

Abstract
Given that the cDNA of F8 is too large to be packaged into adeno-associated virus (AAV) capsids, gene transfer of some versions of B-domain-deleted F8 (BDD-F8) for hemophilia A (HA) treatment has been attempted with promising results. Here, we describe an efficient gene correction via single-stranded-oligodeoxynucleotide (ssODN)-mediated in-frame deletion within the B domain of F8 with CRISPR/Cas9 in HA-patient-derived induced pluripotent stem cells (HA-iPSCs). The expression and activity of FVIII was restored in corrected HA-iPSC-derived induced endothelial progenitor cells (C-iEPCs) in vitro and in vivo. The bleeding phenotype was rescued in HA mice after C-iEPC infusion. Our results demonstrate an efficient approach for in situ gene correction via introduction of a tiny deletion using ssODN and CRISPR/Cas9 to reframe the F8 transcript and restore FVIII function in HA-iPSC-derived EPCs with potential clinical impact in HA gene therapy. For the first time, we demonstrated in vitro and in vivo the FVIII function that is encoded by the endogenous F8 gene with a partially deleted B domain. This work also suggests an applicable strategy for genetic correction of other gene frameshift mutations.
Keywords: hemophilia A, CRISPR/Cas9, ssODN, iPSCs, in-frame deletion, gene therapy
Graphical Abstract
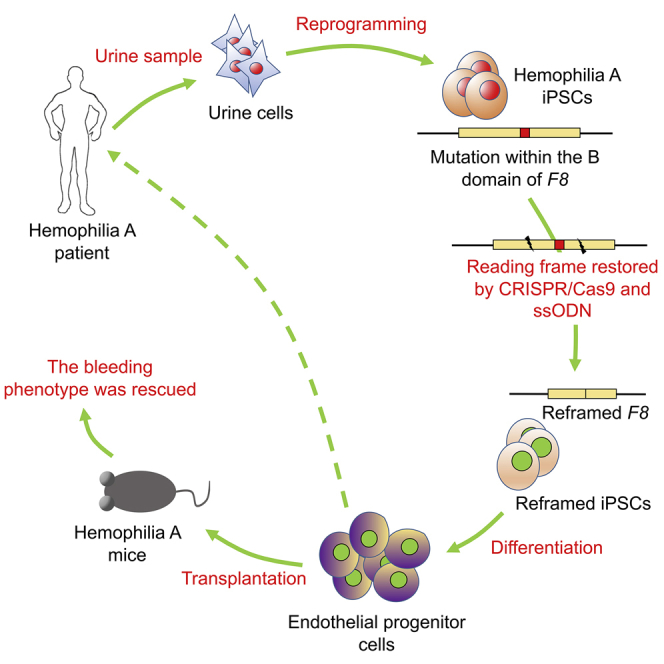
Introduction
Hemophilia A (HA) is an X-linked recessive genetic bleeding disorder (incidence of 1 in 5,000 male births).1 It is caused by genetic mutations in human coagulation factor 8 (F8). The Human Gene Mutation Database (HGMD)2 reports 3,246 mutations (475 in the B domain) of F8 that cause HA. Approximately 90.7% of the B domain mutations result in premature termination.
Traditionally, HA is treated by replacement therapy with repeated infusions of plasma-derived or recombinant factor VIII (FVIII) protein. However, antibody formation and the high cost of repeated infusions limit this method. Gene therapy is a promising option for curing hemophilia. In vivo gene therapy for hemophilia B (HB) has shown therapeutic effects in clinical trials using a recombinant adeno-associated viral (rAAV) vector carrying the F9 gene.3 For gene therapy for HA, which accounts for 80%–85% of all hemophilia cases, the F8 cDNA (7 kb) is too large for packaging into the AAV capsids. Approaches using B-domain-deleted F8 (BDD-F8) have been tested, with some having shown therapeutic effects in animal models4 and some having proceeded to clinical trials.5 However, the potential for immune responses, transduction of nontarget cells, and random insertional mutagenesis continues to hinder its clinical application.6 On the other hand, as genome editing is emerging as an ideal approach for manipulation of genes in situ, studies have focused on ex vivo gene therapy for HA after targeted genetic modification of pluripotent stem cells, including in situ gene correction,7 reversion8, 9 for the F8 inversion mutations, and ectopic integration of BDD-F8 at the rDNA locus,10 some of which have been shown to restore FVIII function in cells or in HA mice.
The CRISPR/Cas9 system has been used for genome editing, including chromosomal segment deletion,11 elimination,12 rearrangement,13, 14 and precise gene mutations.15, 16 Combination of CRISPR/Cas9 with single-stranded oligodeoxynucleotide (ssODN) has been shown to be efficient at inducing gene addition and precise gene mutation.17, 18 However, there has been no report on ssODN-mediated precise deletion with CRISPR/Cas9, whereas efficient ssODN-mediated targeted gene deletion with transcription-activator-like effector nucleases (TALENs) has been described.19
Because of its unlimited proliferation ability and multidirectional differentiation potential, induced pluripotent stem cells (iPSCs) have become ideal targets for ex vivo gene therapy. Liver sinusoidal endothelial cells (LSECs) are highly specialized endothelial cells (ECs) that produce and secret FVIII in humans.20, 21 ECs express von Willebrand factor (vWF), which is stored in Weibel-Palade bodies as a carrier protein to stabilize FVIII.22 Studies have shown that iPSC-derived endothelial progenitor cells (iEPCs) are integrated into liver sinusoids after intrahepatic injection in mice, resulting in therapeutic levels of FVIII production.23
In this study, to avoid invasive sampling, urine cells were isolated from a patient with severe HA (4-bp frameshift deletion of the B domain in exon 14 of F8, which resulted in premature termination of translation), and reprogrammed into iPSCs (HA-iPSCs). Using CRISPR/Cas9 and ssODN, we achieved an efficient reframing of the 4-bp frameshift deletion via targeted deletion of 50 bp within the F8 B domain in HA-iPSCs. FVIII expression and activity were demonstrated in reframed iPSC-derived EPCs (iEPCs) in vitro and in vivo.
Results
Generation and Characterization of HA-iPSCs
First, we genotyped and identified an F8 frameshift mutation of c.3167del CTGA in a 21-year-old male patient with severe HA (Figure 1A). Given that invasive biopsy should be avoided in bleeding disorders, and isolation of urinary cells is simple, cost-effective, and universal,24 we collected urine samples from the patient to expand the urine cells. The urine cells appeared cobblestone-like (Figure S1) and expressed markers of renal tubular epithelial cells, zonula occludens-protein 1 (ZO-1), intermediate filament keratin 7 (KRT7), and β-catenin (Figure S1). These results are consistent with those described in previous reports on the cultivation of human renal tubular epithelial cells from the urinary tract and the renal tubular system.25, 26 We then generated HA-iPSCs from the renal tubular epithelial cells using retroviral vectors carrying four Yamanaka factors (OCT4, Sox2, Klf4, and c-Myc).25, 27 After induction, the human embryonic stem cell (ESC)-like clones were isolated for further characterization (Figure 1B). We examined the pluripotency and genetic stability of the iPSC line, using immunofluorescence, teratoma formation, and karyotyping. The results showed that the HA-iPSCs were karyotypically normal (Figure 1C) and maintained the mutated genotype. Normal iPSCs (N-iPSCs) purchased from ATCC were used as a control (Figure 1D). The HA-iPSCs expressed OCT4, NANOG, and stage-specific embryonic antigen (SSEA)-4, but did not express SSEA-1, which was consistent with the expression observed in human ESCs (hESCs) (Figure 1E). Spontaneous differentiation in the center of the HA-iPSC colony was occasionally observed during iPSCs maintenance, as it is in hESCs. The HA-iPSCs formed teratomas in vivo that contained all three germ layers, including respiratory epithelium (endoderm), cartilage tissue (mesodermal), and neuroepithelial tissue (ectoderm) (Figure 1F).
