

Abstract
The limited availability of rapid and reliable flow cytometry-based assays for ex vivo quantification of autophagy has hampered their clinical applications for studies of diseases pathogenesis or for the implementation of autophagy-targeting therapies. To this aim, we modified and improved the protocol of a commercial kit developed for quantifying the microtubule-associated protein 1A/1B light chain 3B (LC3), the most reliable marker for autophagosomes currently available. The protocol modifications were set up measuring the autophagic flux in neoplastic (THP-1 cells) and primary cells (peripheral blood mononuclear cells; PBMC) of healthy donors. Moreover, PBMC of active tuberculosis (TB) patients were stimulated with the Mycobacterium tuberculosis purified protein derivatives or infected with live Mycobacterium bovis bacillus Calmette-Guerin (BCG). We found that the baseline median fluorescent intensity (MFI) of THP-1 cells changed depending on the time of sample acquisition to the flow cytometer. To solve this problem, a fixation step was introduced in different stages of the assay’s protocol, obtaining more reproducible and sensitive results when a post-LC3-staining fixation was performed, in either THP-1 cells or PBMC. Furthermore, since we found that results are influenced by the type and the dose of the lysosome inhibitor used, the best dose of Chloroquine for LC3 accumulation were set up in either THP-1 cells or PBMC. Finally, applying these experimental settings, we measured the autophagic flux in CD14+ cells from active TB patients’ PBMC upon BCG infection. In conclusion, our data indicate that the protocol modifications here described improve the stability and accuracy of a flow cytometry-based assay for the evaluation of autophagy, thus assuring more standardised cell analyses.
Key words: Autophagy, tuberculosis, BCG, flow cytometry, LC3, PBMC, THP-1
Introduction
Autophagy is a pathway by which cytoplasmic materials, including macromolecules, organelles and pathogens, are delivered to the lysosome for degradation and, eventually, for recycling of intracellular constituents. Based on the mechanisms for lysosome delivery, three types of autophagy have been defined: macroautophagy, microautophagy and chaperonemediated autophagy.1 During macroautophagy (hereinafter referred as autophagy) the materials to be degraded are sequestered and delivered to lysosomes through de novo generation of a double-membrane vesicles, termed autophagosomes.2 Their formation requires, among several proteins encoded by the autophagy-related genes, the activity of the microtubule-associated proteins 1A/1B light chain 3B (LC3), which is considered the most reliable marker of autophagosomes. Under normal conditions, LC3 is mainly diffuse in the cytosol as an unmodified form, denominated LC3-I. Upon autophagy initiation, LC3-I is modified by the addition of a phosphatidylethanolamine moiety through a ubiquitin-like conjugation system in the LC3-II form, which translocates to nascent autophagosomes regulating their formation.3 Autophagic processes are either constitutive or activated in response to different stimuli. Autophagy is involved in various physiological processes and its alterations have been associated with different pathologies such as neurodegenerative diseases, cardiovascular complications, cancers and infectious diseases.4-6 In particular, autophagy has been implicated in the control of microbial infections for its role in the innate and specific immunity, such as pathogen clearance, lymphocyte development, antigen presentation and immunoglobulin production.7-9 Several pathogens have developed strategies to overcome or manipulate the autophagy response to prevent their clearance, allowing them to establish infection. Therefore, it would be important to use sensitive quantitative assays to measure variations in the autophagy flux for the study of several diseases. For instance, in the tuberculosis field, it has been shown that Mycobacterium tuberculosis (Mtb) impairs cell autophagic flux in primary macrophages infected in vitro and that specific T cells rescue this block.10 Therefore, it would be interesting to evaluate in primary cells of active tuberculosis (TB) patients a modulation of the autophagic flux. The most widely approaches used to detect LC3-I to LC3-II conversion, and hence the number of autophagosomes, is the immunoblot analysis, while the LC3 cellular localisation is mainly detected by fluorescence microscopy analysis, which are, however, time consuming procedures not applicable for a clinical purpose. 11 In the attempt to make the autophagy evaluation significantly less burdensome, several efforts have been made to measure LC3 by flow cytometry, which allows rapid analysis that can be combined with other phenotypic and functional markers for the characterization of different cell populations. 12-14 For instance, engineered LC3 fused with fluorescent proteins (i.e. Green or Red Fluorescent Protein or mCherry) has been generated, however, this approach requires cells transfection, a methodology not easily feasible with primary cells and that requires several days of cultures. On the other hand, although several anti-LC3 antibodies have been tested, to the best of our knowledge, no antibody specific for LC3-II form has been developed, which confines the analysis to the measurement of total LC3 with no distinction between LC3- I and LC3-II forms. Recently, this limit has been circumvented by the development of a commercial method that eliminates the cytosolic LC3-I form by a selective cell permeabilisation step, while leaving LC3-II inside the autophagosomes, which is measurable by flow cytometry.15-17
Aim of this study was to improve the accuracy of a commercial cytometric test for the detection of autophagic flux in cell lines and primary cells. To this goal, LC3 levels were measured either in THP-1 cells or in peripheral blood mononuclear cells (PBMC) introducing modifications in the assay’s procedure. Moreover, in an effort to evaluate the autophagic flux modulation during the antigen-specific response or bacterial infection, autophagy was measured in PBMC isolated from active TB patients, either treated with the Mtb purified protein derivatives (PPD) or infected with live Mycobacterium bovis bacillus Calmette- Guerin (BCG).
