

SUMMARY
Ric-8A is a 530-amino acid cytoplasmic molecular chaperone and guanine nucleotide exchange factor (GEF) for i, q, and 12/13 classes of heterortrimeric G protein alpha subunits (Gα). We report the 2.2-Å crystal structure of the Ric-8A Gα-binding domain with GEF activity, residues 1–452, and is phosphorylated at Ser435 and Thr440. Residues 1–429 adopt a superhelical fold comprised of Armadillo (ARM) and HEAT repeats, and the C terminus is disordered. One of the phosphorylated residues potentially binds to a basic cluster in an ARM motif. Amino acid sequence conservation and published hydrogen-deuterium exchange data indicate repeats 3 through 6 to be a putative Gα-binding surface. Normal mode modeling of small-angle X-ray scattering data indicates that phosphorylation induces relative rotation between repeats 1–4, 5–6, and 7–9. 2D 1H-15N-TROSY spectra of [2H,15N]-labeled Gαi1 in the presence of R452 reveals chemical shift perturbations of the C terminus and Gαi1 residues involved in nucleotide binding.
Graphical Abstract
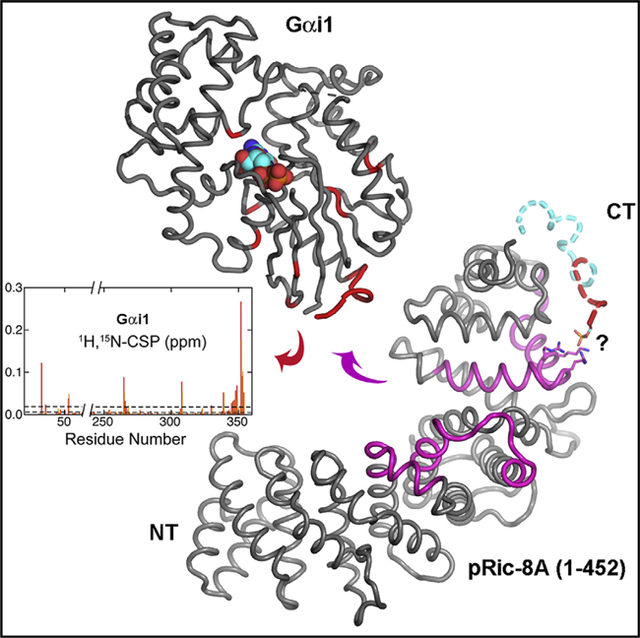
In Brief
Zeng et al. use X-ray crystallography and small-angle scattering, in conjunction with NMR spectroscopy, to reveal the structure of and dynamics of the G protein chaperone and activator Ric-8A and probe its interaction with the G protein alpha subunit i1.
INTRODUCTION
In eukaryotes, heterotrimeric G proteins– –composed of guanine nucleotide binding alpha subunits (Gα) and tightly bound heterodimers of Gβ and Gγ subunits—regulate the activities of intracellular effector enzymes and ion channels that control a variety of biochemical processes such as protein phosphorylation and dephosphorylation, gene transcription, and ion transport through membranes (Cabrera-Vera et al., 2003). These in turn regulate physiological processes that include sensory processing, energy metabolism, neurotransmission, immune activation, and cytokinesis. G proteins, when localized at the intracellular surface of the plasma membrane, are activated by membrane-embedded G protein-coupled receptors (GPCRs) on their stimulation by extracellular agonists. GPCRs activate G proteins by catalyzing the exchange of GDP for GTP at the G protein alpha subunit (Gα), and its subsequent dissociation from Gβγ. Both Gα·GTP and Gβγ are effector regulators or co-regulators. The regulatory cycle is terminated on GTP hydrolysis at the nucleotide binding site of Gα.
G protein activation is not confined to the plasma membrane or other membrane compartments (DiGiacomo et al., 2018; Hinrichs et al., 2012). In a mutational scan of mutants with resistance to inhibitors of cholinesterase in Caenorhabditis elegans, Miller et al. (1996) identified a mutant they cataloged as ric-8. The novel product of this gene, Ric-8 (or Synembryn) was found to be upstream of, or epistatic to, C. elegans homologs of Gαq and Gαo, and RIC-8 defective mutations elicited defects in secretory vesicle priming and mitotic spindle orientation and movement (Miller et al., 2000; Miller and Rand, 2000). Tall et al. (2003) found that rat Ric-8A possesses GEF activity toward Gα subunits of the i, q, and 12/13 families. The in vitro GEF activity toward Gα is independent of and, indeed, inhibited by, Gβγ, in contrast to the action of GPCRs for which G protein heterotrimers are the physiological substrates. In the wake of this discovery, several laboratories uncovered apparent roles for Ric-8A in asymmetric cell division involving Gα guanine nucleotide dissociation inhibitors and GTPase activating proteins (reviewed in Bellaiche and Gotta, 2005; Hampoelz and Knoblich, 2004). Roles for Ric-8A have also been described in cytokinesis abscission (Boularan et al., 2014), and in regulation of synapse number in neurons, where it is subject to inhibition by frequenin (Romero-Pozuelo et al., 2014). These observations suggested models for a functional G protein GTPase regulatory cycle independent of GPCRs or Gβγ and operating at the cell cortex or cytoplasm rather than the plasma membrane (Blumer and Lanier, 2014; Tall, 2013). More recently, mammalian Ric-8A and its Gas class-specific homolog Ric-8B, were shown to act as chaperones for Gα subunits, assisting their acquisition of native structure, transport to the membrane, and resistance to ubiquitination and subsequent degradation (Chan et al., 2013; Gabay et al., 2011; Nagai et al., 2010). Deletion of Ric-8A impairs biogenesis of functional Gα proteins in a variety of cell lines (Papasergi et al., 2015). It has been recently discovered that Ric-8A phosphorylation at five casein kinase II Ser/Thr sites that are highly conserved across Ric-8 phylogeny activates both the GEF and chaperone activity of Ric-8A (Papasergi-Scott et al., 2018). Thus, whereas Gα activation at the membrane is regulated by exogenous GPCR agonists, cytoplasmic activation via Ric-8 is likely subject to regulation by cellular kinases and phosphatases.
Little is understood about the mechanism by which Ric-8 homologs catalyze nucleotide exchange at Gα. As is the case for GPCR-catalyzed activation, the nucleotide exchange reaction proceeds with the formation of a nucleotide-free Ric-8:Gα intermediate (Tall et al., 2003). This intermediate is biochemically stable in the absence of GTP. Biophysical investigations of this complex revealed that nucleotide-free Gαi1 adopts a molten-globule-like state when bound to Ric-8A (Thomas et al., 2011) and is structurally heterogeneous (Van Eps et al., 2015). The secondary structure scaffold that supports the nucleotide binding site in the Gαi1 Ras-like domain becomes accessible to hydrogen-deuterium exchange (HDX) and is therefore likely destabilized (Kant et al., 2016). That Ric-8A promotes a structurally heterogeneous state while inducing long-range conformational changes in Gαi1 suggests a common mechanistic basis for its GEF and chaperone activities.
Clearly, a detailed understanding of the mechanism of Ric-8A cannot be attained without knowledge of the structures of Ric-8 and its complex with Gα. Although the atomic structures of a variety of Gα proteins have been determined in several conformational states (Sprang et al., 2007), only computational models are available for Ric-8A (Figueroa et al., 2009; Kant et al., 2016; Papasergi-Scott et al., 2018). The accuracy of such models is tenuous in view of the absence of reasonably close (>30% amino acid sequence identity) homologs to any members of the Ric-8 family. Here, we describe the high-resolution structure of a fragment of rat Ric-8A composed of the N-terminal 452 residues of the intact, 530-residue protein, which we term R452. This protein retains the two (of five) casein kinase II phosphorylation sites that are critical for GEF stimulation. We show that R452 retains partial GEF activity that is stimulated by phosphorylation, and forms a stable complex with Gαi1. The crystal structure of phosphorylated R452 (pR452), in conjunction with the results of earlier HDX-mass spectrometry (MS) experiments, small-angle X-ray scattering (SAXS) data and evolutionary conservation analysis, provides insight into the mechanism of Ric-8A binding, and the global structural consequences of phosphorylation. We describe heteronuclear NMR experiments that begin to define the structural elements of Gαi1 with which Ric-8A interacts, suggesting commonalities with the mechanism of GPCR activation despite the absence of structural homology between Ric-8 isoforms and members of the GPCR family.
RESULTS
Attempts to generate crystals of intact Ric-8A or the highly active fragment comprising residues 1–491 (R491) did not yield crystals suitable for structure determination. Earlier experiments with C-terminal truncation mutants of Ric-8A that retain partial GEF activity (Thomas et al., 2011), led us to a more thorough investigation of a 452-residue fragment that includes all but the C-terminal 78 residues of Ric-8A (R452). In accordance with the pattern of casein kinase II (CK2) phosphorylation reported for intact Ric-8A, mass analysis shows an increase in mass of 160 Da on treatment of R452 with CK2, consistent with phosphorylation at rat Ric-8A residues Ser435 and Thr440 (Figure S1A). These are the only two CK2 phosphorylation sites within the amino acid sequence range of R452 that are shared with the intact protein (Papasergi-Scott et al., 2018).
