

SUMMARY
Elucidation of the sequence of events underlying the dynamic interaction between transcription factors and chromatin states is essential. Maternal transcription factors function at the top of the regulatory hierarchy to specify the primary germ layers at the onset of zygotic genome activation (ZGA). We focus on the formation of endoderm progenitor cells and examine the interactions between maternal transcription factors and chromatin state changes underlying the cell specification process. Endoderm-specific factors Otx1 and Vegt together with Foxh1 orchestrate endoderm formation by coordinated binding to select regulatory regions. These interactions occur before the deposition of enhancer histone marks around the regulatory regions, and these TFs recruit RNA polymerase II, regulate enhancer activity, and establish super-enhancers associated with important endodermal genes. Therefore, maternal transcription factors Otx1, Vegt, and Foxh1 combinatorially regulate the activity of super-enhancers, which in turn activate key lineage-specifying genes during ZGA.
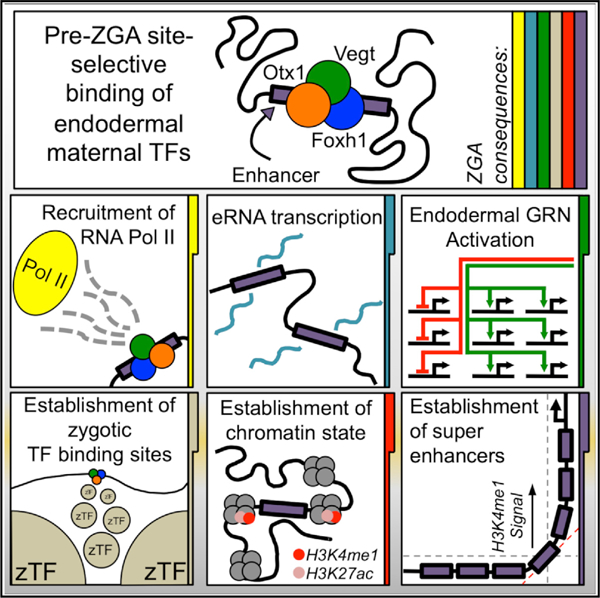
Graphical Abstract
In Brief
How do maternal transcription factors interact with chromatin regions to coordinate the endodermal gene regulatory program? Paraiso et al. demonstrate that combinatorial binding of maternal Otx1, Vegt, and Foxh1 to select enhancers and super-enhancers in the genome controls endodermal cell fate specification during zygotic gene activation.
INTRODUCTION
Metazoan development begins with a single totipotent cell that gives rise to numerous cell types, each expressing lineage-restricted sets of genes. The activation of gene transcription in the embryo relies on maternal transcription factors (TFs), which sit high in the regulatory hierarchy to coordinate the gene regulatory cascades that lead to the stereotypical development of embryos. Zygotic gene activation (ZGA) is a major regulatory event in which control of the zygotic genome is transferred from maternal to zygotic TFs (Jukam et al., 2017). While the timing of the transition differs between species, all undergo ZGA, which is controlled by maternal inputs. With the exception of amniotes, the processes of germ layer specification are coupled to ZGA and depend on unequally distributed maternal determinants present in the egg before fertilization (Minakhina and Steward, 2005). These maternal inputs are the first to specify the differentiation of germ layer cell types.
Maternal TFs presumably coordinate the actions of enhancers, which are cis-regulatory modules (CRMs) that dock numerous TFs, to regulate the activity of gene promoters. Since TFs drive lineage-specific transcription programs by binding CRMs dispersed throughout the genome, a major question that remains to be addressed is how maternal TFs bind specific regions in the chromatin to endow the transcriptional responses that initiate germ layer specification, patterning, and subsequent differentiation. We address this fundamentally important biological question by going back to the earliest stages of embryonic development, when transcription from the embryonic genome has not yet begun, the number of different cell types is small, and the genome appears relatively naive.
Genomic studies of human and mouse embryonic stem cells have suggested a role for the epigenetic priming of CRMs and promoter regions (Calo and Wysocka, 2013; Kim et al., 2018). Priming of these sites is thought to play important roles in the deployment of gene regulatory networks (GRNs). However, examination of the chromatin states of early Xenopus, zebrafish, and Drosophila embryos have not been able to find epigenetic signatures that are frequently associated with active or repressive enhancer states. In Xenopus, chromatin immunoprecipitation (ChIP)-qPCR reveals that histone H3R8me2 activating marks appear on sia1 and nodal3, expressed before the major wave of ZGA (Blythe et al., 2010), but the majority of enhancer chromatin marks appear to arise during the major ZGA phase (Akkers et al., 2009; Gupta et al., 2014; Hontelez et al., 2015). Therefore, before ZGA, cleavage, and early blastula stage, chromatin is free of both the activating enhancer marks H3K4me1 and H3K27ac and permissive H3K4me3, as well as repressive H3K27me3 histone marks. Similarly, in zebrafish, neither H3K4me3 nor H3K27me3 were detected before ZGA; and a study of a larger cohort of histone modifications in Drosophila, which includes H3K4me1, H3K4me3, and H3K27me3, showed that epigenetic states are largely established post-ZGA (Vastenhouw et al., 2010; Li et al., 2014). Furthermore, we previously demonstrated that the maternally expressed forkhead-domain TF Foxh1 binds to the genome during cleavage stages pre-ZGA, before the appearance of activating enhancer marks, suggesting that Foxh1 selectively pre-marks CRMs on mesendodermal genes (Charney et al., 2017a). These findings raise an important question about how active CRMs are chosen and activated during germ layer specification by maternal TFs.
In Xenopus, a subset of maternal RNAs are specifically enriched animally (future ectoderm) or vegetally (future endoderm) in the egg, and these factors are subsequently asymmetrically inherited by different blastomeres. Specification of the endodermal germ layer is regulated by the maternal T-box TF Vegt (Stennard et al., 1996; Lustig et al., 1996; Zhang and King, 1996; Horb and Thomsen, 1997; Zhang et al., 1998; Clements et al., 1999; Xanthos et al., 2001), which sits at the top of the hierarchy of both endodermal and mesodermal gene regulatory cascades (Charney et al., 2017b). These experiments show that Vegt is capable of activating both mesodermal and endodermal genes, implying that other TFs are involved in restricting the function of Vegt to endoderm gene activation in the vegetal pole cells fated to this lineage. This raises important questions. What are the other maternal TFs involved in endoderm specification? How do these TFs interact to activate selective enhancers to coordinate the endodermal gene regulatory program? Here, we report that Otx1 and Vegt act together with Foxh1 to co-bind endoderm CRMs harboring binding motifs for these 3 TFs before the onset of ZGA and to initiate the recruitment of RNA polymerase II (Pol II) to begin endodermal differentiation programs. CRMs pre-marked by Otx1, Vegt, and Foxh1 binding (OVF-CRMs) subsequently serve as docking sites for numerous other zygotically active TFs, including Foxa, Smad2/3, and Gsc. Furthermore, OVF-CRMs tend to cluster near endodermal genes, forming super-enhancers (SEs), which are activated during ZGA. Our findings are consistent with the view that early line-age specification is driven by the coordinated binding of critical maternal TFs to selective OVF-CRMs or CRM clusters that regulate lineage-specific gene expression. This results in the recruitment of epigenetic regulators that modify the chromatin template to further facilitate the assembly of transcriptional machinery as development proceeds.
RESULTS
RNA-Seq Screen for Maternal Regulators of Endodermal Fate
We wished to identify maternal TFs that may play roles in endodermal specification in the early embryo. Therefore, we examined maternal transcript localization in cleavage stage Xenopus tropicalis embryos before the onset of ZGA (Figure 1A). Eight-cell embryos were chosen, as this is the earliest stage at which animal and vegetal hemispheres are segregated into blastomeres by an equatorial cleavage plane and ZGA has not occurred. RT-qPCR and RNA sequencing (RNA-seq) analysis on these samples showed the expected vegetal localization of vegt and gdf1/vg1 RNAs, as well as the animal localization of foxi2 RNA (Figures S1A–S1D). We identified 309 genes that are differentially expressed between the animal and vegetal cells (Figure S1E; Table S1). Functionally, these genes are consistent with the biological processes occurring during early embryogenesis as suggested by Gene Ontology (GO) analysis, revealing terms such as “developmental process” and “multicellular organism development” (germ layer formation); “lipid biosynthetic process” (yolk localization in putative endodermal cells); “cytoskeleton organization” and “microtubule-based process” (animal-vegetal RNA microtubule-based translocation); and “developmental process involved in reproduction” (germ plasm localization in the vegetal pole) (Figure S1F). While hundreds of TFs are expressed in the egg (Blitz et al., 2017), only a small fraction are expressed in a regionalized manner: 9 TFs are enriched vegetally, while 4 TFs are enriched animally (Figure 1B). The vegt and otx1 genes are among the most highly expressed vegetally and are the most differentially expressed (>8-fold) between vegetal and animal blastomeres (Figure 1C). Our findings are consistent with previous RNA-seq-based screens for localized factors, which showed the vegetal enrichment of otx1 (De Domenico et al., 2015; Owens et al., 2017). Previously, otx1 transcripts were detected in the vegetal pole of oocytes (Pannese et al., 2000); however, their expression pattern in early embryos has not been well described. Consistent with our RNA-seq, whole-mount in situ hybridization shows that otx1 is expressed in the vegetal mass (presumptive endoderm) and is excluded from the embryonic marginal region (presumptive mesoderm) (Figures 1D–1F). While Otx1 has not yet been implicated in vertebrate germ layer formation, echinoderm (Hinman et al., 2003; Peter and Davidson, 2010) and ascidian (Wada and Saiga, 1999) Otx TFs control endodermal specification and are similarly expressed in a variety of deuterostomes (Figure S2). We therefore hypothesized that Otx1 is an important regulator of endoderm formation in Xenopus, perhaps functioning together with maternal Vegt.
Figure 1. Screen for Core Maternal Endodermal TFs.

(A) Dissection of six to eight 8-cell stage embryos in 3 biological replicates at 2 h post-fertilization (hpf) to separate animal and vegetal blastomeres for RNA sequencing.
(B) Log2-transformed transcripts per million (TPM) expression of TFs in animal and vegetal blastomeres. Differentially expressed TFs are highlighted in green (vegetal) or orange (animal).
(C) Log2-transformed fold change (animal over vegetal) of expression levels of localized TFs.
(D–F) In situ hybridization showing otx1 expression (cells in blue) in the vegetal mass cells (presumptive endoderm) during early blastula (D), late blastula (E), and early gastrula (F).
Vegt and Otx1 Combinatorially Specify the Endodermal Transcriptome
To gain insight into the role of Otx1 in the endoderm, we expressed Otx1 in animal cap cells (presumptive ectoderm) and assayed for the induction of endodermal genes by RT-qPCR at early gastrula stage 10.5 (Figure 2A). Otx1 is sufficient to activate endodermal markers such as nodal, mixer, and darmin, in addition to foxa2, which is expressed in both endodermal and mesodermal layers.
Figure 2. Vegt and Otx1 Are Dual Function TFs in the Endoderm.

(A) Ectopic expression of otx1 in animal caps shows the induction of endodermal genes by RT-qPCR. dicer1 was used for normalization. Shown is a single representative biological replicate out of 3. Error bars represent variation from 3 technical replicates.
(B) Single and combined ectopic expression of 100 pg otx1 mRNA and 100 pg vegt mRNA shows synergistic and antagonistic co-regulation by RT-qPCR. dicer1 was used for normalization. Shown is a single representative biological replicate out of 3. Error bars represent variation from 3 technical replicates.
(C) Combinatorial ectopic expression of vegt and otx1 in the animal cap using dosage titration to assay for similarly co-regulated genes by RT-qPCR. Yellow diamond indicates the subthreshold concentration of vegt, with which titrating doses of otx1 mRNA are co-injected. RNA doses are expressed in picograms. dicer1 was used for normalization. Shown is a single representative biological replicate out of 3. Error bars represent the variation from 3 technical replicates.