Materials and Methods
Cells
PBMC from individuals, either with pulmonary active TB or healthy donors, were isolated from whole blood by Ficoll density gradient centrifugation.18 Active TB was diagnosed by a positive culture for Mtb from the sputum.19 The study was approved by the ethical committee of the National Institute of Infectious Diseases (INMI) “Lazzaro Spallanzani” (Approvals no. 29/2014; 34/2010; 28/2014), Rome, Italy. All participants were informed about the aims of the study and their written and signed informed consent was obtained. THP-1, a human monocytic leukemia cell line, were cultured as already reported20 and routinely screened for mycoplasma infection (Applied Biological Materials Inc., Richmond, BC, Canada - Cat. No. G238. THP-1 cells and PBMC were cultured in RPMI 1640 supplemented with heat inactivated 10% fetal bovine serum, 2 mM L-glutamine and 1% penicillin/streptomycin solution (Sigma-Aldrich, St. Louis, MA, USA; Cat. No. R0883; F7524; G7513; P0781, respectively) and maintained at 5% CO2, 37°C.
Autophagic flux evaluation
Briefly, cells, either THP-1 or PBMC, were seeded at 1x106/tube and left untreated or treated with autophagy inhibitors for the indicated times (i.e. 4 h or 24 h). Autophagy inhibition was obtained using different compounds: Reagent A (RA) present in the FlowCellect™ Autophagy LC3 Antibodybased Assay Kit (Merck Millipore, Burlington, MA, USA; Cat. No. FCCH100171; hereinafter referred as FlowCellect Kit) accordingly with the manufacturer’s instruction; Bafilomycin A1 (BafA1; 5μM) and Chloroquine (CQ) at the indicated concentrations (Sigma-Aldrich; Cat. No. B1793, C6628, respectively).
PBMC from active TB patients were either infected with BCG using a MOI=1 (1 bacillus/cell) (BCG Pasteur (TMC1011)), kindly provided by Prof. Giovanni Delogu (Fondazione Policlinico Universitario A. Gemelli IRCCS, Università Cattolica del Sacro Cuore, Rome, Italy),21 or stimulated with 25 μg/mL of Mtb PPD (Statens Serum Institut, Copenhagen, Denmark). Treatments with BCG or PPD were carried out for 24 h either alone or in presence of CQ (20 μM) for the last 20 h.
Flow cytometry
The expression of LC3 was evaluated by the FlowCellect Kit, which contains a mouse FITC-conjugated anti-LC3 monoclonal antibody (clone 4E12), using the FACS Canto II flow cytometer (BD Biosciences, Franklin Lake, NJ, USA). To ensure more data consistency over time, cytometer performance was optimized in each experiment using the cytometer setup and tracking beads (BD Biosciences, Cat. No. 642412). Relative fluorescence intensity value and histograms were obtained and analysed with FlowJo software (Tree Star Inc., Ashland, OR, USA). The histograms plots showed the intensity of expression vs the number of events. Each analysis included at least 100,000 events and each experiment was performed at least in triplicates. Briefly, the LC3 staining was conducted either following the manufacturer’s instruction or with the addition of a fixation step using a 4% (v/v) neutral buffered formalin [about 1.6% (v/v) formaldehyde; UCS Diagnostic, Morlupo, Rome, Italy; Cat. No. FRN500] in 1xPBS solution introduced either i) soon after the permeabilisation step before LC3 staining (Fix pre-staining) or ii) after the staining step with the anti-LC3 antibodies (Fix post-staining) as schematized in Table 1 (see also Supplementary Figure 1).
In some experiments, PBMC were costained for CD3 (Miltenyi Biotec Inc., CD3-APC-Vio770; Cat. No. 130-096-610) or CD14 (CD14-PE-Cy7; Cat. No. 557742; BD Biosciences), in addition to anti-LC3. In experiments carried out using more than one fluorescent antibody, we used compensation beads to optimize fluorescence settings for multicolour flow cytometric analyses (BD Biosciences; Cat. No. 552843).
Immunoblotting analysis
Western blotting analysis was carried out as previously reported.22,23 Whole cell extracts (12 μg), obtained using CelLytic™ MT Cell Lysis Reagent (Sigma-Aldrich, Cat. No. C3228), were separated on SDSPAGE 13.5% gels and electroblotted onto polyvinylidene difluoride membrane (Merck Millipore, Cat. No. IPVH20200). Blots were incubated with primary antibodies diluted in 5% (w/v) non-fat dry milk in 1X TBST buffer (tris-buffered saline (TBS), 0,1% Tween20; Sigma-Aldrich, Cat. No. T5030; P1379, respectively). The primary antibodies used in this study were rabbit anti-LC3 (Cell Signaling Technology, Danvers, MA, USA; Cat. No. 2775; diluted 1:1000) and mouse anti-glyceraldehyde-3- phosphate dehydrogenase (GAPDH; Merck Millipore; Cat. No. CB1001; diluted 1:5000). Detection was achieved using horseradish peroxidase-conjugate secondary antibodies (Jackson Immuno- Research Laboratories, West Grove, PA, USA; anti-mouse Cat. No. 715036150, antirabbit Cat. No. 711036152) and visualized with either Luminata crescendo for LC3 or Luminata Classico for GAPDH (Merck Millipore, Cat. No. WBLUR0500, WBLUC0500, respectively) using the BioRad Chemidoc. Quantification of band intensity was performed with Image Lab 5.2.1.
Table 1.