R452 Possesses Significant GEF Activity
R452 guanine nucleotide exchange activity is assayed by its stimulation of the steady-state rate of Gαi1-catalyzed GTP hydrolysis, a process that is limited by the ‘‘off’’-rate of GDP at the active site of Gα: the intrinsic rate of nucleotide exchange is about 10-fold lower than that of GTP hydrolysis (Gilman, 1987). A Ric-8 protein concentration response series was used with 1 μM Gαi1 to show that the stimulatory activity of R452 is ~50% of that of intact Ric-8A (Figure 1). Under the same conditions, the GEF activity of R452 is enhanced by 15% on phosphorylation at Ser435 and Thr440. As indicated by surface plasmon resonance analysis (SRP), both phosphorylated and non-phosphorylated R452 form an equally stable complex with nucleotide-free Gαi1. Gαi1 dissociates from R452 and pR452 slowly at a rate of ~1 × 10−4 s−1 comparable with that of Ric-8A (1–491), a larger fragment of Ric-8A with high GEF activity that is more amenable to SRP analysis than intact Ric-8A (Figure S2). The stable Gαi1:pR452 complex can be isolated by size-exclusion chromatography.
Figure 1. R452 Is an Active Exchange Factor.
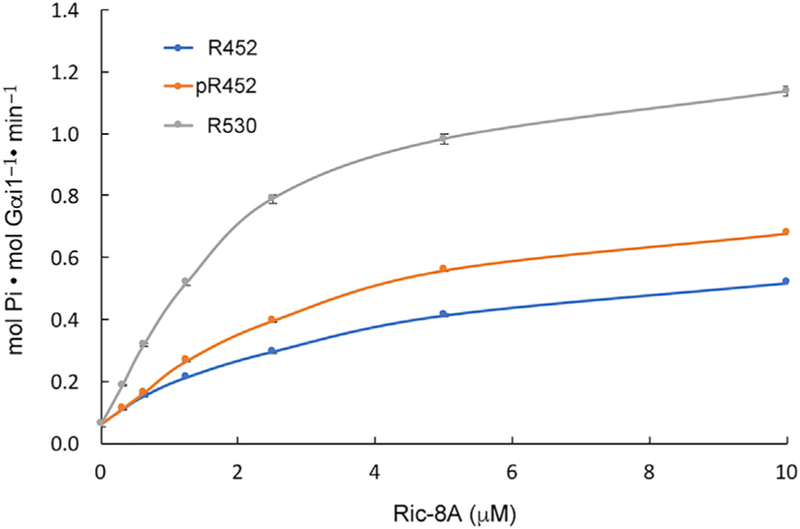
Steady-state rates of GTP hydrolysis catalyzed by 1 μM myristoylated Gαi1 and 30 μM GTP in the presence of varying concentrations of phosphorylated and non-phosphorylated R452 and intact Ric-8A (R530). Data points are represented as mean ± SEM for three replicates. See also Figures S1, S2, and S5B.
pR452 Adopts a Mixed Armadillo/HEAT Repeat Fold
We grew crystals of pR452 in space group P212121 by vapor diffusion from a buffered PEG 3500 solution, forming orthorhombic plates that attained dimensions of 50–150 μm in the longest dimension and 5–20 μm in cross-section after 2–3 weeks. The unphosphorylated protein formed crystals under the same conditions, but these were considerably smaller and less suitable for X-ray data collection. Å Crystallographic phases derived from data measured at λ = 1.77 Å to 3.4 Å resolution were determined by single anomalous dispersion arising from a substructure composed of 40 sulfur atoms. The final atomic model was refined with data measured at λ = 0.979 Å extending to 2.2 Å resolution to working and free R factors of 0.22 and 0.27, respectively. A dataset was also measured for crystals, prepared as described above, that were incubated overnight in paratone-N, a viscous cryoprotectant. Paratone-N-immersed crystals shrank along all three unit cell axes as described below. Diffraction from these crystals was anisotropic, such that data for which I/σ(I) < 2 extended to 1.9 Å along a*, but only to ~2.3 Å along b* and c*. For this reason, a resolution limit of 2.3 Å was imposed for data used in model refinement. Data collection and refinement statistics are recorded in Table 1. Of the two molecules in the asymmetric unit, the reference model described hereinafter is molecule B in the asymmetric unit of the native crystals. In both native and oil-immersed crystals, molecule B exhibits better connectivity and is better ordered within loop regions than molecule A.
Table 1.
X-Ray Data Collection and Crystallographic Refinement Statistics
| Native | Sulfur-SAD | Oil-Immersed | |
|---|---|---|---|
| Data Collection | |||
| Wavelength (Å) | 0.979 | 1.77 | 0.980 |
| Resolution range (Å) | 31.05–2.2 (2.28–2.2) | 29.1–3.41 (3.72–3.40) | 39.65–2.3 (2.382–2.3) |
| Space group | P 21 21 21 | P 21 21 21 | P 21 21 21 |
| Unity cell dimensions | |||
| a, b, c (Å) | 67.0, 103.6, 141.5 | 66.8, 103.4,141.8 | 63.30, 100.1, 130.0 |
| Total reflections | 324,978 (22,568) | 11,088,284 (1,536,621) | 977,649 (98,737) |
| Unique reflections | 50,537 (4,980) | 25,770 (6,848) | 37,176 (3,675) |
| Redundancy | 6.4 (4.5) | 785.9 (803.4) | 26.3 (26.9) |
| Completeness (%) | 99.4 (99.2) | 99.5 (100) | 99.11 (99.19) |
| Mean I/σ(I) | 15.4 (2.2) | 118.3 (67.1) | 30.72 (4.55) |
| Wilson B factor | 32.8 | 83.3 | 41.0 |
| Rmeasa | 0.10 (0.58) | 0.22 (0.37) | 0.09 (1.04) |
| Rp.i.m.a | 0.04 (0.28) | 0.008 (0.013) | 0.02 (0.20) |
| CC½b | 1.0 (0.32) | 1.0 (0.98) | 1 (0.95) |
| Anomalous CCb (%) | 58.4 | ||
| Bijvoet ratioc | 1.22 | ||
| Refinement | |||
| Rworka | 0.230 (0.373) | 0.211 (0.247) | |
| Rfreea | 0.276 (0.404) | 0.260 (0.338) | |
| CCworkb | 0.959 (0.353) | 0.957 (0.901) | |
| CCfreeb | 0.924 (0.263) | 0.930 (0.780) | |
| No. of total atoms | |||
| Protein | 6,671 | 6,445 | |
| Ligands (ions) | 20 | 0 | |
| Solvent | 202 | 96 | |
| Total protein residues | 843 | 813 | |
| Root-mean-square deviation | |||
| Bond lengths (Å) | 0.005 | 0.003 | |
| Bond angles (◦) | 1.01 | 0.58 | |
| Ramachandran plot (%) | |||
| Favoredd | 97 | 98 | |
| Allowedd | 3 | 2 | |
| Outliersd | 0 | 0 | |
| Rotomer outliers (%)d | 0.14 | 0 | |
| Clash scored | 6.95 | 6.51 | |
| Average B factor | |||
| Macromolecules | 47.3 | 50.6 | |
| Sulfate | 75.9 | NA | |
| Water | 42.9 | 49.91 | |
Data for highest-resolution shell are given in brackets.
, where Ii(hkl) is the ith observation of the intensity of the reflection hkl and <I(hkl)> is the mean over n observations.. , where Fobs and Fcalc are the observed and calculated structure factor amplitudes for each reflection hkl. Rfree was calculated for 3% of the diffraction data that were selected randomly and excluded from refinement.
Correlation coefficients: CC = Σi((xi − <x>)(yi − <y>))/(Σi((xi − <x>)2 (yi − <y>)2)½, where xi and yi are the ith of n observations of quantities whose mean values are <x> and <y>; for CC½, xi, and yi correspond to intensity measurements derived from each of two randomly selected half-datasets from the set of unmerged data; For CCwork and CCfree, xi and yi refer to observed structure factor amplitudes and structure factor amplitudes computed from the refined atomic model, respectively, for the working dataset and the set used to compute Rfree. CCano = <Δano Δano,obs>/(<Δ2ano>½ <Δ2ano,obs>½), where Δano and Δano,obs are the anomalous structure factor amplitude differences (F+ − F− ), respectively, computed from the anomalously scattering atomic substructure, and the observed anomalous differences. CCano was calculated using data truncated to dmin = 3.4 Å.
Bijvoet ratio [(<|F+/−|>)/(<F>)] × 100, was calculated using James Holton’s xtalsize server (http://bl831.als.lbl.gov/xtalsize.html).
Calculated using MolProbity (Chen et al., 2010).
pR452 adopts a superhelical fold comprised of nine repeat units (Figures 2A, 2B, and S3). These are not uniform, but conform to either two-helix-bundle HEAT motifs (repeats 1, 2, 6, and 9) or three-helix Armadillo (ARM) motifs (repeats 3, 4, 5, 7, and 8) in which the middle helix serves as a linker between the nearly antiparallel first and last helices of the bundle (Andrade et al., 2001) (Figure 2B). Ric-8A appears to be unusual among helical repeat proteins in that it is composed of both types of repeat units (Andrade et al., 2001). Generally, residues within the concave surface of pR452, formed largely by the αB and α3 helices of HEAT and ARM repeats, respectively, are well conserved among Ric-8A paralogs (Figures S3, 5B). The angular rotation that superposes one repeat to the next is fairly uniform over the first six repeats, ranging from 20° to 40° (Table S1), but the C-terminal repeat pairs are related by larger rotation angles, from ~60° to ~110°. The distance between the centers of mass between repeats is fairly constant, 9.8–13.3 Å, consistent with extensive packing interactions between each pair of repeats. Due to differences in the number of residues that comprise each repeat, the solvent-accessible surface area (SASA) buried by the structural elements of each ranges from 560 to 2,250 Å (Table S1). In contrast, the SASA occluded between adjacent repeats 1 through 8 is relatively constant, ranging from 1,950 to 2,490 Å2. In contrast, the interface between the last two repeats buries only 1,550 Å2. Loop excursions between helical elements, both within and between superhelical repeats, project toward the convex surface of the molecule. The longer of these (>10 residues) join helical elements within, rather than between ARM repeats.
