

Summary
Group 3 innate lymphoid cells (ILC3) play key roles in protective immunity and mucosal barrier maintenance. Here we showed that vitamin D/vitamin D receptor (VDR) signaling regulates gut ILC3. VDR deletion or 1,25-dihydroxyvitamin D deficiency in mice led to a marked reduction in colonic ILC3 populations at steady state and impaired ILC3 responses following Citrobacter rodentium infection, resulting in substantial increases in intestinal bacterial growth and mouse mortality. VDR regulation of ILC3 was independent of T and B lymphocytes or gut microflora. Correction of 1,25-dihydroxyvitamin D deficiency rescued the ILC3 defects. Mechanistically, VDR deletion or 1,25-dihydroxyvitamin D deficiency markedly reduced colonic Ki67+ ILC3 populations, and in vivo and in vitro studies confirmed that vitamin D hormone directly stimulated ILC3 proliferation. Therefore, vitamin D/VDR signaling is required for ILC3-mediated innate immunity through regulation of ILC3 proliferation.
Subject Areas: Biomolecules, Immunology, Immune Response, Components of the Immune System
Graphical Abstract
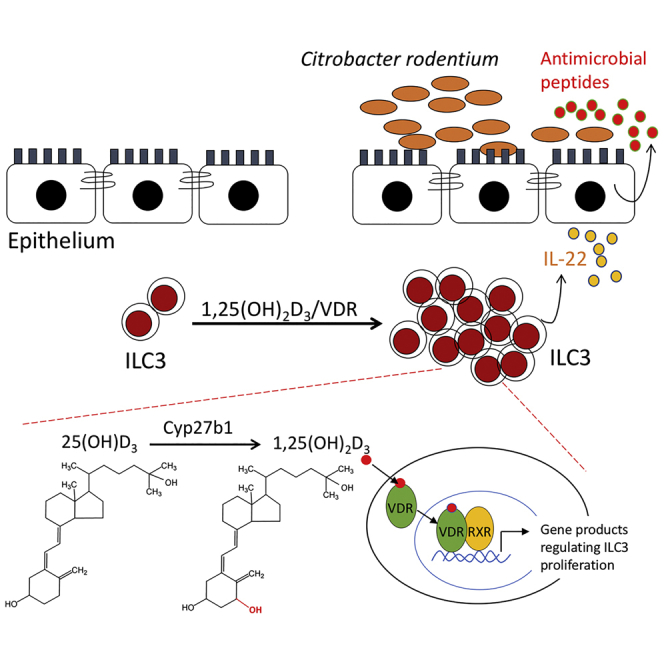
Highlights
-
•
VDR or 1,25(OH)2D3 deficiency reduces ILC3 populations and impairs ILC3 immunity
-
•
Vitamin D/VDR signaling is required for proper ILC3 proliferation
-
•
Vitamin D regulation of ILC3 is independent of T and B cells or gut microflora
Biomolecules; Immunology; Immune Response; Components of the Immune System
Introduction
The vitamin D hormone is a major endocrine system with pleiotropic functions (Bouillon et al., 2008). Physiologically, the majority of the body's vitamin D content is derived from photosynthesis in the skin following UV irradiation, and thus body's vitamin D levels are influenced by geographic locations, seasonal changes, and skin pigmentations (Holick, 2007, Holick, 2018). Vitamin D is converted to the active hormone 1,25-dihydroxyvitamin D (1,25(OH)2D3) by two steps of hydroxylation: 25-hydroxylation in the liver followed by 1α-hydroxylation in the kidney (Holick, 1996). Cyp27b1 (25-hydroxyvitamin D 1α-hydroxylase) is the rate-limiting enzyme required for the synthesis of 1,25(OH)2D3. The biological activities of 1,25(OH)2D3 is mediated by the vitamin D receptor (VDR), a nuclear hormone receptor (Haussler et al., 2013). The gut is one of the tissues in the body that have the most abundant VDR expression (Wang et al., 2012), indicating that it is a major physiological target of vitamin D. It is long established that vitamin D/VDR signaling regulates duodenal transcellular calcium transport (Lee et al., 2015). Recent studies demonstrated that gut epithelial VDR plays a key role in protecting the mucosal barrier integrity (He et al., 2018, Liu et al., 2013). However, little is known about the effect of vitamin D/VDR signaling on the immune components of the gut, including the gut innate immunity.
Innate lymphoid cells (ILCs) are a set of newly discovered innate immune cells emerging as crucial effectors of innate immunity in diverse physiological functions including host defense against infection, tissue remodeling, and maintenance of mucosal barrier integrity (Artis and Spits, 2015, Bernink et al., 2013). The diverse populations of ILC family are characterized by a classic lymphoid cell morphology that lacks expression of somatically rearranged antigen receptors and lineage surface markers characteristic of myeloid origin or adaptive immune system; as such, ILCs do not exhibit any degree of antigen specificity and are lineage marker-negative cells (Artis and Spits, 2015). ILCs are derived from an Id2-dependent common lymphoid progenitor cell population and require interleukin (IL)-7 for their maintenance. ILCs can be grouped based on the transcription factors required for their development and function, and currently there are three major groups: T-bet+ ILC (Group 1, ILC1), GATA3+ ILC (Group 2, ILC2), and RORγt+ ILC (Group 3, ILC3) (Artis and Spits, 2015, Sonnenberg and Artis, 2012).