LC3 staining steps.
| No fix (Manufacturer’s protocol) | Pre-staining fixation | Post-staining fixation | |
|---|---|---|---|
| Permeabilisation (Reagent B) | Yes | Yes | Yes |
| Fixation (1.6% formaldehyde) | / | Yes (5’ at RT) | / |
| Wash (1X PBS) | Yes | Yes | Yes |
| Anti-LC3 Ab s(30’ at R.T.) | Yes | Yes (+0.5% Sap) | Yes |
| Fixation (1.6% formaldehyde) | / | / | Yes (5’ at RT) |
| Wash (1X PBS) | Yes | Yes | Yes |
Sap, saponin
Statistical analysis
Data analysis was performed using GraphPad Prism version 7.0.4 for Windows (GraphPad Software, San Diego, CA, USA; www.graphpad.com) and the Mann Whitney U test was used to analyze unpaired data to compare the groups.
Results
Comparison of different autophagy inhibitors in THP-1 cells
To accurately measure cellular autophagic activity, it is essential to evaluate the so-called “autophagic flux”, which is defined as the amount of autophagosomes degradation in a given period of time. This is usually measured by comparing the levels of LC3-II in the presence or absence of lysosomal inhibitors. In the attempt to optimize the measurement of autophagic flux by flow cytometry, we used the FlowCellect Kit developed by Millipore. To this aim we, firstly, compared the effects on LC3 accumulation of THP-1 cells induced by Reagent A (RA; diluted 1:1000), an undisclosed autophagy-regulating compound(s) provided by the FlowCellect Kit, to those stimulated by either BafA1 (5 μM) or CQ (80 μM), two well characterized autophagy inhibitors that affect lysosome functions and, consequently, block autophagosomes degradation.24 Cells were treated with one of the three compounds or left untreated for 24 h and the LC3 levels evaluated using the FlowCellect Kit, following the manufacture’s instruction, measuring the relative median fluorescence intensity (MFI) (Figure 1). Each sample was measured by flow cytometry at three different times: i) right after the LC3 staining (0 h), ii) after 2 h (data not shown because no differences with the 0 h samples were found) or iii) after one day (24 h). The three compounds display different ability to induce LC3 accumulation in THP-1 cells, at least at the concentration tested, with RA being the most powerful inducer of LC3 accumulation, followed by CQ and BafA1, as measured by flow cytometry (Figure 1 A-E). When samples were acquired right after the end of the assay (0 h) RA, CQ and BafA1 accumulated LC3 14.97±0.35, 5.77±0.51 and 4.21±0.08 fold with respect to that of untreated cells (NT), respectively (Figure 1A, red columns). This ranking was the same also when the samples were acquired one day after the end of LC3-staining procedure (24 h), although with slightly lower values (12.82±0.04; 5.07±0.43 and 3.79±0.04 fold of NT, respectively) (Figure 1A, blue columns). The same trend of LC3-II accumulation was observed by western blot analysis (Figure 1 F,G). Since the levels of LC3-I form are similarly represented independently of the treatments used (with the only exception of the RA-treatment), while the LC3-II amount significantly increases in treated cells, it is likely that the LC3 measured by flow cytometry is mainly LC3-II rather than a mixture of LC3-II/LC3-I forms. Since the permeabilisation step, which is necessary for LC3-I extracellular release, affects significantly the shape of the cells (data not shown), we measured also the baseline MFI of the THP-1 cells analysing the samples that were not stained with LC3 antibodies (unstained, US). As shown in Figure 1 C,D, we found that, regardless of the type of stimulation, the unstained samples showed a different MFI depending on the time of sample acquisition to the flow cytometer. In fact, increased MFI values were measured in cells acquired 24 h after LC3 staining (blue columns) with respect to the same sample acquired right after the end of the assay (0 h) (1.38±0.06; 1.26±0.03; 1.26±0.02 and 1.25±0.00 fold of 0h for NT, RA, BafA1 and CQ, respectively). These differences were substantial when the LC3-stained samples were compared to the paired unstained samples. In fact, as shown in Figure 1E, the ratio between LC3-stained/unstained samples decreased when the cells were acquired 24 h post-assay compared to the same samples acquired right after the LC3 staining procedure (blue vs red columns, respectively). These results suggest, therefore, that a fixation step may be useful to avoid misinterpretation of the results, particularly when samples have to be acquired several hours after the staining procedure. Moreover, as mentioned above, although the RA provided by the kit better induces LC3 accumulation, we chose to work with a widely used autophagy inhibitor, as the CQ, for the procedure setting up.
Setting up a fixation step and characterization of CQ treatment in THP-1 cells
With the aim of avoiding the increased baseline MFI of THP-1 cells, we evaluated the introduction of a cell fixation procedure right after the cell membranes permeabilisation step, before the LC3 staining (Fix prestaining; see materials and methods section). The LC3 levels of THP-1 cells, either untreated (NT) or treated with CQ (80 μM) for 24 h, were measured by flow cytometry comparing the manufacture’s procedure (i.e. No Fix protocol; grey columns and histograms) with our modified method (i.e. Fix Pre-staining procedure; black columns and histograms). As shown in Figure 2 A,B, higher MFI values were measured when the Fix Pre-staining method was used with respect to the No Fix procedure, in all the sample tested, untreated and no LC3- stained (US), untreated and LC3-stained (NT) and CQ-treated LC3-stained (CQ) cells. These differences were more evident in the samples acquired soon after the staining procedures (0 h; Figure 2B, left panels) respect to those acquired one day after the staining procedure (24 h; Figure 2B, right panels). This is mainly due to the fact that in the No Fix-processed samples the MFI increased after 24 h from the staining procedures (Figure 2C, left panels), while the differences between samples acquired at 0 h or at 24 h after LC3 staining are negligible in the fixed samples (Figure 2C left panels).