Figure 2. Annotated Amino Acid Sequence and Structural Features of pR452.

(A) Amino acid sequence of R452 showing the numbering convention for structural repeats. ARM repeats consist of three helices: α1X, α2X, and α3X, where X is the nth repeat of the structure. HEAT repeats consist of two helices: αAX and αBX. α-Helical secondary structure is shown as a series of loops above the amino acid sequence. Helices with 310 hydrogen bonding and geometry are so labeled. Straight-line sections indicate loop segments. No electron density is observed beyond residue 423 (molecule A) and 429 (molecule B). Magenta-tinted overlay on segments of the amino acid sequence indicate regions that are protected from hydrogen-deuterium exchange by Gαi1 (Kant et al., 2016), with color intensity proportional to degree of protection as shown in Figure 4. The two phosphorylation sites, Ser435 and Thr440, are highlighted in yellow. Blue and cyan bars shown below the amino acid sequence indicate residues that are solvent accessible (blue), partially accessible (cyan), or buried (white). Figure modified from output from ESPript 3.0 server (Robert and Gouet, 2014). See also Figures S3 and S4.
(B) Schematic representation of the structure of pR452. Cylinders represent a helices. Rendered in red are the ‘‘A’’ helices of HEAT repeats or the second helix of an ARM triad. ‘‘B’’ helices of HEAT repeats or the third helix of an ARM repeat are colored yellow, and the first helices of ARM repeats are rendered in green. See also Figure S4.
(C) Electrostatic potential (positive, blue; negative, red) at the molecular surface of the C terminus of pR452, viewed from the direction indicated by the arrow in (B), showing secondary structure (ribbons) and side chains of positively charged residues (stick figures) of residues 343–365 (α18) and residues 400–415 (αA9, αB9). Molecular surface and electrostatic potential computed using PyMOL Molecular Graphics System Version1.7 Schrödinger LLC.
No connected electron density is observed beyond residue 423 of molecule A or 429 of molecule B, which include both phosphorylated residues Ser435 and Thr440. MALDI-TOF analysis shows that pR452 is intact in crystals (Figure S1B). The C-terminal-most residues of pR452 molecules A and B are adjacent to solvent channels of sufficient volume to accommodate the disordered residues. In keeping with an amino acid composition rich in glycine, proline, and both charged and polar residues, the C terminus of pR452 has a high probability for adopting an intrinsically disordered state (Dyson, 2016), as indicated by the DISOPRED3 predictor (Jones and Cozzetto, 2015), and also a high potential for serving as a protein binding site (Figure S4).
The electrostatic potential contact map rendered at the molecular surface of pR452 reveals dispersed and non-contiguous regions of positive and negative charge density. A striking exception is the extensive positively charged surface near the C terminus (Figure 2C). The surface charge arises from a constellation of ten arginine and lysine side chains that project from α18 and αA9, all but two of which are highly conserved among Ric-8 homologs (Figure S3). Because the C-terminal residues of R452 are part of an extended disordered region, it is possible that the positively charged surface is solvent-exposed in intact Ric-8A. Thus, the conserved Arg345/Arg348/Lys349 triad, which binds a sulfate ion derived from the crystallization buffer, could serve as a recognition site for one of the C-terminal phosphorylated serine or threonine residues (Figure S5A). Mutation of Arg345 to glutamine or Lys349 to alanine results in the reduction of GEF activity of either pR452(R345Q) or pR452(K349A) to that of unphosphorylated wild-type R452. However, an approximately equal reduction of GEF activity of R452(R345Q) or R452(K349A) relative to wild-type unphosphorylated R452 is also observed (Figure S5B).
The two C-shaped molecules of pRic-8A in the asymmetric unit of the unit cell are intimately packed, such that the α18 and αA9 elements of molecule B are nested into the cavity formed by multiple ARM/HEAT repeats on the convex surface of molecule A (Figure S6). The corresponding surface of molecule B forms similar, but less intimate contacts with the α18 and αA9 motifs of a symmetry-related copy of molecule A. Examination of lattice packing reveals solvent channels that appear to be sufficient volume to accommodate the disordered C termini of pR452. Immersion of crystals in paratone oil before data collection results in shrinkage along the a, b, and c axes of 4, 6.5, and 11.5 Å, respectively (Table 1, Figure S7). Lattice compaction results in little distortion of the structure itself (root-mean-square differences at Cα a positions = 0.63 and Å for chains A and B, respectively, relative to their counterparts in native crystals). Rather, symmetry-related molecules undergo relative translations and reorientations within the lattice that largely preserve the original molecular packing. An exception is the exclusion of the four sulfate anions observed in the parent crystals.
Phosphorylation Induces Global Changes in the Solution Structure of R452
We employed SAXS to determine the low-resolution structure of pR452 and R452 in solution. These experiments reveal fundamental hydrodynamic properties of these molecules, including radius of gyration (Rg), maximum molecular dimension (dmax), and conformational flexibility (Table 2) (Mertens and Svergun, 2010; Trewhella et al., 2017). SAXS profiles were measured for pR452 and R452 after passage through a size-exclusion column (Figure S8A). Averaging of successive frames derived from peak fractions over which the computed radius of gyration (Rg) was invariant afforded redundancy in measurements of scattering intensity data (Figures 3A and S8; Table 2) extending to q = 4πsinθ/λ = 0.435 (d = 2π/qλ = 13.38 Å). Rg was found to be constant over a range of concentrations, consistent with the absence of R452 aggregation or multimerization. Rg values for R452 and pR452 computed from the slope of the linear portion of the Guinier plot (Figure 3A, inset), are within error estimates and agree with values computed from the pair distribution function, P(r) (Figure S8B). The Kratky plot for R452 and pR452 indicates that both are compactly folded in solution (Figure 3B). Estimates of dmax from the pair distribution function (Figure S8B) are consistent with the crystal structure of pR452 (Table 2). The slight rise in the Kratky plot (Figure 3B) at high scattering angle is indicative of the presence of a disordered region, which may correspond to the C-terminal ~25 residues that are not observed in the crystal structure.
Table 2.
Parameters Derived from SAXS Analysis of R452 and pR452
| Parametera | R452 | pR452 |
|---|---|---|
| I(0) (cm−1) from Guinier plot | 84.65 ± 0.081 | 106 ± 0.18 |
| Rg (Å) from Guinier plot | 29.7 ± 0.57 | 29.2 ± 0.53 |
| qRg limit for Guinier analysis | 1.17 | 1.18 |
| I(0) (cm−1) from P(r) | 84.63 | 106.2 |
| Rg (Å ) from P(r) | 30.01 | 29.59 |
| dmax (Å) from P(r) | 101 | 92 |
| Porod volume estimate (Å3) | 69,043 | 69,991 |
I(0), total scattering intensity at zero scattering angle; Rg, radius of gyration; q, momentum transfer, = 4π sin(θ)/λ, where 2θ is the scattering angle, and λ is the wavelength of incident radiation; dmax is the maximum particle dimension.
Figure 3. Small-Angle X-Ray Scattering Shows that pR452 and R452 are Well Folded in Solution.

(A) Scattering data for R452 (closed circles) and pR452 (open circles), Guinier plots for each sample are shown in the insert. Linear fit of ln(I(q)) versus q2 in the range 0.0065 < q < limit (see Table 2) for determination of Rg is shown in yellow.
(B) Kratky plot, q2I(q) versus q, shows a bell-shaped curve indicative of a compact globular particle. Increase in the function at high q suggests presence of unfolded component. See also Figure S8.
Using the crystal structure of pR452 as an initial state, we employed normal mode perturbations, as implemented in the program SREFLEX (Panjkovich and Svergun, 2016), to generate models of pR452 and R452 most consistent with their respective SAXS scattering curves extending to q = 0.3 (d = 20.27 Å), while minimizing steric clashes and chain breaks (see the STAR Methods). Projections along the lowest normal modes gave rise to quasi-rigid body angular displacements of three subdomains of pR452, comprising residues 1–183, 184–287, and 288–429, which correspond roughly to helical repeats 1–4, 5–6, and 7–9 (Figure 2A; Table S1).
