

Summary
MsbA is an essential ATP-binding cassette transporter in gram-negative bacteria that transports lipid A and lipopolysaccharide from the cytoplasmic leaflet to the periplasmic leaflet of the inner membrane. Here we report the X-ray structure of MsbA from Salmonella typhimurium at 2.8 Å resolution in an inward-facing conformation after co-crystallization with lipid A and utilizing a stabilizing facial amphiphile. The structure displays a large amplitude opening in the transmembrane portal, which is likely required for lipid A to pass from its site of synthesis into the protein-enclosed transport pathway. Putative lipid A density is observed further inside the transmembrane cavity, consistent with a trap and flip model. Additional electron density attributed to lipid A is observed near an outer surface cleft at the periplasmic ends of the transmembrane helices. These findings provide new structural insights into the lipid A transport pathway through comparative analysis with existing MsbA structures.
Keywords: ABC transporter, lipid flippase, lipid A, lipopolysaccharide, facial amphiphile, X-ray crystallography
eTOC Blurb
Padayatti et al. determined a crystal structure of the ABC transporter MsbA in the presence of lipid A substrate. Putative lipid A binding sites are found not only inside the protein cavity, but also on an outer surface cleft. This study offers new insights into MsbA-mediated lipid A transport pathway.
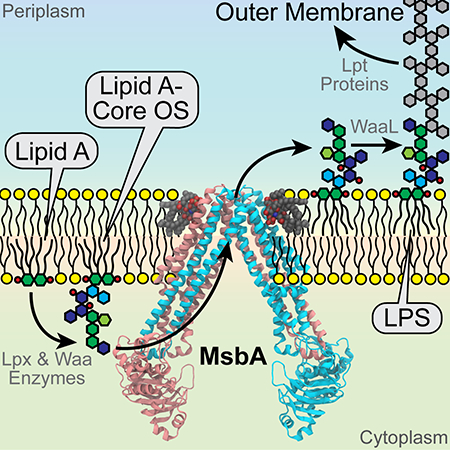
Graphical Abstract
INTRODUCTION
Gram-negative bacteria benefit from resistance to many antibiotics due to the low permeability of their outer cell membranes, which are surrounded by a dense layer of lipopolysaccharide (LPS). LPS is a complex glycolipid composed of a hydrophobic lipid A anchor, a non-repeating core oligosaccharide, and a distal repeating oligosaccharide, termed the O-antigen (Figure 1). Lipid A is in general the most conserved domain of LPS, although its structure varies among bacterial species (Raetz and Whitfield, 2002). Biosynthesis of the lipid A-core subunit of LPS occurs at the interface between the cytosol and cytoplasmic membrane via the so-called Raetz pathway (Raetz and Whitfield, 2002; Whitfield and Trent, 2014). One of the shortest LPS species consists of lipid A with two 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) units. Kdo biosynthesis is normally required for cell growth but can be bypassed in Escherichia coli or Salmonella typhimurium if MsbA, an energy-dependent ATP-binding cassette (ABC) transporter, is overexpressed (Meredith et al., 2006; Reynolds and Raetz, 2009). MsbA is functionally characterized as a lipid flippase that transports lipid A with or without core sugars from the cytoplasmic leaflet to the periplasmic leaflet of the inner membrane (Doerrler et al., 2004; Doerrler and Raetz, 2002; Doerrler et al., 2001; Eckford and Sharom, 2008; Polissi and Georgopoulos, 1996; Reynolds and Raetz, 2009; Wang et al., 2004; Zhou et al., 1998). On the periplasmic side, O-antigen is ligated to the lipid A core (Bi et al., 2018), and further delivery of the completed LPS to the cell surface is accomplished by a complex of seven transport proteins (Lpt A-G) that span from the inner membrane to the outer membrane (Figure 1) (May et al., 2015; Simpson et al., 2015).
Figure 1. LPS trafficking and assembly in gram-negative bacteria.

The lipid A core-subunit of LPS is flipped across the inner membrane (IM) by MsbA after its synthesis on the cytoplasmic side. Following the attachment of O-antigen on the periplasmic surface (not shown), LPS is delivered to the outer membrane (OM) through a complex of LPS transport proteins (Lpt A-G). The chemical structure of the hexa-acylated lipid A from E. coli or S. typhimurium with core sugar units attached to the 6′-hydroxy group of the lipid A glucosamine unit is shown on the right.
Because of its critical role in LPS trafficking and the assembly of the outer cell membrane, MsbA is essential to the viability of most gram-negative pathogens and, hence, a viable target for new antibiotics (Ho et al., 2018; Zhang et al., 2018). MsbA may act also as a flippase of glycerophospholipids (Doerrler et al., 2004; Eckford and Sharom, 2010; Zhou et al., 1998), and a temperature-sensitive mutation of MsbA in E. coli results in accumulation of not only lipid A but also major phospholipids along the inner leaflet of cytoplasmic membranes (Doerrler et al., 2001). In addition to lipids, some drug molecules are also reported substrates for MsbA, suggesting substrate promiscuity similar to that seen among homologous multidrug resistance ABC transporters (Eckford and Sharom, 2008; Reuter et al., 2003; Woebking et al., 2005).
MsbA is a homodimer composed of two transmembrane domains (TMDs), each containing six transmembrane (TM) helices, and two cytosolic nucleotide binding domains (NBDs). This prototypical ABC transporter has been subjected to various biochemical, biophysical and structural studies. X-ray crystallography (Terakado et al., 2010; Ward et al., 2007), electron microcopy (Moeller et al., 2015), computational simulation (Moradi and Tajkhorshid, 2013), cross-linking (Doshi and van Veen, 2013; Doshi et al., 2010), fluorescence or luminescence resonance energy transfer (FRET or LRET) (Borbat et al., 2007; Cooper and Altenberg, 2013; Zoghbi et al., 2016), and electron spin resonance (EPR) spectroscopy (Borbat et al., 2007; Zou et al., 2009), have collectively demonstrated that MsbA samples a wide range of conformations. The different states are coupled to the binding and hydrolysis of ATP in the NBDs, with the TMDs switching between the inward and outward-facing conformations. Despite all of these studies, a detailed substrate transport pathway has remained elusive because most structural studies have been conducted in the absence of lipid A or LPS.
Most recently, a cryo-EM structure of E. coli MsbA embedded in lipid nanodiscs was solved in the absence of nucleotides at an overall resolution of 4.7 Å (4.2 Å for the TMDs). For the first time, a molecule of copurified LPS was observed to be trapped within a TM pocket, with the acyl chains oriented towards the periplasmic end of the TMD (Mi et al., 2017). The inward openness of MsbA (NBD separation distance) was considerably narrower than more open conformations observed in single particle analyses of transporters in detergent micelles, and which also had lower ATPase activity (Mi et al., 2017). The binding of LPS deep inside MsbA was confirmed by a very recent X-ray structure solved at higher resolution (2.9 Å) using a facial amphiphile (FA-3) (Ho et al., 2018). The crystal structure displayed an inward-facing conformation with overall similarity to the cryo-EM structure, although the concurrent binding of a small molecule antagonist shifted MsbA to an inactive state that is not competent for ATP hydrolysis. These structural reports represent major advances to elucidate a detailed mechanism for lipid flipping by MsbA. However, these LPS-bound structures appear to be inconsistent with the large NBD separation that was observed in a previous lower-resolution (5.3 Å) X-ray structure solved without LPS and in detergents (PDB accession 3B5W) (Ward et al., 2007). This structure represents the most wide-open conformer among all ABC transporter structures solved to date and whose physiological relevance has long been debated.