These differences were more evident when the ratio between LC3- stained/unstained samples was measured; in the unfixed samples (i.e. manufacture’s procedure) it decreased evidently when the cells were acquired 24 h post-assay, compared to the same samples acquired right after the staining procedure (Figure 2D, left columns; blue vs red columns, respectively). Notably these differences were not found in the fixed samples (Figure 2D, right columns), thus confirming that the fixation step leads to more stable results, regardless of the time of sample acquisition to the flow cytometer. Western blot analysis suggested that, as mentioned above, the increased levels of LC3 in CQ-treated cells measured by flow cytometry are mainly due to LC3-II accumulation rather than a mixture of LC3- II/LC3-I forms (Figure 2 E,F). On the other hand, we observed that LC3 levels are always higher in the fixed samples than in the unfixed samples. A possible explanation could be that the fixation prevents the complete release of the cytosolic LC3-I form. To address this point, we evaluated the introduction of cell fixation in the protocol after the LC3 staining step (Fix Post-staining) and comparing the two fixation procedures. Moreover, with the aim of setting up the best dose of CQ for inducing LC3 accumulation we treated THP-1 cells with different concentration of this autophagy inhibitor. When the LC3 level was evaluated as fold increase of those of untreated cells, we found that the samples analysed with the post-staining fixation protocol had an increased LC3 signal compared to that measured using the pre-staining fixation method, starting from 60 μM of CQ (Figure 3 A,B). Notably, regardless of the fixation protocols, no significant differences in LC3 MFI were found in samples acquired right after the end of the assay or 24 h later (data not shown), thus confirming that the fixation step prevented significant changes in the MFI of THP-1 cells. Although with different magnitude, a similar LC3 accumulation trend was measured by western blotting (Figure 3 C,D).
Figure 1.
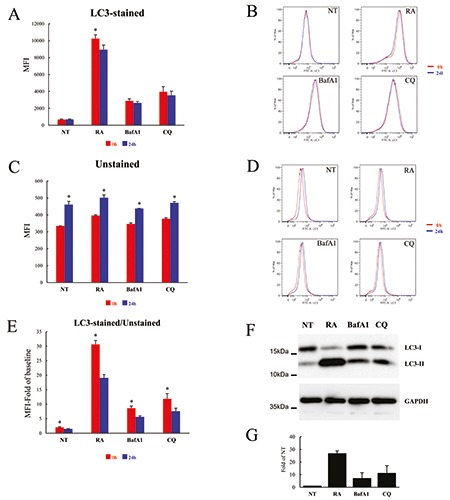
LC3 accumulation induced by different autophagy inhibitors in THP-1 cells. 3A) Cells were treated for 24 h with Reagent A (RA; Diluted 1:1000), BafA1 (5 μM) and CQ (80 μM) or left untreated (NT) and analysed by flow cytometry using the FlowCellect Kit, following the manufacturer’s instructions; the same samples were analysed by flow cytometry both right after the end of the kit’s procedure (0 h; red columns) and one day later (24 h; blue columns); data are expressed as average ±SD (n=4); *P<0.05 RA-0h vs RA- 24h. B) Cells were treated and analysed as in A; representative flow cytometry histograms of LC3 levels analysed at 0 h (red line) or 24 h (blue line) after the end of staining procedure. C) Cells were treated as in A and analysed for the baseline MFI (i.e. cells processed with the kit but not stained with the anti-LC3 Ab); the same samples were analysed by flow cytometry both at the end of the kit’s procedure (0 h; red columns) and one day after (24 h; blue columns); data are expressed as average ±SD (n=4); *P<0.05 NT-0h vs NT-24h; RA-0h vs RA-24h; BafA1-0h vs BafA1-24h; CQ-0h vs CQ-24h. D) Cells were treated as in A and analysed as in C; representative flow cytometry histograms of baseline MFI of cells analysed at 0 h (red line) or 24 h (blue line) after the end of staining procedure; E) Data represent MFI of LC3 levels normalized to baseline MFI of unstained cells (value =1), stimulated as indicated and analysed right after the end of the kit’s procedure (0 h; red columns) and one day later (24 h; blue columns); data are expressed as average ±SD (n=4); *P<0.05 NT-0h vs NT-24h; RA-0h vs RA-24h; BafA1-0h vs BafA1-24h; CQ-0h vs CQ-24h. F) A representative immunoblotting analysis of LC3 in THP-1 cells treated as in A; GAPDH levels were used as loading control. G) Quantification of the ratio of LC3-II to GAPDH band intensities obtained by immunoblotting analysis, as in F, and expressed as average ±SD (n=3).
Figure 2.