The solution structures of pR452 and R452 generated by this procedure afford better fits to their respective scattering curves than the crystal structure of pR452 (Figures 4A and 4D). The top five solution state models of pR452 and R452 produced by SREFLEX were fairly uniform with respect to the quality of fit (χ2) to the corresponding experimentally generated SAXS profiles, but differed in stereochemical quality (Table S2). The most stereochemically reasonable models for pR452 and R452 are depicted in Figures 4B and 4E, respectively. These exemplify the magnitude of relative subdomain rotations that optimize the fit of the model to its scattering curve. Thus, the solution structure of pR452 (model P-05, Table S2) differs from the crystal structure by a rotation of the N- and C-terminal subdomains by 12° and 15°, respectively, relative to the central subdomain (Figure 4B). Taking P-05 as the reference for the solution structure of pR542, the unphosphorylated state is achieved by the N- and C-terminal subdomain counter-rotations of 25° and 5°, respectively (Figure 4E). The contacts between solvent-inaccessible residues at the subdomain interfaces are for the most part hydrophobic (Figure S3), and thus can likely be maintained after a relative rotation of one with respect to the next. The model solution structures of pR452 and R452 fit well to the ab initio molecular envelopes generated from the solution scattering profiles of phosphorylated and unphosphorylated R452 (Figures 4C and 4F). Thus, the SAXS data provide evidence that crystal packing forces may distort the structure of pR452 from its equilibrium solution state, and that phosphorylation induces subdomain-level conformational changes.
Figure 4. Solution Structures of pR452 and R452 Modeled from SAXS.

(A and D) Scattering profiles for pR452 (A, open circles) and R452 (D, closed circles). Computed scattering profiles from the crystallographic coordinates of pR452 and R452 are shown in magenta. Solution scattering profiles generated from normal mode refinement of pR452 crystallographic coordinates with respect to experimental scattering curves are shown for pR452 (A, green), and R452 (D, cyan). The bottom panels of (A) and (D) show the error-weighted residual difference plots ΔI/σ = [Iexperimental(q)– CImodel(q)]/σ(q) versus q, where C is a normalizing scale factor. Normal mode refinement of pR452 coordinates affords improved fits to the experimental scattering curves.
(B) Superposition of the crystal structure pR452 (magenta) onto the model generated by normal mode refinement of the latter with respect to the solution scattering curve for pR452 (green). Models, rendered as cylinders for α-helical segment. Helices at subdomain interfaces and the N and C termini are labeled. The coordinate sets were superimposed using the Cα atomic positions of the central rigid body domain, residues 184–287. See also Table S2.
(E) Superposition of the modeled solution structure of pR452 (green), as shown in (B), onto the modeled solution structure of R452 (cyan).
(C and F) Solution structure models of pR452 (green) and R452 (cyan) fit to their respective ab initio molecular envelopes. See also Figure S8.
Deduction of the Gα Binding Site of R452 from HDX-MS and Sequence Conservation
HDX analysis of a fragment of Ric-8A comprising residues 1–491, a highly active GEF, revealed residues that interact with Gα or are perturbed by that interaction (Kant et al., 2016). Two distinct surfaces, the first formed by residues in structural elements α23 through αA6, and the second by residues in α37 and α18, are protected by Gαi1 (Figures 2A and 5A). Projection of evolutionary conservation scores of Ric-8A homologs onto the structure of pR452 using the CONSURF server (Landau et al., 2005) (Figures 5B and S3) shows that, in addition to the αB and α3 helices that form the concave surface of R452, virtually all of the residues within structural elements from α37 to the C terminus are highly conserved. The contiguous surface encompassed by the V-shaped helical hairpin formed by α37 and α18 has a mean protection factor of −8% (change in deuteration), and an average conservation score exceeding 7 (maximum score is 9). We propose that this surface, which partly overlaps the positively charged region described above, harbors a Gαi1 binding site, or is otherwise occluded by Gαi1 binding. Segments within the second surface, including the N terminus of α16, and C-terminal residues of α34 and α35 are also conserved and moderately protected from HDX by Gαi1 binding, and thus may also form a Gαi1 interaction surface.
Figure 5. Changes in Hydrogen-Deuterium Exchange at R452 and Amino Acid Sequence Conservation Suggest Sites for Gαi1 Binding.

(A) Segment colored magenta are protected by Gαi1 binding; de-protected segments are colored green. Extent of protection/de-protection indicated by color intensity, to maximum/minimum values of ±16% in change in HDX protection on Gαi1 binding.
(B) Amino acid sequence conservation, computed using CONSURF (Ashkenazy et al., 2010) for 150 Ric-8 homologs, mapped on the surface of pR452. Conservation is depicted over a color range from burgundy (most conserved) to cyan (most variable). Black denotes absence of HDX information.
(C) Spatial relationship between central subdomain, with helices rendered as cylinders and flanking subdomains rendered as coils. Segments that exhibit high HDX protection are colored burgundy and positively charged residues in conserved basic surface are shown as blue spheres at Cα positions. See also Figure S3.
Interaction of R452 with Gαi1
To identify the residues within Gαi1 that interact with R452, we prepared uniformly 2H- and 15N-labeled Gαi1Δ31, which lacks the N-terminal 31 residues of intact Gαi1. 2D-[1H,15N]-TROSY experiments were conducted to monitor chemical shift perturbations and resonance broadening in the spectrum of 2H,15N-Gαi1Δ31 on titration with R452 (Figures 6A–6C). The 2D-[1H,15N] –TROSY spectra of Gαi1ΔD31in complex with GTP analogs and GDP were assigned in earlier studies (Goricanec and Hagn, 2019; Goricanec et al., 2016), thus affording identification of many of the residues that are perturbed by interaction with R452. At a 1.6:1 stoichiometric ratio of R452 to Gαi1Δ31, additional signals appear in the random coil region (~8 ppm 1H chemical shift) of the NMR spectrum, indicative of the presence of a second, most likely less structured conformation in slow exchange (microsecond to second timescale), in addition to substantial resonance broadening throughout the spectrum, caused by the formation of a larger complex.
Figure 6. Perturbation of the Structure of Gαi1Δ31 by R452.

(A) 2D-[1H,15N]-TROSY spectra of 100 μM 2H,15N Gαi1Δ31.
(B) As in (A) in the presence of R452 at 0.5 stoichiometric ration with respect to Gαi1Δ31.
(C) As in (A) in the presence of R552 at 1.6 stoichiometric ratio with respect to Gαi1Δ31.
(D) Magnitude of chemical shift perturbation (CSP) of Gαi1Δ31 backbone resonances. Average CSP and average plus 1xσ are marked by broken lines. Orange and red bars indicate CSPs calculated with the spectra at 0.5 or 1.6 molar ratio, respectively.
(E) Residues at which CSPs exceed average values are depicted on a ribbon model Gαi1Δ31 (PDB: 1CIP) as spheres, colored in increasingly deeper shades of red according to magnitude of the CSP. A van der Waals model of GTP is shown to mark the position of the guanine nucleotide binding site, which is not occupied in the complex with R452.
Gαi1Δ31 residues that experience the strongest chemical shift perturbations due to R452 binding are located within α5, the C terminus of Gαi (residues 345–354), the same region that forms the major contact site in Gα complexes with GPCRs (Figures 6D and 6E). Also perturbed are: residues in the α1 helix (e.g., M53) that follows the P loop that interacts with the guanine nucleotide phosphate moieties; the β2β3 hairpin and the β5 strand (265–268) that precedes a loop that forms part of the nucleotide purine binding site; the C terminus of α4 (F307) that forms a hydrophobic interaction with β5 L266, and residues at the N terminus of α5. All of these Gαi1 residues experience an increase in HDX on Ric-8A binding (Kant et al., 2016), consistent with their structural destabilization as a consequence of interaction with Ric8A.
DISCUSSION
Unlike many helical repeat proteins, the N-terminal 423 residues of Ric-8A are composed of both HEAT and ARM repeats to generate a protein fold that is in general agreement with predictions from secondary structure analysis, model building, and circular dichroism spectroscopy (Figueroa et al., 2009; Kant et al., 2016; Papasergi-Scott et al., 2018; Thomas et al., 2011). Although present in the R452 construct, residues beyond the C-terminal ARM repeat are disordered in the crystal structure. This C-terminal ~28 residue sequence has characteristics of an intrinsically disordered region and includes serine 435 and threonine 440, which, when phosphorylated by casein kinase II, induce substantial stimulation of both GEF and Gα chaperone activity (Papasergi-Scott et al., 2018). Both pR452 and R452 bind tightly to nucleotide-free Gαi1 with an off-rate comparable with that of a larger fragment consisting of residues 1–491, which is a more efficacious GEF than intact Ric-8A in both unphosphorylated (Thomas et al., 2011) and phosphorylated states (Papasergi-Scott et al., 2018). That phosphorylation of R491 at Ser335 and Thr340 results in tighter binding of nucleotide-free Gαi1, but phosphorylation of the same residues of R452 does not (Figure S2), suggests that the effect of phosphorylation depends at least partly on residues beyond R452, as indicated by previous HDX-MS studies (Kant et al., 2016). Because R452 possesses significant phosphorylation-stimulated GEF activity we propose that it constitutes the minimal Gα-binding and catalytic domain of Ric-8A. Indeed, truncation of Ric-8A at residue 426, which delimits the ordered ARM/HEAT repeat structure described here, virtually abolishes GEF activity (Thomas et al., 2011).
