenacher M; Mayer G; Gatto L; Campos A; Chalkley RJ; Kraus HJ; Albar JP; Martinez-Bartolome S; Apweiler R; Omenn GS; Martens L; Jones AR; Hermjakob H ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Paik YK; Overall CM; Deutsch EW; Hancock WS; Omenn GS Progress in the Chromosome-Centric Human Proteome Project as highlighted in the Annual Special Issue IV. J. Proteome Res. 2016, 15, 3945–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Deutsch EW; Overall CM; Van Eyk JE; Baker MS; Paik YK; Weintraub S; Lane L; Martens L; Vandenbrouck Y; Kusebauch U; Hancock WS; Hermjakob H; Aebersold R; Moritz RL; Omenn GS Human Proteome Project mass spectrometry data interpretation guidelines 2.1. J. Proteome Res. 2016, 15, 3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lane L; Bairoch A; Beavis RC; Deutsch EW; Gaudet P; Lundberg E; Omenn GS Metrics for the Human Proteome Project 2013–2014 and strategies for finding missing proteins. J. Proteome Res. 2014, 13, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Omenn GS; Lane L; Lundberg EK; Beavis RC; Overall CM; Deutsch EW Metrics for the Human Proteome Project 2016: Progress on Progress on identifying and characterizing the human proteome, including post-translational modifications. J. Proteome Res. 2016, 15, 3951–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jeong SK; Hancock WS; Paik YK GenomewidePDB 2.0: A newly upgraded versatile proteogenomic database for the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 14, 3710–3719. [DOI] [PubMed] [Google Scholar]
- (9).Burton GJ; Jauniaux E What is the placenta? Am. J. Obstet. Gynecol. 2015, 213, S3–5. [DOI] [PubMed] [Google Scholar]
- (10).Costa MA The endocrine function of human placenta: an overview. Reprod. BioMed. Online 2016, 32, 14–43. [DOI] [PubMed] [Google Scholar]
- (11).Goldman-Wohl DS; Yagel S Examination of distinct fetal and maternal molecular pathways suggests a mechanism for the development of preeclampsia. J. Reprod. Immunol. 2007, 76, 54–60. [DOI] [PubMed] [Google Scholar]
- (12).Noris M; Perico N; Remuzzi G Mechanisms of disease: Preeclampsia. Nat. Clin. Pract. Nephrol. 2005, 1, 98–114. [DOI] [PubMed] [Google Scholar]
- (13).Hutcheon JA; Lisonkova S; Joseph KS Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best. Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 391–403. [DOI] [PubMed] [Google Scholar]
- (14).Vatish M; Randeva HS; Grammatopoulos DK Hormonal regulation of placental nitric oxide and pathogenesis of pre-eclampsia. Trends Mol. Med. 2006, 12, 223–233. [DOI] [PubMed] [Google Scholar]
- (15).Kaufmann P; Black S; Huppertz B Endovascular trophoblast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reprod. 2003, 69, 1–7. [DOI] [PubMed] [Google Scholar]
- (16).Sun J; Nishiyama T; Shimizu K; Kadota K TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinformatics 2013, 14, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lee HJ; Jeong SK; Na K; Lee MJ; Lee SH; Lim JS; Cha HJ; Cho JY; Kwon JY; Kim H; Song SY; Yoo JS; Park YM; Kim H; Hancock WS; Paik YK Comprehensive genome-wide proteomic analysis of human placental tissue for the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2013, 12, 2458–2466. [DOI] [PubMed] [Google Scholar]
- (18).Park M; Lim JS; Lee HJ; Na K; Lee MJ; Kang CM; Paik YK; Kim H Distinct protein expression profiles of solid—pseudopapillary neoplasms of the pancreas. J. Proteome Res. 2015, 14, 3007–3014. [DOI] [PubMed] [Google Scholar]