(D) Two biological replicates from (C) were subjected to RNA-seq. Genes that are similarly co-regulated as nodal, mixer, and fgf20 were identified by correlating gene expression in TPM using the Pearson correlation coefficient. Plotted is the spatial expression pattern in the gastrula stage of genes that are positively co-regulated (nodal type and mixer type) or negatively co-regulated (fgf20-type) by vegt and otx1.
(E and F) Vegt and Otx1 morpholinos were injected independently or together to vegetal masses at the 1-cell stage, and vegetal masses were assayed by RNA-seq at stage 10 in biological duplicates. The Venn diagrams show genes that are downregulated (E) or upregulated (F) in different conditions, where regulation is identified as a >2-fold change in gene expression compared to uninjected control.
(G and H) Fold change expression of downregulated (G) or upregulated (H) genes in the RNA-seq datasets in TPM over uninjected control, along with a set of 300 randomly sampled genes. Significance was determined using the Student’s t test.
(I) Expression of mesodermal (fgf20, mespa) genes and endodermal (foxa1, hnf1b) genes in the morpholino RNA-seq experiment in TPM.
(J) Gastrula stage spatial expression of genes that are downregulated and upregulated in the double morpholino experiment, along with a set of 300 randomly sampled genes. TPM is in log2 scale.
Next, we tested whether Otx1 and Vegt collaborate, positively and/or negatively, to regulate endodermal gene expression. We microinjected a single dose of vegt or otx1 mRNA or a cocktail of these 2 mRNAs into the animal pole at the 1-cell stage and assayed for endodermal and mesodermal gene expression in dissected animal caps. As expected, Otx1 and Vegt alone act as activators of the endodermal genes nodal and mixer (Figure 2B). Combinatorially, Otx1 and Vegt additively activate the expression of mixer. We find a strong synergistic induction of nodal in the presence of both mRNAs. Conversely, Otx1 strongly downregulates the induction of the mesodermally expressed gene fgf20 caused by Vegt (Lea et al., 2009). We then performed dose-response experiments, in which a subthreshold concentration of vegt mRNA was kept constant and increasing doses of otx1 were added (Figure 2C). Increasing doses of Otx1 induced nodal and mixer expression, with nodal being more sensitive to Otx1 dose than mixer. fgf20 was strongly repressed even at the lowest Otx1 dose. This finding also proposes a dual function for maternal Otx1-promoting endoderm development vegetally, while repressing mesodermal development in the endoderm by blocking the expression of fgf20.
We examined the genome-wide interaction between Otx1 and Vegt in regulating mesendodermal genes by using the same vegt and otx1 mRNA titration conditions followed by RNA-seq analysis. To identify similarly regulated genes, we searched our RNA-seq expression profiles for the dose-response patterns of nodal, mixer, and fgf20. We identified the 50 most highly correlated genes with similar regulation to the patterns of nodal, mixer, and fgf20 by ranking Pearson correlation coefficients (Table S2). Among the genes that are regulated similarly to the nodal and mixer types are tbx3, lefty, lhx1, snai1, cer, and mix1, which show enriched expression in the endoderm of the early gastrula. Among the fgf20-type regulated genes are the mesodermally expressed genes fgf4 and wnt11b. The gastrula stage expression pattern of nodal and mixer type genes are consistently strongest in the endoderm, while the fgf20-type genes are most strongly expressed in the mesoderm (Figure 2D).
To assess the combinatorial roles of endogenous Otx1 and Vegt, we blocked their activities by injecting translation-blocking antisense morpholino oligonucleotides (MOs) against vegt (Rana et al., 2006) and otx1 (Figures S3A and S3B) into embryos, either independently or in combination. Vegetal masses were dissected at gastrula stage 10.5, followed by RNA-seq. Single knockdowns (KDs) of Vegt or Otx1 affected the expression of 275 and 203 genes, respectively, based on a 2-fold cutoff criterion (Figures 2E and 2F). However, the expression of a much larger cohort of 376 genes is affected in the presence of both MOs. Furthermore, genes in the latter category are influenced by independent KDs of vegt or otx1, although the effect is not as strong as in the double MO (Figures 2G and 2H). This suggests that a 2-fold cutoff is too stringent to identify some Vegt or Otx1 targets and that a double MO KD is useful for detecting the combinatorially regulated genes. Among the genes that are affected are the mesodermally expressed genes fgf20 and mespa, which are both inhibited by Vegt and Otx1 (Figure 2I). The endodermally expressed genes foxa1 and hnf1b are activated by both Vegt and Otx1 (Figure 2I). Spatially, the Vegt and Otx1 negatively co-regulated genes tend to be expressed ectodermally or mesodermally, whereas positively co-regulated genes tend to be expressed endodermally (Figure 2J). Overall, our experiments support the notion that maternal Vegt and Otx1 function in a combinatorial manner to drive the proper expression of endodermal genes while minimizing the expression of genes for alternative cell fates.
Vegt and Otx1 Co-bind to Endodermal cis-Regulatory Modules before the Onset of ZGA
We previously showed that the ubiquitously expressed Foxh1 binds to the genome before ZGA and before the appearance of promoter-bound Pol II and H3K4me1 and Ep300 enhancer marks (Charney et al., 2017a). We therefore asked whether the endoderm-specific TFs Vegt and Otx1 also interact with the genome before the onset of ZGA. Chromatin immunoprecipitation coupled with deep sequencing (ChIP-seq) was used to investigate Vegt and Otx1 binding at early blastula stage 8. The quality of Vegt (Sudou et al., 2012) and Otx1 antibodies was verified (Figures S3C and S3D). We identified 5,151 and 21,711 biologically reproducible Otx1- and Vegt-bound regions, respectively. De novo motif analysis of the bound sequences identified a variation of the Bicoid-type homeodomain (TAATCCCY) (Driever et al., 1989) and T-box half-site motifs (TCACACCT) (Conlon et al., 2001) for Otx1 and Vegt, respectively (Figure S4). Both Vegt and Otx1 bind to mesodermal and endodermal genes such as nodal6, sox17b, mix1, mixer, fgf20, gsc, and hhex, and their binding regions are highly overlapping (Figures 3A and 3B). The set of Vegt and Otx1 overlapping peaks represents 64% of Otx1 peaks and 18% of Vegt peaks. Simulation of a set of peaks of similar size to the Otx1 peak set across the genome shows an average overlap of 56 (1.1%) peaks over 1,000 trials with the 21,711 Vegt peaks, suggesting that the overlap between Vegt and Otx1 chromatin binding is statistically significant (p < 2.2e-16).
Figure 3. Vegt and Otx1 Bind to the Chromatin near Mesendodermal Genes Pre-ZGA.

(A) Venn diagram of Otx1 and Vegt biologically reproducible peak overlaps.
(B) Genome browser view of Otx1 and Vegt ChIP-seq near endodermal (nodal6, sox17b.1, sox17b.2, mixer, mix1), mesodermal (fgf20), and mesendodermal (gsc, hhex) genes.
(C) Genomic annotation of the location of all Vegt, all Otx1, and Vegt+Otx1 overlapping peaks.
(D) Gastrula stage expression pattern of genes associated with all Vegt, all Otx1, and Vegt+Otx1 overlapping peaks. Genes were assigned as “endoderm,” “mesoderm,” or “ectoderm,” based on which germ layer showed the highest expression in TPM.
(E and F) Chromatin binding of Vegt (E) and Otx1 (F) near genes involved in germ layer formation identified using ChIP-qPCR on 32-cell stage embryos. eef1a1 and ins promoters were used as negative control. The error bars represent the variation from 3 technical replicates.
We then examined the distribution of peaks relative to genomic features. Approximately 1.1% of the entire X. tropicalis genome represents a promoter-proximal sequence (within 1 kb upstream of transcription start sites [TSSs]), ~25% is intronic and ~70% is intergenic (Figure 3C). Vegt and Otx1 binding is enriched 2-to 3-fold in promoter-proximal regions and ~1.3-fold in intronic regions at the expense of intergenic regions. Despite this enrichment, >90% of Vegt and Otx1 binding resides in the intronic and intergenic regions, suggesting binding to enhancers over promoters. By assigning these binding regions to the nearest genes, we find that Vegt and Otx1 tend to bind to endodermal and mesodermal genes, rather than ectodermal genes (Figure 3D). Furthermore, by identifying genes with an adjacent ChIP binding peak that are affected by independent morpholino KD from the RNA-seq experiments, we were able to identify a set of 74 Vegt and 21 Otx1 direct target genes (Table S3). By looking at genes affected by the Vegt/Otx1 double MO and that are bound by both TFs, we identified 47 genes that are combinatorially directly regulated by Vegt and Otx1, including the endodermally expressed genes gata4, foxa1, and pnhd.
Next, we asked whether Vegt and Otx1 can engage chromatin during the cleavage stages by ChIP-qPCR on 32-cell stage embryos, 3–4 cell cycles pre-ZGA (Figures 3E and 3F). Strong binding of endogenous Vegt and Otx1 was detected during the pre-ZGA stages on the regulatory regions of mesodermal and endodermal genes. These results, combined with similar observations on Foxh1 (Charney et al., 2017a), suggest that maternal TFs engage chromatin during the transcriptionally quiescent cleavage stages, before the establishment of enhancer marks.
Maternal Otx1 and Vegt Assemble on cis-Regulatory Regions Together with Foxh1
A de novo motif analysis of the regions bound by Otx1 and Vegt was used to identify a set of candidate maternal TFs that co-regulate genes with Otx1 and Vegt. This analysis (Figure S4) identified their respective motifs and the Fox motif. This finding suggests that Vegt and Otx1 co-bind in the genome together with the ubiquitously expressed TF Foxh1. A comparison of Otx1 and Vegt peaks to Foxh1 peaks at blastula stage 8 shows that >70% of Otx1 peaks and >25% of Vegt peaks overlap with Foxh1 peaks (Figures 4A and 4B). To ensure co-occupancy in the same endodermal cells, we performed sequential ChIP-qPCR. ChIP was performed using the Otx1 antibody, followed by dissociation, and then a second round of ChIP was performed using Vegt, Foxh1, or FLAG (as a negative control) antibodies. Regions bound by Otx1 are positive for Foxh1 and Vegt binding, but not FLAG (Figures 4B and 4C). Co-occupancy possibly favors the binding of TFs on these CRMs over other regions of the genome. To test this model, Foxh1 expression was knocked down using a morpholino (Chiu et al., 2014) and assessed as to whether Otx1 and Vegt binding requires the presence of Foxh1. In the absence of Foxh1, the association of both Otx1 and Vegt in multiple regions is decreased (Figure 4D). These observations suggest that these 3 maternal TFs co-occupy CRMs in the genome as an assembly.
Figure 4. Otx1, Vegt, and Foxh1 Form an Assembly in the Chromatin.

(A) Venn diagram of Otx1, Vegt, and Foxh1 biologically reproducible peak overlaps.
(B) Genome browser view of Otx1, Vegt, and Foxh1 binding, highlighting 4 regions of peak overlaps near the bpm7.2, pou5f3.3, fgf20, and nodal6 genes.
(C) Sequential ChIP-qPCR using anti-Otx1 followed by anti-FLAG, anti-Vegt, or anti-Foxh1 antibody in regions of peak overlap highlighted in (B). ins and id3 promoters were used as negative controls. The error bars represent variation from 3 technical replicates.
(D) Foxh1 morpholino KD followed by Otx1 or Vegt ChIP-qPCR performed on stage 8 embryos. eef1a1 and odc1 promoters were used as negative control. Shown is a single representative biological replicate. The error bars represent the variation from 3 technical replicates.