RORγt+ ILC3 is the major ILC in the gut that predominantly produces IL-22 and IL-17 and plays critical roles in protective immunity against bacteria (Sonnenberg and Artis, 2012). It is known that IL-22 stimulates epithelial cells to secret anti-microbial peptides, including RegIIIγ, lipocalin-2, and β-defensins, to constrain pathogenic bacterial growth (Wolk et al., 2004, Zheng et al., 2008). IL-22 also induces mucin expression and colonic epithelial cell proliferation to maintain the integrity of the mucosal barrier (Sugimoto et al., 2008). ILC3 is heterogeneous in mice and humans. In mice, two subsets can be distinguished based on the expression of chemokine receptor CCR6. CCR6+ ILC3 include CD4+ and CD4- lymphoid tissue inducer (LTi) cells, namely, LTi4 and LTi0, and CCR6- ILC3 encompass cells that express natural cytotoxicity receptor NKp46, namely, NKp46+ (or NK22) cells. The development of ILC3 is regulated by a number of nuclear hormone receptors, including RORγt+, RARα, and AHR (Artis and Spits, 2015, Qiu et al., 2012, Sawa et al., 2010, Spencer et al., 2014). Moreover, the development of ILC2 and ILC3 in the gut is deeply influenced by micronutrients such as vitamin A (Spencer et al., 2014).
Vitamin D as a micronutrient is well known for its immune-modulatory activity (Hart et al., 2011, Mora et al., 2008). A body of research has demonstrated the importance of vitamin D in the regulation of classic innate immune cells (e.g., monocytes, macrophages, dendritic cells) (Liu et al., 2006, Penna and Adorini, 2000) and adaptive immune cells (e.g., TH1, TH17, Treg, B cells) (Joshi et al., 2011, Lemire et al., 1985, Mora et al., 2008, Penna et al., 2005, Rigby et al., 1984, Shirakawa et al., 2008, von Essen et al., 2010); however, few studies have directly assessed the role of the vitamin D/VDR signaling in ILC biology. Here we present evidence to demonstrate that this signaling pathway is an important regulator of ILC3 development and is required for ILC3-mediated innate immunity in the gut.
Results
Reduced Colonic ILC3 Populations in VDR−/− Mice
Our interest in ILC3 initially derived from an observation that gut ILC3 expressed a high level of VDR. We quantified by qRT-PCR the baseline Vdr transcript level in colonic ILC3 (CD3−CD90hiCD45lo) (Guo et al., 2014) and TH17 cells (CD3+CD4+TCRβ+GFP(RORγt)+) that were sorted from colonic lamina propria (LP) cells by fluorescence-activated cell sorting (FACS) and found that ILC3 Vdr transcript was higher compared with sorted TH17 cells or purified intestinal epithelial cells (Figure S1), suggesting a role of VDR in ILC3 biology. To address whether VDR regulates ILC3, we compared the colonic mucosal profiles of ILC3 and the sub-populations between VDR−/− and wild-type (WT) mice by FACS analyses (see Figure S2 for the gating strategy). The FACS data showed that RORγt+ ILC3 (Figures 1A and 1D), as well as NKp46+ (Figures 1B and 1E), LTi4, and LTi0 (Figures 1C and 1F) subsets, were all markedly decreased in VDR−/− mice at steady state compared with WT mice, independent of sexes.
Figure 1.

Global VDR Deletion Impairs Gut ILC3 Development in Mice
Colonic lamina propria (LP) cells were isolated from WT and VDR−/− mice, and the cells were analyzed by FACS for ILC3 populations.
(A–C) Representative FACS plots for analysis of RORγt+ILC3 (A), NKp46+ cells (B), and LTi4 and LTi0 cells (C) in WT and VDR−/− mice at steady state.
(D–F) Quantitation based on FACS data of RORγt+ILC3 (D), NKp46+ cells (E), and LTi4 and LTi0 cells (F) in WT and VDR−/− mice. The data were presented as percentage of the gated population and absolute cell number. **p < 0.01; ***p < 0.001. n = 4 each genotype. Data are represented as mean ± SEM.
To eliminate the potential confounding influence of T and B lymphocytes on ILC3 in the context of VDR ablation, we generated Rag1−/−VDR+/+ and Rag1−/−VDR−/− mice that lacked mature T and B lymphocytes (Mombaerts et al., 1992). FACS analyses confirmed that the colonic CD3−RORγt+ ILC3 population, as well as LTi4 and LTi0 subsets, but not NKp46+ cells, remained reduced in Rag1−/−VDR−/− mice compared with Rag1−/−VDR+/+ controls (Figures S3A–S3F), indicating that the effect of VDR deletion on gut ILC3 abundance, particularly the LTi4 and LTi0 cells, is completely independent of T and B cells.
In contrast to gut RORγt+ T cells (e.g., TH17), the development of gut RORγt+ ILC3 is pre-programmed and does not require gut commensal bacteria (Sawa et al., 2010). To address whether gut commensal bacteria affects gut ILC3 in the absence of VDR, we depleted gut microbiota from WT and VDR−/− mice by 4-week antibiotic treatment as described previously (Mukherji et al., 2013, Rakoff-Nahoum et al., 2004). Fecal bacterial 16S rRNA gene was almost undetectable by PCR following antibiotic treatment, confirming that the antibiotics had effectively depleted gut bacteria (Figure S4A). FACS data showed that colonic ILC3 profiles, including CD3−RORγt+ ILC3 and the NKp46+, LTi4 and LTi0 subpopulations, remained suppressed in VDR−/− mice compared with WT controls in the context of the microbiota depletion (Figures S4B–S4G), suggesting that VDR regulation of gut ILC3 is independent of the commensal bacteria.