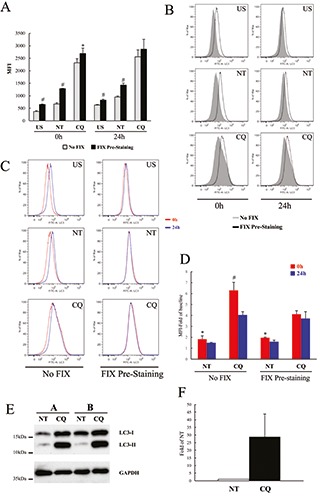
LC3 levels measured introducing a fixation step in THP-1 cells. A) Cells were treated for 24 h with and CQ (80 μM) or left untreated (NT) and analysed by flow cytometry using the FlowCellect Kit, following either the manufacturer’s instructions (No Fix; grey columns) or introducing cellular fixation right after cell permeabilisation (Fix Pre-staining; black columns); MFI of samples not stained with LC3 antibodies (US) were also measured; the same samples were analysed by flow cytometry both right after the end of the kit’s procedure (0 h) and one day after (24 h) as indicated; data are expressed as average ±SD (n=6); #P<0.005 Fix-US vs No Fix-US; Fix-NT vs No Fix-NT; *P<0.05 Fix-CQ vs No Fix-CQ. B) Cells were treated and analysed as in A; representative flow cytometry histograms of LC3 levels of THP-1 cells analysed with either No Fix (grey filled lines) or Fix Pre-staining (black lines). C) Cells were treated and analysed as in A. Representative flow cytometry histograms of cells, either No Fixed or Pre-staining fixed as indicated, analysed at 0 h (red line) or 24 h (blue line) after the end of staining procedure. D) Data represent MFI of LC3 levels normalized to baseline MFI of unstained cells, stimulated as indicated and analysed right after the end of the kit’s procedure (0 h; red columns) and one day after (24 h; blue columns). Data are expressed as average ±SD (n=6); #P<0.005; CQ-0h vs CQ-24h; *P<0.05 NT-0h vs NT-24h. E) A representative immunoblotting analysis of LC3 in THP-1 cells treated as in A. GAPDH levels were used as loading control. F) Quantification of the ratio of LC3-II to GAPDH band intensities obtained by immunoblotting analysis, as in E, and expressed as average ±SD (n=6).
Figure 3.
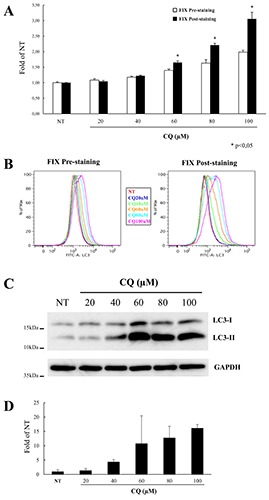
LC3 accumulation induced by different doses of CQ in THP-1 cells. A) Cells were treated for 24 h with different doses of CQ, as indicated, or left untreated (NT) and analysed by flow cytometry using the FlowCellect Kit in which a fixation step was introduced either before (Fix Pre-staining; white columns) or after LC3 labelling (Fix Post-staining; black columns); data represent MFI of LC3 levels normalized to that of NT cells analysed using the pre-staining fixation protocol and expressed as average ±SD (n=4); *P<0.05 Fix-Pre-staining vs Fix-Post-staining. B) Representative flow cytometry histograms of LC3 levels of THP-1 cells treated and analysed as in A. C) A representative immunoblotting analysis of LC3 in THP-1 cells treated as in A; GAPDH levels were used as loading control. D) Quantification of the ratio of LC3-II to GAPDH band intensities obtained by immunoblotting analysis, as in C, and expressed as average ±SD (n=4).
Figure 4.
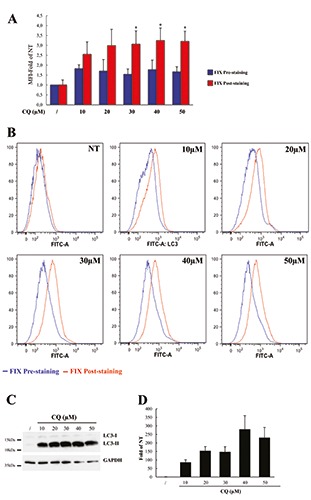
LC3 accumulation induced by different CQ treatments in human PBMC. A) Human PBMC were isolated from healthy donors and treated with different doses of CQ, as indicated, or left untreated (NT) for 24 h; LC3 levels were measured by flow cytometry using the FlowCellect Kit in which a fixation step was introduced either before (Fix Pre-staining; blue columns) or after LC3 labelling (FIX Post-staining; red columns); data represent MFI of LC3 levels normalized to MFI of untreated cells analysed using the pre-staining fixation protocol and expressed as average ±SD (n=4); *P<0.05 Fix-Pre-staining vs Fix-Post-staining. B) Flow cytometry histograms show representative intracellular LC3 expression of cells unstimulated (NT) or stimulated with different doses of CQ as indicated for 24 h and analysed for LC3 levels using the pre-staining (blue lines) or the post-staining (red lines) procedure. C) A representative immunoblotting analysis of LC3 in THP-1 cells treated as in A; GAPDH levels were used as loading control. D) Quantification of the ratio of LC3-II to GAPDH band intensities obtained by immunoblotting analysis, as in D, and expressed as average ±SD (n=4).
Characterization of CQ treatment and setting up the fixation step in human PBMC
Our final goal was to find a procedure to evaluate reliably the autophagy flux of human PBMC. To this aim, LC3 levels were measured in PBMC stimulated with different concentrations of CQ using the modified protocols set up in THP-1 cells.