Pre-marking of Endodermal CRMs by Maternal Otx1, Vegt, and Foxh1 Assembly
As ChIP-seq identified thousands of possible regulatory regions, we wished to determine which TF bound sites are active. We hypothesized that the Otx1, Vegt, and Foxh1 co-bound CRMs (OVF-CRMs) correspond to functional cis-regulatory modules. We looked at whether these sites are potential binding sites for zygotically endodermally active TFs Ctnnb1/β-catenin, Foxa, Gsc, Otx2, and Smad2/3 (Chiu et al., 2014; Yasuoka et al., 2014; Nakamura et al., 2016; Charney et al., 2017a). OVF-CRMs flanking endodermally expressed genes such as foxa1, pnhd, and admp show extensive co-binding of zygotic TFs (Figure 5A). Genome-wide analysis supports this finding, as ChIP-seq signals of zygotic TFs overlap with the location of OVF-CRMs (Figure 5B). Consistent with our hypothesis, CRMs bound by either 1- or 2-way combinations of Otx1, Vegt, or Foxh1 showed weaker zygotic TF ChIP read density than OVF-CRMs (Figures 5B and S5A). These observations suggest that the maternally established OVF-CRMs are preferential sites for subsequent assembly of zygotically active TF recruitment.
Figure 5. Combinatorial Otx1, Vegt, and Foxh1 Binding Pre-marks Zygotic cis-Regulatory Modules.

(A) Genome browser of maternal Otx1, Vegt, and Foxh1 binding with Pol II binding, mesendodermal zygotic TF binding (b-catenin, Foxa, Gsc, Otx2, and Smad2/3), and enhancer marks (Ep300, H3K27ac, and H3K4me1) near the genes foxa4, pnhd, and admp.
(B) Zygotically active TF ChIP-seq signal of b-catenin, Foxa, Gsc, Otx2, and Smad2/3 in OVF-CRMs and 1- to 2-TF CRMs during the gastrula stage.
(C) Heatmap of H3K4me3 promoter mark ChIP-seq signal in OVF-CRMs, 1–2 TF CRMs, and gene promoters as a positive control.
(D–F) Heatmap of enhancer marks H3K4me1 (D), Ep300 (E), and H3K27ac (F) ChIP-seq signal in OVF-CRMs and 1-to 2-TF CRMs.
(G) Fold change of Pol II ChIP-qPCR signal at gastrula stage 10.5 from animal caps ectopically expressing otx1 and vegt mRNA assaying for association with OVF-CRMs, compared to uninjected animal caps. Shown is a single representative biological replicate. The error bars represent the variation from 3 technical replicates.
(H) Fold change of Pol II ChIP-qPCR signal at stage 10.5 from vegetal masses in Otx1, Vegt, and Foxh1 triple morpholino KD conditions assaying for association with OVF-CRMs, compared to uninjected vegetal masses. Shown is a single representative biological replicate. The error bars represent the variation from 3 technical replicates.
(I) RT-qPCR assay for eRNA transcription from OVF-CRMs in uninjected, and otx1 and vegt mRNA injected animal caps at stage 10.5. Genomic DNA contamination is quantified using an RT-qPCR control reaction with no reverse transcriptase (–RT). Shown is a single representative biological replicate. The error bars represent the variation from 3 technical replicates.
The experiments for (G)–(I) were repeated at least twice.
While CRMs that act as enhancers typically display epigenetic marks, in early frog, fish, and Drosophila embryos, enhancer marks are not present until post-ZGA (Gupta et al., 2014; Hontelez et al., 2015; Vastenhouw et al., 2010; Li et al., 2014). We therefore wished to determine whether OVF-CRMs are subsequently decorated with the enhancer marks H3K4me1, H3K27ac, and Ep300 (Gupta et al., 2014; Hontelez et al., 2015). As a negative control, when H3K4me3 was plotted in regions centered around maternal TFs, we were unable to detect enrichment of this promoter mark (Figure 5C). In contrast, the general enhancer mark H3K4me1 correlates well with the OVF-CRMs (Figure 5D). This finding is consistent with the earlier finding that Otx1 and Vegt tend to bind in intergenic and intronic regions of the genome. The general enhancer mark H3K4me1 is absent at stage 8, while Otx1, Vegt, and Foxh1 binding occurs well before this stage (Figure 3). Our results show that the H3K4me1 mark is largely established in OVF-CRMs by stage 9 and that this mark appears to persist through later embryonic developmental stages (Figure 5D). Ep300 peaks are not detectable before stage 8 (Hontelez et al., 2015) and become detectable after blastula stage 9, after ZGA has begun. Post-ZGA Ep300 displays a strong signal at the regions of maternal TF co-binding (Figure 5E). The H3K27ac active enhancer mark is weakly detectable at stages 8 and 9; however, the bimodal distribution pattern of this mark around the OVF-CRMs becomes more pronounced at stage 10 (Figure 5F). These marks are particularly enriched in the OVF-CRMs when compared to other regions having either only 1 or 2 of Otx1, Vegt, or Foxh1 binding (Figures 5D–5F and S5B). These datasets, taken together with the detection of Otx1, Vegt, and Foxh1 binding as early as the 32-cell stage (Figures 3E and 3F), suggest that maternal TF assembly pre-marks OVF-CRMs and that OVF-bound CRMs function at later developmental stages as enhancer elements that recruit other zygotically expressed TFs.
Otx1, Vegt, and Foxh1 Regulate Pol II Recruitment to OVF-CRMs
To further demonstrate the enhancer regulatory function of OVF-CRMs, we examined the recruitment and activity of Pol II on these elements. Active enhancers are known to interact, via DNA looping, with promoters of their target genes and to recruit Pol II to their promoters. Therefore, Pol II should be detectable on active enhancer regions. We therefore hypothesized that Otx1, Vegt, and Foxh1 regulate Pol II recruitment to OVF-CRMs near endodermal genes that are regulated by these TFs. Ectopic expression of Otx1 and Vegt in animal caps (Foxh1 is ubiquitously expressed) increased Pol II association with OVF-CRMs (Figure 5G). Conversely, simultaneous morpholino KD of Otx1, Vegt, and Foxh1 drastically reduces Pol II association to these regions in vegetal tissue (Figure 5H), indicating that the OVF TFs form an active complex with Pol II.
Enhancer RNAs (eRNAs) represent a class of relatively short non-coding RNA molecules transcribed from the DNA sequence of enhancer regions. The expression of eRNAs is frequently correlated with the chromatin marks of active enhancers (Kim et al., 2010; Lam et al., 2014). We therefore tested whether OVF-CRMs produce non-coding eRNAs, By ectopic expression of Otx1 and Vegt in animal caps, we detected increased levels of eRNA transcription from OVF-CRMs in comparison to uninjected control animal caps (Figure 5I), suggesting that OVF-CRMs respond to Otx1 and Vegt inputs. Overall, based on zygotic TF binding, epigenetics changes, Pol II recruitment, and eRNA expression, we conclude that OVF-CRMs are active CRMs regulated by Otx1, Vegt, and Foxh1.
Endodermal Super Enhancers Are Epigenetically Distinct from Regular Enhancers and Are Hubs of TF Binding Sites
During our analysis of endodermally enriched maternal TF binding, we noticed that not only do these maternal TFs co-bind but also their CRMs tend to form clusters near endodermally expressed genes. This CRM clustering is reminiscent of super-enhancers (SEs), a cluster of enhancers with unusually high epigenetic marking established near key cell identity genes (Lové n et al., 2013; Whyte et al., 2013). From a gene regulatory standpoint, this class of enhancers is particularly appealing as (1) they are much more likely than regular enhancers (REs) to regulate their closest neighboring gene and (2) they are stronger activators of gene expression than REs (Whyte et al., 2013). Due to these special properties of SEs, we wished to explore whether OVF TFs associate with endodermal SEs established during germ layer formation. To identify SEs, we performed H3K4me1 ChIP-seq at the gastrula stage on endodermal cells from dissected vegetal masses. After read mapping and peak calling, we obtained 18,380 peaks. We excluded peaks in the promoter regions, and the remaining 15,836 peaks were used for SE analysis. We used previously described protocols to distinguish between REs and SEs (Lové n et al., 2013; Whyte et al., 2013).
From this analysis, we identified 441 SEs (Table S4) and 7,468 REs (Figure 6A). As expected, SEs are much longer (Figure 6B) and contain more constituent enhancers than REs (Figure 6C). The embryonic endodermal SEs were identified near genes among which code for regulators of endodermal cell fate genes such as admp, pnhd, foxa4, sox17, hhex, frzb, gata6, and foxa2 (Figures 6D and S6A).
Figure 6. Otx1, Vegt, and Foxh1 Establish SEs near Key Endodermal Genes.

(A) Categorization of H3K4me1 enhancer regions as regular enhancers (REs) and super-enhancers (SEs) using the rank order of super-enhancers (ROSE) algorithm based on H3K4me1 signal.
(B) Length of RE and SE in kilobases.
(C) Number of constituent enhancers (H3K4me1 peaks) within REs and SEs.
(D) Genome browser view of SEs in the pnhd, admp, foxa4, and sox17 locus, which are populated with Otx1, Vegt, and Foxh1 ChIP-seq peaks.
(E) Comparison of epigenetic marks in REs and SEs focusing on heterochromatin, enhancer, and polycomb marks. Plotted is the fold change of the median ChIP read density in SE over RE.
(F) Maternal TF ChIP read density in REs and SEs.
(G) Overlap of 1-TF, 2-TF, or 3-TF CRMs (OVF-CRMs) peaks with the H3K4me1-identified REs or SEs.
(H) GO analysis of genes associated with SEs.
(I) Temporal expression of genes associated with REs and SEs, along with randomly selected genes, normalized to the gene expression at 0 hpf.
(J) Gastrula stage spatial expression of genes associated with REs and SEs, along with randomly selected genes in log2(TPM).
(K) Log2-fold change of gene expression in Vegt + Otx1 MO and Foxh1 MO over uninjected control of genes associated with REs and SEs, along with randomly selected genes.
Significance for (E) and (F) was determined using the Wilcoxon rank-sum test by comparing the signals in the set of SEs (N = 441) to the set of REs (N = 7,468). The significance for (K) was determined using the Wilcoxon rank-sum test whereby the number of random genes was sampled to the number of SE genes (N = 490) from the set of genes not associated with REs or SEs. The number of RE genes was sampled down to the number of SE genes.
Epigenetically, SEs are distinct from REs as they contain a much stronger signal of the H3K4me1 mark. To compare the epigenetics between SEs and REs, we used published bulk embryo datasets: H3K4me1, Ep300, and H3K27ac as markers of enhancers; H3K27me3, Jarid, and Ezh2 as polycomb markers; and H3K9me2 and H4K20me3 as markers of hetero-chromatin. Our data support the idea that these SEs are important regulators of cell fate specification as they are enriched with enhancer marks across early development (Figures 6E and S6B). In addition, they are enriched for polycomb marks, which suggest that SEs, while acting as cis-activators of gene expression, could be used for repression in alternative cell fates. On the contrary, heterochromatin marks ChIP read densities are similar between REs and SEs.
As we previously identified Otx1, Vegt, and Foxh1 as regulators of endodermal fate, we asked whether these maternal TFs are important in the establishment of the endodermal SEs. By comparing the ChIP read density, we find that all 3 maternal TFs are enriched in SEs (Figure 6F). In particular, Foxh1 appears to have the strongest enrichment, suggesting a key role for Foxh1 in establishing SEs in the endoderm. We then asked whether combinations of TF binding are critical in establishing SEs. We compared the SE and RE occupancy of OVF-CRMs, 2-TF bound CRMs (2-way combinations of Otx1, Vegt, and Foxh1), and 1-TF bound CRMs. OVF-CRMs are much more likely to overlap with H3K4me1 peaks and are enriched in both REs and SEs (Figure 6G). In addition to maternal TF binding, SEs are also enriched for endodermally active zygotic factors (Figure S6C). These analyses suggest that endodermal SEs are epigenetically distinct from REs and are hubs for endodermal maternal and zygotic TF binding.