VDR−/− Mice Develop Impaired Immunity against Citrobacter rodentium
To assess the immunological implication of gut ILC3 reduction in VDR−/− mice, we analyzed their immune response to Citrobacter rodentium infection, a model widely used to assess ILC3-mediated immunity (Koroleva et al., 2015). Following oral gavage of C. rodentium, VDR−/− mice showed marked weight loss compared with WT mice (Figure 2A), and about 40% VDR−/− mice died during the infection course (about 30% died within 5 days post infection) (Figure 2B). We quantified fecal C. rodentium using MacConkey agar plates (Bouladoux et al., 2017), on which C. rodentium colonies formed distinctive morphology characterized by a pink center with white rim (Figure S5A). Furthermore, we also validated the identity of C. rodentium by PCR amplification of the virulence eae gene, which is DBS100 strain specific (Petty et al., 2010, Schauer and Falkow, 1993) (Figure S5B). Following infection, the fecal C. rodentium counts increased daily before reaching the peak by day 7, followed by bacterial clearance. The peak bacterial count of VDR−/− mice was >4 times higher compared with that of WT mice. In fact, in VDR−/− mice the increase in fecal bacterial loads was one to two order of magnitude higher each day before reaching the peak, yet the bacterial clearance rate was substantially slower in comparison with WT counterparts (Figure 2C). FACS analyses detected a robust ILC3 response in WT mice, as total ILC3 population as well as IL-22+ILC3, NKp46+, LTi4, and LTi0 subsets were all markedly increased on day 5 after C. rodentium infection (Figures 2D–2J). In contrast, VDR−/− mice showed a clearly impaired ILC3 response; the induction of ILC3, IL-22+ILC3, NKp46+, LTi4, and LTi0 cells were all markedly attenuated (Figures 2D–2J). Particularly, IL-22+ ILC3 was almost not induced (Figures 2E and 2H). Thus, impaired ILC3 response, particularly attenuated IL-22 production, is likely the cause for the dramatic increase in bacterial growth in the gut of VDR−/− mice leading to their high mortality.
Figure 2.

Global VDR Deletion Leads to Impaired Gut Immunity against C. rodentium
WT and VDR−/− mice were gavaged with C. rodentium. The mice were monitored daily for 16 days post infection. The mice were killed on day 5 following infection for FACS analyses of colonic LP cells.
(A–C) (A) Body weight changes (n = 10), (B) survival curve (n = 10–11), and (C) fecal daily bacterial counts (CFU) per gram of feces in WT and VDR−/− mice under C. rodentium infection (n = 6–7). p < 0.001 by log rank test.
(D–G) Representative FACS plots for the analysis of RORγt+ILC3 (D), IL-22+ILC3 (E), NKp46+ (F), and LTi4 and LTi0 cells (G) in WT and VDR−/− mice at steady state (Ctrl) and under C. rodentium infection.
(H–J) Quantitation RORγt+ILC3 and IL-22+ILC3 (H), NKp46+ cells (I), and LTi4 and LTi0 cells (J) in WT and VDR−/− mice at baseline and under C. rodentium infection. The data were presented as percentage of the gated population and absolute cell number. *p < 0.05; **p < 0.01; ***p < 0.001. n = 4 each group. Data are represented as mean ± SEM.
Deficiency in 1,25(OH)2D3 Impairs Gut ILC3 Development and ILC3-Mediated Immunity
Cyp27b1−/− mice are completely deficient in 1,25(OH)2D3 production, as they carry a genetic deletion in the 1α-hydroxylase gene (Panda et al., 2001), the rate-limiting enzyme required for 1,25(OH)2D3 biosynthesis. To assess whether the regulatory effect of VDR on ILC3 is ligand dependent, we compared the colonic ILC3 profiles between WT and Cyp27b1−/− mice at steady state and under C. rodentium infection. FACS analyses revealed marked decreases in CD3−RORγt+ ILC3 populations at the steady state in Cyp27b1−/− mice compared with WT mice (Figures S6A and S6B, and Figures 3C–3I). When infected with C. rodentium, Cyp27b1−/− mice displayed much more severe body weight loss (Figure 3A) and higher fecal bacterial loads on days 2 and 5 (Figure 3B). FACS analyses showed that ILC3-mediated immunity was clearly impaired in Cyp27b1−/− mice. On day 5 after infection, ILC3 responses, including the increase in CD3−RORγt+ ILC3, NKp46+, LTi4, and LTi0 cells, were all markedly attenuated in Cyp27b1−/− mice compared with WT controls (Figures 3C–3I). Importantly, the induction of the IL-22+ILC3 population was severely compromised in Cyp27b1−/− mice (Figures 3D and 3G). These data are similar to those of the ILC3 phenotype seen in VDR−/− mice, indicating that either VDR deletion or VDR ligand deficiency can lead to gut ILC3 deficiency and impaired ILC3-mediated immunity.
Figure 3.

Deficiency in 1,25(OH)2D3 Synthesis Impairs ILC3 Development and Its Immunity against C. rodentium
WT and Cyp27b1−/− mice were gavaged with C. rodentium. The mice were killed on day 5 following infection for FACS analyses of colonic LP cells.
(A and B) (A) Body weight changes; (B) fecal bacterial counts per gram of feces in WT and Cyp27b1−/− mice on days 2 and 5 following C. rodentium infection. ***p < 0.001 vs. WT. n = 4–5 each group.
(C–F) Representative FACS plots for the analysis of RORγt+ILC3 (C), IL-22+ILC3 (D), NKp46+ (E), and LTi4 and LTi0 cells (F) in WT and Cyp27b1−/− mice at steady state (Ctrl) and under C. rodentium infection.
(G–I) Quantitation RORγt+ILC3 and IL-22+ILC3 (G), NKp46+ cells (H), and LTi4 and LTi0 cells (I) in WT and Cyp27b1−/− mice at baseline and under C. rodentium infection.
The data were presented as percentage of the gated population and absolute cell number. *p < 0.05; **p < 0.01; ***p < 0.001. n = 4 each group. Data are represented as mean ± SEM.
To confirm that 1,25(OH)2D3 is required for ILC3 development, we reconstituted Cyp27b1−/− mice with 1,25(OH)2D3 through daily intraperitoneal injection for one week. FACS analyses showed that 1,25(OH)2D3 reconstitution was able to partially correct gut ILC3 deficiency in Cyp27b1−/− mice. The total ILC3 population was substantially recovered following 1 week of 1,25(OH)2D3 injection. Specifically, LTi4 and LTi0 subsets, but not NKp46+ cells, were partially increased by 1,25(OH)2D3 treatment (Figure S7). Therefore, both 1,25(OH)2D3 and VDR are required for normal ILC3 development and function in the gut, especially for the LTi cells.