Figure 1.

Generation and Characterization of c.3167del CTGA HA-iPSCs
(A) Molecular diagnosis of the F8 frameshift mutation c.3167del CTGA using Sanger sequencing. The arrow indicates the location of the deletion. (B) Bright field image of a representative iPSC clone. Scale bar, 500 μm. (C) Karyotyping of HA-iPSCs is shown. (D) Sanger sequencing analysis of HA-iPSCs. N-iPSCs were used as the normal control. (E) Immunofluorescence showed that iPSCs expressed the markers NANOG, OCT4, and SSEA-4 and did not express the differentiation marker SSEA-1. (F) H&E staining of a teratoma derived from HA-iPSCs that included three germ layers: endoderm (respiratory epithelium), mesoderm (cartilage), and ectoderm (neural tissue). Scale bar, 50 μm.
CRISPR/Cas9 and ssODN-Mediated Targeted Deletion in F8 of HA-iPSCs
CRISPR/Cas9 and ssODN were used to create an in-frame deletion. The designation of single-guide RNA (sgRNA) is restricted by the protospacer adjacent motif (PAM) sequence. In this case, the minimal deletion that could be achieved is 54 bp, including the 4-bp frameshift deletion of the B domain in exon 14 of the F8 gene (Figure 2A). The nearest dual-sgRNAs mapping to the target sites were designed,28 constructed, and verified to possess cleavage activity via sequencing and the T7 endonuclease I (T7E1) assay (Figure S2; Table S1). The cleavage activities of F8-E14-sg1 and F8-E14-sg2 were 41.30% and 18.92%, respectively (Figure 2B). The corresponding ssODN template was synthesized directly by Sangon Biotech. To perform gene targeting, plasmids expressing sgRNAs and the CRISPR/Cas9 complex and ssODN were nucleofected into the HA-iPSCs. After nucleofection with CRISPR/Cas9 and ssODN, the HA-iPSCs were dissociated into single cells, plated on murine embryonic fibroblast (MEF) feeder cells without drug selection, and then picked and expanded. These clones were initially screened via PCR. Then, the in situ gene-corrected iPSCs (C-iPSCs) were verified by Sanger sequencing. Of the 34 analyzed clones, 5 were identified as the corrected clones, indicating high targeting efficiency (14.7%) without screening, but only 1 positive clone was found among the 48 clones in the non-ssODN group (Table S2). We randomly selected two stably corrected clones—2-6-iPSC and 2-46-iPSC—for further research, in which the targeted deletion of 50 bp was verified by PCR (Figure 2C) and Sanger sequencing (Figure 2D). In addition, the 2-6-iPSCs and 2-46-iPSCs were shown to be pluripotent by immunofluorescence (Figure 2E) and were karyotypically normal (Figure 2F). Spontaneous differentiation in the center of the C-iPSC colony was observed during iPSC maintenance, as in the HA-iPSCs. The percentage of differentiated colonies was lower than 5% in the HA-iPSCs, N-iPSCs, and C-iPSCs, as more than 95% of the cell population was positive for the stem cell marker OCT4. A premature F8 mRNA was detected, indicating that there was no nonsense-mediated mRNA decay (NMD) of the reframed mRNA in the HA-iPSCs and C-iPSCs (Figure S3A), which is consistent with a previous report.29 FVIII expression was detected in cell lysates, but almost none was detected in the supernatant of the HA-iPSCs, C-iPSCs, and N-iPSCs via ELISA, indicating that FVIII was not secreted at the iPSC stage (Figure S3B). It has been reported that the complex formed by lectin mannose-binding 1 (LMAN1) and multiple coagulation factor deficiency 2 (MCFD2) mediates the secretion of FVIII, and mutation of LMAN1 or MCFD2 leads to a lack of both FV and FVIII.30 We detected LMAN1 by western blot analysis and found that the LMAN1 protein was absent in human iPSCs but was expressed in human iECs (Figure S3C), indicating that there was no secretion of FVIII at the iPSC stage.
Figure 2.

Generation of Corrected HA-iPSCs Mediated by ssODN with CRISPR/Cas9
(A) Schematic representation of CRISPR/Cas9-mediated precise deletion. The design of two sgRNAs for F8 exon 14 was shown. The red dotted line indicates c.3167del CTGA. Green letters indicate the sgRNA sequences, and blue letters indicate the protospacer adjacent motif (PAM). The red arrow represents the predicted CRISPR/Cas9 cleavage sites. The reframed F8 exhibits a precise 50-bp deletion and correction of the reading frame of frameshift mutations in this region. The vertical line indicates the upstream and downstream connections. Yellow-shaded bases are left homologous sequences, and gray-shaded bases are right homologous sequences. (B) Sanger sequencing of the indels induced by F8-E14-sg1 or F8-E14-sg2 in HA-iPSCs. WT, wild-type; Δ, deletion; +, insertion; ×, times. The black arrow indicates the position of insertion. The blue-labeled bases are the PAM sequences of CRISPR/Cas9, the target sequence of sgRNA is shown in green, and red indicates inserted bases. (C) PCR-based screening of C-iPSCs. The sizes of the PCR products were 448 bp for the 50-bp deletion, 502 bp for the N-iPSCs, and 498 bp for the HA-iPSCs, amplified using the primer pair F8-E14-F/R. (D) The PCR products were sequenced by Sanger sequencing. (E) Each C-iPSC line (2-6-iPSCs and 2-46-iPSCs) expressed SSEA-4, NANOG, and OCT4 and did not express SSEA-1, as determined by immunofluorescence. DAPI was used for nuclear staining. (F) Karyotypes of C-iPSC (2-6-iPSC and 2-46-iPSC) lines are shown.
No Off-Target Sites Were Observed Among the Predicted Sites in C-iPSCs
To analyze the off-target activity of CRISPR/Cas9, genomic DNA was isolated from the HA-iPSCs and C-iPSCs (2-6-iPSCs and 2-46-iPSCs). We selected and designed primers to amplify the five potential off-target sites for F8-E14-sg1 and F8-E14-sg2, respectively, that were predicted by the Optimized CRISPR Design website (Tables S3, S4, and S5).28 Then, the DNA of the C-iPSCs was amplified by PCR and sequenced, and no indels were observed upon comparison with the HA-iPSCs (Figures S4 and S5). Meanwhile, unintended mutations after CRISPR/Cas9 editing in the C-iPSCs (2-6-iPSCs and 2-46-iPSCs) were investigated by comparison with the HA-iPSCs, using whole-exome sequencing (WES). We focused on indels rather than single nucleotide variants (SNVs) based on the finding that programmable nucleases rarely generate point mutations.31, 32 First, we identified indels in the C-iPSCs but not in the HA-iPSCs; there were 167 unique indels in the 2-6-iPSCs and 189 in the 2-46-iPSCs. Next, potential off-target sites for mismatches with on-target sites of CRISPR/Cas9 of up to five nucleotides (Table 1) were searched for by using Cas-OFFinder (http://www.rgenome.net/).32 Finally, no off-target indels were identified upon comparison of the potential off-target sites with the indel locations identified by WES (Table 2). Consistent with a previous report,33 these results indicated that there were no indel off-target mutations caused by CRISPR/Cas9 in the C-iPSCs in this study.