SEs Are Associated with Endodermally Expressed Genes Activated during ZGA
While SEs are located near known endodermal genes (Figures 6D and S6A), we wanted to obtain a whole-genome view of genes regulated by endodermal SEs (Table S4). By performing GO analysis on the set of 490 genes associated with SEs, we obtained terms related to embryonic development and TF activity, suggesting that these genes are regulators of cell fate (Figure 6H). In general, these genes are activated during ZGA (between 4 and 5 hours post-fertilization), unlike genes associated with REs or randomly selected genes that are not associated with REs or SEs (Figure 6I). Genes associated with SEs tend to be more highly expressed spatially than RE-associated genes or random genes, regardless of germ layer tissue expression. While RE-associated genes tend to be similarly expressed across the 3 germ layers, the SE-associated genes are enriched in the endoderm relative to the ectoderm (Figure 6J). SE-associated gene expression is also enriched in the mesoderm over the ectoderm, which is consistent with the appearance of genes expressed in both the mesoderm and endoderm such as foxa2 and foxa4. Overall, our analyses suggest that SEs are established near genes coding for the key regulators of endodermal fate activated during ZGA.
Otx1, Vegt, and Foxh1 Regulate the Expression of SE-Associated Genes
While SEs are likely regulated by numerous TFs (Lovén et al., 2013; Whyte et al., 2013), we wondered whether the maternal TFs Otx1, Vegt, and Foxh1 are required for the regulation of genes associated with endodermal SEs. Genes that are associated with SEs are downregulated by the combinatorial KD of Otx1 and Vegt (Figure 6K). Similarly, Foxh1 KD (Chiu et al., 2014) also decreased the expression of SE-associated genes. On the contrary, genes associated with REs do not show a statistically significant downregulation in any of the KD experiments. Our analysis suggests that Otx1, Vegt, and Foxh1 are highly active in SE regions and are important regulators of the genes associated with these regions.
DISCUSSION
Here, we identify a role for maternal Otx1 in vertebrate endodermal specification and explore its relation with 2 other maternal TFs, Vegt and Foxh1. Based on our findings, we propose the following model. The genome in early embryonic cells is compact, relatively inaccessible, and largely lacking in the chromatin marks associated with enhancer activation (or repression) until ZGA. Otx1, Vegt, and Foxh1 together gain entry to the chromatin and bind to CRMs at least as early as the 32-cell stage. Since these TFs can bind to DNA target sites, presumably in relatively closed chromatin (Almouzni and Wolffe, 1995; Amodeo et al., 2015), before the activation of enhancers and gene expression modulation, these maternal TFs act as pioneer TFs that initiate the establishment of functional cis-regulatory regions in the embryo (Zaret and Carroll, 2011). Following the initial gene activation, these CRMs would remain functional platforms for TF recruitment, as they are later occupied by zygotically expressed TFs produced shortly after ZGA. Enhancer marks appear around the onset of ZGA and strengthen thereafter, suggesting that these marks are deposited in response to gene activation or repression rather than regulating maternal TF association to the genome. The highest signal for these enhancer marks occurs on CRMs that are clustered, forming SEs around developmentally relevant genes, and these SEs are enriched in Otx1, Vegt, and Foxh1 binding. The roles of maternal TFs in regulating zygotic gene expression are likely to be many. At least one role is the enhanced recruitment of Pol II (leading to eRNA expression). It is tempting to speculate that maternal TFs also recruit histone-modifying factors to establish the earliest zygotic chromatin states at and after the onset of ZGA.
Interplay of Maternal TFs and Chromatin States
The sequence of events leading to the appearance of open chromatin, epigenetic marks, and TF binding is under intensive investigation. The early metazoan embryonic (pre-ZGA) genome lacks enhancer marks, and both permissive and repressive histone modifications (H3K4me3 and H3K27me3, respectively) are established during late blastula and gastrula stages (Akkers et al., 2009; Gupta et al., 2014; Hontelez et al., 2015; Vastenhouw et al., 2010; Li et al., 2014). We find that combinatorial binding of the maternal TFs Otx1, Foxh1, and Vegt occurs pre-ZGA and before the appearance of H3K4me1, Ep300, and Pol II recruitment. This suggests that chromatin binding by maternal TFs is not controlled by epigenetics, but rather by TF binding preferences for DNA motifs influenced by protein-protein interactions on CRMs.
The maternal TFs may also be involved in the recruitment of the writers of epigenetic marks. An example of epigenetic writer recruitment by TFs during Xenopus dorsal-ventral patterning is the facilitation by Ctnnb1/β-catenin of Prmt2 (protein arginine methyltransferase 2) binding to the sia1 regulatory region (Blythe et al., 2010). Prmt2 then deposits H3R8me2 marks, resulting in the transcriptional activation of sia1. Inhibition of zygotic transcription through α-amanitin treatment showed that maternal factors are involved in the majority of H3K4me3 promoter marks and H3K27me3 polycomb repressive marks and a subset of Ep300 recruitment through late gastrula stages (Hontelez et al., 2015). While it is unclear how these maternal TFs interact with chromatin modifiers, we believe that they are the major drivers of site-selective chromatin modifications in the early embryo. Thus, a better understanding of the interplay between the maternal TFs and chromatin modifiers will be essential to comprehend the molecular mechanism of ZGA.
While our evidence suggests that the pioneering activity of maternal TFs is critical for establishing epigenetic states and Pol II function, it is possible that the same maternal TFs are important for establishing open chromatin (Zaret and Carroll, 2011). Early Xenopus embryos contain high levels of nucleosomes, which results in relatively close chromatin, and these nucleosomes are therefore viewed as repressors of gene expression pre-ZGA (Almouzni and Wolffe, 1995; Amodeo et al., 2015). The basal TF Tbp, which recruits Pol II to gene promoters, competes with the nucleosomes to initiate gene transcription (Prioleau et al., 1994). Maternal TFs possibly perform a similar function in enhancer regions where clusters of DNA binding motifs are located. This mechanism has been observed during ZGA in the case of maternally expressed Zelda in Drosophila (Foo et al., 2014; Schulz et al., 2015) and Pou5f3 in zebrafish (Joseph et al., 2017), whereby these TFs are required for chromatin opening in their respective binding sites. This is a testable mechanism using techniques such as assay for transposase-accessible chromatin sequencing (ATAC-seq) and DNase-seq, and whether Xenopus maternal TFs open chromatin is a critical question that must be addressed to understand the full repertoire of maternal TF function.
Enhancers and SEs in Development
SEs have been understudied in the context of early embryogenesis. Via interrogation of genome-wide binding, we find numerous regions co-occupied by the maternal TFs Otx1, Vegt, and Foxh1, suggesting that these factors collaborate to regulate gene behavior. While maternal TFs bind to tens of thousands to hundreds of thousands of sites in the genome, we find that the co-bound regions are functional regulatory regions by epigenetic marking and Pol II activity. Regions where these TFs co-bind (OVF-CRMs) are subsequently decorated with epigenetic enhancer marks and become docking sites for numerous zygotically active TFs. We find that a set of SEs that are marked with an unusually high signal of active chromatin marks and zygotic TF binding are clustering around OVF-CRMs and are associated with endodermal specification genes activated during ZGA. It is tempting to speculate that the TFs Otx1, Vegt, and Foxh1 establish a paradigm for the assembly of SEs by maternal TFs, suggesting that maternally controlled SEs regulate key zygotic germ layer-specifying genes during early embryonic development. While we have so far identified an assembly of three maternal TFs on enhancers and SEs, other maternal TFs are likely to be involved in this process. Some candidate maternally expressed TFs include vegetally localized Sox7 (Figure 1; Owens et al., 2017; De Domenico et al., 2015) and ubiquitously expressed TFs such as Zic2, Sox3, and Pou5f3/Oct60, motifs of which are enriched under Vegt and Otx1 peaks (Figure S5). Although the precise roles of SEs are not well established, the constituent enhancers within these enhancer clusters could function through the combinations of gene expression amplitude modulation and the specification of correct spatial and temporal localization in early embryonic tissues. Sea urchin endo16 (Yuh et al., 1998) and Drosophila eve (Goto et al., 1989; Harding et al., 1989) are two well-studied early developmental genes that provide examples for complex regulation by clusters of enhancers, and thus may have similar SE features.
Otx1 in the Context of Endodermal Evo-Devo
How does the role of Otx1 in early frog embryos apply to the endoderm in other organisms? Otx genes are most well known for their roles in embryonic anterior specification, a function that pre-dates the evolutionary split between deuterostomes and protostomes. The only functional studies on otx genes in the initial specification steps of endoderm come from echinoderms, where vegetally localized otx activates the expression of multiple endodermal genes (Hinman et al., 2003; Peter and Davidson, 2010), and ascidians, where otx represses mesodermal derivatives (Wada and Saiga, 1999). Therefore, we mined publically available in situ hybridization and transcriptomic datasets to determine whether otx1 orthologs are expressed maternally and vegetally. Otx1 orthologs seem to play a role in endoderm formation across deuterostomes, with the exception of amniote embryos. Our analysis shows that Otx1 directly activates gata4 and foxa1 in Xenopus (Table S3), similar to the activation of gatae and foxa orthologs by Otx in the sea urchin (Peter and Davidson, 2010), suggesting the conservation of gene regulatory networks. While much is known about the expression pattern of Otx1 homologs, further functional analysis is needed to uncover their roles in diverse endodermal gene regulatory programs.
Dual Function of Lineage-Specific TFs
By combining ChIP-seq peaks with the findings from the KD experiments, we identified a set of Vegt-regulated, Otx1-regulated, and co-regulated genes during zygotic gene activation (Table S3), further expanding our understanding of the endodermal gene regulatory program (Charney et al., 2017b). From our list, both Vegt and Otx1 appear to act as direct activators of endodermal gene expression and repressors of mesodermal gene expression.
The function of maternal Vegt has been extensively characterized as an activator of endodermal and mesodermal genes (Stennard et al., 1996; Lustig et al., 1996; Zhang et al., 1998; Clements et al., 1999; Xanthos et al., 2001). However, when we analyzed RNA expression in the vegetal mass of Vegt morphants, mesodermal genes were induced, corroborating the previous finding that the putative mesoderm shifts to the vegetal mass in Vegt KDs (Zhang et al., 1998). Combined with the chromatin binding, our data support a model whereby maternal Vegt acts as a direct repressor of mesodermal genes vegetally. This finding poses a novel inhibitory role for Vegt and suggests that Vegt acts as a dual function TF. This would add Vegt to the list of T-box TFs, which can act as repressors. The list includes mouse Tbx20 (Sakabe et al., 2012), Xenopus Tbx3 (He et al., 1999), zebrafish Tbx24 (Kawamura et al., 2008), Caenorhabditis elegans Tbx2 (Milton and Okkema, 2015), and Drosophila Midline (Formaz-Preston et al., 2012). Mechanistically, the repressive roles of Tbx20, Tbx24, and Midline have been attributed to the interaction with the general co-repressor Tle/Groucho (Kawamura et al., 2008; Formaz-Preston et al., 2012; Kaltenbrun et al., 2013). Alternatively, the switch from the activator to the repressive role of Tbx2 has been attributed to the small ubiquitin-like modifier (SUMO)ylation of this protein (Milton and Okkema, 2015). These mechanisms from other T-box family members present some attractive hypotheses in studying the possible mechanism for the repressive function of Vegt.