Critical Roles of ILC3-intrinsic VDR Signaling in the Regulation of ILC3
The effect of global VDR deletion in VDR−/− mice on ILC3 could be influenced by many confounding factors in the body. To specifically delineate the role of ILC3-intrinsic VDR signaling in ILC3 regulation, we generated VDRflox/flox;RORγt-Cre mice that carried VDR deletion in ILC3. VDRflox/flox;RORγt-Cre mice showed a marked and significant reduction in steady-state colonic CD3−RORγt+ILC3 population compared with VDRflox/flox mice (Figures 4C and 4G). The baseline IL-22+ILC3 population was slightly higher, which may reflect an intrinsic feedback mechanism to maintain IL-22 expression (Figures 4D and 4G). Specifically, the LTi4 and LTi0 subsets, but not NKp46+ cells, were significantly decreased in VDRflox/flox;RORγt-Cre mice at the steady state (Figures 4E, 4F, 4H, and 4I), confirming a critical role of ILC3-intrinsic VDR in the basal development of ILC3, particularly the LTi subsets. When VDRflox/flox and VDRflox/flox;RORγt-Cre mice were infected with C. rodentium, VDRflox/flox;RORγt-Cre mice showed more severe body weight loss and much higher fecal bacterial counts on day 4 compared with VDRflox/flox mice (Figures 4A and 4B). ILC3 responses, including the increases in CD3−RORγt+ ILC3 and LTi4 and LTi0 subsets (not NKp46+) were markedly attenuated in VDRflox/flox;RORγt-Cre mice (Figures 4C–4I). Particularly, the IL-22+ILC3 population failed to be induced in VDRflox/flox;RORγt-Cre mice (Figures 4D and 4G). Thus, ILC3-intrinsic VDR deletion impairs the ILC3 baseline development as well as ILC3-mediated immunity, indicating a critical role of ILC3-intrinsic vitamin D/VDR signaling in ILC3 biology.
Figure 4.

ILC3-specific Deletion of VDR Impairs ILC3 Development and Its Immunity against C. rodentium Infection
VDRflox/flox and VDRflox/flox;RORγt-Cre mice were gavaged with C. rodentium. The mice were killed on day 4 following infection.
(A and B) (A) Body weight changes; (B) fecal bacterial counts per gram of feces in VDRflox/flox and VDRflox/flox;RORγt-Cre mice on day 4 after C. rodentium infection. ***p < 0.001 vs. VDRflox/flox. n = 4–5 each genotype.
(C–F) Representative FACS plots for the analysis of RORγt+ILC3 (C), IL-22+ILC3 (D), NKp46+ (E), and LTi4 and LTi0 cells (F) in VDRflox/flox and VDRflox/flox;RORγt-Cre mice at steady state (Ctrl) and under C. rodentium infection.
(G–I) Quantitation RORγt+ILC3 and IL-22+ILC3 (G), NKp46+ cells (H), and LTi4 and LTi0 cells (I) in VDRflox/flox and VDRflox/flox;RORγt-Cre mice at baseline and under C. rodentium infection.
The data were presented as percentage of the gated population and absolute cell number. *p < 0.05; **p < 0.01; ***p < 0.001. n = 4 each group. Data are represented as mean ± SEM.
RORγt is the master transcription factor in ILC3 and TH17 lineages, but it may also express in other tissues; as such, RORγt-Cre could be leaky to other cells/tissues. To confirm that it was VDR deletion in ILC3 that led to ILC3 defects, we transplanted Rag1−/− mice with bone marrow (BM) cells obtained from VDRflox/flox or VDRflox/flox;RORγt-Cre mice using procedures as previously reported (Szeto et al., 2012). The success of the BM transplantation was validated in a parallel control experiment in which CD45.2 VDR−/− BM cells were transplanted to CD45.1 recipient mice (Figure S8). As shown in Figure S8, 8 weeks after transplantation, CD45.2+ cells repopulated the blood and colonic lamina propria of the CD45.1 recipients. Importantly, the recipient's RORγt+ cells (which include ILC3) were now CD45.2+. Interestingly, 8 weeks after transplantation, Rag1−/− mice receiving VDRflox/flox;RORγt-Cre BM cells still had significantly lower CD3−RORγt+ILC3 population as well as lower LTi4 and LTi0 subpopulations compared with Rag1−/− mice receiving VDRflox/flox BM cells (Figures 5A and 5B). These results confirmed that the ILC3-intrinsic VDR signaling indeed directly regulates ILC3 development, particularly the LTi subsets.
Figure 5.

Bone Marrow (BM) Transplantation Confirms VDR Deletion in ILC3 Leading to Impaired ILC3 Development
BM cells obtained from VDRflox/flox or VDRflox/flox/RORγt-Cre mice were transplanted to recipient Rag1−/− mice 6 h after receiving lethal γ-irradiation. The recipient mice were analyzed 8 weeks after transplantation.
(A and B) (A) Representative FACS plots for RORγt+ILC3 analyses; (B) quantitation of colonic RORγt+ILC3, NKp46+, and LTi4 and LTi0 cells in recipient mice transplanted with VDRflox/flox or VDRflox/flox;RORγt-Cre BM cells. The data were presented as percentage of the gated population and absolute cell number. *p < 0.05, **p < 0.01. n = 3–4 each group. Data are represented as mean ± SEM.