- (19).Brenner S The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim S; Lee HJ; Hahm JH; Jeong SK; Park DH; Hancock WS; Paik YK Quantitative profiling identifies potential regulatory proteins involved in development from dauer stage to l4 stage in Caenorhabditis elegans. J. Proteome Res. 2016, 15, 531–539. [DOI] [PubMed] [Google Scholar]
- (21).Kim KY; Joo HJ; Kwon HW; Kim H; Hancock WS; Paik YK Development of a method to quantitate nematode pheromone for study of smallmolecule metabolism in Caenorhabditis elegans. Anal Chem. 2013, 85, 2681–2688. [DOI] [PubMed] [Google Scholar]
- (22).Kim MS; Pinto SM; Getnet D; Nirujogi RS; Manda SS; Chaerkady R; Madugundu AK; Kelkar DS; Isserlin R; Jain S; Thomas JK; Muthusamy B; Leal-Rojas P; Kumar P; Sahasrabuddhe NA; Balakrishnan L; Advani J; George B; Renuse S; Selvan LD; Patil AH; Nanjappa V; Radhakrishnan A; Prasad S; Subbannayya T; Raju R; Kumar M; Sreenivasamurthy SK; Marimuthu A; Sathe GJ; Chavan S; Datta KK; Subbannayya Y; Sahu A; Yelamanchi SD; Jayaram S; Rajagopalan P; Sharma J; Murthy KR; Syed N; Goel R; Khan AA; Ahmad S; Dey G; Mudgal K; Chatterjee A; Huang TC; Zhong J; Wu X; Shaw PG; Freed D; Zahari MS; Mukherjee KK; Shankar S; Mahadevan A; Lam H; Mitchell CJ; Shankar SK; Satishchandra P; Schroeder JT; Sirdeshmukh R; Maitra A; Leach SD; Drake CG; Halushka MK; Prasad TS; Hruban RH; Kerr CL; Bader GD; Iacobuzio-Donahue CA; Gowda H; Pandey A A draft map of the human proteome. Nature 2014, 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wilhelm M; Schlegl J; Hahne H; Moghaddas Gholami A; Lieberenz M; Savitski MM; Ziegler E; Butzmann L; Gessulat S; Marx H; Mathieson T; Lemeer S; Schnatbaum K; Reimer U; Wenschuh H; Mollenhauer M; Slotta-Huspenina J; Boese JH; Bantscheff M; Gerstmair A; Faerber F; Kuster B Mass-spectrometrybased draft of the human proteome. Nature 2014, 509, 582–587. [DOI] [PubMed] [Google Scholar]
- (24).Ahmadi Rastegar D; Sharifi Tabar M; Alikhani M; Parsamatin P; Sahraneshin Samani F; Sabbaghian M; Sadighi Gilani MA; Mohammad Ahadi A; Mohseni Meybodi A; Piryaei A; Ansari-Pour N; Gourabi H; Baharvand H; Salekdeh GH Isoform-level gene expression profiles of human Y chromosome Azoospermia factor genes and their X chromosome paralogs in the testicular tissue of non—obstructive Azoospermia patients. J. Proteome Res. 2015, 14, 3595–3605. [DOI] [PubMed] [Google Scholar]
- (25).Alm T; von Feilitzen K; Lundberg E; Sivertsson A; Uhlen M A chromosomecentric analysis ofantibodies directed toward the human proteome using Antibodypedia. J. Proteome Res. 2014, 13, 1669–1676. [DOI] [PubMed] [Google Scholar]
- (26).Sun YY; Lu M; Xi XW; Qiao QQ; Chen LL; Xu XM; Feng YJ Regulation of epithelial-mesenchymal transition by homeobox gene DLX4 in JEG-3 trophoblast cells: a role in preeclampsia. Reprod. Sci 2011, 18, 1138–1145. [DOI] [PubMed] [Google Scholar]
- (27).Glasser SR; Julian J; Munir MI; Soares MJ Biological markers during early pregnancy: trophoblastic signals of the periimplantation period. Environ. Health Perspect. 1987, 74, 129–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ji L; Brkic J; Liu M; Fu G; Peng C; Wang YL Placental trophoblast cell differentiation: physiological regulation and pathological relevance to preeclampsia. Mol. Aspects Med. 2013, 34, 981 –1023. [DOI] [PubMed] [Google Scholar]
- (29).Prutsch N; Fock V; Haslinger P; Haider S; Fiala C; Pollheimer J; Knofler M The role of interleukin-1β in human trophoblast motility. Placenta 2012, 33, 696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Muhlhauser J; Crescimanno C; Kasper M; Zaccheo D; Castellucci M Differentiation of human trophoblast populations involves alterations in cytokeratin patterns. J. Histochem. Cytochem. 1995, 43, 579–589. [DOI] [PubMed] [Google Scholar]