Here, we report a 2.8 Å-resolution structure of MsbA from S. typhimurium solved by cocrystallization with lipid A (and without bound antagonist), utilizing MsbA prepared in FA-3 in an active state. The crystal structure shows MsbA in an intermediate inward-facing conformation with the separation between the NBDs falling between the previously reported X-ray and EM structures, yet wide enough to allow LPS/lipid A to access the protein-enclosed transport pathway. Putative electron densities for lipid A are revealed not only inside MsbA, but also in a cleft on MsbA’s outer surface at the periplasmic face of the bilayer, which we hypothesize as a possible post-transport lipid A docking site. The relevance of the current structure is discussed in the context of a dynamic conformational pathway by comparison with previous structures, thereby offering fresh insights into MsbA-mediated lipid A transport pathway.
RESULTS AND DISCUSSION
Cocrystallization of MsbA and Lipid A in Facial Amphiphiles.
X-ray structure determination at high-resolution of ABC transporters with bound transport substrates or agonists is challenging given the typically weak binding affinity and brief residence time of these ligands and the significant conformational flexibility of the transport machineries. A crystal structure for MsbA beyond 3 Å resolution was only achieved recently (Ho et al., 2018), and entailed binding of a potent antagonist (G907) along with the use of a structurally unique amphiphile, FA-3 (Figure 2A), which was initially developed by our group (Lee et al., 2013). We selected FAs for crystallization of MsbA with lipid A substrate based on an evaluation of more than 200 new detergents that we have synthesized over the past few years. Preparations of MsbA in FA-3 exhibited ATPase activity in the range of 6–10 μmol ATP/min/mg protein, which is comparable to that determined in lipid nanodiscs and significantly higher than in most other detergents that we have assessed, including popular commercial detergents such as n-dodecyl-β-D-maltoside (DDM) and n-undecyl-β-D-maltoside (UDM) (Figure 2B). The ATPase activity of MsbA in FA-3 exhibited a Km value of 0.31 ± 0.05 mM, similar to that previously reported for MsbA in nanodiscs (Figure 2C) (Mi et al., 2017). Addition of lipid A to MsbA had only a small effect on Km but increased the Vmax of hydrolysis by ~25%. Inhibition of the catalytic ATPase activity by vanadate further validated active MsbA in the presence of FA-3 (Figure 2D). The use of FAs not only helped to preserve the high activity of MsbA, but also enhanced its stability. MsbA exhibited a higher melting temperature (Tm) in FA-3 (63.3 °C) than in UDM and lauryl maltose neopentyl glycol (LMNG) (53.0 and 58.6 °C, respectively), as measured using a thermal unfolding assay that probes the accessibility of an engineered single cysteine (A30C) to a fluorescent reagent (see Methods) (Figure 2E).
Figure 2. Activity and stability of MsbA.

(A) Structure of FA-3 used in the crystallization of MsbA. (B) ATPase activity of MsbA solubilized in DDM, UDM, FA-3 and MSP1D1 nanodiscs composed of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) lipids. (C) ATPase activity of MsbA/FA-3 (with or without lipid A, 160 μM) measured as function of ATP concentration (0–5 mM). Data are fit to the Michaelis-Menten equation. (D) Inhibition of the ATPase activity of MsbA by vanadate (200 μM). (E) Representative fluorescence thermal unfolding curves for MsbA (A30C mutant) in FA-3, UDM or LMNG. Data in (B-D) are averaged from triplicate measurements with standard error bars.
Our early trials with FAs demonstrated their feasibility of crystallizing MsbA and several other membrane proteins (Lee et al., 2013). However, those MsbA crystals grown in the absence of nucleotides diffracted only to > 4.5 Å resolution. The difficulty in improving these crystals may be ascribed to the substantial conformational flexibility of MsbA as visualized in our single particle EM analysis, where we showed that MsbA occupied a continuum of open and closed conformations of the NBD (Moeller et al., 2015). Better-diffracting crystals were consistently obtained when lipid A was incorporated, suggesting that the binding of lipid A might further stabilize the protein conformation. The highest resolution dataset was processed to 2.8 Å and revealed an open-inward state of the protein (Figure 3A). Data collection and refinement statistics are outlined in Table 1 for crystals obtained in the presence and absence of lipid A. Unless otherwise specified, the structure features that we describe here are derived from the higher resolution structure solved for MsbA with lipid A present.
Figure 3. MsbA crystal structure with lipid A.

(A) The inward-facing MsbA structure captured in the presence of lipid A. The monomers forming a functional dimer are colored blue and yellow respectively. TM4 and TM5 are situated so as to interact with TM1′, TM2′, TM3′ and TM6′ in the opposing monomer, and with NBD2 through a short coupling helix (IH2) in between. Putative lipid A binding sites are observed on the outer surface (positions indicated by red arrows) of MsbA in addition to the canonical binding site (red box region) inside the TMD dimer. These exterior binding sites are further described in Figures 6 and S3. Horizontal black lines represent approximate boundaries of the lipid bilayer. (B) Placement of the current structure within a conformational spectrum of MsbA defined by X-ray and cryo-EM structures (top panel). The lower panel displays an ensemble of MsbA conformers visualized in the absence of nucleotides or under steady-state ATP hydrolysis conditions by low-resolution EM (Moeller et al., 2015). See also Figures S1–S3.
Table 1.