Vitamin D/VDR Signaling Regulates ILC3 Proliferation
To address how vitamin D/VDR signaling regulates ILC3 development, we assess the effect of VDR deletion on ILC3 apoptosis and proliferation, two processes that potentially determine the abundance of gut ILC3. To assess ILC3 apoptosis, we generated VDR−/−Rorcgfp/+ mice whose ILC3 is marked by GFP. FACS analyses found no differences between VDR+/+Rorcgfp/+ and VDR−/−Rorcgfp/+ mice in Annexin V+/7-AAD+ and Annexin V+/7-AAD- cells among all the gut ILC3 populations (Figure S9), suggesting that ILC3 defects caused by VDR deletion is not due to excessive ILC3 apoptosis. We then quantified ILC3 proliferation by Ki67 staining. Remarkably, Ki67+ cells in the CD3−RORγt+ILC3 population as well as in LTi4 and LTi0 subsets were clearly decreased at steady state in VDR−/− mice (Figures 6A and 6B), VDRflox/flox;RORγt-Cre mice (Figures 6C and 6D), and Cyp27b1−/− mice (Figures 6E and 6F) compared with VDR+/+ mice, VDRflox/flox mice, and Cyp27b1+/+ mice, respectively. Under C. rodentium infection, the Ki67+ ILC3 cells, including CD3−RORγt+ cells and LTi4 and LTi0 subsets, were dramatically induced in VDR+/+ mice and VDRflox/flox mice, but the induction was greatly attenuated in VDR−/− mice and VDRflox/flox;RORγt-Cre mice (Figures 6A–6D). In these three mouse models, however, there were almost no changes in Ki67+NKp46+ cells (Figures 6A–6F). These observations suggest that the vitamin D/VDR signaling plays a key role in the regulation of gut ILC3 expansion, mainly the LTi cells.
Figure 6.

Deletion of VDR or Cyp27b1 Reduces ILC3 Proliferation
Colonic LP cells were isolated from WT or VDR−/− mice at steady state (Ctrl) or under C. rodentium infection for 5 days (A and B), from VDRflox/flox or VDRflox/flox;RORγt-Cre mice at steady state or under C. rodentium infection for 5 days (Cand D), or from WT and Cyp27b1−/− mice at steady state (E and F). Ki67+ ILC3 and ILC3 subsets were quantified by FACS. (A, C, and E) Representative FACS plots for Ki67+ RORγt+ILC3; (B, D, and F) quantitation of Ki67+RORγt+ILC3 and Ki67+NKp46+, Ki67+LTi4, and Ki67+LTi0 subsets. *p < 0.05; **p < 0.01; ***p < 0.001. n = 4–5 each group. Data are represented as mean ± SEM.
To confirm this notion, we further examined the effect of VDR activation by its ligand on ILC3 proliferation. As shown in Figure 7, reconstitution of Cyp27b1−/− mice with 1,25(OH)2D3 by 1-week daily injection was able to mostly restore the Ki67+ ILC3 populations (CD3−RORγt+, LTi4, LTi0) in the colonic mucosa (Figure 7A). Moreover, daily treatment of WT mice with a low calcemic vitamin D analogue, paricalcitol, for 1 week was able to substantially stimulate colonic Ki67+ ILC3 populations (CD3−RORγt+, LTi4 and LTi0) (Figure 7B). Consistently, there were little effects from these treatments on Ki67+ NKp46+ cells (Figures 7A and 7B).
Figure 7.

Ligand Activation of VDR Stimulates ILC3 Proliferation
(A) FACS quantitation of colonic Ki67+RORγt+ILC3 and Ki67+NKp46+, Ki67+LTi4, and Ki67+LTi0 subpopulations in WT, Cyp27b1−/− or Cyp27b1−/− mice treated with 1,25(OH)2D3 for 1 week; (B) FACS quantitation of colonic Ki67+RORγt+ILC3 and Ki67+NKp46+, Ki67+LTi4, and Ki67+LTi0 subsets in WT untreated or treated with paricalcitol for 1 week; (C) Lin−Thy1.2hiKLRG1−IL-7Rα+ cells were sorted by FACS and cultured in the presence or absence of 1,25(OH)2D3 for 3 days, and Ki67+RORγt+ILC3 population was analyzed by FACS. Shown are representative FACS plots. *p < 0.05, **p < 0.01; ***p < 0.001. n = 4 each group. Data are represented as mean ± SEM.
To address whether vitamin D/VDR signaling directly regulates ILC3 proliferation, we FACS-sorted Lin−Thy1.2hiKLRG1−IL-7Rα+ cells from colonic LP cells from Rag1−/− mice as described previously (Spencer et al., 2014) (see Figure S10 for the sorting strategy) and then cultured these cells for 3 days in a medium containing IL-7 and stem cell factor (SCF) in the presence or absence of 1,25(OH)2D3 before quantifying the Ki67+ ILC3 population by FACS. We found that 1,25(OH)2D3 treatment was able to increase the Ki67+RORγt+ cell population by >56% in vitro (Figure 7C). This is compelling evidence demonstrating that the vitamin D/VDR signaling indeed directly stimulates ILC3 proliferation.
Discussion
In this report we presented several lines of evidence to demonstrate that the vitamin D/VDR signaling pathway critically regulates ILC3 development and function in the gut. We showed that VDR deletion caused a marked reduction in gut ILC3 populations and compromises ILC3-mediated innate immunity against bacterial infection. This regulatory action of VDR on ILC3 is independent of T and B lymphocytes and gut commensal bacteria. We also showed that the deficiency of 1,25(OH)2D3, the VDR ligand, basically phenocopied VDR deficiency in terms of affecting gut ILC3 development and function, indicating that the regulatory action of VDR on ILC3 is ligand dependent. Given that the majority of VDR actions is known to be dependent on 1,25(OH)2D3, the similar ILC3 phenotypes seen in VDR−/− and Cyp27b1−/− mice actually provide a strong confirmation for the reliability of our findings. By specifically deleting VDR from ILC3 we further demonstrated that the ILC3-intrisinc VDR signaling is required for normal development and function of ILC3. Our data suggest that promoting ILC3 proliferation and differentiation is an important mechanism whereby the vitamin D/VDR signaling regulates ILC3. Together these findings extend our understanding of the immunoregulatory activities of the vitamin D/VDR signaling in innate immunity.