The two phosphorylation sites, of which Thr440 is particularly important (Papasergi-Scott et al., 2018), confer potent regulatory activity on the residues that extend beyond the HEAT/ARM repeat domain. Further toward the Ric-8A C terminus, between residues 452 and 474, HDX-MS and mutagenic scanning experiments (Kant et al., 2016), revealed three closely spaced hotspots for GEF activity. The ab initio molecular envelopes generated from SAXS data, supported by dmax values derived from pair distribution functions, suggest that pR452 is a more compact structure than its dephosphorylated counterpart. The ion-pairs formed between sulfate ions in the crystallization buffer and Arg345, Arg348, and Lys349 in the positively charged cluster within the last HEAT repeat may mimic possible contacts with pSer435 or pThr440. However, mutation of either of two of these basic amino acids to neutral residues reduces the GEF activity of both R452 and pR452. Hence it is possible that the sulfate-binding triad interacts with negatively charged residues elsewhere in R452, for example, the glutamate- and aspartate-rich cluster within the disordered segment surrounding the phosphorylated residues extending from residues 429 to 452, or residues of Gα itself. In the former instance these and other residues within the basic patch (Figures 2C, S3, and 5C) might serve to orient and stabilize putative Gα binding sites located between residues 452 and ~490 near the C terminus of Ric-8A (Kant et al., 2016), or within the disordered segment itself. Such interactions might formed only in the complex with Gα. DISOPRED3 analysis indicates that the segment between ~450 and 490 is likely to be ordered.
Although the interfaces between the ARM/HEAT repeats of R452 are well-packed, normal mode analysis identifies three blocks of residues (roughly, repeats 1–3, 4–7, and 8–9) that constitute domains that exhibit uniform collective motion. Global structural deformations that relate the crystal and solution structures (via SAXS) of pR452 can be modeled as rotational displacements between these subdomains. It is remarkable that phosphorylation at Ser435 and Thr440 in a flexible region of the molecule appears to induce long-range structural changes that also can be modeled as relative rotations between the same subdomains. Because several members of a family of relative domain rotations of the same set of subdomains can account for the SAXS data, it is not possible to speculate on the mechanism by which Ric-8A phosphorylation might induce rotational displacements of this type.
The results of HDX experiments (Kant et al., 2016), examined in the light of amino acid sequence conservation in the Ric-8 family, are consistent with an extended Gα binding surface. Global structural rearrangements induced by phosphorylation could increase the potency and efficacy of GEF activity by optimizing the spatial complementarity of these non-contiguous (in amino acid sequence) interaction sites with Gα. Indeed, structural segments of Ric-8A that are protected by Gαi1 from HDX overlap with the boundaries of subdomains that are predicted to rotate with respect to each other on phosphorylation (Figures 5C and S3).
Ric-8A adopts a fold that is quite different from that of members of the GPCR family. Nevertheless, the NMR data presented here, together with earlier studies, suggest some similarities in the mechanism by which the two types of GEFs catalyze nucleotide exchange at Gα. Notably, as suggested by other studies (Thomas et al., 2011), the NMR titration experiments provide direct evidence that the C terminus of Gα, which includes residues of the α5 helix, is a Ric-8A binding site. Other Gαi1 sites perturbed by Ric-8A include α1, which may disrupt interactions with α5 and also result in structural changes at the guanine nucleotide phosphate binding loop (P loop), the β2β3 loop, α4, and β6. In view of its interactions with these sites, it appears that Ric-8A employs a mechanism similar to that used by GPCRs to disrupt the network of interactions that retain nucleotides in the active site of Gα (Flock et al., 2015; Kant et al., 2016; Rasmussen et al., 2011; Thaker et al., 2012). On the other hand, deformation of the β5-αG loop, which dictates specificity for guanine nucleotides, is not observed in crystal structures of GPCR-bound Gα, and may be a unique feature of Ric-8A exchange activity. Whether observed chemical shift perturbations are due to direct binding of Ric-8A or are induced by binding, cannot be determined. It is noteworthy that the chemical shift perturbation observed require only Ric-8A residues N-terminal to residue 452. It remains to be seen whether the C-terminal 78 residues of Ric-8A afford additional interactions with Gαi1, as suggested by HDX-MS and mutagenic scans (Kant et al., 2016) or only modulate the strength or efficacy of interactions formed by the N-terminal Gα binding domain.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen R. Sprang (Stephen.sprang@umontana.edu).
METHOD DETAILS
Protein Expression, Purification and Mutagenesis
Rat Ric-8A 1–452 (R452) was expressed as an N-terminal Hexahistidine (His)6 fusion protein from a pET-28A vector and purified as described (Thomas et al., 2011) with some alterations. Briefly, the N-terminal hexa-histidine-tagged protein construct was expressed in E. coli BL21 (DE3)-RIPL cells in TB media containing kanamycin (100mg/L) and induced with 50μM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at 20°C. Subsequent purification steps were conducted at 5°C. After cell lysis and clarification by centrifugation in lysis buffer (50mM Tris, pH 8.0, 250mM NaCl, 5% Glycerol, 2mM β-mercaptoethanol, 2mM phenylmethylsulfonyl fluoride (PMSF)), protein was loaded onto a Profinity IMAC (Bio-Rad) column and eluted by gravity with elution buffer (50mM Tris, pH 8.0; 250mM NaCl; 5% Glycerol; 2mM β-mercaptoethanol; 2mM PMSF; 300mM Imidazole). Tobacco Etch Virus (TEV) protease was added to (His)6-Ric-8A at a 1:15 (w/w) TEV:Ric-8A ratio and the proteolysis reaction was allowed to proceed for 15 to 20 hours at 4 degrees-C. The reaction product was subjected to two rounds of dialysis into Q-buffer (5mM β-mercaptoethanol, 50mM NaCl, 5% glycerol) and then applied to IMAC column as above to recover TEV-cleaved Ric-8A. The IMAC flow-through was then loaded onto a HiTrap Q XL anion exchange column (GE Healthcare) and eluted in Q-buffer with a NaCl gradient (0mM to 500mM). After concentration, the protein was applied to a Superdex 200 10/300 GL size exclusion column (GE Healthcare) and eluted with storage/crystallization buffer (50mM HEPES, pH 8.0, 150mM NaCl and 1mM tris(2-carboxyethyl) phosphine (TCEP)).
Phosphorylated R452 (pR452) was prepared essentially as described (Yu et al., 2019). Briefly, 10 mg R452 was incubated in 1.1 ml reaction mixture containing 1mM ATP, 20 mM Tris-HCl pH 7.5, 50 mM KCl, 10 mM MgCl2, 50mM HEPES, pH 8.0, 150mM NaCl and 1mM TCEP, and 3000 units of holo casein kinase II (New England Biolabs) and incubated overnight at 25°C. Phosphorylated protein was purified by anion exchange and size exclusion chromatography as described above. Yield of doubly-phosphorylated R452 is > 90% as judged by triple quadrupole time-of-flight mass spectrometry.
Myristoylated Gαi1 (myrGαi1) was prepared, with modifications, as described (Mumby and Linder, 1994). Internally hexa-histidine tagged rat Gαi1 encoded in a pQE60 expression vector was co-expressed in JM109 cells with yeast N-myristoyltransferase encoded in a pBB131 vector. After cell lysis using an EmulsiFlex-C5 cell disruptor (Avestin) in lysis buffer (50mM Tris pH 8.0, 100mM NaCl, 5mM β-mercaptoethanol, 10μM GDP, 2mM PMSF) and clarified by centrifugation, His6-tagged myrGαi1 was applied to a nickel-NTA (Qiagen) column and eluted with 50mM Tris pH 8.0, 100mM NaCl, 5mM β-mercaptoethanol, 10μM GDP, 2mM PMSF,150mM imidazole, and further purified by ion exchange chromatography on a HiTrap Q Sepharose FF column (GE Healthcare) and size exclusion chromatography as described above.
Site directed mutations of R452, Arg335Gln and Lys349A were carried out using the QuikChange II-XL mutagenesis kit (Agilent) following the manufacturer’s protocols. Mutagenesis primers (Integrated DNA Technologies) were designed using the Agilent QuikChange Primer Design Tool using the pET29a R452 wild type plasmid as the template. The PCR products were transformed into Top10 competent E. coli for amplification. Mutations were were confirmed by DNA sequencing performed by Eurofins Genomics.
Assay of Ric-8A-Stimulated Gαi1 Nucleotide Exchange Activity
Steady-state GTPase reactions catalyzed by Gαi1 in the presence and absence of Ric-8A were assayed by the production of 32Pi from γ−32P-GTP as described (Ross, 2002). Reactions were conducted in assay buffer (50mM HEPES, pH 8.0,100mM NaCl, 1mM EDTA, 1mM TCEP, and 10mM MgCl) containing R452, pR452 or intact Ric-8A (R530) at concentrations ranging from 0 to 10μM, and 30μM GTP doped with γ−32P-GTP (6000 Ci/mmol, PerkinElmer) to a specific activity of 1000 cpm/pmol. GTPase reactions were initiated with addition of myrGαi1 to a final concentration of 1μM ina 20μL reaction volume and allowed to proceed for 5 minutes at 30°C, then quenched by addition of 180μL cold 50mM NaH2PO4, pH 4.0 and mixed with 800μL 5% w/v Norit charcoal suspension in the same buffer. After clarification by centrifugation, 150μL of clarified supernatant was added to 10ml of 3a70B scintillation cocktail (RPI) and beta emission was counted in a scintillation counter. Reactions were conducted in triplicate. A negative control with 10μM Ric-8A species and no myrGαi1, was included with each Ric-8A concentration series to account for residual GTPase or casein kinase II contamination.