Crystallographic data collection and refinement statistics for MsbA in the presence and absence of lipid A
| Data Collection | ||
|---|---|---|
| With lipid A | Apo | |
| Beamline | SSRL BL7–1 | SSRL BL11–1 |
| Wavelength | 1.12708 | 0.97945 |
| Space group | P21212 | P21212 |
| Unit cell parameters (Å) | 82.0, 154.8, 228.9 | 82.3, 153.2, 232.5 |
| Resolution range (Å) | 48.0 – 2.80a | 46.8 – 4.46 |
| Total observationsb | 306,422 (11,670) | 42,949 (1957) |
| Unique reflections | 60,957 (2847) | 15,470 (683) |
| Redundancy | 5.0 (4.1) | 2.8 (2.9) |
| Completeness (%) | 84.9 (81.5) | 85.0 (75.8) |
| < />/<σ/> | 10.0 (0.5) | 10.0 (0.6) |
| Rmerge (%)c | 13.6 (317) | 11.4 (166) |
| Rmeas (%)d | 14.8 (358) | 13.8 (201) |
| Rpim (%)e | 5.7 (161) | 7.7 (111) |
| Wilson B value (Å2) | 68 | 194 |
| CC1/2f | 89.3 (44.0) | 95.0 (85.8) |
| MR model | PDB-3B5W | PDB-6BL6 |
| Refinement | ||
| Resolution range (Å) | 48.0–2.80 (2.85–2.80) | 46.8–4.46 (4.81–4.46) |
| Reflections(work, free) | 54303 (3269) | 14246 (719) |
| Rwork (%) | 26.9 (53.6) | 30.7 (39.9) |
| Rfree (%) | 30.1 (52.6) | 33.9 (44.2) |
| Non-hydrogen atoms | 8894 | 8894 |
| Protein residues | 1150 | 1150 |
| RMS (bonds, Å) | 0.009 | 0.003 |
| RMS (angles) | 1.37 | 0.779 |
| Ramachandran values | ||
| favored (%) | 93.5 | 93.5 |
| allowed (%) | 5.4 | 5.4 |
| outliers (%) | 1.1 | 1.1 |
| Clashscoreg | 1.7 | 2.7 |
| Average B-value (Å2) | 116 | 272 |
| PDB ID code | 6BL6 | 6O30 |
Effective resolution 2.96 Å. Resolution where (<I>/<σI>=2) 3.08–3.15 Å
Numbers in parentheses are for highest resolution shell
Rmerge = Σhkl Σi=1,n|Ii(hkl)− <I(hkl)>| / Σhkl Σi=1,n Ii(hkl)
Rmeas = Σhkl √(n/n−1) Σi=1,n|Ii(hkl)− <I(hkl)>| / Σhkl Σi=1,n Ii(hkl)
Rpim = Σhkl √(1/n−1) Σi=1,n|Ii(hkl)− <I(hkl)>| / Σhkl Σi=1,n Ii(hkl)
CC1/2 = Pearson Correlation Coefficient between two random half datasets
Number of unfavorable all-atom steric overlaps ≥ 0.4Å per 1000 atoms
Considering that endogenous LPS was co-purified with MsbA in the recent structural reports (Ho et al., 2018; Mi et al., 2017), we also measured the LPS content in our MsbA preparations using a Limulus amoebocyte lysate (LAL) chromogenic endotoxin quantification assay. Interestingly, the LPS level from our measurement is extremely low and almost negligible compared to the protein (~0.04 ng LPS/1 mg MsbA, < 1:105 in molar ratio), likely as a result of the varying detergents and protocols that we used in our MsbA preparations (see Methods).
MsbA Structure in an Intermediate Inward-facing Conformation.
The MsbA structure here was solved in an inward-facing conformation that displays a different degree of openness compared to previously reported MsbA structures (Mi et al., 2017; Ward et al., 2007) (Figure 3B). The distance measured between the Cα positions of T561 located at the center tip of each NBD was 75.6 Å. This NBD separation is ~10 Å narrower than in the previous lower-resolution crystal structure of E. coli MsbA, and comparable to the separation seen in several other reported ABC transporter structures (Figure S1A). The higher resolution here enables a better description of the overall membrane domain, with clearer density for the backbone and many side chains (Figures S2A). The crystal packing reveals major lattice contacts through NBDs (Figure S1B), which may help to stabilize the single conformation captured in our crystal structure. Using the CHARMM36m force field, the interaction energy between two flanking MsbA dimers (full transporters) is calculated at −45.7 kcal/mol, showing a favorable crystallographic contact that indeed can stabilize the MsbA conformation; however, this interaction is small compared to the energy between the two MsbA monomers of the same transporter (−953.6 kcal/mol and −867.4 kcal/mol in each dimer of the asymmetric unit).
Of note, the openness of our crystal structure is still considerably wider than the recent cryo-EM structure determined in lipid nanodiscs (PDB accession 5TV4), for which the two NBDs are in close contact (separation distance between T561 Cα positions is 35 Å) (Figure 3B). Nanodiscs represent good membrane mimetics, but their diameter is determined by the particular membrane scaffold protein (MSP) (Bayburt and Sligar, 2010), raising the possibility that it might constrain lateral opening of MsbA. Indeed, the 75 Å separation on the inner leaflet side in our crystal structure approaches the internal diameter of MSP1D1 nanodiscs (76 Å not including the MSP (Bayburt and Sligar, 2010)) primarily used for the EM structure determination (Figure S1C). However, that study demonstrated similar separation distance for MsbA in MSPE3D1 nanodiscs with a 108 Å diameter, which tends not to support lateral constraint of MsbA openness by MSP. In contrast, negative-stain EM of MsbA that had been reconstituted into lipid nanoparticles surrounded by short β-strand peptides exhibited a broader conformational spectrum (Moeller et al., 2015). This X-ray structure falls within this conformational range, as do other reported MsbA structures (Figure 3B). We have shown that virtually all MsbA particles, including a broad range of open MsbA conformers, can be converted to the outward facing state under stabilizing nucleotide-bound conditions (Moeller et al., 2015). Therefore, we assume that this open, inward MsbA crystal structure represents an active intermediate state in the transport cycle. Consistent with this assumption, a large range of structural fluctuations of MsbA was observed using molecular dynamics simulations in an explicit phospholipid bilayer (Figure S1D). The separation of NBDs (T561 Cα distance) varies between 41–85 Å with a median at ~65 Å. EPR studies for MsbA either in liposomes or in detergents have shown a largely comparable range of conformational motions (Borbat et al., 2007; Zou et al., 2009). Similar conclusions were drawn from a recent single molecule FRET study, which also showed that the range of MsbA conformations is constrained even in large MSP1E3D1 nanodiscs compared to that measured in detergents and liposomes (Liu et al., 2018). However, conflicting data have also been reported in two LRET-based studies of MsbA conformations (Cooper and Altenberg, 2013; Zoghbi et al., 2016).
Membrane Portals for Lipid A Access.
The inward-facing MsbA structure shows two portals on opposite sides, formed by TM4 and TM6 of each monomer, which are open to the inner membrane leaflet and likely allow entry of the amphiphilic lipid A or LPS molecule by lateral diffusion from the site of synthesis (Figure 4). The portals are lined near and above the boundary of the bilayer with a row of positively charged and polar residues, including Lys194, Arg190, Lys187, and Arg183 on TM4, and Arg310, Gln309, Asn305, Asn303, and Lys299 on TM6 (Figure 4A). These residues may interact with the negatively charged sugar groups in LPS, providing substrate recognition and helping to guide LPS into the MsbA-enclosed transport pathway. Multidentate ionic interactions are well known to provide selective binding of LPS in LPS-binding proteins (Eckert et al., 2013), toll-like receptor 4 and myeloid differentiation factor 2 complex (Park et al., 2009), and polymyxins (Velkov et al., 2013). Earlier EM and crystal structures of MsbA showing trapped LPS (5TV4 and 6BPL) identified several positively charged residues (e.g. Arg78, Arg148, Arg296) as key to the binding of the phosphorylated glucosamine groups in the lipid A moiety (Ho et al., 2018; Mi et al., 2017), but these cationic amino acids are buried more deeply in the protein. As a result, the LPS molecule captured in these structures is shifted ~15 Å upward from the membrane boundary, with the alkyl chains approaching the periplasmic side of the cavity. Those structures featured only small enclosed membrane portals and could not be easily reconciled with the initial entry of bulky LPS (Figure 4B). The more open conformation adopted in the X-ray structure here may thus represent an essential earlier stage in the transport of LPS.
Figure 4. Lipid A entry and early transport pathway.