Table 1.
Potential Off-Target Sites with up to Five Mismatches Identified by CAS-OFFinder
| Target Sequence | Mismatch | Number of Targets Identified |
|---|---|---|
| GGCATTCTGTCATGAATCAANRG | 1 | 1 |
| GGCATTCTGTCATGAATCAANRG | 2 | 3 |
| GGCATTCTGTCATGAATCAANRG | 3 | 26 |
| GGCATTCTGTCATGAATCAANRG | 4 | 329 |
| GGCATTCTGTCATGAATCAANRG | 5 | 3,125 |
| GTGAGAATAGTCCATCAGTCNRG | 3 | 11 |
| GTGAGAATAGTCCATCAGTCNRG | 4 | 150 |
| GTGAGAATAGTCCATCAGTCNRG | 5 | 1,803 |
Table 2.
Variants of iPSCs Identified Using WES
| Clone | HA-iPSCs | 2-6-iPSCs | 2-46-iPSCs |
|---|---|---|---|
| All variants | 31,245 | 30,842 | 31,381 |
| All indels | 1,741 | 1,655 | 1,750 |
| Reframed clone-specific indels | N/A | 167 | 189 |
| Possible off-target sites after alignment with predicted off-target sites | N/A | 0 | 0 |
N/A, not applicable.
FVIII Expression Was Restored in C-iPSC-Derived ECs
The 50-bp targeted deletion was expected to reframe the 4-bp frameshift deletion in the F8 B domain and restore the function of the FVIII protein. Because human ECs can secrete FVIII, the C-iPSCs (2-6-iPSCs and 2-46-iPSCs), HA-iPSCs, and N-iPSCs were differentiated into C-iECs, HA-iECs, and N-iECs (Figure 3A) via the optimized OP9 coculture method, to examine the recovery of FVIII secretion.34 A deleted F8 transcript was detected, indicating that there was no NMD of the 54-bp deletion mRNA in the C-iECs (2-6-iECs and 2-46-iECs) (Figure 3B). ELISA results showed that FVIII secretion in the C-iECs (2-6-iECs and 2-46-iECs) was higher than that in the C-iPSCs, similar to that in the N-iECs, and higher than that in the HA-iECs (Figure 3C). Meanwhile, FVIII activity was measured in the culture supernatant of the C-iECs (2-6-iECs and 2-46-iECs), but not in that of the HA-iECs (Figure 3D), indicating that FVIII function was restored in the C-iECs.
Figure 3.

Restoration of FVIII Expression in C-iECs
(A) Immunofluorescence staining of iECs expressing markers for CD31 (red) and CD144 (green). DAPI was used to visualize the nucleus. (B) RT-PCR analysis of F8 expression in iECs. Primers based on exon 14 (E14) and primers based on exons 23 and 26 (E23–E26) were used to detect F8 expression. GAPDH was used as a loading control. (C) FVIII antigen levels in ECs derived from iPSCs were measured by ELISA. Bars represent the mean ± SEM (n = 3, independent cultures). (D) FVIII activity in the supernatants of differentiated ECs. The FVIII activity of the HA-iECs was lower than the detection limit of the instrument. No significant difference in FVIII activity was observed between C-iECs (2-6-iECs and 2-46-iECs) and N-iECs. Bars represent the mean ± SEM (n = 3, independent cultures). ns, not significant, compared with N-iECs.
Restoration of FVIII Expression and Secretion in C-iPSC-Derived EPCs
Using a previously described method,35 we differentiated C-, HA-, and N-iPSCs into C-iEPCs, HA-,iEPCs and N-iEPCs via small-molecule-mediated activation of WNT signaling; flow cytometric analysis showed that the differentiation efficiency was between 10% and 20% (Figure 4A). The iEPCs were sorted by a CD31 microbead-based antibody; characterized using immunostaining of CD34 (sc-74499; Santa Cruz), CD31 (P8590l; Sigma-Aldrich), and CD144 (ab33168; Abcam) (Figure 4B); and functionally verified by angiogenesis (Figure S6A) and Dil-acetylated-low-density-lipoprotein (Dil-AcLDL) endocytosis (Figure S6B). RT-PCR was used to distinguish the premature F8 transcript from the 4-bp-deleted F8, indicating that there was no NMD in the HA-iEPCs (Figure 4C). ELISA results showed that the 2-6- and 2-46-iEPCs secreted FVIII at levels of 1.21 ng/106 cells and 0.66 ng/106 cells, respectively, which were higher than the levels in the HA-iEPCs (0.24 ng/106 cells) and similar to those in the N-iEPCs (0.46 ng/106 cells) (Figure 4D). After cultivation for 1 week, the sorted iEPCs expressed vWF (ab6994; Abcam), indicating that mature iECs were formed from the iEPCs (Figure 4E). FVIII in the C-iECs (2-6-iECs and 2-46-iECs) and the N-iECs was detected by both an N-terminal FVIII antibody (sc-27649; Santa Cruz) (Figure 4F) and a C-terminal FVIII antibody (ESH-8; Sekisui Diagnostics) (Figure 4G), whereas the truncated FVIII in the HA-iEPCs was detected by only the N-terminal FVIII antibody (Figure 4F).
Figure 4.

FVIII Expression Was Restored in C-iPSC-Derived EPCs
(A) FACS analysis of the expression of the iEPC markers CD31 and CD34 on day 5 after differentiation. (B) Immunofluorescence staining of CD34 (red), CD31 (red), and CD144 (green) in C-iEPCs. Scale bar, 25 μm. (C) RT-PCR analysis of F8 expression in iEPCs. Primers based on exon 14 (E14) and primers based on exons 23 and 26 (E23–26) were used to detect F8 expression. GAPDH was used as a loading control. (D) FVIII antigen in iEPCs differentiated from hiPSCs was detected by ELISA. Bars represent the mean ± SEM (n = 3, independent cultures). (E) Immunostaining of endothelial markers CD31 and vWF in the mature iECs from iEPCs. (F and G) F8 expression in mature ECs derived from HA-iPSCs (HA-iECs), C-iPSCs (2-6-iECs and 2-46-iECs), and normal controls (N-iECs) was detected via immunostaining by an (F) N-terminal or (G) C-terminal FVIII antibody. DAPI was used for nuclear staining. FVIII-N, F8 protein, N-terminal. FVIII-C, F8 protein, C-terminal. Scale bar, 25 μm.