The role of maternal Otx1 in germ layer specification has not previously been investigated. We identified otx1 mRNA as vegetally localized, which is consistent with previous studies on Xenopus oocytes and early embryos (De Domenico et al., 2015; Owens et al., 2017; Pannese et al., 2000), and sought to understand its role in endoderm specification. Otx1 collaborates with Vegt to activate endodermal gene expression and to repress mesoderm formation. Repression of mesodermal gene expression appears to occur at least in part through Otx1 downregulation of mesodermal Fgf signaling, which inhibits endodermal formation in both Xenopus and zebrafish (Cha et al., 2004, 2008; Poulain et al., 2006). The mechanism for toggling the dual functions of Otx1 on different genes remains unclear, but 2 mechanisms have been proposed. In the first, activation versus repression depends on pairing with other co-bound TFs: Otx2 together with Lhx1 function as an activator, while Otx2 together with Gsc functions as a repressor (Yasuoka et al., 2014). A second mechanism suggests that the phosphorylation of Otx2 plays a role in its repressor function (Satou et al., 2018) to modulate binding Tle/Groucho (Puelles et al., 2004). Since these phosphorylation sites are also conserved in Otx1, it is possible that the dual role of Otx1 may be similarly regulated.
In addition to Otx1 and Vegt, Foxh1 has been shown to be a dual function TF (Chiu et al., 2014; Reid et al., 2016). In this study, we have focused on the relation between these maternal TFs and activating enhancer marks. However, it is presently unclear how these TFs inhibitory functions lead to the establishment of repressive chromatin marks. Future work examining the relation between these maternal TFs and chromatin modifying enzymes involved in both activation and repression is needed.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ken W.Y. Cho (kwcho@uci.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Xenopus tropicalis males and females were obtained from NASCO (University of Virginia stock) or raised in the laboratory; and were maintained in accordance with the University of California, Irvine Institutional Animal Care Use Committee (IACUC). X. tropicalis females were injected with 10 units of Chorulon HCG (Merck and Co.) 1–3 nights before embryo collection and 100 units of HCG on the day of embryo collection. Eggs were collected in a dish coated with 0.1% BSA in 1/9x MMR. The eggs were in vitro fertilized with sperm suspension in 0.1% BSA in 1/9x MMR (Ogino et al., 2006). The embryos were dejellied with 3% cysteine in 1/9x MMR, pH 7.8, 10 minutes after fertilization and were then ready for manipulation. Embryos were staged using the Nieukwoop-Faber developmental table (Nieuwkoop and Faber, 1958; Khokha et al., 2002; Owens et al., 2016). Microinjections and microdissection of Xenopus embryos are described below specific for each experiment.
METHOD DETAILS
RNA ectopic expression (animal caps)
The Xenopus tropicalis vegt and otx1 open reading frames were cloned into the BamHI site of the pCS2+ vector using Gibson cloning (Gibson et al., 2009). Similarly, the Otx1 open reading frame was cloned into the BamHI site of pCS2+3xFLAG vector to generate an epitope-tagged Otx1. The plasmids were then digested with HpaI and mRNA was generated using by in vitro transcription using mMessage mMachine SP6 Transcription Kit (Thermo Fisher Scientific). Ectopic expression was performed by injecting in two opposite sites in the animal cap region at 1–2 cell stage. For experiments that required no embryo fixation, animal caps were dissected an hour prior to harvest. For embryos that were fixed, whole embryos were fixed at the stage of harvest (see ChIP assays and analysis), and animal caps were dissected after fixation.
Morpholino knockdown and rescue (vegetal masses)
otx1 translation blocking morpholino was designed by GeneTools, Inc., against X. tropicalis otx1 (50-ATGACATCATGCTCAAGGCTG GACA-3′). The vegt (5′-TGTGTTCCTGACAGCAGTTTCTCAT-3′) (Rana et al., 2006) and the foxh1 (Sequence: 5′-TCATCCTGAG GCTCCGCCCTCTCTA-3′) (Chiu et al., 2014) translation blocking morpholinos were previously tested. otx1 morpholino rescue was performed using the FLAG-otx1 construct. Morpholino knockdown was performed by injecting directly into the vegetal pole at 1-cell stage. For experiments that required no embryo fixation, vegetal masses were dissected an hour prior to harvest. For embryos that were fixed, whole embryos were fixed at the stage of harvest (see ChIP assays and analysis), and vegetal masses were dissected after fixation.
Whole mount in situ hybridization
The protocol of Harland (1991) with modifications as previously described (Blitz and Cho, 1995) was used. Template for otx1 riboprobes was obtained following PCR amplification from cDNA prepared by Smart-seq2 (Picelli et al., 2014) reverse transcription. PCR product contained bacteriophage T3 and T7 promoters (underlined below) for the synthesis of sense and antisense probe, respectively. The forward and reverse primers for amplification were:
5′-GCAGCAATTAACCCTCACTAAAGGTTCAGCGGGGTGGATTGCAG-3′
5′-GCAGCTAATACGACTCACTATAGGacacagggacaaacagagccaa-3′.
RNA assays
RNA isolation: RNA was collected from embryo and embryo fragments as previously described (Chomczynski and Sacchi, 1987). RT-qPCR Primers: See Table S5. RT-qPCR: RNA samples were reverse transcribed using the MMLV reverse transcriptase. qPCR was performed using Roche Lightcycler 480 II using the Roche SYBR green I master mix with the default SYBR green protocol. RNA-seq: RNA-seq libraries were generated using Smart-seq2 cDNA synthesis followed by tagmentation (Picelli et al., 2014), quality-tested using an Agilent Bioanalyzer 2100, quantified using KAPA qPCR and sequenced using Illumina sequencers at the UC Irvine Genomics High Throughput Facility. Enhancer RNA Primers: See the ChIP-qPCR section of Table S5. Enhancer RNA detection: After RNA was isolated, as described above, the resuspended nucleic acids were treated with TURBO DNase at 37°C for 30 minutes to eliminate genomic DNA contamination. RNA was then isolated by phenol/chloroform extraction and isopropanol precipitation. RNA samples were then subjected to reverse transcription, along with a no reverse transcriptase (-RT) control, and qPCR was performed as described above.
Chromatin immunoprecipitation (ChIP) assays
Antibodies: The anti-Vegt (Sudou et al., 2012), anti-Foxh1 (Chiu et al., 2014), anti-Pol II (Bio-Legend MMS-126R; Charney et al., 2017a), and anti-H3K4me1 (Abcam; Gupta et al., 2014; Hontelez et al., 2015) antibodies were previously tested. The rabbit polyclonal anti-Otx1 peptide antibody was generated (GenScript USA Inc.) against the peptide sequence GYTGTGLPFNSSDC (AA 288–301 of the Xenopus tropicalis Otx1 protein). ChIP: ChIP on X. tropicalis embryos was performed as previously described (Chiu et al., 2014). ChIP-qPCR Primers: See Table S5. ChIP-qPCR: qPCR was performed using Roche Lightcycler 480 II using the Roche SYBR green I master mix with the default SYBR green protocol. ChIP-seq: ChIP-seq libraries were generated using Nextflex ChIP-seq kit (Bioo Scientific), analyzed using an Agilent Bioanalyzer 2100, quantified using KAPA qPCR and sequenced using Illumina instruments at the UC Irvine Genomics High Throughput Facility. Sequential ChIP: The initial steps of ChIP were performed as previously described. After incubation with the first antibody, elution was performed using 1x TE, pH 8.0, with 10mM DTT, 500 mM NaCl and 0.1% SDS at 37°C for 30 minutes. After the incubation, the eluate was diluted 10x with RIPA. The diluted eluate was incubated with the second antibody and the rest of the ChIP was performed as previously described (Geisberg and Struhl, 2004).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA analysis
RT-qPCR: Fold change in gene expression was calculated using the ΔΔCp approach and the error among technical replicates was calculated using error propagation and the first approximation of the Taylor expansion. RNA-seq: Reads were aligned using RSEM v.1.2.12 (Li and Dewey, 2011) and Bowtie 2 v2.2.7 (Langmead and Salzberg, 2012) to the Xenopus tropicalis genome version 9.0 (Hellsten et al., 2010; Karimi et al., 2018), generating expression values in transcripts per million (TPM) and normalized read counts. Differential Expression: Differential expression of genes in normalized read counts between samples was performed using the EBseq v1.8.0 (Leng et al., 2013) function EBTest on R v3.1.0 (R Core Team, 2014). Pattern matching for similarly regulated genes: In the combinatorial ectopic expression of Vegt and Otx1 experiment followed by RNA-seq, we first identified genes that were differentially expressed compared to the uninjected control in any of the treatments. From this set of genes, we pattern matched the expression in TPM with the TPM expression of nodal, mixer and fgf20 with the cor function in R using the Pearson method to identify similarly expressed genes. We then ranked the correlation metric of the entire genome to each of the patterns and selected the 50 most correlated genes (R Core Team, 2014). Gastrula stage gene expression analysis: Gastrula expression in TPM in five tissue fragments (i.e., animal cap, ventral marginal zone, lateral marginal zone, dorsal marginal zone, and vegetal mass) were obtained from NCBI GEO accession number GSE81458 (Blitz et al., 2017), and the gastrula stage expression pattern of gene lists were identified using these TPM values. Animal cap expression was used for ectoderm, vegetal mass expression was used for endoderm and the average expression of ventral-, lateral- and dorsal-marginal zones was used for mesoderm. Genes that are maternally-expressed (TPM > 1 in 0 hpf of the time course RNA-seq (Owens et al., 2016), which could mask spatial expression pattern of zygotic genes, are removed from the analysis. For assignment of genes to specific germ layers (Figure 3D), we assigned each gene to the germ layer by which it has the strongest gene expression in TPM. Time course gene expression analysis: Gene expression from a ribosomal RNA depleted RNA-seq time-course were obtained from NCBI GEO accession number GSE65785 (Owens et al., 2016). The expression in TPM was generated by aligning the reads to the X. tropicalis genome as outlined above, and temporal expression of gene lists were identified using these TPM values. Enhancer RNA detection: Genomic DNA at 2.5, 25 and 250 pg was assayed using the same qPCR primers to generate a standard curve. The eRNA levels were quantified from the genomic DNA standard curve. The error among technical replicates was calculated using error propagation and the first approximation of the Taylor expansion.