(A) Stereo view of the MsbA structure in the presence of lipid A shows a front membrane portal formed by TM4 and TM6, which is open to the cytoplasmic leaflet of the inner membrane. A row of basic and polar residues (green sticks) on TM4 and TM6 are arrayed to promote entry of the negatively charged lipid A or LPS in combination with a lateral inward motion of TMs as shown in (C and D). Yellow sticks represent an LPS molecule manually modeled at the corresponding positions in 5TV4 and 6BPL, with key residues Arg78 (TM2), Arg148 (TM3), and Arg296 (TM6) (purple sticks) more deeply buried for selective interaction with the two phosphorylated glucosamine units of lipid A at a later stage of transport. A red arrow is drawn to describe a rough path of lipid A entry and halfway through its transport. (B) Electrostatic surface representation of MsbA structures (blue, +10 kT/e; red, −10 kT/e). A decreased opening of the membrane portal is highlighted by the green triangle from the inward open (left) to inward closed (right) conformation. (C) Structure alignment with 5TV4 (dark grey) and 6BPL (light grey). Only a monomer is shown, and the NBDs are omitted for clarity. The inward motion of TM4 and TM5 towards TM6 results in gradual closure of the membrane portal as seen in (B). (D) Cytosolic view of the lateral movements occurring between the current structure (left) and the cryo-EM structure (right) bound with lipid A or LPS. Key lipid A binding residues (Arg78, Arg148 and Arg296) shift closer to the center of the lipid channel, allowing tighter binding and possibly the upward movement of lipid A that bridges the two TMDs.
Shuttling Lipid A to the Cavity Inside MsbA.
Alignment of this new structure with the LPS-trapped cryo-EM and crystal structures indicates that TM4 and TM5 form a dynamic gate enabling access to the TMD opening while TM1, TM2, TM3 and TM6 form a conformationally stable group (Figure 4C). The movement of TM4 and TM5 is linked with the associated NBD2 through a short intracellular helix (IH2), and this whole group pivots ~22° inward on a flexible hinge formed by extracellular loops 2 and 3. This inward motion of TMDs likely produces the tighter binding of LPS upon its translocation toward the periplasmic side as observed in the narrowly open MsbA structures (5TV4 and 6BPL). Indeed, the key residues of the interior lipid A binding site (Arg78, Arg148, Arg296) lie farther from the opposing monomer in our crystal structure than in 5TV4 and 6BPL, suggesting that lipid A binding at this interior site may be weaker in the more open conformation (Figure 4D). We observed extra electron density inside the cavity of MsbA near this interior lipid A binding site (Figure S3A). However, the density was fragmentary, presumably due to weak and flexible lipid A binding in the widened cavity. Additionally, the internal density was distributed along the 2-fold, non-crystallographic symmetry axis of the MsbA dimer, making it difficult to model the asymmetric lipid A molecule. As a result, we cannot convincingly model lipid A at this site. Notably, an elongated density was also found in the same region of the apo MsbA structure (with FA-3 but without externally added lipid A; processed to 4.46 Å) where it bridges the two symmetric halves of the MsbA dimer (Figure S3B). For similar reasons outlined above, we cannot ascertain the identity of what this density corresponds to either.
Implications for the Antagonist Binding-Induced Conformational Change.
The inward swing of TM4 and TM5 towards TM6 also helps to interpret the binding of antagonist G907 as observed in the MsbA/LPS cocrystal structures (Ho et al., 2018) (Figure 5). Our crystal structure shows an inward motion of TM4 and TM5 is necessary to enclose the allosteric site for G907 binding which, in the more closed conformation, is wedged between TM4, TM5 and TM6 (Figures 5A and 5B). Interestingly, TM4 is a continuous long helix that is slightly arched, while the binding of G907 to the key Arg190 in TM4 appears to induce the unwinding of one turn of this helix (residues 190–193) (Figure 5C). A local alignment of TM4 and TM5 of the two structures reveals a change in conformation of TM4 in the G907-bound structure such that the two ends of this helix above and below the unwound segment tilt in opposite directions (Figure 5C). This antagonist-induced conformational fit in TM4 has been linked to an uncoupling and displacement of the associated NBDs, resulting in their inability to properly dimerize and hydrolyze ATP (Ho et al., 2018). B-values derived from crystallographic observations are higher for TM4 than for the other TM helices, which also indicate an inherent structural flexibility (Figure S2B). We note that TM4 flexibility and its ligand-induced local conformational change have been observed in homologous P-glycoprotein structures (Alam et al., 2018; Kodan et al., 2014; Szewczyk et al., 2015), implying that these may represent common structural features in ABC transporters.
Figure 5. Conformational changes that accompany antagonist binding.

(A) Side-by-side comparison of the MsbA structure in the presence of lipid A (left) with the LPS and G907-bound MsbA structure (6BPL, right). The inward motion of TM4 and TM5 towards TM6 encloses the binding site for G907 (green sphere) with a salt bridge formed between Arg190 and the carboxylate of G907. LPS is omitted for clarity but shown in (B) (yellow stick). (B) View of the conformational change from the cytosolic side of the two structures. (C) Alignment of TM4 and TM5 to further illustrate the G907-induced local conformational perturbation in TM4, including the unwinding of a helix turn (190–193). TM4 and TM5 in 6BPL are depicted in dark grey.
Putative Lipid A Binding Site on the Periplasmic Surface of MsbA.
In addition to the extra density found inside MsbA, we consistently observed two large bodies of extra density (symmetrically represented in each TMD half) along the periplasmic boundary on the outer protein surface in different datasets (Figure S3A). In contrast, crystal data for the apo MsbA structure showed no similar density at these positions (Figure S3B). Density-based docking strongly favored modeling of the lipid A head group over other components present in the crystallization solution, such as FA-3 (Figures S3C and S3D) or potentially copurified phospholipids, although it did not allow us to unambiguously determine the orientation of the head group with respect to the protein. As a result, we tentatively modeled lipid A at this position (Figure 6A). Of note, this putative lipid A binding site is situated above a shallow surface groove formed at the periplasmic ends of TM1, TM2 and TM3 (Figures S3A and 6B). Several aromatic and polar residues potentially interact with lipid A bound at this site (Figures 6B and S4). Similarly located sites for ligand binding have been found in other ABC transporters, such as in a P-glycoprotein homolog for binding of a cyclopeptide inhibitor (Kodan et al., 2014) and in PglK for binding of its lipid substrate (Figure S5). Of note, this surface groove is formed upon the closure of a V-shaped opening between TM1 and TM3 as MsbA resets from the outward-facing to the inward-facing conformation, and the opening between TM1 and TM3 in outward-facing MsbA is a putative exit portal for lipid A substrate (Ward et al., 2007) (Figure 6C). We hypothesize that lipid A may dock for a short time on the surface groove just after exit from MsbA’s transport path. The transient binding of lipid A on the outer surface of MsbA is expected to be weak in order to permit its subsequent processing by enzymes and transporters downstream in the LPS biogenesis pathway (Figure 1), for which we still have very little understanding.
Figure 6. Putative lipid A binding site on the outer surface of MsbA.