PBMC isolated from healthy donors were left untreated (NT) or treated for 24 h with different doses of CQ (from 10 to 50 μM) and LC3 levels measured by the modified FlowCellect kit procedure. A higher LC3 accumulation was found in the poststaining fixed samples, with an average of 2.5-fold rise compared to NT samples, at 10 μM concentration, and about 3.0-fold for the other concentrations (Figure 4A, red columns; Figure 4B, red line). Lower levels of LC3 were measured in the pre-staining fixed samples with an average of about 1.7- fold rise for all CQ concentrations compared to the untreated samples (Figure 4A, blue columns; Figure 4B, blue line), thus suggesting that in these cells the fixation procedures may affect LC3 detection. Although with lower values, the results obtained by flow cytometry with the post-staining fixation procedure present a similar trend of LC3-II accumulation as measured by western blot analysis (Figure 4 C,D). These results suggest, again, that the LC3 measured by flow cytometry is mainly LC3-II and confirm the different sensitivity of the two assays in evaluating the autophagic flux. Next, we evaluated whether CQ treatment for shorter time was sufficient to induce LC3 levels measurable by flow cytometry. To this aim, PBMC were treated for 4 h with different doses of CQ and LC3 levels measured as above. Only the post-staining fixing procedure was able to measure an increase in LC3 levels with respect to the untreated samples (Supplementary Figure 2A; red columns), although a substantial amount of LC3-II was accumulated in cells, as measured by western blotting (Supplementary Figure 2B and 2C), thus confirming the different sensitivity of the two procedures.
Figure 5.
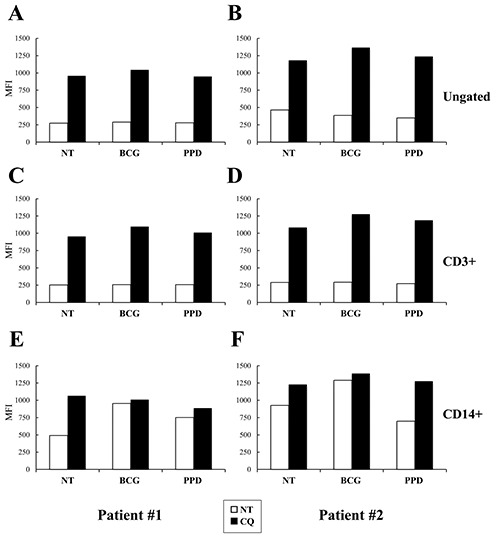
Measurement of LC3 levels induced by BCG or PPD treatments in human PBMC of patients with active TB. PBMC were isolated from two patients (panels A,C,E and B,D,F, respectively) with active TB and stimulated with live BCG (MOI=1:1) and PPD (25 μm/mL) or left untreated (NT) for 24 h either in absence (white columns) or in presence of CQ (20 μM; black columns). LC3 levels were measured by flow cytometry using the post-staining protocol in which CD3 and CD14 staining were also performed. LC3 MFI of the whole PBMC (A,B), CD3+ (C,D) and CD14+ (E,F) cells are reported.
BCG stimulation induces autophagy in the CD14+ cells of active TB patients
We finally aimed to evaluate whether with our modified fix post-staining flow cytometric method is possible to measure the autophagic flux in PBMC stimulated with Mtb-specific antigens. Therefore, we tested whether either live BCG or the Mtb PPD modulated LC3 levels in PBMC isolated from patients with active TB (Figure 5).
Since BCG and PPD are known to stimulate different cell types, including T cells and macrophages,25-30 we labelled PBMC also for CD14 and CD3 in addition to LC3. PBMC from two patients with active TB were stimulated for 24 h with BCG or PPD either alone or in presence of CQ (20 μM) for the last 20 h. No significant differences in the LC3 levels were measured in the ungated population (Figure 5 A,B), CD3+ cells (Figure 5 C,D) or CD14-/CD3- (data not shown) in response to either BCG or PPD. Differently, we found an increased LC3 level in CD14+ cells in response to BCG infection (Figure 5 E,F). However, these increased levels were no further augmented by CQ addition, thus suggesting that BCG infection induces an accumulation of autophagosomes due to a decreased rate of degradation within lysosomes. Altogether, these results indicate that the autophagy flux could be measured by this method also in primary cells infected by Mycobateria.
Discussion
Autophagic activity is tightly regulated in many physiological conditions. Alterations of autophagy have been associated to numerous pathological disorders, such as cancer, neurodegenerative and heart diseases or pathological outcomes of immune responses. 4-6 Moreover, several human pathogens are degraded by autophagy, including viruses and bacteria (e.g., group A streptococcus, Mtb, Shigella flexneri, Salmonella enterica, Listeria monocytogenes and Francisella tularensis). 7-9,31-34
An improved capability to measure autophagy in pathological conditions may allow to evaluate the relevance of this process in the pathogenesis and/or outcome of different diseases and to identify potential targets for diagnostic and therapeutic approaches. It is, therefore, important from a clinical point of view to find tools to measure autophagy in a more routinely diagnostic procedure. Flow cytometry has been fundamental for clinical research, allowing the multiparametric high-throughput phenotypic and functional analysis of individual cells.16
This study aimed to improve the protocol of a commercial kit for the evaluation of the autophagy flux in human PBMC by flow cytometry. Among different available kits, we chose the FlowCellect Kit developed by Millipore because it measures the autophagosome-related LC3 (LC3-II form), allowing the removal of the cytosolic LC3 (LC3-I form) by a selective cell permeabilisation procedure. Relative changes in MFI was used for LC3 quantitation and, therefore, as correlation with the number of autophagosomes present in the cell.
We propose a cell fixation step as main change in the assay’s procedure based mainly on two considerations. Firstly, the unfixed cells increased their baseline MFI when analysed hours after the LC3 staining procedure, which renders the autophagy measurement unstable and, therefore, less reliable. Secondly, in flow cytometry experiments antibodies directed against various surface markers are essential for the autophagy evaluation of specific cell subpopulations. Without a fixation step, such antibodies could be lost during the assay’s procedure, particularly in this setting in which cells are permeabilised and, consequently, have cellular structures seriously affected. This variation of specific signal may lead to unreliable results, particularly when the samples are analysed hours after the antibodies staining procedure.