Rescue of FVIII Deficiency and the Bleeding Phenotype Via C-iEPC Transplantation in HA Mice
To validate the therapeutic effects in vivo, HA-iEPCs, C-iEPCs (2-6-iEPCs and 2-46-iEPCs), and N-iEPCs were infused into HA mice via the retro-orbital vein. Two weeks after infusion, a tail-clip assay was performed, as previously described9; the evaluation duration of the tail-clip challenge experiment is 48 h. All the untreated HA mice (n = 9), as well as those receiving transplanted HA-iEPCs (n = 9), died soon after the tail-clip challenge (Figure 5A). The average survival time was 5.73 h (range, 1.97–10.67 h) and 5.42 h (range 1.66–8.38 h), respectively (Figure 5B). Although the average survival time in HA mice treated with the cells derived from the C-iPSCs (2-6-iEPCs and 2-46-iEPCs) was 16.83 h (range, 7.67–29.50 h) and 15.70 h (7.47–30.63 h), which exhibited a significant increase compared to non-treated HA mice (Figure 5B). Notably, 4 of the 12 HA mice with 2-6-iEPCs and 4 of the 10 HA mice with 2-46-iEPCs were alive 2 days after the tail-clip challenge, which was the endpoint of this experiment. Then, the mouse plasma was collected and assayed for FVIII activity, which is reported here as a percentage of the activity in wild-type mice. The FVIII activity in HA mice with C-iEPCs was 12.74% and 14.09%, which was significantly higher than that in HA mice (6.41%) (Figure 5C). The livers of the mice with 2-46-iEPCs that survived the challenge were collected on day 7 after the tail-clip challenge (liver collection was performed during week 3 after the 2-46-iEPC injection) to detect human cells by using immunofluorescence, and the results showed that anti-human vWF-positive cells homed to the livers of HA mice injected with 2-46-iEPCs but not in livers of HA mice with Dulbecco’s phosphate-buffered saline (DPBS). Liver collection was performed during week 2 after DPBS injection. (Figures 5D and 5E). Anti-human vWF-positive cells were also detected in the lungs, but not in the spleens, hearts, kidneys, or brains of HA mice with 2-46-iEPCs (Figure 5E).
Figure 5.

Functional Rescue of FVIII Deficiency in HA Mice by C-iEPCs
(A) Proportions of surviving mice after the tail-clip challenge. HA mice, mice with hemophilia A (n = 9); HA-iEPCs, mice with hemophilia A, receiving transplanted cells derived from HA-iPSCs (n = 9); 2-6-iEPCs, mice with hemophilia A, receiving transplanted cells derived from 2-6-iPSCs (n = 12); 2-46-iEPCs, mice with hemophilia A, receiving transplanted cells derived from 2-46-iPSCs (n = 10); N-iEPCs, mice with hemophilia A, receiving transplanted cells derived from N-iPSCs (n = 9). ns, not significant compared with HA mice; ***p < 0.001 compared with the HA-iEPC group (log-rank test). (B) Average survival time of mice after the tail-clip challenge. Notably, 4 of the 12 HA mice that received 2-6-iEPC transplants and 4 of the 10 HA mice that received 2-46-iEPC transplants and survived the challenge were excluded from this analysis. Bars represent the mean ± SEM; ns, not significant compared with HA mice. ***p < 0.001 and **p < 0.01 compared with the HA-iEPCs. (C) Relative FVIII activity was determined in plasma from HA mice with no transplant (n = 6) and HA mice with a transplant of Dulbecco’s phosphate-buffered saline (DPBS) (n = 6), HA-iEPCs (n = 6), N-iEPCs (n = 8), or C-iEPCs (2-6-iEPCs and 2-46-iEPCs) (n = 8). Bars represent the mean ± SEM. ***p < 0.001 compared with HA-iEPCs. (D) Livers of HA mice with 2-46-iEPCs that survived the tail-clip challenge were collected and analyzed using tissue immunofluorescence with an anti-human vWF antibody, and the results indicated that the transplanted human cells homed to the mouse liver. No signal was detected in the negative control (DPBS) group. CD31 (red); vWF (green). DAPI was used for nuclear staining. (E) Tissue immunofluorescence analysis of main organs of mice with 2-46-iEPCs, showing that anti-human vWF-positive cells were detected in the livers and lungs, but not in the spleens, hearts, kidneys, or brains. CD31 (red); vWF (green). DAPI was used for nuclear staining.
Discussion
The function of the B domain of the F8 gene remains unclear.36 Previous studies have suggested that the F8 B domain is associated with intracellular synthesis,37 secretion,38, 39, 40 activation,41, 42 inactivation,43 and clearance44 of FVIII. However, recombinant B domain-deleted FVIII (BDD-FVIII) has been demonstrated to be effective in clinical applications.45, 46 Gene addition of BDD-F8 cDNA rescued the phenotype in HA model mice,47, 48 and a phase I/II clinical trial demonstrated the effectiveness of AAV5-BDD-F8 gene transfer.5 In the present study, a 4-bp frameshift deletion within the B domain in exon 14 of F8 resulted in premature termination of FVIII translation or FVIII truncation, leading to a severe phenotype in the patient. Theoretically, in-frame deletion (in multiples of 3) of base pairs that include the 4-bp deletion within the B domain will reframe the F8 frameshift mutation and restore FVIII function. We therefore precisely induced a 54-bp deletion that included the intrinsic 4-bp deletion within the B domain to reframe the F8 open reading frame (ORF) in HA-iPSCs using ssODN and CRISPR/Cas9. FVIII expression and secretion were restored in C-iECs and C-iEPCs, and FVIII activity and the bleeding phenotype were rescued in HA mice after transplantation of C-iEPCs. Our findings indicated that deletion of 54 bases (18 amino acids) in the B domain did not affect FVIII function. We used the Swiss Model to predict protein structure,49, 50 showing that the predicted N-terminal and C-terminal structures of the FVIII protein from the 54-bp-deleted F8 gene were both identical to those of the wild-type FVIII protein, further supporting the effectiveness of our strategy.
HA has long been identified as an ideal target for gene therapy because 1%–5% of normal plasma FVIII can significantly improve the quality of life of patients. In situ gene correction enables expression of the corrected gene under the regulation of endogenous promoter and cis-acting elements.51, 52 Some studies53, 54 have shown that knocking out some exons of the DMD gene in iPSCs derived from Duchenne muscular dystrophy (DMD) patients effectively reduced DMD phenotypes at the cellular and whole-mouse levels. In this study, our strategy to reframe a frameshift mutation by deleting some tiny sequence was not only an effective in situ genetic repair mechanism, but also greatly improved the targeting efficiency, exhibiting potential for future in vivo applications. Although it addresses only specific cases, this study provides a reference for the repair of other gene mutations that lead to premature termination of translation, further expanding the range of applications.