Chromatin immunoprecipitation analysis
ChIP-qPCR (percent input): ChIP DNA using 1 embryo equivalent, and ChIP input DNA at 0.1, 0.01 and 0.001 embryo equivalents assayed in triplicates by qPCR. Percent input of ChIP DNA was calculated by generating a linear model of input DNA embryo equivalents and qPCR Cp values. ChIP-qPCR (fold-change): When comparing ChIP-qPCR signal between two conditions (knockdown and ectopic expression conditions compared to uninjected wild-type as seen in Figures 4D, 5G–H), fold change in ChIP signal was calculated using the ΔΔCp approach and the error among technical replicates was calculated using error propagation and the first approximation of the Taylor expansion. The normalization control region is indicated in each figure in gray with a fold change of 1. ChIP-seq peak calling and IDR: Reads were aligned to the X. tropicalis genome v9.0 (Hellsten et al., 2010; Karimi et al., 2018) using Bowtie 2 v2.2.7 (Langmead and Salzberg, 2012) with default options, and peaks were called against stage 8 input DNA (Charney et al., 2017a) using MACS2 v2.0.10 (Zhang et al., 2008) with the option -p 0.001 but otherwise default options. ENCODE based irreproducibility discovery rate (IDR) was performed using the following p value thresholds for the following comparisons: 0.01 for original biological replicates, 0.02 for pseudoreplicates of each biological replicate and 0.0025 for pseudoreplicates generated from pooled reads of biological replicates (Li et al., 2011). Peak location annotation: Peaks were annotated as ‘promoter’, ‘intron’, ‘exon’, ‘intergenic’ or ‘transcription termination site (TTS)’ using HOMER v4.7 using the function annotatePeaks.pl (Heinz et al., 2010). Motif analysis: Motif analysis was performed using DREME (Bailey, 2011) and matching identified motifs to databases was performed using TOMTOM (Gupta et al., 2007). Genome browser visualization: Using Samtools v0.1.19, aligned reads in SAM format were converted to BAM format (samtools view -bS) and duplicates were removed (samtools rmdup) (Li et al., 2009). Bedtools v2.19.1 (Quinlan and Hall, 2010) was used to convert BAM files to BED format. IGVtools functions sort (default options) and count (-w 25 -e 250) were used to generate the WIG or TDF files loaded into IGV v2.3.20 (Robinson et al., 2011). Accession of published X. tropicalis ChIP-seq datasets: Datasets were downloaded from NCBI GEO using the accession numbers: GSE41161 for Jarid and Ezh2 stage 9 (van Heeringen et al., 2014); GSE85273 for Pol II and stage 10 Foxa (Charney et al., 2017a); GSE67974 for H3K9me2, H4K20me3, H3K4me1, Ep300 and H3K4me3 (Hontelez et al., 2015); GSE56000 for H3K4me1 and H3K27ac (Gupta et al., 2014); GSE72657 for stage 10 Ctnnb1/β-catenin (Nakamura et al., 2016); and GSE53654 for stage 10 Smad2/3 (Chiu et al., 2014). The stage 10 Gsc and Otx2 ChIP-seq, and their respective input DNA datasets were obtained from the DDBJ Sequence Read Archive using the accession numbers DRA000508, DRA000510, DRA000576 and DRA000577 (Yasuoka et al., 2014). ChIP signal heatmaps: The ChIP signal of histone modifications, Ep300, and TFs near Vegt, Otx1 and Foxh1 peaks were generated as follows. Using the TF summit file generated by MACS2 after peak calling, 2500 bp was added to the 5′ and 3′ sides of the summit to generate a 5000 bp peak window using Python v2.6.6. Heatmaps of ChIP signal were then generated against these locations where first, the ChIP datasets were processed as in the ‘Genome browser visualization’ section all the way through the IGVtools sort function (see above). After sorting, the BED files from this analysis were overlaid into the TF ChIP 5000 bp peak window using the Bedtools v2.19.1 (Quinlan and Hall, 2010) function coverageBed. The coverageBed output is pre-processed to generate ChIP signal in matrix form, and the signal matrix is plotted in R v3.1.0 using the heatmap function (R Core Team, 2014). Comparison of ChIP read density: The ChIP datasets were processed as in the ‘Genome browser visualization’ section all the way through the IGVtools sort function (see above). The total number of ChIP reads under peaks was quantified using the Bedtools v2.19.1 (Quinlan and Hall, 2010) function coverageBed and was then normalized to the length of each peak. This was defined as the ChIP read density. The ChIP read density between two populations of peaks were compared using the Wilcoxon rank sum test. Super enhancer identification: The sequences from the H3K4me1 ChIP-seq biological duplicates were concatenated into one file, which was then used for the super enhancer analysis. The H3K4me1 reads were aligned to the X. tropicalis genome v9.0 (Hellsten et al., 2010; Karimi et al., 2018) using Bowtie 2 v2.2.7 (Langmead and Salzberg, 2012) with default options, and then peak called against the stage 10 input DNA (Charney et al., 2017a) using MACS2 v2.0.10 (Zhang et al., 2008) with the cutoff of p value < 10−9. The peaks that correspond to promoters were then excluded for further analysis. These regions were defined as peaks that overlap with 500 bp upstream or downstream of the transcription start site. Super enhancers were then identified using previously published methods (Lovén et al., 2013; Whyte et al., 2013) where first, the peaks were stitched while allowing for 12.5 kilobases of distance in-between. The H3K4me1 ChIP signal within the constituent enhancers inside these stitched regions were quantified as the total number of reads per million. These regions are then ranked, and the point where the slope of the H3K4me1 ChIP signal curve hits 45° was the point that distinguished ‘regular enhancers’ from ‘super enhancers’ using the Ranking of Super Enhancers (ROSE) algorithm. Gene ontology: Gene ontology was performed using The Gene Ontology consortium online tool assaying for enrichment for biological processes or molecular function (Ashburner et al., 2000; The Gene Ontology Consortium, 2017)
Maternal gene expression of non-Xenopus otx genes
Most early expression and functional information of otx genes were readily available from the following references: Wasp/Nasonia vitripennis (Lynch et al., 2006); Flour beetle/Tribolium castaneum (Schröder, 2003); Fruit fly/Drosophila melanogaster (Levin et al., 2016); Sponge/Amphimedon queenslandica (Levin et al., 2016); Sea anemone/Nematostella vectensis (Helm et al., 2013); Star-fish/Asterina miniata (Hinman et al., 2003); Sea urchin/Strongylocentrotus purpuratus (Mitsunaga-Nakatsubo et al., 1998; Peter and Davidson, 2010); Acorn worm/Ptychodera flava (Harada et al., 2000); Sea pineapple/Halocynthia roretzi (Wada et al., 1996; Wada and Saiga, 1999); Ciona/Ciona savignyi (Satou et al., 2001); Ciona/Ciona intestinalis (Matsuoka et al., 2013); Amphioxus/Branchiostoma belcheri (Yang et al., 2016); Skate/Leucoraja erinacea (Suda et al., 2009); Bichir/Polypterus senegalus (Suda et al., 2009); Fugu/Fugu niphobles (Suda et al., 2009); Goby/Leucopsarion petersii (Kamimoto et al., 2003); Zebrafish/Danio rerio (Mori et al., 1994; Aanes et al., 2011); Axolotl/Ambystoma mexicanum (Jiang et al., 2017); Chick/Gallus gallus (Hwang et al., 2018); and Human/Homo sapiens (Wu et al., 2018). For those not readily available, the early embryonic RNA-seq datasets were obtained from NCBI GEO using the accession numbers GSE22830 for zebrafish (Aanes et al., 2011) and GSE86592 for chick (Hwang et al., 2018) and were aligned to their respective genomes (Howe et al., 2013; Warren et al., 2017) using RSEM v.1.2.12 (Li and Dewey, 2011) and Bowtie 2 v2.2.7 (Langmead and Salzberg, 2012).
DATA AND SOFTWARE AVAILABILITY
Raw and processed RNA-seq and ChIP-seq datasets generated for this study are available at NCBI Gene Expression Omnibus using the accession GEO: GSE118024.`
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Pol II 8WG16, ascites | Bio-Legend | MMS-126R |
| Mouse monoclonal anti-Flag | Sigma-Aldrich | Cat#F3165 |
| Rabbit polyclonal anti-Vegt | Sudou et al., 2012 | N/A |
| Rabbit polyclonal anti-Foxh1 | Chiu et al., 2014 | N/A |
| Rabbit polyclonal anti-Otx1, designed against the peptide sequence GYTGTGLPFNSSDC (AA 288–101 of the Xenopus tropicalis Otx1 protein) | GenScript; This Paper | N/A |
| Rabbit polyclonal anti-H3K4me1 | Abcam | Cat#ab8895 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dynabeads Protein G | Life Technologies | Cat#10003D |
| Critical Commercial Assays | ||
| mMessage mMachine Sp6 Transcription Kit | Thermo Fisher Scientific | Cat#AM1340 |
| SYBR Green I Master | Roche | Cat#04707516001 |
| NEXTflex ChIP-seq kit | Bioo Scientific | Cat#NOVA-5143–01 |
| Superscript II | Life Technologies | Cat#18064014 |
| KAPA HiFi HotStart ReadyMix (2x) | Kapa Biosystems | Cat#KK2601 |
| Agencourt AMPure XP beads | Beckman Coulter | Cat#A63881 |
| Nextera DNA Library Prep Kit | Illumina | Cat#FC-121–1030 |
| TURBO DNase | Thermo Fisher | Cat#AM2238 |
| Deposited Data | ||
| X. tropicalis RNA-seq and ChIP-seq | This Paper | GEO:GSE118024 |
| X. tropicalis genome version 9.0 | Hellsten et al., 2010; Karimi et al., 2018 | RRID:SCR_003280; URL: http://www.xenbase.org/entry/ |
| X. tropicalis gastrula stage (Stage 10.5) dissected fragments, RNA-seq | Blitz et al., 2017 | GEO:GSE81458 |
| X. tropicalis wild type embryo temporal profiling, RNA-seq | Owens et al., 2016 | GEO:GSE65785 |
| X. tropicalis Stage 8 ChIP-seq input DNA | Charney et al., 2017a | GEO:GSE85273 |
| X. tropicalis Foxh1 Stage 8, ChIP-seq | Charney et al., 2017a | GEO:GSE85273 |
| X. tropicalis Pol II Stage 7–10, ChIP-seq | Charney et al., 2017a | GEO:GSE85273 |
| X. tropicalis Foxa Stage 10, ChIP-seq | Charney et al., 2017a | GEO:GSE85273 |
| X. tropicalis H3K9me2 Stage 9–30, ChIP-seq | Hontelez et al., 2015 | GEO:GSE67974 |
| X. tropicalis H4K20me3 Stage 9–30, ChIP-seq | Hontelez et al., 2015 | GEO:GSE67974 |
| X. tropicalis H3K4me1 Stage 9–30, ChIP-seq | Hontelez et al., 2015 | GEO:GSE67974 |
| X. tropicalis Ep300 Stage 8–30, ChIP-seq | Hontelez et al., 2015 | GEO:GSE67974 |
| X. tropicalis H3K4me3 Stage 8–30, ChIP-seq | Hontelez et al., 2015 | GEO:GSE67974 |
| X. tropicalis H3K4me1 Stage 8–10, ChIP-seq | Gupta et al., 2014 | GEO:GSE56000 |
| X. tropicalis H3K27ac Stage 8–10, ChIP-seq | Gupta et al., 2014 | GEO:GSE56000 |
| X. tropicalis Ctnnb1 Stage 10, ChIP-seq | Nakamura et al., 2016 | GEO:GSE72657 |
| X. tropicalis Smad2/3 Stage 10, ChIP-seq | Chiu et al., 2014 | GEO:GSE53654 |
| X. tropicalis Gsc Stage 10, ChIP-seq | Yasuoka et al., 2014 | DRA:DRA000576; DRA000577 |
| X. tropicalis Otx2 Stage 10, ChIP-seq | Yasuoka et al., 2014 | DRA:DRA000508; DRA000510 |
| X. tropicalis Jarid Stage 9, ChIP-seq | van Heeringen et al., 2014 | GEO:GSE41161 |
| X. tropicalis Ezh2 Stage 9, ChIP-seq | van Heeringen et al., 2014 | GEO:GSE41161 |
| X. tropicalis Foxh1 MO, RNA-seq | Chiu et al., 2014 | GEO:GSE53654 |
| Experimental Models: Organisms/Strains | ||
| X. tropicalis, out-bred Nigerian | University of Virginia, NASCO | URL: https://www.enasco.com/ |
| Oligonucleotides | ||
| Template switching oligo | Picelli et al., 2014 | N/A |
| ISPCR primers | Picelli et al., 2014 | N/A |
| Indexing primers | Buenrostro et al., 2013 | N/A |
| Primers for RT-qPCR and ChIP-qPCR (See Table S5) | Integrated DNA Technologies | N/A |
| Otx1 MO 5′- ATGACATCATGCTCAAGGCTGGACA-3′ | GeneTools; This Paper | N/A |
| Vegt MO 5′-TGTGTTCCTGACAGCAGTTTCTCAT-3′ | GeneTools; Rana et al., 2006 | N/A |
| Foxh1 MO 5′-TCATCCTGAGGCTCCGCCCTCTCTA-3′ | GeneTools; Chiu et al., 2014 | N/A |
| Recombinant DNA | ||
| pCS2+otx1 (X. tropicalis otx1 ORF) | This paper | N/A |
| pCS2+vegt (X. tropicalis vegt ORF) | This paper | N/A |
| pCS2+3xFLAG-otx1 (X. tropicalis otx1 ORF) | This paper | N/A |
| Software and Algorithms | ||
| RSEM v.1.2.12 | Li and Dewey, 2011 | RRID:SCR_013027; URL: http://deweylab.biostat.wisc.edu/rsem/ |
| Bowtie 2 v2.2.7 | Langmead and Salzberg, 2012 | RRID:SCR_016368; URL: http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| EBseq v1.8.0 | Leng et al., 2013 | RRID:SCR_003526;URL: https://www.biostat.wisc.edu/~kendzior/EBSEQ/ |
| R v3.1.0 | R Core Team, 2014 | RRID:SCR_001905; URL:http://www.r-project.org/ |
| MACS2 v2.0.10 | Zhang et al., 2008 | RRID:SCR_013291;URL: https://github.com/taoliu/MACS |
| Irreproducibility Discovery Rate | Li et al., 2011 | URL: https://sites.google.com/site/anshulkundaje/projects/idr |
| HOMER v4.7 | Heinz et al., 2010 | RRID:SCR_010881;URL: http://homer.ucsd.edu/homer/ |
| DREME | Bailey, 2011 | RRID:SCR_001783; URL: http://meme-suite.org/ |
| TOMTOM | Gupta et al., 2007 | RRID:SCR_001783; URL: http://meme-suite.org/ |
| Samtools v0.1.19 | Li et al., 2009 | RRID:SCR_002105; URL: http://samtools.sourceforge.net/ |
| Bedtools v2.19.1 | Quinlan and Hall, 2010 | RRID:SCR_006646; URL: https://github.com/arq5x/bedtools2 |
| IGV v2.3.20 | Robinson et al., 2011 | RRID:SCR_011793; URL: http://software.broadinstitute.org/software/igv/ |
| Sratoolkit v2.8.1 | NCBI NIH | URL: https://www.ncbi.nlm.nih.gov/sra/docs/toolkitsoft/ |
| Gene Ontology | Ashburner et al., 2000; The Gene Ontology Consortium, 2017 | RRID:SCR_002143;URL: http://geneontology.org/ |
| Rank Ordering of Super-Enhancers (ROSE) | Lové n et al., 2013; Whyte et al., 2013 | URL: http://younglab.wi.mit.edu/super_enhancer_code.html |
Highlights.