Fluorimetric guanine nucleotide exchange assays were conducted as described in (Kant et al., 2016). Exchange reactions were conducted in buffer (50mM HEPES pH 8.0, 1 mMTCEP, 150 mMNaCL and 10 mMMgCl2) containing 10 mM guanosine 5’-[γ-thio] triphosphate (GTPγS), 2μM R452 or pR452 or mutants thereof as indicated, and 2μM Gαi1 at a reaction temperature of 25° Reaction volume was 500ml. Reaction was initiated by addition of Gαi1·GDP to R452 mixture and Gαi1·GTPγS measured by flouorescence emission at 345nm (excitation wavelength 295nm) in an LS55 luminescence spectrometer (PerkinElmer Life Sciences). Three to 5 replicates were taken for each data set and significance of differences was estimated by a Student’s T Test.
Crystallization of pR452, X-Ray Data Collection and Structure Determination
Crystals of pR452 were obtained by vapor diffusion from a 1:1 mixture of 0.5μl 20mg/ml protein in crystallization buffer (see above) and reservoir solution containing 0.2 mM Li2SO4 and 25–30% w/v PEG 3350, 0.1 mM HEPES, pH 7.5, set against the same reservoir solution. Crystals were cryoprotected either in 20% w/v PEG400 in mother liquor, or immersed in Paratone-N (Parabar 10312, Hampton Research). Crystals were harvested with 20μm nylon Cryoloops (Hampton Research) or MicroLoops Eloops (MiTeGen) and flash-frozen in liquid nitrogen for data collection.
Data collection and structure determination using native Sulfur anomalous scattering are described in detail in a manuscript in preparation (T.-C. Mou, unpublished data), and are described briefly here. For sulfur-anomalous phasing, eighteen datasets from randomly orientated crystals were recorded at 100K and a wavelength of 1.7712 Å using the helical data collection method on the micro-focusing FMX beamline at National Synchrotron Light Source II (NSLS-II) equipped with an Eiger16M pixel array detector with a 133Hz framing rate. Diffraction data for each crystal were recorded in 0.1–0.2° oscillation images over a range of 360 – 5760° per crystal at 20% transmission at 0.05 – 0.1 s exposure time/image. Diffraction data were integrated and initial scaling performed with XDS (Kabsch, 2010a, b). Final scaling calculations were carried out using the phenix.scaled-and-merged script in the PHENIX program suite (Terwilliger et al., 2016). Datasets recorded from crystals cryoprotected in PEG-400 and Paratone-N were measured using X-rays at a wavelength of 0.979Å and 0.980Å from the APS 19BM and SSRL BL9–2 Beamlines, respectively. Diffraction data were taken in conventional (non-helical) mode from a single PEG-400 protected crystal over a 360° range with 0.5° oscillation steps and exposure time of 5 s. with an ADSC Q210r detector. Data were measured from a single Paratone-N soaked crystal over a 7200degree range with 0.2° oscillation images and an exposure time of 2 s with a Dectris Pilatus 6M detector. Data from PEG-400 protected crystals were integrated and scaled using HKL2000 (Otwinowski and Minor, 1997). Data from Paratone-N-protected crystal were integrated and scaled using XDS (Kabsch, 2010a) implemented by use of the autoxds script (A. Gonzalez and Y Tsai, http://smb.slac.stanford.edu/facilities/software/xds#autoxds_script).
Initial crystallographic phases were determined by use of anomalous differences in scattering from sulfur atoms in pR452. The sulfur substructure was determined using the SHELXC/D/E program suite (Sheldrick, 2010). Preliminary crystallographic phases were determined and refined from the sulfur substructure using the AutoSol wizard in PHENIX (Terwilliger et al., 2009). An initial atomic model was constructed at 3.4Å using AutoBuild (Terwilliger et al., 2008) and by manual fitting of a sigma-weighted 2mFo-DFc map using Coot (Emsley and Cowtan, 2004; Emsley et al., 2010). Phases were extended to 2.2Å resolution using the 0.979Å wavelength data by iterative cycles of manual rebuilding and refinement using the phenix.refine module in PHENIX(Afonine et al., 2012). The refined atomic model was used to determine the structure of pR452 from crystals immersed in Paratone-N by molecular replacement using the Phaser (McCoy et al., 2007) GUI in PHENIX, followed by manual rebuilding and refinement, which, at final stages was guided by inspection of information generated by MolProbity structure evaluation software (Chen et al., 2010; Davis et al., 2007). Refined models and structure factors are deposited in the RCSB Protein Data Bank (Berman et al., 2000) (www.wwwpdb.org) with ID numbers 6NMG (PEG-400 cryoprotected crystals) and 6NMJ (Paratone-N cryoprotected crystals).
Small Angle X-Ray Scattering Data Collection, Analysis and Modeling
Synchrotron radiation X-ray scattering data from the SEC–SAXS experiments were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) beamline BL 4–2 using Blueice (McPhillips et al., 2002) software with a focused 12 keV X-ray source (0.3 × 0.3mm beam size) and recorded on a Pilatus3 X 1M detector at a sample-to-detector distance of 1.7m and over a range of momentum transfer 0.0065<q<0.42Å−1 [ q=4πsin(θ)/λ]. Protein sample (approximately 1 mg/ml in 50mM HEPES, pH 8.0, 150mM NaCl and 1mM TCEP) was allowed to through a 1.5 mm quartz capillary sample cell as the eluate from Superdex 200 Increase 1.3/300 size exclusion column mounted on an Akta-FPLC system (GE Healthcare) at a flow rate at 0.5 ml/min. Scattering data frames were taken with 1 second exposures. Intensity data were radially averaged to produce one-dimensional profiles of scattering intensity vs. q. Data were corrected for background scattering by subtracting the buffer curve from sample curves. Data reduction and analysis were performed using the beamline software SAStool and PRIMUS of the ATSAS suite (Franke et al., 2017) The program AutoGNOM was used to generate Guinier curves and compute the pair distribution function, P(r), to determine the maximum dimension Dmax and radius of gyration Rg from the scattering intensity curve (I(q) versus q) in an automatic, unbiased manner, and rounds of manual fitting in GNOM (Svergun, 1992) were used to verify these values. Ab initio molecular envelopes were computed by the programs DAMMIN (Svergun, 1999). Ten bead models were reconstructed in DAMMIF (Franke and Svergun, 2009), which were aligned and averaged in DAMAVER (Volkov and Svergun, 2003) with no rejections and a normalized spatial discrepancy of 0.486 ± 0.015 and 0.490 ± 0.019 for R452 and pR452 respectively. Molecular envelopes were visualized, and atomic models fit to molecular envelopes using Chimera (Pettersen et al., 2004).
The conformational flexibility of pR452 and R452 was modeled by coarse-grained fitting with respect to experimental SAXS data using SREFLEX program in the ATSAS software package. Normal mode analysis was conducted with automatic determination of rigid body units. The final disposition of rigid body units after application of normal mode projections was determined by rigid body refinement with respect to the computed SAXS profile (Panjkovich and Svergun, 2016). CRYSOL software from the ATSAS software package was used to model scattering profiles from atomic coordinates.
Heteronuclear NMR Data Collection and Analysis
Uniformly 2H,15N isotope-labeled Gαi1Δ31 was expressed and purified as described (Goricanec et al., 2016). 2D-TROSY NMR spectra of 2H,15N Gαi1Δ31 alone or in the presence of R452 at 0.5 and 1.6 stoichiometric ratio to Gαi1Δ31, were recorded with 1024 and 128 complex points in the 1H and indirect 15N dimension, respectively, at 303K on a 600 MHz Bruker spectrometer equipped with a cryogenic TXI probe. Samples contained 100 μM 2H,15N Gαi1Δ31 in 20mM Sodium Phosphate pH 7.0, 50 mM NaCl, 5 mM MgCl2 and 5 mM dithiothreitol, 300 μM GDP. Backbone assignments are available for 73 % of the resonances in the 2D-[1H,15N] -TROSY spectrum of GDP-bound Gαi1Δ31(Goricanec and Hagn, 2019). Chemical shift perturbations were calculated using the empirical formula Δδav = (ΔδH2 + (ΔδN/5)2)½. Data analysis was carried out with NMRFAM Sparky software (Lee et al., 2015) based on Sparky4 (T.D. Goddard and D.G. Kneller, University of California, San Francisco) and structures were visualized with UCSF Chimera (Pettersen et al., 2004).
Mass Spectrometry
Protein sample (400 μl at 1mg/ml) was subjected to chromatography on a 1.0mm × 100 mm Phenomenex Onyx monolithic C18 column (Torrance, CA) with a mobile phase gradient (A= water, 0.1% formic acid; B=acetonitrile, 0.1% formic Acid: 0’−1’, 10%B; 1’−7’, 10% to 70%B; 7’−8’, 70% to 90%B; 8’−9’, 90%B; 9’−10’, 90% to 10%B) in line to a Brucker microTOF focus II at a flow rate of 400 μl/min, scanning in the positive mode over a scan range of 200 – 3000 m/z at a scan rate of 2 Hz. Data analysis was conducted with Bruker DataAnalysis software v 4.2 provided with the instrument, using the Maximum Entropy deconvolution algorithm.