The human LSEC is the main cell type that produces and secretes FVIII.20, 21 For HA therapy, transplantation of mature murine LSECs into HA mice via the portal vein has been reported to improve FVIII activity.55 Mature ECs cannot proliferate to maintain long-term therapeutic effects in vivo after cell transplantation. Given that EPCs can either proliferate or mature into ECs that secrete FVIII and that no tumorigenesis from adult EPCs engraftments has been reported,56, 57 EPCs are clearly a promising cell type for ex vivo HA therapy. In addition, human leukocyte antigen (HLA) matching is very strict for EPCs; therefore, transplantation of autologous EPCs and/or ECs as individualized treatments also leads to satisfactory therapeutic effects in clinical applications.58 In the present study, we intravenously infused C-iEPCs into HA mice and demonstrated therapeutic effectiveness in vivo. Interestingly, the infused human iEPCs exhibited homing to the livers of mice. Although the number of homed C-iEPCs was relatively small, the bleeding phenotype in HA mice was rescued, which was consistent with a previous report in which substantial levels of human factor IX (hFIX) were obtained, even though hF9 mRNA accounted for a small fraction (0.5%) of the total wild-type mouse albumin (mAlb) transcript.59
Materials and Methods
Generation of HA-iPSCs
Sterile urine was collected from a 21-year-old male patient who was diagnosed with severe HA. Cells from the urine sample were isolated and expanded. Briefly, after centrifugation at 170 × g for 10 min at room temperature, the urine sample was washed twice with DPBS, and the cells from the urine sample were plated on gelatin-coated 12-well plates using primary medium (DMEM/F12 supplemented with 10% fetal bovine serum [FBS]; SingleQuot Kit; Lonza, Alpharetta, GA, USA) and 1% penicillin-streptomycin). After 4 days of cultivation with primary medium, the urine cells were expanded in renal epithelial basal medium (Lonza) supplemented with Renal Epithelial Growth Medium (REGM) from the SingleQuot Kit. The urine cells were stained with β-catenin (BD Transduction Laboratories, Lexington, KY, USA), KRT7 (Gene Tex, Irvine, CA, USA), and ZO-1 (Thermo Fisher Scientific, Waltham, MA, USA). The cells were then reprogrammed to iPSCs by virus containing pMXs-hSox2, pMXs-hOCT4, pMXs-hKlf4, pMXs-hc-Myc, and pCL-Eco (all from Addgene, Cambridge, MA, USA). The detailed procedures for viral packaging were the same as those described in a previous report.7 In brief, HEK293T cells were seeded on six-well plates at a density of 80,000/cm2. When confluence reached 80%, the cells were transfected with pMXs vectors (pMXs-hSox2, pMXs-hOCT4, pMXs-hKlf4, pMXs-hc-Myc, and pCL-Eco), using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA); 2.5 μg of the pCL-Eco packaging vector and 2.5 μg of the corresponding pMXs vector were used per well. On the day before infection, renal tubular epithelial cells were seeded on six-well plates at a density of 6,000/cm2 and infected with viral supernatants that were collected 48 and 72 h after transfection in the presence of 8 μg/mL polybrene (Sigma-Aldrich, Burlington, MA, USA). After incubated for 24 h at 37°C, 5% CO2, the renal tubular epithelial cells were infected with viral supernatants for a second infection. Then, the cells were trypsinized and seeded at 100,000 cells per 10-cm dish containing feeders in ESC medium 4 days after infection. After 2 weeks, ESC-like clones were picked up and expanded for further characterization. All experimental protocols were approved by the Ethics Committee of the Center for Medical Genetics of Central South University, and the participant in this study provided and signed written informed consent. The N-iPSCs were purchased from ATCC (DYR0100, https://www.atcc.org/Products/All/ACS-1011.aspx).
Characterization of iPSCs
iPSC surface markers were identified by immunofluorescent staining. Cells were seeded on chamber slides and fixed in 4% paraformaldehyde for 15 min and then permeabilized with 0.1% Triton X-100 in DPBS for 15 min. The cells were blocked in 5% BSA for 30 min and then incubated with primary antibody diluted 1:100 in blocking buffer (OCT4; ab181557; Abcam), NANOG (ab109250; Abcam), SSEA-1 (SCR001; Merck Millipore), SSEA-4 (SCR001; Merck Millipore) at room temperature for 1 hour. After a thorough washing, the cells were blocked for 30 min again and then treated with secondary antibodies for 1 hour. Nuclei were stained with DAPI. Stained cells were photographed with a fluorescence microscope (Leica DM IRB, Wetzlar, Germany). Teratoma formation experiments were performed to test the multilineage differentiation potential of the cells. Five million cells were dissociated with 0.05% trypsin/EDTA (Gibco, Grand Island, NY, USA), resuspended in 140 μL DMEM/F12 and 70 μL Matrigel (Corning, Corning, NY, USA) and injected into the groins of nude mice. The formed teratoma were harvested approximately 8 weeks later, fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, and stained with H&E. All procedures regarding the care and use of animals were performed in accordance with institutional guidelines. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Center for Medical Genetics of Central South University.
Karyotyping
After being treated with 0.1 μg/mL colcemid (Sigma-Aldrich, Burlington, MA) for 4 h, cells were trypsinized, pelleted, and incubated in 0.075 M KCl for 10 min at 37°C. After fixation with Carnoy fixative, metaphase chromosome spreads were prepared by using the air-drying method. The chromosomes were G-banded by Giemsa staining (Sigma-Aldrich) after appropriate baking at 75°C and digestion with trypsin.
Construction of CRISPR/Cas9 Plasmids and Detection of Cleavage Activity
Two sgRNAs were designed to target human F8 exon 14 using Optimized CRISPR Design (http://zlab.bio/guide-design-resources/)28 and then verified by using the CHOPCHOP website (http://chopchop.cbu.uib.no/)60 and CRISPR RGEN tool (http://www.rgenome.net/cas-designer/).61 The synthetic sgRNAs were synthesized by Sangon Biotech (Shanghai, China), then cloned into pX330, which was obtained from Feng Zhang’s laboratory (Addgene No. 42230). Briefly, the complementary sgRNA oligos were annealed and then ligated with the BbsI (New England Biolabs, Ipswich, MA, USA) digested plasmid using T4 DNA ligase (Thermo Fisher Scientific, Waltham, MA, USA). To detect cleavage activity, HA-iPSCs were nucleofected using Nucleofector II (Lonza) set at program B016 according to the manufacturer’s instructions. Genomic DNA was extracted 48 h after transfection. The genomic region containing the targeting locus was amplified and cloned, and the construct was used to transform bacteria. For F8-E14-sg1, 46 colonies were picked and sequenced. For F8-E14-sg2, 37 clones were picked and sequenced. Gene disruption was also evaluated by the T7E1 assay. In brief, the PCR products were purified and digested with mismatch-sensitive T7E1. After electrophoresis, the cleaved and uncleaved fragments were quantified by using a gel imaging system.
Gene Targeting of the F8 Locus in HA-iPSCs
HA-iPSCs cultured on Matrigel were detached with TrypLE Select (Life Technologies, Grand Island, NY, USA). The dissociated single cells were resuspended with 100 μL of Human Stem Cell Nucleofector Kit 2 (Lonza,) and nucleofected using Nucleofector II (Lonza) set at program B016. For 1 × 106 cells, 2.5 μg of each CRISPR/Cas9 plasmid was used, with or without 50 pmol ssODN synthesized by Sangon Biotech (Shanghai, China). The transfected cells were seeded on Matrigel-coated wells in mTeSR1 (Stem Cell Technologies, Vancouver, BC, Canada) containing 10 μM Y27632. After 2 days, the cells were detached with TrypLE Select (Life Technologies), and 10,000 cells were seeded on MEF feeders in ESC medium containing 10 μM Y27632 in a 100-mm culture dish.62 Approximately 2 weeks later, clones were picked and expanded. To detect gene deletion events, DNA from individual colonies was extracted and subjected to PCR. PCR was performed using the primers F8-E14-forward (F): 5′-GGTGGACCTCTGAGCTTGAGTGA-3′ and F8-E14-reverse (R): 5′-GCATCTTAAAGAACGACATATCTGGAT-3′. The PCR products were identified by agarose gel electrophoresis. The 448-bp product was sequenced by Sanger sequencing.