The effects of fixation were evaluated in two different steps of the assay, one right after the cell permeabilisation (i.e. Fix Prestaining), the other at the end of the procedure after LC3 staining (i.e. Fix Post-staining) (summarised in supplementary Figure 1). The post-staining fixation procedure led to better results in both THP-1 cells and human PBMC, in terms of fold of LC3 accumulation upon CQ treatment, when compared to the untreated samples. This difference may be explained mainly in two ways that are not mutually exclusive: i) fixation with formalin can affect the antibody recognition of LC3, ii) the antibodies used to detect the autophagic flux can be lost during the assay’s procedure because of cell permeabilisation. The observation that unfixed THP-1 cells show a lower LC3 MFI than the fixed samples supports this second hypothesis. In addition, the lower LC3 MFI of unfixed samples may be also explained by the loss of intracellular structures, thus reinforcing the idea that a fixation procedure is very useful.
A second variation that we applied in the procedure was the substitution of RA provided by the kit with CQ. We decided to substitute RA mainly because its composition is not known, therefore making impossible the comparison with previous results obtained either in ours or in other studies. We optimised the CQ concentrations for both THP-1 cells and PBMC. In THP-1 cells the best CQ concentration for LC3 accumulation was 80 μM. On the other hand, in the human PBMC, we found that CQ at 20 μM concentration induced a good LC3 accumulation without major toxic effects. It has to be underlined that concentrations higher than 50 μM cannot be used for 24 h because of toxicity (data not shown).
Since the LC3 levels trend measured by flow cytometry is very similar to LC3-II quantified by western blot analysis, it is very likely that the LC3 measured by flow cytometry is mainly the LC3-II isoform. Although our results confirmed the substantial difference in sensitivity between flow cytometry and western blot analysis, they suggest that flow cytometry can be applied to the study of cellular autophagy.
Our final goal was the development of an assay to evaluate LC3 modulation in primary cells, in particular in PBMC of patients with TB. To this goal, we evaluated the autophagic flux of PBMC stimulated with either live BCG or PPD, by flow cytometry using our modified protocol. In the two samples from active TB patients, we measured a live BCG-induced increased LC3 levels in cells expressing CD14, a human monocyte-associated marker, thus suggesting that this protocol is feasible for the ex vivo assessment of autophagy flux variation induced by specific stimuli.
Overall, we described modifications of a flow cytometry-based assay that improved the measure of ex vivo autophagy. This modified procedure may be useful for the routinely monitoring of the impact of different stimuli and/or clinical conditions on the cellular autophagic flux, opening new research perspectives in the field.
Acknowledgments
The authors are grateful to Gilda Cuzzi for patients’ enrolment.
Funding Statement
Funding: This work was supported by grants from the European Union (643381- TBVAC2020-H2020-PHC-2014-2015), the National Institute of Health of USA (NIH 1R21AI127133-01), the Italian Ministry of Health (“Ricerca Finalizzata”: RF-GR54 and “Ricerca Corrente”) and from the Italian Ministry of University and Research (MIUR) - PRIN 2015 No. 20152CB22L.
References
- 1.Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J 2017;36:1811-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klionsky DJ, Eskelinen EL, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes... wait, I’m confused. Autophagy 2014;10:549-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonioli M, Di Rienzo M, Piacentini M, Fimia GM. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci 2017;42:28-41. [DOI] [PubMed] [Google Scholar]
- 4.Papandreou ME, Tavernarakis N. Autophagy and the endo/exosomal pathways in health and disease. Biotechnol J. 2017;12 Doi: 10.1002 /biot.201600175. [DOI] [PubMed] [Google Scholar]
- 5.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013;13:722-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorburn A. Autophagy and disease. J Biol Chem 2018;293:5425-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seveau S, Turner J, Gavrilin MA, Torrelles JB, Hall-Stoodley L, Yount JS, et al. Checks and balances between autophagy and inflammasomes during infection. J Mol Biol 2018;430:174-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bah A, Vergne I. Macrophage autophagy and bacterial infections. Front Immunol 2017;8:1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castrejon-Jimenez NS, Leyva-Paredes K, Hernandez-Gonzalez JC, Luna-Herrera J, Garcia-Perez BE. The role of autophagy in bacterial infections. Biosci Trends 2015;9:149-59. [DOI] [PubMed] [Google Scholar]
- 10.Ernst JD. Mechanisms of M. tuberculosis immune evasion as challenges to TB vaccine design. Cell Host Microbe 2018;24:34-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1-222. Erratum in: Autophagy 2016;12:443. doi: 10.1080/15548627.2016.1147886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warnes G. Flow cytometric assays for the study of autophagy. Methods 2015;82:21-8. [DOI] [PubMed] [Google Scholar]