For analysis by MALDI-TOF MS, crystals of pR452 (red) were rinsed in reservoir solution three times and dissolved in 0.1% trifluoroacetic acid (TFA) to create a ~10 pmol/μL pR452 solution. Purified R452 and pR452 in solution were also diluted to ~10pmol/μL using 0.1% TFA prior to sample preparation. MALDI matrices (Bruker) were prepared according to manufacturer’s directions. Briefly, a 2’,6’-dihydroxyacetophenone (DHAP) solution was prepared from 7.6 mg of DHAP in 375 μl ethanol and 125 μl of aqueous diammonium hydrogen citrate (DAHC) solution (18 mg/ml). One part sample solution was mixed with one part of 2 % TFA and one part of DHAP solution; then 1μL or 2 μl of sample mixture were spotted on a MSP 96 target polished steel BC microScout Target plate (Bruker) and dried at room temperature. Mass spectrometry experiments were performed using a microflex MALDI-TOF (Bruker) with flexControl (Bruker) software provided with the instrument. Protein samples in the DHAP matrices were ionized at 37%−50% laser power and ion masses scanned over a 10–70 kDa range. Data was analyzed using flexAnalysis (Bruker) software provided with the instrument.
Surface Plasmon Resonance Binding Analysis
All proteins were buffer-exchanged by elution though a Superdex-200 column (GE Healthcare) into running buffer (50mM HEPES pH 8.0, 150mM NaCl, 1mM TCEP). N-terminally hexa-histidine-tagged Ric-8A ligands (R452, pR452, R491, pR491) at 0.1μM in running buffer were anchored to an Ni-NTA sensor chip surface for 5 minutes followed by a 7-minute wash, or until the baseline response level was stable, with buffer to remove unbound/loosely bound ligand molecules. For binding phase, Gαi1 analyte at each of a range of concentrations (0.625 μM, 1.25μM, 2.5μM) of was applied to the ligand-coated sensor chip surface for 3 minutes, followed by a 6-minute wash with running buffer, to complete the dissociation phase. All steps were conducted at room temperature (~25°C) at a flow rate of 30μL/min. After each analyte binding/dissociation step, the Ni-NTA sensor chip surface was regenerated by first stripping with 350mM Ethylenediaminetetraacetic acid (EDTA), followed by doubly distilled water (ddH2O), followed by 0.5mM NiCl2, 3mM EDTA, and finally, running buffer. Using BiaCore X100 evaluation software (GE Healthcare) provided with the instrument, all binding and dissociation comprising a single data set were globally fit to a 1:1 binding model to generate a single kon, koff, Rmax, for the binding phase, R(t), the relative response at time t, = (1 – exp -(kon ·C+koff)t)Req and for the dissociation phase, R(t) = (exp -(koff ·t)Req, where Req=(kon ·C/(kon ·C+koff))Rmax, where Rmax is the maximum value of the response unit if all ligand at concentration C, is occupied by analyte, kon is an apparent pseudo-first order association rate (M−1s−1), and koff is the dissociation rate (s−1).
QUANTIFICATION AND STATISTICAL ANALYSIS
Figure 1: Data points are shown as mean +/− SEM for three replicates. Replicates represent three independent measurements of labeled reaction products from the same reaction volume.
Figures 4A and 4D: Agreement between experimental and modeled scattering intensities is shown as the residual divided by the standard deviation of measurement (ΔI/σ).
Figure S2: Sensograms were determined once for each sample at three analyte concentrations. Standard error for fit of kinetic parameters determined by a global nonlinear fit of observed sensogram data to the models described in STAR Methods.
Figure S6B: Three to 5 replicates were taken for each data set and significance of differences was estimated by a Student’s T Test. Each replicate derives from a progress curve generated by mixture of reaction components from the same preparation.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21 (DE3)-RIPL | Agilent Technologies | Fisher Cat # NC9122855 |
| E. coli JM109 | Promega | Cat # L1001 |
| E. coli TOP10 | ThermoFisher | Cat # C404010 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Casein Kinase II | New England Biolabs | Cat # P6010L |
| γ–32P-GTP | PerkinElmer | Cat # BLU004Z250UC |
| Guanosine 5’-[γ-thio]triphosphate tetralithium salt | Roche | Cat # 10220647001 |
| Guanosine 5’-[γ-thio]triphosphate | Jena Bioscience | Cat # NU-412–10 |
| Paratone-N (Parabar 10312) | Hampton Research | Cat # HR2–643 |
| Critical Commercial Assays | ||
| QuikChange II-XL mutagenesis kit | Agilent | Cat # 200521 |
| Deposited Data | ||
| Atomic Coordinates and Structure Factors, native pR452 structure | RCSB Protein Databank www.wwwpdb.org | 6NMG |
| Atomic Coordinates and Structure Factors, paratone-immersed structure | RCSB Protein Databank www.wwwpdb.org | 6NMJ |
| Small Angle X-ray Scattering Data, R452 | Small Angle Scattering Biological Data Bank https://www.sasbdb.org/ | SASDFA5 |
| Small Angle X-ray Scattering Data, pR452 | Small Angle Scattering Biological Data Bank https://www.sasbdb.org/ | SASDFB5 |
| Oligonucleotides | ||
| Forward and reverse primers for Arg345Gln | Integrated DNA Technologies | n.a. |
| Mutation of R452 | ||
| 5’-gaacttcctggcaggctggtgcatgcgggcac-3’ | ||
| 5’-gtgcccgcatgcaccagcctgccaggaagttc-3’ | ||
| Forward and reverse primers for Arg345Gln | Integrated DNA Technologies | n.a. |
| Mutation of R452 | ||
| 5’-gggccttcaggaacgccctggcaggacggt-3’ | ||
| 5’-accgtcctgccagggcgttcctgaaggccc-3’ | ||
| Recombinant DNA | ||
| Rat His(6)-Ric-8A(1–452) in pET-28A vector | Thomas et al., 2011 | n.a. |
| Rat His(6)-Ric-8A(1–491) in pET-28A vector | Thomas et al., 2011 | n.a. |
| Rat His(6)-Ric-8A (full-length) in pET21a vector | Yu et al., 2019 | n.a. |
| Rat internally His(6)-tagged Gαi1 in pQE60 vector | Mumby and Linder, 1994 | n.a. |
| Yeast N-myristoyltransferase in pBB131 vector | Mumby and Linder, 1994 | n.a. |
| Software and Algorithms | ||
| PHENIX suite | https://www.phenix-online.org/ | n.a. |
| HKL2000 | http://www.hkl-xray.com/ | n.a. |
| autoxds script | http://smb.slac.stanford.edu/facilities/software/xds#autoxds_script | |
| SHELXC/D/E program suite | http://www.ccp4.ac.uk/download/http://shelx.uni-goettingen.de/download.php | |
| COOT | https://www2.mrc-lmb.cam.ac.uk/Personal/pemsley/coot/ | n.a. |
| MolProbity | http://molprobity.biochem.duke.edu/ | n.a. |
| ATSAS suite | https://www.embl-hamburg.de/biosaxs/download.html | n.a. |
| SAStool | https://www-ssrl.slac.stanford.edu/smb-saxs/content/software | n.a. |
| NMRFAM Sparky | https://nmrfam.wisc.edu/nmrfam-sparky-distribution/ | n.a. |
| UCSF Chimera | https://www.cgl.ucsf.edu/chimera/ | n.a. |
| PyMOL 1.7 | https://pymol.org | n.a. |
| ESPript 3.0 | http://espript.ibcp.fr | n.a. |
| Disopred 3.0 | http://bioinf.cs.ucl.ac.uk/psipred/?disopred=1 | n.a. |
| Agilent QuikChange primer design tool | https://www.agilent.com/store/primerDesignProgram.jsp | n.a. |
| CLUSTALW | https://www.ebi.ac.uk/Tools/msa/clustalo/ | n.a. |
| UniProtKb | https://www.uniprot.org/. | n.a. |
Highlights.
The G protein α subunit binding domain of Ric-8A is an Armadillo/HEAT repeat fold
Hydrogen-deuterium exchange data suggest a Gα a binding surface on Ric-8A
Ric-8A binds elements of Gα also recognized by G protein-coupled receptors
The Ric-8A fold harbors a putative binding site for its C-terminal phosphoserine
ACKNOWLEDGMENTS
We thank Drs. Wuxian Shi and Martin Fuchs at the National Synchrotron Light Source II (NSLS II) FMX beamline, and the staff at Advanced Photon Source (APS)/Structural Biology Center (SBC)-CAT 19-BM and Stanford Synchrotron Laboratory (SSRL) beamline 9-1 for excellent assistance with X-ray crystallographic data collection and from Dr. Tsutomu Matsui at the SSRL 4-2 beamline and Cindee Yates-Hansen for assistance with FPLC-SAXS data collection, and Dr. Celestine Thomas for his assistance during earlier phases of the project. The FMX (17-ID-2) beamline is supported by NIH grant P41GM111244 and the Department of Energy (DOE), KP1605010. SSRL is supported by the DOE under contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE and by NIH grant P41GM103393. The SBC-CAT at APS is supported by DOE under contract DE-AC02-06CH11357. This research was supported by NIH R01GM105993 (to S.R.S.), the Helmholtz Society and the Helmholtz Zentrum München grant VH-NG-1039 and DFG grant HA6105/3-1 (to F.H.), NIH R01-GM088242 (to G.G.T.), RHHMI ‘‘Med-into-Grad’’ Fellowship, NIH T32-GM06841 and pre-doctoral fellowship from the PhRMA Foundation (to M.P.-S.). The Macromolecular X-ray Diffraction Core at the University of Montana Center for Biomolecular Structure and Dynamics is supported by NIH COBRE award P20GM103546.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Atomic coordinates and structure factors for native and paratone-immersed pR425 crystals are deposited in the RCSP Protein Data Bank (https://www.rcsb.org) with accession numbers 6NMG and 6NMJ, respectively.