Analysis of Off-Target Mutations Induced by CRISPR/Cas9
To analyze the off-target activity of CRISPR/Cas9, we used Sanger sequencing and WES for HA- and C-iPSCs. Genomic DNA of HA- and C-iPSCs (2-6- and 2-46-iPSCs) was isolated. For Sanger sequencing, the DNA from C-iPSCs (2-6- and 2-46-iPSCs) and HA-iPSCs were PCR amplified for five potential off-target sites for F8-E14-sg1 and F8-E14-sg2 predicted by the Optimized CRISPR Design website and sequenced; indels were used to evaluate off-target effects in C-iPSCs. For WES, genomic DNA isolated from HA-iPSCs and C-iPSCs were fragmented and ligated with adaptors to construct libraries, and then, the libraries were subjected to WES using Novaseq 6000 (Illumina, San Diego, CA, USA). The potential off-target sites of CRISPR/Cas9 were searched for by using Cas-OFFinder, which is accessible at http://www.rgenome.net.
Differentiation of hiPSC-Derived ECs
The differentiation of hiPSCs to ECs was carried out as previously described.34 Briefly, hiPSCs were seeded on Matrigel with OP9-diff-conditioned medium (OP9-diff-CM). OP9-diff-CM was collected from the 5- to 10-day-old OP9 cells cultured with OP9-diff medium (α-MEM supplemented with 10% FBS and monothioglycerol; Sigma-Aldrich) and filtered. The medium was refreshed daily. On day 9, the differentiated cells were harvested and sorted with a CD31 MicroBead Kit (Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer’s instructions. The sorted ECs were characterized by immunostaining of endothelial markers CD31 and CD144. Primary antibodies against CD31 (P8590-.2ML; Sigma-Aldrich) and CD144 (ab33168; Abcam) were used.
Derivation of EPCs from hiPSCs
Human iPSCs were differentiated to EPCs via small-molecule-mediated activation of WNT signaling, as previously described.35 Cells were dissociated with Accutase (Thermo Fisher Scientific) and seeded on Matrigel-coated cell culture wells at 50,000 cells/cm2 in mTeSR1 containing 10 μM Y27632. Cells were cultured in mTeSR1 for 3 days and cultured in LaSR basal medium (Advanced/F12, 2.5 mM GlutaMAX and 60 mg/mL ascorbic acid; Sigma-Aldrich) supplemented with 6 μM CHIR99021 (Selleck Chemicals, Houston, TX, USA) for 2 days. After 2 days, the cells were maintained in LaSR basal medium without CHIR99021 for an additional 3 days. The cells were characterized by flow cytometry and an immunofluorescence assay. The differentiated cells were sorted with a CD31 MicroBead Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Then the sorted EPCs were cultured on collagen IV-coated cell culture dishes (Corning) in endothelial growth medium (EGM-2; Lonza) for further differentiation to ECs.
RT-PCR
Total RNA was extracted with TRIzol reagent (Sigma-Aldrich) and treated with DNase for 30 min. Then the RNA was reverse transcribed using random primers and Oligo(dT)15 (Vazyme, Nanjing, China). Two pairs of primers were used to detect the transcripts of F8. One primer was based on exon 14, which contained the deleted 54-bp fragment, and the other was based on exons 23 and 26. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was amplified as an endogenous control. Primer sequences are shown in Table S1.
FVIII Assay of Culture Supernatants and Cell Lysates
The culture supernatants were harvested from 12-well plates 24 h after medium replacement. Then the cells were dissociated with Accutase and counted. After washing with DPBS, the pelleted cells were resuspended in 500 μL of sample diluent for ELISA (Cedarlane, Burlington, ON, Canada) and lysed by three freeze-thaw cycles. All samples were collected in triplicate. ELISA was performed using Paired Antibodies for ELISA-Factor VIII:C (Cedarlane, Burlington, ON, Canada) according to the manufacturer’s instructions. Reference curves were constructed using serial dilutions of normal pooled plasma, with a correlation coefficient (R2) of at least 0.990 using a semilog fit. For the FVIII activity assay, 24-hour-old cell-free supernatant was collected. To calculate FVIII activity, coagulation factor VIII-deficient plasma (Siemens Healthcare Diagnostics, Marburg, Germany) and a Destiny Max hemostasis analyzer (Tcoag, Lemgo, Germany) were used to examine the activated partial thromboplastin time (aPTT). All manipulations were performed according to the manufacturer’s instructions.
Vascular Tube Formation and AcLDL Uptake
To evaluate the function of iECs, 5 × 104 iECs were plated on Matrigel-coated, 48-well culture plates with EGM-2 medium (Lonza). Tube formation was observed using light microscopy after 16 h of incubation.
For the AcLDL uptake assay, the iECs were washed twice with DPBS and then cultured in EGM-2 supplemented with 2.4 μg/mL Dil-AcLDL (Thermo Fisher Scientific) for 4 h at 37°C, washed with DPBS and observed using fluorescence microscopy.
Transplantation of iEPCs into HA Mice
The use and care of animals complied with the guidelines of the Ethics Committee of the Center for Medical Genetics of Central South University.
For in vivo transplantation, 6-week-old HA mice (strain: B6; 129S4-F8 tm1Kaz/J; Jackson Laboratory,)63 were assigned equally in each group. And the same numbers of 6-week-old C57BL/6 mice were used as wild-type controls. The same number of males and females were in each group. Each mouse was anesthetized with Avertin (Sigma-Aldrich) and then infused with 2 × 106 iEPCs derived from HA-iPSCs, C-iPSCs (2-6-iPSCs and 2-46-iPSCs), or N-iPSCs by orbital vein injection. After transplantation, FK506 (1 mg/kg in 100 μL corn oil; Selleck Chemicals ) treatment was performed via intraperitoneal injection once every other day. Two weeks after transplantation, a tail-clip challenge was carried out. Mice were anesthetized, and tail-clip assays were performed as previously described.9 Briefly, the distal part of the mouse tail with a diameter of 1.5 mm was sheared and allowed to bleed for 5 min. The survival time was recorded after applying firm pressure on the tail for 1 minute. Tissue from the surviving mice was harvested and analyzed by using tissue immunofluorescence. Meanwhile, some mice were anesthetized, and blood samples were collected from their eyes to isolate plasma for the FVIII activity assay. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Center for Medical Genetics of Central South University.
All of the mapped data are available from the SRA: SRP145757.
Statistical Analysis
Data were analyzed using ANOVA for more than two groups. The survival curve was analyzed with the log-rank test. All values in each group are presented as the mean ± SEM. All statistical analyses were performed using GraphPad Prism 5.0 software.
Author Contributions
D.L. and L.W. designed and supervised the research and edited the manuscript. Z.H., M.Z., and Y.W. performed the experiments. Z.H. drafted the manuscript. M.Z. prepared the figures and edited the manuscript. Y.W., Z.L. and X.L. assisted in manuscript preparation. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Wei Yin for feeding the animals, Ting Liang for the FVIII activity assay, Minyuan Peng for flow cytometry, and Yun Xie for biochemical detection. This work was supported by grants from the National Natural Science Foundation of China (31571313 and 81770200) and the National Key Research and Development Program of China (2016YFC0905102).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2019.05.019.
Contributor Information
Lingqian Wu, Email: wulingqian@sklmg.edu.cn.
Desheng Liang, Email: liangdesheng@sklmg.edu.cn.