(A) Lipid A (yellow sticks) is modeled into extensive electron density (green mesh, Fo–Fc difference map contoured at 2.0 σ without inclusion of lipid A in the refinement) outside MsbA. This density only appears in maps from crystals grown in the presence of lipid A (Figures S3A and S3B). (B) The putative lipid A binding site is near a surface groove (green) harbored by the periplasmic ends of TM1–3. Residues located in this region that potentially interact with lipid A are labeled. (C) A potential path of lipid A release to the periplasmic leaflet of the inner membrane. Structures in the left and right represent, respectively, the outward (PDB accession 3B60) and inward-facing MsbA. Lipid A (yellow sphere) is manually docked in 3B60 for illustration purposes. It may be released through the gap that opens between TM1and TM3 (rendered in green helices) and come to rest on the nearby surface groove illustrated in (B) in concert with the conformational change. See also Figures S3–S6.
To further validate this periplasmic lipid A binding site in MsbA and investigate its biological significance, detailed biochemical, biophysical and structural studies are still required. Here, we conducted 200 ns of molecular dynamic simulation in lipid bilayers, starting from lipid A modeled in a position as shown in Figure 6A. Of the two surface-bound lipid A, we observed that one remained associated with MsbA, albeit slithering along its surface, while the other started to dissociate from the protein surface after ~100 ns (Figure S6A). Interestingly, after the departure of lipid A from the periplasmic binding sites, the sites remained hydrated instead of being filled by adjacent phospholipid molecules (Figure S6B and Video S1). Careful examination of contact frequencies of water or lipid revealed that this putative lipid A binding site is an abnormally hydrophilic surface in the hydrophobic section of the membrane (Figure S6B). Averaging the lipid occupancy around the protein over the simulation trajectory also confirmed a void of phospholipid here (thus a gap between the protein surface and the averaged density of phospholipids). Maintaining hydration at this region is suggestive of a functional role of the sites that may be related to the hypothesized lipid A export and accompanied with conformational changes (Figure 6C).
We caution that errors might be introduced at the initial model of our molecular dynamics simulations since we cannot unambiguously assign coordinates to the lipid A molecule in our crystal structure. However, we reason that the inaccuracy in lipid A modeling will not significantly affect its equilibrium behavior in simulations given sufficient simulation length, especially in regard to its interactions with the lipid bilayer. During our analysis, lipid A displayed strong electrostatic interactions with the POPE headgroups through H-bonds and phosphate-ethanolamine salt bridges. The tail mixing between lipid A and POPE also provided favorable hydrophobic (van der Waals) interactions over time albeit less substantial than the polar interactions. Consequently, the lipids conferred strong “solvating” power for lipid A dissociation from MsbA. It is possible that lipid A stays at the periplasmic binding site in the crystal structure here because FA-3 does not provide strong dissolving power as phospholipids, due to its lack of both electrostatic interactions and long aliphatic chains.
CONCLUSIONS
Here, a high-resolution MsbA structure has been captured in an open, inward-facing conformation in the presence of lipid A substrate without bound nucleotide. With the goal of capturing ABC transporters in an early stage of the transport cycle for mechanistic studies, most structural studies have been conducted in the absence of nucleotides, which ought nevertheless to be present in the cellular milieu. Although dissociated NBDs in an inward-facing conformation of ABC transporters can bind ATP or other nucleotide analogues (Esser et al., 2017; Shintre et al., 2013), the presence of nucleotides is known to shift the conformational equilibrium toward NBD-dimerized, outward-facing states. Of note, MsbA bound with ADP, an intermediate state occurring post-ATP hydrolysis, has been characterized as adopting a similar range of conformations as the apo state (Mi et al., 2017; Moeller et al., 2015). In this study, we show that MsbA solubilized in FA-3 can actively hydrolyze ATP at rates comparable to that measured in lipid bilayers, suggesting that the crystallized structure, even without bound nucleotides, is likely to represent an active intermediate state in the transport cycle. This new structure complements and extends findings from the previous MsbA structures and other biochemical and biophysical studies, thus adding essential new insights into MsbA conformations associated with lipid A transport.
Based on comparative analysis with existing MsbA structures, sequential conformational changes along with a lipid transport pathway can be proposed. First, lateral opening of the membrane portals as shown in the current structure allows the entry of a bulky lipid A molecule into the protein-enclosed transport pathway (Figure 4A). Positively charged residues aligned on the portal surface of inward-facing MsbA may provide recognition and guidance of lipid A, with opening and closure of the membrane portal serving as a selective uptake mechanism. The inward motion of the two TMDs is proposed to increase affinity for the lipid A bound within MsbA. Meanwhile, preferential binding of lipid A to a ring of positively charged residues near the center of the membrane bilayer helps to shuttle the lipid toward the apex of the closed cavity, nearer the periplasmic side. Concurrent or sequential binding of ATP will dimerize the NBDs, switching the protein to an occluded or open outward-facing conformation (Ward et al., 2007). Ultimately, ATP hydrolysis and subsequent release of ADP and phosphate reset the transporter from the outward to inward facing conformation (Figure 6C). It remains unclear how lipid A is translocated or whether orientation of lipid A will flip at some point during transport, since both nucleotide and lipid A-bound structures in these intermediate states are still lacking. Our proposal that lipid A may dock temporarily on the outer surface of MsbA near its export portal, after the protein has reset from the outward to an inward-facing conformation, is based on the discovery of a possible secondary lipid A binding site on the periplasmic protein surface in our structural investigations. As such, the inward-facing MsbA conformation may represent both a starting and an end state during one complete transport cycle.
Structure determinations of MsbA at high resolution and in different states may help computational simulation and modeling efforts, which are especially valuable in light of the very recent validation of MsbA as a target for the discovery of new antibiotics. In two separate studies reported last year (Ho et al., 2018; Zhang et al., 2018), small molecules either inhibiting or stimulating the ATPase activity of MsbA were characterized as having potent bactericidal effect. As a prototype of ABC transporters, structural studies on MsbA may continue to facilitate a deep understanding of both the transport mechanism and mode of inhibitor action for this important class of proteins.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Qinghai Zhang, Email: qinghai@scripps.edu. The MTA and other corporate partnerships at The Scripps Research Institute is handled directly through the Office of the Technology Development (http://www.scripps.edu/research/technology).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The complete operon from Salmonella typhimurium of MsbA was sub-cloned into plasmid pET-19b vector downstream of a lac inducible operon and induced using IPTG (2mM). The bacterial strains used for sub-cloning and expression are regular hosts like DH5α and BL21DE3.
METHOD DETAILS
Expression and purification of MsbA.
MsbA from S. typhimurium containing an N-terminal His-tag was expressed as described previously (Ward et al., 2007). Briefly, bacterial cell (BL21DE3) membranes expressing MsbA were solubilized for 1 hour at 4 °C in a solution containing 20 mM Tris (pH 8.0), 20 mM NaCl, 1% (wt/vol) undecyl β-D-maltoside (UDM), 10% (vol/vol) glycerol, 0.1 mg/mL Dnase I, and a cocktail of protease inhibitors (Roche). After ultracentrifugation at 38,000 × g (45 min), the supernatant containing solubilized MsbA was sequentially purified by nickel-affinity and size-exclusion chromatography to homogeneity. The purified MsbA was pooled in a buffer containing 20 mM Tris (pH 7.5), 20 mM NaCl and 0.1% UDM. The purification of MsbA in different detergents (2–3 × critical micelle concentration) was performed similarly; alternatively, for the assessment of protein activity and stability, detergent exchange was carried out by cycles of concentration and dilution using 100 kD cutoff centrifugal filters. FA-3 (3α-hydroxy-7α,12α-di-((O-ß-D-maltopyranosyl)ethyloxy)-cholane) is available as Facade-EM from Avanti Polar Lipids.