- (31).Hagiwara K; Nagamori S; Umemura YM; Ohgaki R; Tanaka H; Murata D; Nakagomi S; Nomura KH; Kage-Nakadai E; Mitani S; Nomura K; Kanai Y NRFL-1, the C. elegans NHERF orthologue, interacts with amino acid transporter 6 (AAT-6) for age-dependent maintenance of AAT-6 on the membrane. PLoS One 2012, 7, e43050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xie Z; Yang Z; Druey KM Phosphorylation of RGS13 by the cyclic AMPdependent protein kinase inhibits RGS13 degradation. J. Mol. Cell Biol. 2010, 2, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Johnson EN; Druey KM Functional characterization of the G protein regulator RGS13. J. Biol. Chem. 2002, 277, 16768–16774. [DOI] [PubMed] [Google Scholar]
- (34).Xie Z; Geiger TR; Johnson EN; Nyborg JK; Druey KM RGS13 acts as a nuclear repressor of CREB. Mol. Cell. 2008, 31, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bansal G; DiVietro JA; Kuehn HS; Rao S; Nocka KH; Gilfillan AM; Druey KM RGS13 controls g protein-coupled receptor-evoked responses of human mast cells. J. Immunol. 2008, 181, 7882–7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Iwaki S; Lu Y; Xie Z; Druey KM p53 negatively regulates RGS13 protein expression in immune cells. J. Biol. Chem. 2011, 286, 22219–22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Reczek D; Berryman M; Bretscher A Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J. Cell Biol. 1997, 139, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kreimann EL; Morales FC; de Orbeta-Cruz J; Takahashi Y; Adams H; Liu TJ; McCrea PD; Georgescu MM Cortical stabilization of beta-catenin contributes to NHERF1/EBP50 tumor suppressor function. Oncogene 2007, 26, 5290–5299. [DOI] [PubMed] [Google Scholar]
- (39).Pan Y; Weinman EJ; Dai JL Na+/H+ exchanger regulatory factor 1 inhibits platelet-derived growth factor signaling in breast cancer cells. Breast Cancer Res. 2008, 10, R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Li J; Poulikakos PI; Dai Z; Testa JR; Callaway DJ; Bu Z Protein kinase C phosphorylation disrupts Na+/H+ exchanger regulatory factor 1 autoinhibition and promotes cystic fibrosis transmembrane conductance regulator macromolecular assembly. J. Biol. Chem. 2007, 282, 27086–27099. [DOI] [PubMed] [Google Scholar]
- (41).Culetto E; Sattelle DB A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum. Mol. Genet. 2000, 9, 869–877. [DOI] [PubMed] [Google Scholar]
- (42).Mello CC; Kramer JM; Stinchcomb D; Ambros V Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991, 10, 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Eisenmann DM; Kim SK Protruding vulva mutants identify novel loci and Wnt signaling factors that function during Caenorhabditis elegans vulva development. Genetics 2000, 156, 1097–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shenolikar S; Voltz JW; Minkoff CM; Wade JB; Weinman EJ Targeted disruption of the mouse NHERF-1 gene promotes internalization ofproximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc. Natl. Acad. Sci. USA. 2002, 99, 11470–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Morales FC; Takahashi Y; Kreimann EL; Georgescu MM Ezrin-radixinmoesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc. Natl. Acad. Sci. USA. 2004, 101, 17705–17710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.