Supplemental Information
References
- 1.Chuah M.K., Nair N., VandenDriessche T. Recent progress in gene therapy for hemophilia. Hum. Gene Ther. 2012;23:557–565. doi: 10.1089/hum.2012.088. [DOI] [PubMed] [Google Scholar]
- 2.Stenson P.D., Mort M., Ball E.V., Evans K., Hayden M., Heywood S., Hussain M., Phillips A.D., Cooper D.N. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017;136:665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McIntosh J., Lenting P.J., Rosales C., Lee D., Rabbanian S., Raj D., Patel N., Tuddenham E.G., Christophe O.D., McVey J.H. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood. 2013;121:3335–3344. doi: 10.1182/blood-2012-10-462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 6.Chandler R.J., Sands M.S., Venditti C.P. Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Hum. Gene Ther. 2017;28:314–322. doi: 10.1089/hum.2017.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y., Hu Z., Li Z., Pang J., Feng M., Hu X., Wang X., Lin-Peng S., Liu B., Chen F. In situ genetic correction of F8 intron 22 inversion in hemophilia A patient-specific iPSCs. Sci. Rep. 2016;6:18865. doi: 10.1038/srep18865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park C.Y., Kim J., Kweon J., Son J.S., Lee J.S., Yoo J.E., Cho S.R., Kim J.H., Kim J.S., Kim D.W. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. USA. 2014;111:9253–9258. doi: 10.1073/pnas.1323941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park C.Y., Kim D.H., Son J.S., Sung J.J., Lee J., Bae S., Kim J.H., Kim D.W., Kim J.S. Functional Correction of Large Factor VIII Gene Chromosomal Inversions in Hemophilia A Patient-Derived iPSCs Using CRISPR-Cas9. Cell Stem Cell. 2015;17:213–220. doi: 10.1016/j.stem.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Pang J., Wu Y., Li Z., Hu Z., Wang X., Hu X., Wang X., Liu X., Zhou M., Liu B. Targeting of the human F8 at the multicopy rDNA locus in Hemophilia A patient-derived iPSCs using TALENickases. Biochem. Biophys. Res. Commun. 2016;472:144–149. doi: 10.1016/j.bbrc.2016.02.083. [DOI] [PubMed] [Google Scholar]
- 11.Xiao A., Wang Z., Hu Y., Wu Y., Luo Z., Yang Z., Zu Y., Li W., Huang P., Tong X. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuo E., Huo X., Yao X., Hu X., Sun Y., Yin J., He B., Wang X., Shi L., Ping J. CRISPR/Cas9-mediated targeted chromosome elimination. Genome Biol. 2017;18:224. doi: 10.1186/s13059-017-1354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blasco R.B., Karaca E., Ambrogio C., Cheong T.C., Karayol E., Minero V.G., Voena C., Chiarle R. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep. 2014;9:1219–1227. doi: 10.1016/j.celrep.2014.10.051. [DOI] [PubMed] [Google Scholar]
- 14.Maddalo D., Manchado E., Concepcion C.P., Bonetti C., Vidigal J.A., Han Y.C., Ogrodowski P., Crippa A., Rekhtman N., de Stanchina E. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–427. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H., Wang H., Shivalila C.S., Cheng A.W., Shi L., Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshimi K., Kunihiro Y., Kaneko T., Nagahora H., Voigt B., Mashimo T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 2016;7:10431. doi: 10.1038/ncomms10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivera-Torres N., Kmiec E.B. A Standard Methodology to Examine On-site Mutagenicity As a Function of Point Mutation Repair Catalyzed by CRISPR/Cas9 and SsODN in Human Cells. J. Vis. Exp. 2017:56195. doi: 10.3791/56195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X., Wang Y., Huang H., Chen B., Chen X., Hu J., Chang T., Lin R.J., Yee J.K. Precise gene modification mediated by TALEN and single-stranded oligodeoxynucleotides in human cells. PLoS ONE. 2014;9:e93575. doi: 10.1371/journal.pone.0093575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fomin M.E., Zhou Y., Beyer A.I., Publicover J., Baron J.L., Muench M.O. Production of factor VIII by human liver sinusoidal endothelial cells transplanted in immunodeficient uPA mice. PLoS ONE. 2013;8:e77255. doi: 10.1371/journal.pone.0077255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shahani T., Covens K., Lavend’homme R., Jazouli N., Sokal E., Peerlinck K., Jacquemin M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J. Thromb. Haemost. 2014;12:36–42. doi: 10.1111/jth.12412. [DOI] [PubMed] [Google Scholar]
- 22.Wagner D.D. The Weibel-Palade body: the storage granule for von Willebrand factor and P-selectin. Thromb. Haemost. 1993;70:105–110. [PubMed] [Google Scholar]
- 23.Xu D., Alipio Z., Fink L.M., Adcock D.M., Yang J., Ward D.C., Ma Y. Phenotypic correction of murine hemophilia A using an iPS cell-based therapy. Proc. Natl. Acad. Sci. USA. 2009;106:808–813. doi: 10.1073/pnas.0812090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou T., Benda C., Dunzinger S., Huang Y., Ho J.C., Yang J., Wang Y., Zhang Y., Zhuang Q., Li Y. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012;7:2080–2089. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]
- 25.Zhou T., Benda C., Duzinger S., Huang Y., Li X., Li Y., Guo X., Cao G., Chen S., Hao L. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011;22:1221–1228. doi: 10.1681/ASN.2011010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dörrenhaus A., Müller J.I., Golka K., Jedrusik P., Schulze H., Föllmann W. Cultures of exfoliated epithelial cells from different locations of the human urinary tract and the renal tubular system. Arch. Toxicol. 2000;74:618–626. doi: 10.1007/s002040000173. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 28.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zimmermann M.A., Oldenburg J., Müller C.R., Rost S. Expression studies of mutant factor VIII alleles with premature termination codons with regard to inhibitor formation. Haemophilia. 2014;20:e215–e221. doi: 10.1111/hae.12388. [DOI] [PubMed] [Google Scholar]
- 30.Zheng C., Liu H.H., Yuan S., Zhou J., Zhang B. Molecular basis of LMAN1 in coordinating LMAN1-MCFD2 cargo receptor formation and ER-to-Golgi transport of FV/FVIII. Blood. 2010;116:5698–5706. doi: 10.1182/blood-2010-04-278325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim Y., Kweon J., Kim J.S. TALENs and ZFNs are associated with different mutation signatures. Nat. Methods. 2013;10:185. doi: 10.1038/nmeth.2364. [DOI] [PubMed] [Google Scholar]
- 32.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veres A., Gosis B.S., Ding Q., Collins R., Ragavendran A., Brand H., Erdin S., Cowan C.A., Talkowski M.E., Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Z., Wu Y., Zhou M., Wang X., Pang J., Li Z., Feng M., Wang Y., Hu Q., Zhao J. Generation of reporter hESCs by targeting EGFP at the CD144 locus to facilitate the endothelial differentiation. Dev. Growth Differ. 2018;60:205–215. doi: 10.1111/dgd.12433. [DOI] [PubMed] [Google Scholar]