ATPase assay.
ATPase activity of MsbA was measured using a coupled enzyme assay described elsewhere (Urbatsch et al., 1995). Briefly, ~1 μg MsbA was added at 37 °C to 100 μL LE buffer containing 10 mM ATP unless otherwise noted, 12 mM MgCl2, 6 mM phosphoenolpyruvate, 1 mM NADH, 10 units of lactate dehydrogenase, 10 units of pyruvate kinase and 50 mM Tris-HCl (pH 7.5). Reaction rate was measured by monitoring the decrease in absorbance of NADH at 340 nm using a FilterMax F4 microplate reader (Molecular Devices). Vanadate (200 μM) was added as a control to inhibit the ATPase activity of MsbA.
Thermal stability assay.
Thermal stability of MsbA was assessed using a modified procedure incorporating a thiol-reactive fluorophore, N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM), which undergoes an increase in fluorescence emission upon reaction with cysteine residues exposed by protein unfolding (Alexandrov et al., 2008). A single cysteine mutant of MsbA was generated for this assay by mutating two native cysteine residues (C88 and C315) to alanine and introducing a unique Cys residue A30C. Reactions contained 2 μM CPM and ~0.2 μM protein solubilized in different detergents. Fluorescence emission (λex = 387 nm; λem = 463 nm) was measured at 1 °C intervals in a microcuvette on a Cary Eclipse Fluorescence Spectrophotometer while ramping from 25 – 85 °C at a rate of 2 °C/min. Midpoints of the thermal transitions (Tm) were obtained using a least squares non-linear regression analysis (GraphPad Prism) of fluorescence signal versus temperature plots. Tm values for each sample were measured at least twice and replicates typically agreed within 1 °C.
LPS quantification.
A Limulus amoebocyte lysate chromogenic endotoxin quantification kit (Pierce) was used to measure the LPS content in MsbA samples and to generate a standard curve (Ho et al., 2018). One endotoxin unit equals approximately 0.1 to 0.2 ng of LPS. Duplicate measurements were conducted on two separate MsbA preparations purified in UDM.
Protein Crystallization.
For repetitive crystallization experiments, MsbA was purified in UDM and the detergent exchanged to FA-3 (0.04 wt %, 0.36 mM), as described above, along with the supplement of lipid A (5 μM), and was concentrated to ~12 mg/mL. MsbA crystals were grown using the hanging drop method at 4 °C by combining protein with precipitants consisting of 0.1 M Tris - 0.2 M tri-sodium citrate (pH 6.34–7.0), 0.12 M LiSO4, and 32.5% PEG 300 at a ratio between 1:1 and 1:0.7. The crystallization conditions were the same with or without lipid A. Crystals appeared in five days and continued to grow to a full size of 200–500 μm in approximately two months.
Data collection and refinement.
X-ray diffraction data were collected for crystals grown in the presence of lipid A at SSRL beam line 7–1 (equipped with ADSC Q315R CCD detector). The data were indexed and scaled using HKL-2000 (Otwinowski and Minor, 1997) to a resolution limit of 2.8 Å. Overall completeness was 85% so that the effective resolution of the data is 2.96 Å. The structure was determined by molecular replacement with Phaser (McCoy et al., 2007), using as a search model an all Ala model obtained using Cα coordinates from PDB accession 3B5W. The asymmetric unit contains a dimer of MsbA with a high solvent content of ~78% and a calculated Matthews’ coefficient of 5.65. The protein structure was refined with Refmac (Murshudov et al., 2011) to Rcryst and Rfree values of 26.9% and 30.1% respectively. Modeling lipid A at the periplasmic boundary reduced Rcryst and Rfree to 23.8% and 25.1%, respectively. For deposition in the RCSB Protein Data Bank (accession code 6BL6), we removed the ligands as the orientation of the modeled lipid A is not well defined. An additional 3.2 Å resolution data set was also processed, which confirmed the extra densities for putative lipid A binding.
Data from a crystal grown without added lipid A were collected at SSRL beam line 11–1 (Dectris PILATUS 6M detector). Data were indexed and scaled using HKL-2000 to 4.46Å with an overall completeness of 85%. These crystals are isomorphous to those grown in the presence of lipid A, and those higher resolution coordinates were used for a starting phasing model (PDB 6BL6). Coordinates were refined by rigid body refinement, followed by refinement with restraints to the higher resolution model to Rwork and Rfree of 26.9% and 30.1%, respectively.
Molecular Dynamics Simulations.
The simulation system was built based on the solved crystal structure of MsbA, using procedures similar to previously described for P-glycoprotein (Wen et al., 2013). The internally hydrated MsbA dimer (using DOWSER (Zhang and Hermans, 1996) and Solvate (Grubmuller, 1996)) with the two periplasmic-bound lipid A molecules were placed in a patch of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE) in a periodic boundary box of 160×160×140 Å3 containing 0.1 M NaCl solution. The resulting simulation system contains a total of 304,351 atoms, including 68,428 water molecules, 134 Na+ and 128 Cl− ions, with 327 and 314 POPE molecules in the periplasmic and cytoplasmic leaflets, respectively. Prior to the equilibration of the system, a short simulation of 0.5 ns was performed with all non-acyl atoms constrained, in order to mix and “melt” the orderly placed lipid tails. The system was then equilibrated for 10 ns as an isothermal-isobaric (NPT) ensemble where harmonic restraints at 5 kcal/mol Å2 magnitude were applied to all protein heavy atoms (except the modeled N- and C-termini residues). The harmonic restraints were removed after the equilibration and the simulation continues for another 200 ns as the production run.
Molecular dynamics simulations were performed using NAMD 2.12 (Phillips et al., 2005), with the CHARMM36m force field (Huang et al., 2017) used for protein, CHARMM36 force fields for POPE (Klauda et al., 2010) and lipid A (Wu et al., 2013), and TIP3 model for water molecules (Jorgensen et al., 1983). The NPT ensemble was maintained at 1.01325 bar and 310 K using Langevin dynamics and the Nosé-Hoover Lagevin piston method (Feller et al., 1995; Martyna et al., 1994) with a damping coefficient γ = 0.5 ps−1. An anisotropic pressure coupling scheme is applied due to the simulation of lipid bilayer, that the pressure is only coupled between the x and y dimensions of the system but not z (membrane normal). Non-bonded interactions were calculated within 12 Å cutoff and the switching function applied between 10–12 Å, whereas long range electrostatic interactions were computed using the particle mesh Ewarld (PME) method (Darden et al., 1993). Simulations were integrated in 2 fs steps, except for the first 0.5 ns “tail-melting” performed in 1fs steps.