- 13.Zappavigna S, Lombardi A, Misso G, Grimaldi A, Caraglia M. Measurement of autophagy by flow cytometry. Methods Mol Biol 2017;1553:209-16. [DOI] [PubMed] [Google Scholar]
- 14.Wang F, Li B, Schall N, Wilhelm M, Muller S. Assessing autophagy in mouse models and patients with systemic autoimmune diseases. Cells 2017; 6(3). pii: E16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozako T, Suzuki T, Yoshimitsu M, Uchida Y, Kuroki A, Aikawa A, et al. Novel small-molecule SIRT1 inhibitors induce cell death in adult T-cell leukaemia cells. Sci Rep 2015;5:11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizrahi O, Ish Shalom E, Baniyash M, Klieger Y. Quantitative flow cytometry: Concerns and recommendations in clinic and research. Cytometry B Clin Cytom 2018;94:211-8. [DOI] [PubMed] [Google Scholar]
- 17.Siggs OM, Stockenhuber A, Deobagkar-Lele M, Bull KR, Crockford TL, Kingston BL, et al. Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc Natl Acad Sci USA 2016;113:E3706-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petruccioli E, Chiacchio T, Pepponi I, Vanini V, Urso R, Cuzzi G, et al. First characterization of the CD4 and CD8 Tcell responses to QuantiFERON-TB Plus. J Infect 2016;73:588-97. [DOI] [PubMed] [Google Scholar]
- 19.Petrone L, Bondet V, Vanini V, Cuzzi G, Palmieri F, Palucci I, et al. First description of agonist and antagonist IP-10 in urine of patients with active TB. Int J Infect Dis 2019;78:15-21 [DOI] [PubMed] [Google Scholar]
- 20.Tempestilli M, Elisei F, Cimini E, D’Avolio A, Grassi G, Nicastri E, et al. Low-density lipoprotein and ritonavir: an interaction between antiretrovirals and lipids mediated by P-glycoprotein. J Antimicrob Chemother 2014;69:1760-6. [DOI] [PubMed] [Google Scholar]
- 21.Sali M, Di Sante G, Cascioferro A, Zumbo A, Nicolo C, Dona V, et al. Surface expression of MPT64 as a fusion with the PE domain of PE_PGRS33 enhances Mycobacterium bovis BCG protective activity against Mycobacterium tuberculosis in mice. Infect Immun 2010;78:5202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grassi G, Di Caprio G, Santangelo L, Fimia GM, Cozzolino AM, Komatsu M, et al. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting snail degradation. Cell Death Dis 2015;6:e1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Refolo G, Ciccosanti F, Di Rienzo M, Basulto Perdomo A, Romani M, Alonzi T, et al. Negative regulation of mitochondrial antiviral signaling proteinmediated antiviral signaling by the mitochondrial protein LRPPRC during hepatitis C virus infection. Hepatology 2019;69:34-50. [DOI] [PubMed] [Google Scholar]
- 24.Pasquier B. Autophagy inhibitors. Cell Mol Life Sci 2016;73:985-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sargentini V, Mariotti S, Carrara S, Gagliardi MC, Teloni R, Goletti D, et al. Cytometric detection of antigen-specific IFN-gamma/IL-2 secreting cells in the diagnosis of tuberculosis. BMC Infect Dis 2009;9:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gioia C, Agrati C, Goletti D, Vincenti D, Carrara S, Amicosante M, et al. Different cytokine production and effector/memory dynamics of alpha beta+ or gamma delta+ T-cell subsets in the peripheral blood of patients with active pulmonary tuberculosis. Int J Immunopathol Pharmacol 2003;16: 247-52. [DOI] [PubMed] [Google Scholar]
- 27.Smith SG, Lalor MK, Gorak-Stolinska P, Blitz R, Beveridge NE, Worth A, et al. Mycobacterium tuberculosis PPDinduced immune biomarkers measurable in vitro following BCG vaccination of UK adolescents by multiplex bead array and intracellular cytokine staining. BMC Immunol 2010;11:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stern JN, Keskin DB, Romero V, Zuniga J, Encinales L, Li C, et al. Molecular signatures distinguishing active from latent tuberculosis in peripheral blood mononuclear cells, after in vitro antigenic stimulation with purified protein derivative of tuberculin (PPD) or Candida: a preliminary report. Immunol Res 2009;45:1-12. [DOI] [PubMed] [Google Scholar]
- 29.Cosmi L, Maggi L, Santarlasci V, Liotta F, Frosali F, Angeli R, et al. Detection by flow cytometry of ESAT-6- and PPD-specific circulating CD4+ T lymphocytes as a diagnostic tool for tuberculosis. Int Arch Allergy Immunol 2007;143:1-9. [DOI] [PubMed] [Google Scholar]
- 30.Petruccioli E, Romagnoli A, Corazzari M, Coccia EM, Butera O, Delogu G, et al. Specific T cells restore the autophagic flux inhibited by Mycobacterium tuberculosis in human primary macrophages. J Infect Dis 2012;205: 1425-35. [DOI] [PubMed] [Google Scholar]
- 31.Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol 2018;16:341-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goletti D, Petruccioli E, Romagnoli A, Piacentini M, Fimia GM. Autophagy in Mycobacterium tuberculosis infection: a passepartout to flush the intruder out? Cytokine Growth Factor Rev 2013;24: 335-43. [DOI] [PubMed] [Google Scholar]
- 33.Romagnoli A, Petruccioli E, Palucci I, Camassa S, Carata E, Petrone L, et al. Clinical isolates of the modern Mycobacterium tuberculosis lineage 4 evade host defense in human macrophages through eluding IL-1betainduced autophagy. Cell Death Dis 2018;9:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Etna MP, Sinigaglia A, Grassi A, Giacomini E, Romagnoli A, Pardini M, et al. Mycobacterium tuberculosisinduced miR-155 subverts autophagy by targeting ATG3 in human dendritic cells. PLoS Pathog 2018;14:e1006790. [DOI] [PMC free article] [PubMed] [Google Scholar]