Although it is well established that the vitamin D endocrine system regulates immune responses (Hart et al., 2011, Mora et al., 2008), few studies have explored the effect of vitamin D and VDR on innate lymphoid cells. A previous study by Chen et al. reported that global VDR deletion caused dysbiosis, which conferred resistance to C. rodentium infection to VDR knockout mice because of increased ILC3 and antimicrobial peptides; however, elimination of gut microbiota with antibiotics rendered VDR knockout mice more susceptible to C. rodentium than control mice. These findings are inconsistent with our results. However, this same study also showed that elimination of T and B lymphocytes from VDR knockout mice (Rag1−/−VDR−/− mice) increased their susceptibility to C. rodentium infection when compared with controls (Chen et al., 2015), which is consistent with our findings reported here, confirming the notion that the VDR regulation of ILC3 is independent of functional T and B lymphocytes. This prior report, although interesting, provided no clear answers as to whether and how VDR influences ILC3. Additionally, there are two other reports by Ryz et al. that studied the relationship between vitamin D and C. rodentium infection but not ILC3. One paper showed that vitamin D deficiency increased mouse's susceptibility to C. rodentium mostly because of increased production of pro-inflammatory cytokines (Ryz et al., 2015), and the other showed that 1,25-dihydroxyvitamin D treatment also increased mouse's susceptibility to C. rodentium mostly because of the suppression of TH17 response (Ryz et al., 2012). Together these unsettled and seemingly contradictory studies provide a strong rationale to explore the roles of the vitamin D/VDR signaling pathway in ILC3 regulation and in C. rodentium infection. Therefore, in the current study we employed a series of genetic mouse models to tackle these important issues.
As the biological action of the vitamin D endocrine system depends on both the vitamin D hormone and its receptor, we studied genetic models that lack either the receptor (VDR knockout) or the ability to synthesize the vitamin D hormone (Cyp27b1 knockout). We also examined VDR knockout mice that lack mature T and B lymphocytes (Rag1−/−/VDR−/− mice) or commensal microflora to eliminate the potentially confounding effects of the adaptive immune system and microbiota on the results. Because VDR knockout mice develop many abnormalities (Bouillon et al., 2008) that may interfere with data interpretation, we further studied VDRflox/flox;RORγt-Cre mice that lack VDR signaling in ILC3 as well as lethally irradiated mice that are repopulated with VDRflox/flox;RORγt-Cre bone marrow cells. These cell-specific deletion and bone marrow transplantation models generated compelling evidence for a direct and critical role of VDR in ILC3 biology. In addition to ILC3, RORγt is also expressed in TH17 and Treg cells; thus VDRflox/flox;RORγt-Cre mice may carry VDR deletion in ILC3 as well as in TH17 and Treg cells. However, because we had already demonstrated that VDR deletion in T cells did not influence the effect of VDR on ILC3, particularly the LTi cells (see Figure S3, Rag1−/−/VDR−/− mice), the VDRflox/flox;RORγt-Cre mice thus allowed us to study and elucidate the intrinsic role of ILC3 VDR in ILC3 regulation. Indeed, the intrinsic stimulatory effect of vitamin D/VDR on ILC3 was confirmed in the in vitro ILC3 culture (Figure 7C). Furthermore, in this study we analyzed not only the CD3−RORγt+ cells but also the NKp46+ and LTi0/4 subsets. The data from these models all firmly support the notion that the vitamin D/VDR signaling is required for the development and anti-bacterial function of gut ILC3. In the absence of 1,25(OH)2D3 or VDR, gut ILC3 populations markedly decreased and failed to proliferate leading to impaired immunity against C. rodentium. Particularly, the IL-22-producing ILC3 population failed to expand in response to C. rodentium infection, which likely accounts for the severe bacterial growth and high mortality seen in the mutant mice, because IL-22 released from LTi4 cells in the first 6 days following C. rodentium infection is the major IL-22 source to counter the infection (Sonnenberg et al., 2011). In fact, the results from all our mouse models, as well as from in vitro ILC3 culture, are very consistent to show that the vitamin D/VDR signaling promotes the proliferation of LTi cells, but the effect on NKp46+ cells is inconsistent. That is, our data strongly suggest that the mechanism responsible for vitamin D/VDR regulation of ILC3-mediated innate immunity lies in its stimulation of proliferation of LTi0 and LTi4 cells, but not NKp46+ cells. Since NKp46+ and LTi cells are derived from different precursors (Artis and Spits, 2015), it is not surprising that the vitamin D/VDR activity has different effects on these cells. In fact, whether vitamin D regulates NKp46+ cells or not should not have a meaningful impact on vitamin D-regulated ILC3 immunity, as NKp46+ cells or NKp46-derived IL-22 only plays a minor role in innate immunity against C. rodentium (Cella et al., 2009). Therefore, given the importance of LTi cells, future studies should be focused on how vitamin D/VDR regulates LTi cell proliferation and LTi-mediated immunity.
IL-22 is a key cytokine produced by ILC3 that plays key roles in host defense against infection as well as in tissue remodeling and maintenance of mucosal barrier integrity (Artis and Spits, 2015, Bernink et al., 2013). The latter is overlapped with the mucosal barrier-protecting activity of the vitamin D/VDR signaling (Du et al., 2015, He et al., 2018, Liu et al., 2013). It is conceivable that vitamin D/VDR may also protect the integrity of the mucosal barrier through regulating IL-22 production from ILC3. Here our data suggest that vitamin D/VDR regulates IL-22 synthesis through controlling ILC3 proliferation, but it is also possible that VDR directly regulates IL-22 gene expression, as seen in other nuclear receptors such as RORγt+ and AHR (Qiu et al., 2012, Sawa et al., 2010). A most recent study reported that vitamin D/VDR downregulates IL-23 receptor-mediated pathway in human NKp44+ ILC3 (mouse NKp46+ equivalent), suppressing IL-22 expression (Konya et al., 2018). This in vitro observation in human ILC3 appears in contrast to our in vivo finding, but it is irrelevant because our studies have shown that vitamin D/VDR has little effects on NKp46+ cells in mice. The reason behind the discrepancy between humans and mice in terms of vitamin D regulation of NKp44+/NKp46+ cells is unclear.