Simulation trajectory was analyzed using VMD (Humphrey et al., 1996) between t = 100 ns and t = 200 ns of the production run at 5 ps interval. Protein-water contacts and polar contacts between protein and lipid head groups were calculated using 2.5 Å cutoff distance, whereas hydrophobic contacts between protein and lipids were calculated using 3.0 Å cutoff to aliphatic hydrogen atoms of POPE. Interaction energy was calculated using the NAMD Energy plug-in of VMD, where only non-bonded energy terms (van der Waals and electrostatic interactions) were calculated between crystallographic neighboring MsbA dimers, as well as between lipid A and MsbA, and between lipid A and POPE molecules of the entire bilayer.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical analysis of the assay data of standard deviations are obtained from the GraphPad Prism 5 built in features
DATA AND SOFTWARE AVAILABILITY
The co-ordinates for the structures reported here are deposited in the PDB with IDs 6BL6 and 6O30.
Supplementary Material
Video S1. Molecular dynamics simulation of lipid A dissociation from the periplasmic surface of MsbA, related to Figure S6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| None | ||
| Bacterial and Virus Strains | ||
| DH5 alpha High efficiency competent cells | Invitrogen | Cat No. 18258012 |
| BL21DE3 | New England Biolabs | Cat No. C 25271 |
| Biological Samples | ||
| None | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| n-Undecyl-β-D-Maltopyranoside, Anagrade | Anatrace | Cat No. U300LA |
| Façade®-EM 3α-hydroxy-7α,12α-di-((O-β-D-maltosyl)-2-hydroxyethoxy)-cholane | Avanti Polar Lipids | Cat No. 850552 |
| Lipid A, diphosphoryl from E. coli F583 | Sigma Aldrich | Cat No. L5399 |
| ATP Adenosine 5’-triphosphate (ATP) disodium salt hydrate | Sigma Aldrich | Cat No. FLAAS |
| β-Nicotinamide adenine dinucleotide hydrate | Sigma Aldrich | Cat No. N 7004 |
| L-Lactic dehydrogenase from rabbit muscle | Sigma Aldrich | Cat No. L 2500 |
| Sodium orthovanadate | Sigma Aldrich | Cat. No. S6508 |
| CPM (7-Diethylamino-3-(4’-Maleimidylphenyl)-4-Methylcoumarin) | Thermo Fisher | Cat No. D 346 |
| Critical Commercial Assays | ||
| None | ||
| Deposited Data | ||
| MsbA 2.8 Å crystal structure | This paper | 6BL6 |
| MsbA 4.46 Å crystal structure | This paper | 6O30 |
| Experimental Models: Cell Lines | ||
| None | ||
| Experimental Models: Organisms/Strains | ||
| None | ||
| Oligonucleotides | ||
| None | ||
| Recombinant DNA | ||
| pET19b | Novagen | Cat No. 71146 |
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad | N/A |
| Other | ||
| PDB files used in this study | https://www.rcsb.org/ | 3B60, 3B5X, 3B5W, 5TV4, 6BPL |
Highlights.
MsbA structure solved in an open inward-facing conformation at 2.8 Å-resolution
Structure enabled by cocrystallization with lipid A and using facial amphiphiles
Putative lipid A binding sites found both inside and outside MsbA
Offered new insights into the lipid A transport pathway
ACKNOWLEDGEMENTS
We thank Dr. Andrew Ward for helpful discussions and comments and the staff at Stanford Synchrotron Radiation Lightsource (SSRL beamlines 7–11 and 11–1) for help with crystallographic data collection. The SSRL Structural Molecular Biology Program is supported by the US department of Energy (DOE), the DOE Office of Biological and Environmental Research, National Institutes of Health (NIH), and National Institute of General Medical Sciences (NIGMS). This work was supported by NIH Grants (R01 GM118594 and R01 GM098538) to Q.Z and (R01-GM123455, U54-GM087519, and P41-GM104601) to E.T. Molecular dynamics simulation was performed using the Blue Waters supercomputer of the National Center for Supercomputing Applications at the University of Illinois.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: Authors declare no competing interests.
REFERENCES
- Alam A, Kung R, Kowal J, McLeod RA, Tremp N, Broude EV, Roninson IB, Stahlberg H, and Locher KP (2018). Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 115, E1973–E1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov AI, Mileni M, Chien EY, Hanson MA, and Stevens RC (2008). Microscale fluorescent thermal stability assay for membrane proteins. Structure 16, 351–359. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, and Sligar SG (2010). Membrane protein assembly into Nanodiscs. FEBS Lett. 584, 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Y, Mann E, Whitfield C, and Zimmer J (2018). Architecture of a channel-forming O-antigen polysaccharide ABC transporter. Nature 553, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbat PP, Surendhran K, Bortolus M, Zou P, Freed JH, and McHaourab HS (2007). Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 5, e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RS, and Altenberg GA (2013). Association/dissociation of the nucleotide-binding domains of the ATP-binding cassette protein MsbA measured during continuous hydrolysis. J. Biol. Chem 288, 20785–20796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, and Pedersen L (1993). Particle mesh Ewald - an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092. [Google Scholar]
- Doerrler WT, Gibbons HS, and Raetz CR (2004). MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J. Biol. Chem 279, 45102–45109. [DOI] [PubMed] [Google Scholar]
- Doerrler WT, and Raetz CR (2002). ATPase activity of the MsbA lipid flippase of Escherichia coli. J. Biol. Chem 277, 36697–36705. [DOI] [PubMed] [Google Scholar]
- Doerrler WT, Reedy MC, and Raetz CR (2001). An Escherichia coli mutant defective in lipid export. J. Biol. Chem 276, 11461–11464. [DOI] [PubMed] [Google Scholar]
- Doshi R, and van Veen HW (2013). Substrate binding stabilizes a pre-translocation intermediate in the ATP-binding cassette transport protein MsbA. J. Biol. Chem 288, 21638–21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi R, Woebking B, and van Veen HW (2010). Dissection of the conformational cycle of the multidrug/lipidA ABC exporter MsbA. Proteins 78, 2867–2872. [DOI] [PubMed] [Google Scholar]
- Eckert JK, Kim YJ, Kim JI, Gurtler K, Oh DY, Sur S, Lundvall L, Hamann L, van der Ploeg A, Pickkers P, et al. (2013). The crystal structure of lipopolysaccharide binding protein reveals the location of a frequent mutation that impairs innate immunity. Immunity 39, 647–660. [DOI] [PubMed] [Google Scholar]
- Eckford PD, and Sharom FJ (2008). Functional characterization of Escherichia coli MsbA: interaction with nucleotides and substrates. J. Biol. Chem 283, 12840–12850. [DOI] [PubMed] [Google Scholar]
- Eckford PD, and Sharom FJ (2010). The reconstituted Escherichia coli MsbA protein displays lipid flippase activity. Biochem. J 429, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser L, Zhou F, Pluchino KM, Shiloach J, Ma J, Tang WK, Gutierrez C, Zhang A, Shukla S, Madigan JP, et al. (2017). Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity. J. Biol. Chem 292, 446–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller SE, Zhang YH, Pastor RW, and Brooks BR (1995). Constant-pressure molecular-dynamics simulation - the Langevin piston method. J. Chem. Phys 103, 4613–4621. [Google Scholar]
- Grubmuller H (1996). SOLVATE v. 1.0. Theoretical Biophysics Group, Institute for Medical Optics, Ludwig-Maximilians University, Munich. [Google Scholar]