Otx1 and Vegt drive endoderm fate as dual-function transcription factors (TFs)
Otx1, Vegt, and Foxh1 co-occupy enhancers and control zygotic gene activation (ZGA)
TF co-occupancy precedes chromatin dynamics in enhancers and super-enhancers
Super-enhancer-associated genes are endodermal factors activated during ZGA
ACKNOWLEDGMENTS
This work was made possible, in part, through access to the Genomic High-Throughput Facility Shared Resource of the Cancer Center Support Grant (P30CA-062203) at the University of California, Irvine, and NIH shared instrumentation grants 1S10RR025496-01, 1S10OD010794-01, and S10OD021718-01. We thank Xenbase for genomic and community resources (http://www.xenbase.org/entry, RRID: SCR_003280), in addition to their bioinformatic assistance; and the University of California, Irvine High Performance Computing Cluster (https://hpc.oit.uci.edu/) for their valuable resources and helpful staff. We would like to thank Margaret Fish, Rebekah Charney, Elmira Forouzmand, Elizabeth Chamu, Yuuri Yasuoka, and previous and current members of the Ken Cho lab for experimental assistance, valuable advice, and critical comments. This research was funded by the following grants awarded to K.W.Y.C.: NIH R01GM126395 and National Science Foundation (NSF) 1755214. K.D.P. was a recipient of NIH T32-HD60555.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.013.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aanes H, Winata CL, Lin CH, Chen JP, Srinivasan KG, Lee SG, Lim AY, Hajan HS, Collas P, Bourque G, et al. (2011). Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res 21, 1328–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkers RC, van Heeringen SJ, Jacobi UG, Janssen-Megens EM, Françoijs KJ, Stunnenberg HG, and Veenstra GJ (2009). A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus embryos. Dev. Cell 17, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almouzni G, and Wolffe AP (1995). Constraints on transcriptional activator function contribute to transcriptional quiescence during early Xenopus embryogenesis. EMBO J 14, 1752–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodeo AA, Jukam D, Straight AF, and Skotheim JM (2015). Histone titration against the genome sets the DNA-to-cytoplasm threshold for the Xenopus midblastula transition. Proc. Natl. Acad. Sci. USA 112, E1086–E1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. ; The Gene Ontology Consortium (2000). Gene ontology: tool for the unification of biology. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL (2011). DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz IL, and Cho KW (1995). Anterior neurectoderm is progressively induced during gastrulation: the role of the Xenopus homeobox gene orthodenticle. Development 121, 993–1004. [DOI] [PubMed] [Google Scholar]
- Blitz IL, Paraiso KD, Patrushev I, Chiu WTY, Cho KWY, and Gilchrist MJ (2017). A catalog of Xenopus tropicalis transcription factors and their regional expression in the early gastrula stage embryo. Dev. Biol 426, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe SA, Cha SW, Tadjuidje E, Heasman J, and Klein PS (2010). beta-Catenin primes organizer gene expression by recruiting a histone H3 arginine 8 methyltransferase, Prmt2. Dev. Cell 19, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, and Wysocka J (2013). Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha SW, Hwang YS, Chae JP, Lee SY, Lee HS, Daar I, Park MJ, and Kim J (2004). Inhibition of FGF signaling causes expansion of the endoderm in Xenopus. Biochem. Biophys. Res. Commun 315, 100–106. [DOI] [PubMed] [Google Scholar]
- Cha SW, Lee JW, Hwang YS, Chae JP, Park KM, Cho HJ, Kim DS, Bae YC, and Park MJ (2008). Spatiotemporal regulation of fibroblast growth factor signal blocking for endoderm formation in Xenopus laevis. Exp. Mol. Med 40, 550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney RM, Forouzmand E, Cho JS, Cheung J, Paraiso KD, Yasuoka Y, Takahashi S, Taira M, Blitz IL, Xie X, and Cho KW (2017a). Foxh1 Occupies cis-Regulatory Modules Prior to Dynamic Transcription Factor Interactions Controlling the Mesendoderm Gene Program. Dev. Cell 40, 595–607.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney RM, Paraiso KD, Blitz IL, and Cho KWY (2017b). A gene regulatory program controlling early Xenopus mesendoderm formation: network conservation and motifs. Semin. Cell Dev. Biol 66, 12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WT, Charney Le R, Blitz IL, Fish MB, Li Y, Biesinger J, Xie X, and Cho KW (2014). Genome-wide view of TGFb/Foxh1 regulation of the early mesendoderm program. Development 141, 4537–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, and Sacchi N (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Clements D, Friday RV, and Woodland HR (1999). Mode of action of VegT in mesoderm and endoderm formation. Development 126, 4903–4911. [DOI] [PubMed] [Google Scholar]
- Conlon FL, Fairclough L, Price BM, Casey ES, and Smith JC (2001). Determinants of T box protein specificity. Development 128, 3749–3758. [DOI] [PubMed] [Google Scholar]
- De Domenico E, Owens ND, Grant IM, Gomes-Faria R, and Gilchrist MJ (2015). Molecular asymmetry in the 8-cell stage Xenopus tropicalis embryo described by single blastomere transcript sequencing. Dev. Biol 408, 252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever W, Thoma G, and Nüsslein-Volhard C (1989). Determination of spatial domains of zygotic gene expression in the Drosophila embryo by the affinity of binding sites for the bicoid morphogen. Nature 340, 363–367. [DOI] [PubMed] [Google Scholar]
- Foo SM, Sun Y, Lim B, Ziukaite R, O’Brien K, Nien CY, Kirov N, Shvartsman SY, and Rushlow CA (2014). Zelda potentiates morphogen activity by increasing chromatin accessibility. Curr. Biol 24, 1341–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formaz-Preston A, Ryu JR, Svendsen PC, and Brook WJ (2012). The Tbx20 homolog Midline represses wingless in conjunction with Groucho during the maintenance of segment polarity. Dev. Biol 369, 319–329. [DOI] [PubMed] [Google Scholar]
- Geisberg JV, and Struhl K (2004). Quantitative sequential chromatin immunoprecipitation, a method for analyzing co-occupancy of proteins at genomic regions in vivo. Nucleic Acids Res 32, e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Goto T, Macdonald P, and Maniatis T (1989). Early and late periodic patterns of even skipped expression are controlled by distinct regulatory elements that respond to different spatial cues. Cell 57, 413–422. [DOI] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, and Noble WS (2007). Quantifying similarity between motifs. Genome Biol 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Wills A, Ucar D, and Baker J (2014). Developmental enhancers are marked independently of zygotic Nodal signals in Xenopus. Dev. Biol 395, 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada Y, Okai N, Taguchi S, Tagawa K, Humphreys T, and Satoh N (2000). Developmental expression of the hemichordate otx ortholog. Mech. Dev 91, 337–339. [DOI] [PubMed] [Google Scholar]
- Harding K, Hoey T, Warrior R, and Levine M (1989). Autoregulatory and gap gene response elements of the even-skipped promoter of Drosophila. EMBO J 8, 1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland RM (1991). In situ hybridization: an improved whole-mount method for Xenopus embryos. Methods Cell Biol 36, 685–695. [DOI] [PubMed] [Google Scholar]
- He Ml., Wen L, Campbell CE, Wu JY, and Rao Y (1999). Transcription repression by Xenopus ET and its human ortholog TBX3, a gene involved in ulnar-mammary syndrome. Proc. Natl. Acad. Sci. USA 96, 10212–10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V, Ovcharenko I, Putnam NH, Shu S, Taher L, et al. (2010). The genome of the Western clawed frog Xenopus tropicalis. Science 328, 633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm RR, Siebert S, Tulin S, Smith J, and Dunn CW (2013). Characterization of differential transcript abundance through time during Nematostella vectensis development. BMC Genomics 14, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman VF, Nguyen AT, and Davidson EH (2003). Expression and function of a starfish Otx ortholog, AmOtx: a conserved role for Otx proteins in endoderm development that predates divergence of the eleutherozoa. Mech. Dev 120, 1165–1176. [DOI] [PubMed] [Google Scholar]
- Hontelez S, van Kruijsbergen I, Georgiou G, van Heeringen SJ, Bogdanovic O, Lister R, and Veenstra GJC (2015). Embryonic transcription is controlled by maternally defined chromatin state. Nat. Commun 6, 10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horb ME, and Thomsen GH (1997). A vegetally localized T-box transcription factor in Xenopus eggs specifies mesoderm and endoderm and is essential for embryonic mesoderm formation. Development 124, 1689–1698. [DOI] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang YS, Seo M, Lee BR, Lee HJ, Park YH, Kim SK, Lee HC, Choi HJ, Yoon J, Kim H, and Han JY (2018). The transcriptome of early chicken embryos reveals signaling pathways governing rapid asymmetric cellularization and lineage segregation. Development 145, dev157453. [DOI] [PubMed] [Google Scholar]
- Jiang P, Nelson JD, Leng N, Collins M, Swanson S, Dewey CN, Thomson JA, and Stewart R (2017). Analysis of embryonic development in the unsequenced axolotl: waves of transcriptomic upheaval and stability. Dev. Biol 426, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SR, Pálfy M, Hilbert L, Kumar M, Karschau J, Zaburdaev V, Shevchenko A, and Vastenhouw NL (2017). Competition between histone and transcription factor binding regulates the onset of transcription in zebrafish embryos. eLife 6, e23326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukam D, Shariati SAM, and Skotheim JM (2017). Zygotic Genome Activation in Vertebrates. Dev. Cell 42, 316–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbrun E, Greco TM, Slagle CE, Kennedy LM, Li T, Cristea IM, and Conlon FL (2013). A Gro/TLE-NuRD corepressor complex facilitates Tbx20-dependent transcriptional repression. J. Proteome Res 12, 5395–5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimoto M, Suzuki T, Ito M, Miyake A, Nakatsuji N, and Nakatsuji T (2003). Unequal distribution of otx1 mRnas among cleavage stage blastomeres in the teleost, Leucopsarion petersii (shiro-uo). Int. J. Dev. Biol 47, 77–80. [PubMed] [Google Scholar]