Small Angle Scattering data for R452 and pR452 are deposited in the Small Angle Scattering Biological Data Base (SASBDB) https://www.sasbdb.org/ with accession identifiers SASDFA5 and SASDFB5, respectively.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.str.2019.04.013.
REFERENCES
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade MA, Petosa C, O’Donoghue SI, Muller CW, and Bork P (2001). Comparison of ARM and HEAT protein repeats. J. Mol. Biol 309, 1–18. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Erez E, Martz E, Pupko T, and Ben-Tal N (2010). ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38, W529–W533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellaiche Y, and Gotta M (2005). Heterotrimeric G proteins and regulation of size asymmetry during cell division. Curr. Opin. Cell Biol 17, 658–663. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000). The protein data bank. Nucleic Acids Res 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumer JB, and Lanier SM (2014). Activators of G protein signaling exhibit broad functionality and define a distinct core signaling triad. Mol. Pharmacol 85, 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boularan C, Kamenyeva O, Cho H, and Kehrl JH (2014). Resistance to inhibitors of cholinesterase (Ric)-8A and Galphai contribute to cytokinesis abscission by controlling vacuolar protein-sorting (Vps)34 activity. PLoS One 9, e86680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, and Hamm HE (2003). Insights into G protein structure, function, and regulation. Endocr. Rev 24, 765–781. [DOI] [PubMed] [Google Scholar]
- Chan P, Thomas CJ, Sprang SR, and Tall GG (2013). Molecular chaperoning function of Ric-8 is to fold nascent heterotrimeric G protein alpha subunits. Proc. Natl. Acad. Sci. U S A 110, 3794–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, et al. (2007). MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiacomo V, Marivin A, and Garcia-Marcos M (2018). When heterotrimeric G proteins are not activated by G protein-coupled receptors: structural insights and evolutionary conservation. Biochemistry 57, 255–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson HJ (2016). Making sense of intrinsically disordered proteins. Biophys. J 110, 1013–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa M, Hinrichs MV, Bunster M, Babbitt P, Martinez-Oyanedel J, and Olate J (2009). Biophysical studies support a predicted superhelical structure with armadillo repeats for Ric-8. Protein Sci 18, 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flock T, Ravarani CN, Sun D, Venkatakrishnan AJ, Kayikci M, Tate CG, Veprintsev DB, and Babu MM (2015). Universal allosteric mechanism for Galpha activation by GPCRs. Nature 524, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, et al. (2017). ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr 50, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, and Svergun DI (2009). DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr 42, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay M, Pinter ME, Wright FA, Chan P, Murphy AJ, Valenzuela DM, Yancopoulos GD, and Tall GG (2011). Ric-8 proteins are molecular chaperones that direct nascent G protein alpha subunit membrane association. Sci. Signal 4, ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG (1987). G-proteins. Annu. Rev. Biochem 56, 615–649. [DOI] [PubMed] [Google Scholar]
- Goricanec D, and Hagn F (2019). NMR backbone and methyl resonance assignments of an inhibitory G-alpha subunit in complex with GDP. Biomol. NMR Assign 13, 131–137. [DOI] [PubMed] [Google Scholar]
- Goricanec D, Stehle R, Egloff P, Grigoriu S, Pluckthun A, Wagner G, and Hagn F (2016). Conformational dynamics of a G-protein alpha subunit is tightly regulated by nucleotide binding. Proc. Natl. Acad. Sci. U S A 113, E3629–E3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampoelz B, and Knoblich JA (2004). Heterotrimeric G proteins: new tricks for an old dog. Cell 119, 453–456. [DOI] [PubMed] [Google Scholar]
- Hinrichs MV, Torrejon M, Montecino M, and Olate J (2012). Ric-8: different cellular roles for a heterotrimeric G-protein GEF. J. Cell. Biochem 113, 2797–2805. [DOI] [PubMed] [Google Scholar]
- Jones DT, and Cozzetto D (2015). DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010a). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010b). Xds. Acta Crystallogr. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant R, Zeng B, Thomas CJ, Bothner B, and Sprang SR (2016). Ric-8A, a G protein chaperone with nucleotide exchange activity induces long-range secondary structure changes in Galpha. Elife 5, 10.7554/eLife.19238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, and Ben-Tal N (2005). ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33, W299–W302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Tonelli M, and Markley JL (2015). NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, et al. (2002). Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat 9, 401–406. [DOI] [PubMed] [Google Scholar]
- Mertens HD, and Svergun DI (2010). Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol 172, 128–141. [DOI] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, and Rand JB (1996). A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. U S A 93, 12593–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Emerson MD, McManus JR, and Rand JB (2000). RIC-8 (Synembryn): a novel conserved protein that is required for G(q)alpha signaling in the C. elegans nervous system. Neuron 27, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, and Rand JB (2000). A role for RIC-8 (Synembryn) and GOA-1 (G(o)alpha) in regulating a subset of centrosome movements during early embryogenesis in Caenorhabditis elegans. Genetics 156, 1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumby SM, and Linder ME (1994). Myristoylation of G-protein alpha subunits. Methods Enzymol 237, 254–268. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Nishimura A, Tago K, Mizuno N, and Itoh H (2010). Ric-8B stabilizes the alpha subunit of stimulatory G protein by inhibiting its ubiquitination. J. Biol. Chem 285, 11114–11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Panjkovich A, and Svergun DI (2016). Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis. Phys. Chem. Chem. Phys 18, 5707–5719. [DOI] [PubMed] [Google Scholar]
- Papasergi MM, Patel BR, and Tall GG (2015). The G protein alpha chaperone Ric-8 as a potential therapeutic target. Mol. Pharmacol 87, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasergi-Scott MM, Stoveken HM, MacConnachie L, Chan PY, Gabay M, Wong D, Freeman RS, Beg AA, and Tall GG (2018). Dual phosphorylation of Ric-8A enhances its ability to mediate G protein alpha subunit folding and to stimulate guanine nucleotide exchange. Sci. Signal 11, 10.1126/scisignal.aap8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011). Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Pozuelo J, Dason JS, Mansilla A, Banos-Mateos S, Sardina JL, Chaves-Sanjuan A, Jurado-Gomez J, Santana E, Atwood HL, Hernandez-Hernandez A, et al. (2014). The guanine-exchange factor Ric8a binds to the Ca(2)(+) sensor NCS-1 to regulate synapse number and neurotransmitter release. J. Cell Sci 127, 4246–4259. [DOI] [PubMed] [Google Scholar]
- Ross EM (2002). Quantitative assays for GTPase-activating proteins. Methods Enzymol 344, 601–617. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2010). Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr 66, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprang SR, Chen Z, and Du X (2007). Structural basis of effector regulation and signal termination in heterotrimeric Galpha proteins. Adv. Protein Chem 74, 1–65. [DOI] [PubMed] [Google Scholar]
- Svergun DI (1992). Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr 25, 495–503. [Google Scholar]
- Svergun DI (1999). Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J 76, 2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall GG (2013). Ric-8 regulation of heterotrimeric G proteins. J. Recept. Signal Transduct. Res 33, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall GG, Krumins AM, and Gilman AG (2003). Mammalian Ric-8A (synembryn) is a heterotrimeric Galpha protein guanine nucleotide exchange factor. J. Biol. Chem 278, 8356–8362. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, and Hung LW (2009). Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr 65, 582–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Bunkoczi G, Hung LW, Zwart PH, Smith JL, Akey DL, and Adams PD (2016). Can I solve my structure by SAD phasing? Planning an experiment, scaling data and evaluating the useful anomalous correlation and anomalous signal. Acta Crystallogr. D Struct. Biol 72, 359–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, and Adams PD (2008). Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr 64, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker TM, Kaya AI, Preininger AM, Hamm HE, and Iverson TM (2012). Allosteric mechanisms of G protein-coupled receptor signaling: a structural perspective. Methods Mol. Biol 796, 133–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CJ, Briknarova K, Hilmer JK, Movahed N, Bothner B, Sumida JP, Tall GG, and Sprang SR (2011). The nucleotide exchange factor Ric-8A is a chaperone for the conformationally dynamic nucleotide-free state of Galphai1. PLoS One 6, e23197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trewhella J, Duff AP, Durand D, Gabel F, Guss JM, Hendrickson WA, Hura GL, Jacques DA, Kirby NM, Kwan AH, et al. (2017). 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: an update. Acta Crystallogr. D Struct. Biol 73, 710–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N, Thomas CJ, Hubbell WL, and Sprang SR (2015). The guanine nucleotide exchange factor Ric-8A induces domain separation and Ras domain plasticity in Galphai1. Proc. Natl. Acad. Sci. U S A 112, 1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, and Svergun DI (2003). Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr 86, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Yu M, Papasergi-Scott MM, and Tall GG (2019). Production of Phosphorylated Ric-8A proteins using protein kinase CK2. Protein Expr. Purif 154, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.