- 35.Lian X., Bao X., Al-Ahmad A., Liu J., Wu Y., Dong W., Dunn K.K., Shusta E.V., Palecek S.P. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small-molecule activation of WNT signaling. Stem Cell Reports. 2014;3:804–816. doi: 10.1016/j.stemcr.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pipe S.W. Functional roles of the factor VIII B domain. Haemophilia. 2009;15:1187–1196. doi: 10.1111/j.1365-2516.2009.02026.x. [DOI] [PubMed] [Google Scholar]
- 37.Pipe S.W., Morris J.A., Shah J., Kaufman R.J. Differential interaction of coagulation factor VIII and factor V with protein chaperones calnexin and calreticulin. J. Biol. Chem. 1998;273:8537–8544. doi: 10.1074/jbc.273.14.8537. [DOI] [PubMed] [Google Scholar]
- 38.Cunningham M.A., Pipe S.W., Zhang B., Hauri H.P., Ginsburg D., Kaufman R.J. LMAN1 is a molecular chaperone for the secretion of coagulation factor VIII. J. Thromb. Haemost. 2003;1:2360–2367. doi: 10.1046/j.1538-7836.2003.00415.x. [DOI] [PubMed] [Google Scholar]
- 39.Miao H.Z., Sirachainan N., Palmer L., Kucab P., Cunningham M.A., Kaufman R.J., Pipe S.W. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103:3412–3419. doi: 10.1182/blood-2003-10-3591. [DOI] [PubMed] [Google Scholar]
- 40.Zhang B., Kaufman R.J., Ginsburg D. LMAN1 and MCFD2 form a cargo receptor complex and interact with coagulation factor VIII in the early secretory pathway. J. Biol. Chem. 2005;280:25881–25886. doi: 10.1074/jbc.M502160200. [DOI] [PubMed] [Google Scholar]
- 41.Pittman D.D., Alderman E.M., Tomkinson K.N., Wang J.H., Giles A.R., Kaufman R.J. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood. 1993;81:2925–2935. [PubMed] [Google Scholar]
- 42.Pittman D.D., Marquette K.A., Kaufman R.J. Role of the B domain for factor VIII and factor V expression and function. Blood. 1994;84:4214–4225. [PubMed] [Google Scholar]
- 43.Khrenov A.V., Ananyeva N.M., Saenko E.L. Role of the B domain in proteolytic inactivation of activated coagulation factor VIII by activated protein C and activated factor X. Blood Coagul. Fibrinolysis. 2006;17:379–388. doi: 10.1097/01.mbc.0000233368.95733.3c. [DOI] [PubMed] [Google Scholar]
- 44.Bovenschen N., Rijken D.C., Havekes L.M., van Vlijmen B.J., Mertens K. The B domain of coagulation factor VIII interacts with the asialoglycoprotein receptor. J. Thromb. Haemost. 2005;3:1257–1265. doi: 10.1111/j.1538-7836.2005.01389.x. [DOI] [PubMed] [Google Scholar]
- 45.Gringeri A., Tagliaferri A., Tagariello G., Morfini M., Santagostino E., Mannucci P., ReFacto-AICE Study Group Efficacy and inhibitor development in previously treated patients with haemophilia A switched to a B domain-deleted recombinant factor VIII. Br. J. Haematol. 2004;126:398–404. doi: 10.1111/j.1365-2141.2004.05058.x. [DOI] [PubMed] [Google Scholar]
- 46.Lusher J.M., Roth D.A. The safety and efficacy of B-domain deleted recombinant factor VIII concentrates in patients with severe haemophilia A: an update. Haemophilia. 2005;11:292–293. doi: 10.1111/j.1365-2516.2005.01099.x. [DOI] [PubMed] [Google Scholar]
- 47.Ishiwata A., Mimuro J., Kashiwakura Y., Niimura M., Takano K., Ohmori T., Madoiwa S., Mizukami H., Okada T., Naka H. Phenotype correction of hemophilia A mice with adeno-associated virus vectors carrying the B domain-deleted canine factor VIII gene. Thromb. Res. 2006;118:627–635. doi: 10.1016/j.thromres.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 48.Brown H.C., Wright J.F., Zhou S., Lytle A.M., Shields J.E., Spencer H.T., Doering C.B. Bioengineered coagulation factor VIII enables long-term correction of murine hemophilia A following liver-directed adeno-associated viral vector delivery. Mol. Ther. Methods Clin. Dev. 2014;1:14036. doi: 10.1038/mtm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benkert P., Biasini M., Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryu S.-M., Koo T., Kim K., Lim K., Baek G., Kim S.-T., Kim H.S., Kim D.-e., Lee H., Chung E., Kim J.-S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 52.Zhou M., Hu Z., Qiu L., Zhou T., Feng M., Hu Q., Zeng B., Li Z., Sun Q., Wu Y. Seamless Genetic Conversion of SMN2 to SMN1 via CRISPR/Cpf1 and Single-Stranded Oligodeoxynucleotides in Spinal Muscular Atrophy Patient-Specific Induced Pluripotent Stem Cells. Hum. Gene Ther. 2018;29:1252–1263. doi: 10.1089/hum.2017.255. [DOI] [PubMed] [Google Scholar]
- 53.Young C.S., Hicks M.R., Ermolova N.V., Nakano H., Jan M., Younesi S., Karumbayaram S., Kumagai-Cresse C., Wang D., Zack J.A. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell. 2016;18:533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y., Long C., Li H., McAnally J.R., Baskin K.K., Shelton J.M., Bassel-Duby R., Olson E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017;3:e1602814. doi: 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Follenzi A., Benten D., Novikoff P., Faulkner L., Raut S., Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J. Clin. Invest. 2008;118:935–945. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corselli M., Parodi A., Mogni M., Sessarego N., Kunkl A., Dagna-Bricarelli F., Ibatici A., Pozzi S., Bacigalupo A., Frassoni F., Piaggio G. Clinical scale ex vivo expansion of cord blood-derived outgrowth endothelial progenitor cells is associated with high incidence of karyotype aberrations. Exp. Hematol. 2008;36:340–349. doi: 10.1016/j.exphem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 57.Eldesoqi K., Henrich D., El-Kady A.M., Arbid M.S., Abd El-Hady B.M., Marzi I., Seebach C. Safety evaluation of a bioglass-polylactic acid composite scaffold seeded with progenitor cells in a rat skull critical-size bone defect. PLoS ONE. 2014;9:e87642. doi: 10.1371/journal.pone.0087642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qin M., Guan X., Wang H., Zhang Y., Shen B., Zhang Q., Dai W., Ma Y., Jiang Y. An effective ex-vivo approach for inducing endothelial progenitor cells from umbilical cord blood CD34+ cells. Stem Cell Res. Ther. 2017;8:25. doi: 10.1186/s13287-017-0482-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharma R., Anguela X.M., Doyon Y., Wechsler T., DeKelver R.C., Sproul S., Paschon D.E., Miller J.C., Davidson R.J., Shivak D. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–1784. doi: 10.1182/blood-2014-12-615492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Montague T.G., Cruz J.M., Gagnon J.A., Church G.M., Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–W407. doi: 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park J., Bae S., Kim J.S. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015;31:4014–4016. doi: 10.1093/bioinformatics/btv537. [DOI] [PubMed] [Google Scholar]
- 62.Tang Z.H., Chen J.R., Zheng J., Shi H.S., Ding J., Qian X.D., Zhang C., Chen J.L., Wang C.C., Li L. Genetic Correction of Induced Pluripotent Stem Cells From a Deaf Patient With MYO7A Mutation Results in Morphologic and Functional Recovery of the Derived Hair Cell-Like Cells. Stem Cells Transl. Med. 2016;5:561–571. doi: 10.5966/sctm.2015-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bi L., Lawler A.M., Antonarakis S.E., High K.A., Gearhart J.D., Kazazian H.H., Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat. Genet. 1995;10:119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.