There have been increasing interests in the anti-infectious activity of vitamin D. A good example is a study that demonstrated how vitamin D stimulates anti-microbial activity in macrophages against Mycobacterium tuberculosis (Liu et al., 2006). Here we show that vitamin D also stimulates potent innate immunity against C. rodentium infection via gut ILC3. The results from these investigations suggest that the vitamin D endocrine system has intrinsic anti-infectious and anti-bacterial activity to serve the host defense system. In this regard, maintaining appropriate vitamin D levels in the body is important for an effective host defense against infection.
Limitations of the Study
In this study we presented strong evidence that the vitamin D/VDR signaling is required for proper proliferation and function of ILC3, particularly the LTi0 and LTi4 cells at baseline and under pathogenic bacterial infection; however, exactly how the vitamin D/VDR signaling regulates ILC3 proliferation remains unclear. Future studies are needed to elucidate the underlying molecular mechanism. Moreover, germ-free models are needed to confirm the effect of gut microbiota on vitamin D regulation of ILC3.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank David Goltzman (McGill University) for providing the Cyp27b1 knockout mouse line and Wanyin Deng and B. Brett Finlay (University of British Columbia) for providing Citrobacter rodentium strains and technical assistance. This work was supported in part by National Institutes of Health grants CA180087 and NIDDK P30DK42086.
Author Contributions
Conceptualization: Y.C.L.; Methodology and Investigation: L.H. and M.Z.; Formal analysis: L.H. and Y.C.L.; Writing – Original Draft: L.H. and Y.C.L.; Writing – Review & Editing: Y.C.L.; Fund Acquisition: Y.C.L.; Supervision: Y.C.L.
Declaration of Interests
The authors declare no competing interests.
Published: July 26, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.06.026.
Supplemental Information
References
- Artis D., Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- Bernink J., Mjosberg J., Spits H. Th1- and Th2-like subsets of innate lymphoid cells. Immunol. Rev. 2013;252:133–138. doi: 10.1111/imr.12034. [DOI] [PubMed] [Google Scholar]
- Bouillon R., Carmeliet G., Verlinden L., van Etten E., Verstuyf A., Luderer H.F., Lieben L., Mathieu C., Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr. Rev. 2008;29:726–776. doi: 10.1210/er.2008-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouladoux N., Harrison O.J., Belkaid Y. The mouse model of infection with Citrobacter rodentium. Curr. Protoc. Immunol. 2017;119:19 15 11–19 15 25. doi: 10.1002/cpim.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M., Fuchs A., Vermi W., Facchetti F., Otero K., Lennerz J.K., Doherty J.M., Mills J.C., Colonna M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Waddell A., Lin Y.D., Cantorna M.T. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal. Immunol. 2015;8:618–626. doi: 10.1038/mi.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J., Chen Y., Shi Y., Liu T., Cao Y., Tang Y., Ge X., Nie H., Zheng C., Li Y.C. 1,25-Dihydroxyvitamin D protects intestinal epithelial barrier by regulating the myosin light chain kinase signaling pathway. Inflamm. Bowel Dis. 2015;21:2495–2506. doi: 10.1097/MIB.0000000000000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Qiu J., Tu T., Yang X., Deng L., Anders R.A., Zhou L., Fu Y.X. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. 2014;40:25–39. doi: 10.1016/j.immuni.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart P.H., Gorman S., Finlay-Jones J.J. Modulation of the immune system by UV radiation: more than just the effects of vitamin D? Nat. Rev. Immunol. 2011;11:584–596. doi: 10.1038/nri3045. [DOI] [PubMed] [Google Scholar]
- Haussler M.R., Whitfield G.K., Kaneko I., Haussler C.A., Hsieh D., Hsieh J.C., Jurutka P.W. Molecular mechanisms of vitamin D action. Calcif. Tissue Int. 2013;92:77–98. doi: 10.1007/s00223-012-9619-0. [DOI] [PubMed] [Google Scholar]
- He L., Liu T., Shi Y., Tian F., Hu H., Deb D.K., Chen Y., Bissonnette M., Li Y.C. Gut epithelial vitamin D receptor regulates microbiota-dependent mucosal inflammation by suppressing intestinal epithelial cell apoptosis. Endocrinology. 2018;159:967–979. doi: 10.1210/en.2017-00748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holick M.F. Vitamin D: photobiology, metabolism, mechanism of action, and clinical application. In: Favus M.J., editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Third Edition. Lippincott-Raven; 1996. pp. 74–81. [Google Scholar]
- Holick M.F. Vitamin D deficiency. N. Engl. J. Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- Holick M.F. Photobiology of vitamin D. In: Feldman D., editor. Vitamin D. Fourth Edition. Academic Press; 2018. pp. 45–55. [Google Scholar]
- Joshi S., Pantalena L.C., Liu X.K., Gaffen S.L., Liu H., Rohowsky-Kochan C., Ichiyama K., Yoshimura A., Steinman L., Christakos S. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol. Cell Biol. 2011;31:3653–3669. doi: 10.1128/MCB.05020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konya V., Czarnewski P., Forkel M., Rao A., Kokkinou E., Villablanca E.J., Almer S., Lindforss U., Friberg D., Hoog C. Vitamin D downregulates the IL-23 receptor pathway in human mucosal group 3 innate lymphoid cells. J. Allergy Clin. Immunol. 2018;141:279–292. doi: 10.1016/j.jaci.2017.01.045. [DOI] [PubMed] [Google Scholar]