- Karimi K, Fortriede JD, Lotay VS, Burns KA, Wang DZ, Fisher ME, Pells TJ, James-Zorn C, Wang Y, Ponferrada VG, et al. (2018). Xenbase: a genomic, epigenomic and transcriptomic model organism database. Nucleic Acids Res 46 (D1), D861–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura A, Koshida S, and Takada S (2008). Activator-to-repressor conversion of T-box transcription factors by the Ripply family of Groucho/TLE-associated mediators. Mol. Cell. Biol 28, 3236–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokha MK, Chung C, Bustamante EL, Gaw LW, Trott KA, Yeh J, Lim N, Lin JC, Taverner N, Amaya E, et al. (2002). Techniques and probes for the study of Xenopus tropicalis development. Dev. Dyn 225, 499–510. [DOI] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Tan Y, Ma W, Merkurjev D, Destici E, Ma Q, Suter T, Ohgi K, Friedman M, Skowronska-Krawczyk D, and Rosenfeld MG (2018). Pluripotency factors functionally premark cell-type-restricted enhancers in ES cells. Nature 556, 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Li W, Rosenfeld MG, and Glass CK (2014). Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci 39, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea R, Papalopulu N, Amaya E, and Dorey K (2009). Temporal and spatial expression of FGF ligands and receptors during Xenopus development. Dev. Dyn 238, 1467–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, and Kendziorski C (2013). EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M, Anavy L, Cole AG, Winter E, Mostov N, Khair S, Senderovich N, Kovalev E, Silver DH, Feder M, et al. (2016). The mid-developmental transition and the evolution of animal body plans. Nature 531, 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Sub-group (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752–1779. [Google Scholar]
- Li XY, Harrison MM, Villalta JE, Kaplan T, and Eisen MB (2014). Establishment of regions of genomic activity during the Drosophila maternal to zygotic transition. eLife 3, e03737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig KD, Kroll KL, Sun EE, and Kirschner MW (1996). Expression cloning of a Xenopus T-related gene (Xombi) involved in mesodermal patterning and blastopore lip formation. Development 122, 4001–4012. [DOI] [PubMed] [Google Scholar]
- Lynch JA, Brent AE, Leaf DS, Pultz MA, and Desplan C (2006). Localized maternal orthodenticle patterns anterior and posterior in the long germ wasp Nasonia. Nature 439, 728–732. [DOI] [PubMed] [Google Scholar]
- Matsuoka T, Ikeda T, Fujimaki K, and Satou Y (2013). Transcriptome dynamics in early embryos of the ascidian, Ciona intestinalis. Dev. Biol 384, 375–385. [DOI] [PubMed] [Google Scholar]
- Milton AC, and Okkema PG (2015). Caenorhabditis elegans TBX-2 Directly Regulates Its Own Expression in a Negative Autoregulatory Loop. G3 (Bethesda) 5, 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakhina S, and Steward R (2005). Axes formation and RNA localization. Curr. Opin. Genet. Dev 15, 416–421. [DOI] [PubMed] [Google Scholar]
- Mitsunaga-Nakatsubo K, Akasaka K, Sakamoto N, Takata K, Matsumura Y, Kitajima T, Kusunoki S, and Shimada H (1998). Differential expression of sea urchin Otx isoform (hpOtxE and HpOtxL) mRNAs during early development. Int. J. Dev. Biol 42, 645–651. [PubMed] [Google Scholar]
- Mori H, Miyazaki Y, Morita T, Nitta H, and Mishina M (1994). Different spatio-temporal expressions of three otx homeoprotein transcripts during zebrafish embryogenesis. Brain Res. Mol. Brain Res 27, 221–231. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, de Paiva Alves E, Veenstra GJ, and Hoppler S (2016). Tissue- and stage-specific Wnt target gene expression is controlled subsequent to β-catenin recruitment to cis-regulatory modules. Development 143, 1914–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, and Faber J (1958). Normal Table of Xenopus Laevis (Daudin) (Routledge)
- Ogino H, McConnell WB, and Grainger RM (2006). High-throughput transgenesis in Xenopus using I-SceI meganuclease. Nat. Protoc 1, 1703–1710. [DOI] [PubMed] [Google Scholar]
- Owens NDL, Blitz IL, Lane MA, Patrushev I, Overton JD, Gilchrist MJ, Cho KWY, and Khokha MK (2016). Measuring Absolute RNA Copy Numbers at High Temporal Resolution Reveals Transcriptome Kinetics in Development. Cell Rep 14, 632–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DA, Butler AM, Aguero TH, Newman KM, Van Booven D, and King ML (2017). High-throughput analysis reveals novel maternal germline RNAs crucial for primordial germ cell preservation and proper migration. Development 144, 292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannese M, Cagliani R, Pardini CL, and Boncinelli E (2000). Xotx1 maternal transcripts are vegetally localized in Xenopus laevis oocytes. Mech. Dev 90, 111–114. [DOI] [PubMed] [Google Scholar]
- Peter IS, and Davidson EH (2010). The endoderm gene regulatory network in sea urchin embryos up to mid-blastula stage. Dev. Biol 340, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Poulain M, Fürthauer M, Thisse B, Thisse C, and Lepage T (2006). Zebrafish endoderm formation is regulated by combinatorial Nodal, FGF and BMP signalling. Development 133, 2189–2200. [DOI] [PubMed] [Google Scholar]
- Prioleau MN, Huet J, Sentenac A, and Méchali M (1994). Competition between chromatin and transcription complex assembly regulates gene expression during early development. Cell 77, 439–449. [DOI] [PubMed] [Google Scholar]
- Puelles E, Annino A, Tuorto F, Usiello A, Acampora D, Czerny T, Brodski C, Ang SL, Wurst W, and Simeone A (2004). Otx2 regulates the extent, identity and fate of neuronal progenitor domains in the ventral midbrain. Development 131, 2037–2048. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2014). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing)
- Rana AA, Collart C, Gilchrist MJ, and Smith JC (2006). Defining synphe-notype groups in Xenopus tropicalis by use of antisense morpholino oligonucleotides. PLoS Genet 2, e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CD, Steiner AB, Yaklichkin S, Lu Q, Wang S, Hennessy M, and Kessler DS (2016). FoxH1 mediates a Grg4 and Smad2 dependent transcriptional switch in Nodal signaling during Xenopus mesoderm development. Dev. Biol 414, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe NJ, Aneas I, Shen T, Shokri L, Park SY, Bulyk ML, Evans SM, and Nobrega MA (2012). Dual transcriptional activator and repressor roles of TBX20 regulate adult cardiac structure and function. Hum. Mol. Genet 21, 2194–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satou Y, Imai KS, and Satoh N (2001). Early embryonic expression of a LIM-homeobox gene Cs-lhx3 is downstream of beta-catenin and responsible for the endoderm differentiation in Ciona savignyi embryos. Development 128, 3559–3570. [DOI] [PubMed] [Google Scholar]
- Satou Y, Minami K, Hosono E, Okada H, Yasuoka Y, Shibano T, Tanaka T, and Taira M (2018). Phosphorylation states change Otx2 activity for cell proliferation and patterning in the Xenopus embryo. Development 145, dev159640. [DOI] [PubMed] [Google Scholar]
- Schröder R (2003). The genes orthodenticle and hunchback substitute for bicoid in the beetle Tribolium. Nature 422, 621–625. [DOI] [PubMed] [Google Scholar]
- Schulz KN, Bondra ER, Moshe A, Villalta JE, Lieb JD, Kaplan T, McKay DJ, and Harrison MM (2015). Zelda is differentially required for chromatin accessibility, transcription factor binding, and gene expression in the early Drosophila embryo. Genome Res 25, 1715–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennard F, Carnac G, and Gurdon JB (1996). The Xenopus T-box gene, Antipodean, encodes a vegetally localised maternal mRNA and can trigger mesoderm formation. Development 122, 4179–4188. [DOI] [PubMed] [Google Scholar]
- Suda Y, Kurokawa D, Takeuchi M, Kajikawa E, Kuratani S, Amemiya C, and Aizawa S (2009). Evolution of Otx paralogue usages in early patterning of the vertebrate head. Dev. Biol 325, 282–295. [DOI] [PubMed] [Google Scholar]
- Sudou N, Yamamoto S, Ogino H, and Taira M (2012). Dynamic in vivo binding of transcription factors to cis-regulatory modules of cer and gsc in the stepwise formation of the Spemann-Mangold organizer. Development 139, 1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium (2017). Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45 (D1), D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heeringen SJ, Akkers RC, van Kruijsbergen I, Arif MA, Hanssen LL, Sharifi N, and Veenstra GJ (2014). Principles of nucleation of H3K27 methylation during embryonic development. Genome Res 24, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, Rinn J, and Schier AF (2010). Chromatin signature of embryonic pluripotency is established during genome activation. Nature 464, 922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada S, and Saiga H (1999). Vegetal cell fate specification and anterior neuroectoderm formation by Hroth, the ascidian homologue of orthodenticle/otx. Mech. Dev 82, 67–77. [DOI] [PubMed] [Google Scholar]
- Wada S, Katsuyama Y, Sato Y, Itoh C, and Saiga H (1996). Hroth an orthodenticle-related homeobox gene of the ascidian, Halocynthia roretzi: its expression and putative roles in the axis formation during embryogenesis. Mech. Dev 60, 59–71. [DOI] [PubMed] [Google Scholar]
- Warren WC, Hillier LW, Tomlinson C, Minx P, Kremitzki M, Graves T, Markovic C, Bouk N, Pruitt KD, Thibaud-Nissen F, et al. (2017). A New Chicken Genome Assembly Provides Insight into Avian Genome Structure. G3 (Bethesda) 7, 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Xu J, Liu B, Yao G, Wang P, Lin Z, Huang B, Wang X, Li T, Shi S, et al. (2018). Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature 557, 256–260. [DOI] [PubMed] [Google Scholar]
- Xanthos JB, Kofron M, Wylie C, and Heasman J (2001). Maternal VegT is the initiator of a molecular network specifying endoderm in Xenopus laevis. Development 128, 167–180. [DOI] [PubMed] [Google Scholar]
- Yang KY, Chen Y, Zhang Z, Ng PK, Zhou WJ, Zhang Y, Liu M, Chen J, Mao B, and Tsui SK (2016). Transcriptome analysis of different developmental stages of amphioxus reveals dynamic changes of distinct classes of genes during development. Sci. Rep 6, 23195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuoka Y, Suzuki Y, Takahashi S, Someya H, Sudou N, Haramoto Y, Cho KW, Asashima M, Sugano S, and Taira M (2014). Occupancy of tissue-specific cis-regulatory modules by Otx2 and TLE/Groucho for embryonic head specification. Nat. Commun 5, 4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh CH, Bolouri H, and Davidson EH (1998). Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science 279, 1896–1902. [DOI] [PubMed] [Google Scholar]
- Zaret KS, and Carroll JS (2011). Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25, 2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and King ML (1996). Xenopus VegT RNA is localized to the vegetal cortex during oogenesis and encodes a novel T-box transcription factor involved in mesodermal patterning. Development 122, 4119–4129. [DOI] [PubMed] [Google Scholar]
- Zhang J, Houston DW, King ML, Payne C, Wylie C, and Heasman J (1998). The role of maternal VegT in establishing the primary germ layers in Xenopus embryos. Cell 94, 515–524. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed RNA-seq and ChIP-seq datasets generated for this study are available at NCBI Gene Expression Omnibus using the accession GEO: GSE118024.`