- Ho H, Miu A, Alexander MK, Garcia NK, Oh A, Zilberleyb I, Reichelt M, Austin CD, Tam C, Shriver S, et al. (2018). Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 557, 196–201. [DOI] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmuller H, and MacKerell AD Jr. (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935. [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr., and Pastor RW (2010). Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodan A, Yamaguchi T, Nakatsu T, Sakiyama K, Hipolito CJ, Fujioka A, Hirokane R, Ikeguchi K, Watanabe B, Hiratake J, et al. (2014). Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. USA 111, 4049–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Bennett BC, Hong WX, Fu Y, Baker KA, Marcoux J, Robinson CV, Ward AB, Halpert JR, Stevens RC, et al. (2013). Steroid-based facial amphiphiles for stabilization and crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA 110, E1203–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu Y, He LB, Zhao Y, and Zhang XC (2018). Single-molecule fluorescence studies on the conformational change of the ABC transporter MsbA. Biophys. Rep 4, 153–165. [Google Scholar]
- Martyna GJ, Tobias DJ, and Klein ML (1994). Constant-pressure molecular-dynamics algorithms. J. Chem. Phys 101, 4177–4189. [Google Scholar]
- May JM, Sherman DJ, Simpson BW, Ruiz N, and Kahne D (2015). Lipopolysaccharide transport to the cell surface: periplasmic transport and assembly into the outer membrane. Philos. Trans. R. Soc. Lond. B Biol. Sci 370, 20150027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith TC, Aggarwal P, Mamat U, Lindner B, and Woodard RW (2006). Redefining the requisite lipopolysaccharide structure in Escherichia coli. ACS Chem. Biol 1, 33–42. [DOI] [PubMed] [Google Scholar]
- Mi W, Li Y, Yoon SH, Ernst RK, Walz T, and Liao M (2017). Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 549, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A, Lee SC, Tao H, Speir JA, Chang G, Urbatsch IL, Potter CS, Carragher B, and Zhang Q (2015). Distinct conformational spectrum of homologous multidrug ABC transporters. Structure 23, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi M, and Tajkhorshid E (2013). Mechanistic picture for conformational transition of a membrane transporter at atomic resolution. Proc. Natl. Acad. Sci. USA 110, 18916–18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D, Biol. Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, and Lee JO (2009). The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195. [DOI] [PubMed] [Google Scholar]
- Perez C, Gerber S, Boilevin J, Bucher M, Darbre T, Aebi M, Reymond JL, and Locher KP (2015). Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature 524, 433–438. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polissi A, and Georgopoulos C (1996). Mutational analysis and properties of the msbA gene of Escherichia coli, coding for an essential ABC family transporter. Mol.Microbiol. 20, 1221–1233. [DOI] [PubMed] [Google Scholar]
- Raetz CR, and Whitfield C (2002). Lipopolysaccharide endotoxins. Annu. Rev. Biochem 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter G, Janvilisri T, Venter H, Shahi S, Balakrishnan L, and van Veen HW (2003). The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities. J. Biol. Chem 278, 35193–35198. [DOI] [PubMed] [Google Scholar]
- Reynolds CM, and Raetz CR (2009). Replacement of lipopolysaccharide with free lipid A molecules in Escherichia coli mutants lacking all core sugars. Biochemistry (Mosc) 48, 9627–9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintre CA, Pike AC, Li Q, Kim JI, Barr AJ, Goubin S, Shrestha L, Yang J, Berridge G, Ross J, et al. (2013). Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. USA 110, 9710–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson BW, May JM, Sherman DJ, Kahne D, and Ruiz N (2015). Lipopolysaccharide transport to the cell surface: biosynthesis and extraction from the inner membrane. Philos. Trans. R. Soc. Lond. B. Biol. Sci 370, 20150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk P, Tao H, McGrath AP, Villaluz M, Rees SD, Lee SC, Doshi R, Urbatsch IL, Zhang Q, and Chang G (2015). Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. Section D, Biol. Crystallogr 71, 732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terakado K, Kodan A, Nakano H, Kimura Y, Ueda K, Nakatsu T, and Kato H (2010). Deleting two C-terminal alpha-helices is effective to crystallize the bacterial ABC transporter Escherichia coli MsbA complexed with AMP-PNP. Acta Crystallogr. Section D, Biol. Crystallogr 66, 319–323. [DOI] [PubMed] [Google Scholar]
- Urbatsch IL, Sankaran B, Weber J, and Senior AE (1995). P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem 270, 19383–19390. [DOI] [PubMed] [Google Scholar]
- Velkov T, Roberts KD, Nation RL, Thompson PE, and Li J (2013). Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol. 8, 711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Karbarz MJ, McGrath SC, Cotter RJ, and Raetz CR (2004). MsbA transporter-dependent lipid A 1-dephosphorylation on the periplasmic surface of the inner membrane: topography of francisella novicida LpxE expressed in Escherichia coli. J. Biol. Chem 279, 49470–49478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Reyes CL, Yu J, Roth CB, and Chang G (2007). Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA 104, 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PC, Verhalen B, Wilkens S, McHaourab HS, and Tajkhorshid E (2013). On the origin of large flexibility of P-glycoprotein in the inward-facing state. J. Biol. Chem 288, 19211–19220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield C, and Trent MS (2014). Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem 83, 99–128. [DOI] [PubMed] [Google Scholar]
- Woebking B, Reuter G, Shilling RA, Velamakanni S, Shahi S, Venter H, Balakrishnan L, and van Veen HW (2005). Drug-lipid A interactions on the Escherichia coli ABC transporter MsbA. J. Bacteriol 187, 6363–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EL, Engstrom O, Jo S, Stuhlsatz D, Yeom MS, Klauda JB, Widmalm G, and Im W (2013). Molecular dynamics and NMR spectroscopy studies of E. coli lipopolysaccharide structure and dynamics. Biophys. J 105, 1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Baidin V, Pahil KS, Moison E, Tomasek D, Ramadoss NS, Chatterjee AK, McNamara CW, Young TS, Schultz PG, et al. (2018). Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors. Proc. Natl. Acad. Sci. USA 115, 6834–6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, and Hermans J (1996). Hydrophilicity of cavities in proteins. Proteins 24, 433–438. [DOI] [PubMed] [Google Scholar]
- Zhou Z, White KA, Polissi A, Georgopoulos C, and Raetz CR (1998). Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem 273, 12466–12475. [DOI] [PubMed] [Google Scholar]
- Zoghbi ME, Cooper RS, and Altenberg GA (2016). The lipid bilayer modulates the structure and function of an ATP-binding cassette exporter. J. Biol. Chem 291, 4453–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Bortolus M, and McHaourab HS (2009). Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J. Mol. Biol 393, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Molecular dynamics simulation of lipid A dissociation from the periplasmic surface of MsbA, related to Figure S6.
Data Availability Statement
The co-ordinates for the structures reported here are deposited in the PDB with IDs 6BL6 and 6O30.