- Koroleva E.P., Halperin S., Gubernatorova E.O., Macho-Fernandez E., Spencer C.M., Tumanov A.V. Citrobacter rodentium-induced colitis: a robust model to study mucosal immune responses in the gut. J. Immunol. Methods. 2015;421:61–72. doi: 10.1016/j.jim.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Lee S.M., Riley E.M., Meyer M.B., Benkusky N.A., Plum L.A., DeLuca H.F., Pike J.W. 1,25-Dihydroxyvitamin D3 controls a cohort of vitamin D receptor target genes in the proximal intestine that is enriched for calcium-regulating components. J. Biol. Chem. 2015;290:18199–18215. doi: 10.1074/jbc.M115.665794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire J.M., Adams J.S., Kermani-Arab V., Bakke A.C., Sakai R., Jordan S.C. 1,25-Dihydroxyvitamin D3 suppresses human T helper/inducer lymphocyte activity in vitro. J. Immunol. 1985;134:3032–3035. [PubMed] [Google Scholar]
- Liu P.T., Stenger S., Li H., Wenzel L., Tan B.H., Krutzik S.R., Ochoa M.T., Schauber J., Wu K., Meinken C. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- Liu W., Chen Y., Golan M.A., Annunziata M.L., Du J., Dougherty U., Kong J., Musch M., Huang Y., Pekow J. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J. Clin. Invest. 2013;123:3983–3996. doi: 10.1172/JCI65842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P., Iacomini J., Johnson R.S., Herrup K., Tonegawa S., Papaioannou V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Mora J.R., Iwata M., von Andrian U.H. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat. Rev. Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji A., Kobiita A., Ye T., Chambon P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell. 2013;153:812–827. doi: 10.1016/j.cell.2013.04.020. [DOI] [PubMed] [Google Scholar]
- Panda D.K., Miao D., Tremblay M.L., Sirois J., Farookhi R., Hendy G.N., Goltzman D. Targeted ablation of the 25-hydroxyvitamin D 1alpha -hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc. Natl. Acad. Sci. U S A. 2001;98:7498–7503. doi: 10.1073/pnas.131029498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna G., Adorini L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J. Immunol. 2000;164:2405–2411. doi: 10.4049/jimmunol.164.5.2405. [DOI] [PubMed] [Google Scholar]
- Penna G., Roncari A., Amuchastegui S., Daniel K.C., Berti E., Colonna M., Adorini L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood. 2005;106:3490–3497. doi: 10.1182/blood-2005-05-2044. [DOI] [PubMed] [Google Scholar]
- Petty N.K., Bulgin R., Crepin V.F., Cerdeno-Tarraga A.M., Schroeder G.N., Quail M.A., Lennard N., Corton C., Barron A., Clark L. The Citrobacter rodentium genome sequence reveals convergent evolution with human pathogenic Escherichia coli. J. Bacteriol. 2010;192:525–538. doi: 10.1128/JB.01144-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J., Heller J.J., Guo X., Chen Z.M., Fish K., Fu Y.X., Zhou L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S., Paglino J., Eslami-Varzaneh F., Edberg S., Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rigby W.F., Stacy T., Fanger M.W. Inhibition of T lymphocyte mitogenesis by 1,25-dihydroxyvitamin D3 (calcitriol) J. Clin. Invest. 1984;74:1451–1455. doi: 10.1172/JCI111557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryz N.R., Lochner A., Bhullar K., Ma C., Huang T., Bhinder G., Bosman E., Wu X., Innis S.M., Jacobson K. Dietary vitamin D3 deficiency alters intestinal mucosal defense and increases susceptibility to Citrobacter rodentium-induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015;309:G730–G742. doi: 10.1152/ajpgi.00006.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryz N.R., Patterson S.J., Zhang Y., Ma C., Huang T., Bhinder G., Wu X., Chan J., Glesby A., Sham H.P. Active vitamin D (1,25-dihydroxyvitamin D3) increases host susceptibility to Citrobacter rodentium by suppressing mucosal Th17 responses. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;303:G1299–G1311. doi: 10.1152/ajpgi.00320.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa S., Cherrier M., Lochner M., Satoh-Takayama N., Fehling H.J., Langa F., Di Santo J.P., Eberl G. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. 2010;330:665–669. doi: 10.1126/science.1194597. [DOI] [PubMed] [Google Scholar]
- Schauer D.B., Falkow S. The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect. Immun. 1993;61:4654–4661. doi: 10.1128/iai.61.11.4654-4661.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa A.K., Nagakubo D., Hieshima K., Nakayama T., Jin Z., Yoshie O. 1,25-Dihydroxyvitamin D3 induces CCR10 expression in terminally differentiating human B cells. J. Immunol. 2008;180:2786–2795. doi: 10.4049/jimmunol.180.5.2786. [DOI] [PubMed] [Google Scholar]
- Sonnenberg G.F., Artis D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity. 2012;37:601–610. doi: 10.1016/j.immuni.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg G.F., Monticelli L.A., Elloso M.M., Fouser L.A., Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer S.P., Wilhelm C., Yang Q., Hall J.A., Bouladoux N., Boyd A., Nutman T.B., Urban J.F., Jr., Wang J., Ramalingam T.R. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science. 2014;343:432–437. doi: 10.1126/science.1247606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K., Ogawa A., Mizoguchi E., Shimomura Y., Andoh A., Bhan A.K., Blumberg R.S., Xavier R.J., Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto F.L., Reardon C.A., Yoon D., Wang Y., Wong K.E., Chen Y., Kong J., Liu S.Q., Thadhani R., Getz G.S. Vitamin D receptor signaling inhibits atherosclerosis in mice. Mol. Endocrinol. 2012;26:1091–1101. doi: 10.1210/me.2011-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Essen M.R., Kongsbak M., Schjerling P., Olgaard K., Odum N., Geisler C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat. Immunol. 2010;11:344–349. doi: 10.1038/ni.1851. [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhu J., DeLuca H.F. Where is the vitamin D receptor? Arch. Biochem. Biophys. 2012;523:123–133. doi: 10.1016/j.abb.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Wolk K., Kunz S., Witte E., Friedrich M., Asadullah K., Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Zheng Y., Valdez P.A., Danilenko D.M., Hu Y., Sa S.M., Gong Q., Abbas A.R., Modrusan Z., Ghilardi N., de Sauvage F.J. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.