

Summary
The genome is organized into topologically associated domains (TAD) that enclose smaller subTADs. Here we identify and characterize an enhancer that is located in the middle of the V gene region of the immunoglobulin kappa light chain (Igκ) locus that becomes active preceding the stage at which this locus undergoes V(D)J recombination. This enhancer is a hub of long-range interactions connecting subTADs in the V gene region with the recombination center at the J genes. Deletion of this element results in a highly altered long-range interaction pattern across the locus and, importantly, affects individual V gene utilization locus-wide. These results indicate the existence of an enhancer-dependent framework in the Igκ locus, and further suggest that the composition of the diverse antibody repertoire is regulated in a subTAD-specific manner. This enhancer thus plays a structural role orchestrating the proper folding of the Igκ locus in preparation for V(D)J recombination.
Keywords: V(D)J recombination, long-range chromatin interactions, 3D-structure, antibody repertoire, subTAD, enhancer, Igκ, B cells, chromatin conformation capture, nuclear topology
Introduction
Adaptive immunity relies on the antigen receptor (AgR) diversity on B and T cells to effectively fight a wide array of pathogens. This variety results from the assembly of variable (V), diversity (D) and joining (J) gene segments to form these receptors by V(D)J recombination. Although the Igh and Igκ loci each have over 100 functional V segments, they rearrange with vastly different frequencies, and the reasons for this are not well understood (Aoki-Ota et al., 2012; Bolland et al., 2016; Choi et al., 2013; Williams et al., 2001). The recombination signal sequence (RSS) quality, the epigenetic environment, and bound transcription factors (TF) near each V segment can contribute to its usage, but a specific feature or group of features are not sufficient to account for this differential usage (Bolland et al., 2016; Choi et al., 2013; Gopalakrishnan et al., 2013; Matheson et al., 2017)
V genes in the Igh and Igκ loci are spread over 2.5–3 Mb. Three-dimensional (3D) DNA fluorescence in situ hybridization (3D-FISH) and chromatin conformation capture experiments indicate that V genes move closer to the J genes in a process called locus contraction prior to rearrangement at each AgR locus, providing spatial proximity (Fuxa et al., 2004; Jhunjhunwala et al., 2008; Roldan et al., 2005; Sayegh et al., 2005). Long-range chromatin interactions (LRCI) play a fundamental role in locus contraction but the mechanisms regulating these interactions are not fully understood. Hi-C experiments reveal that there are several areas with high density of LRCI throughout the Igκ locus, and these LRCI are not randomly distributed (Lin et al., 2012). However, the role of these hubs of LRCI in the Igκ locus has not been investigated.
Throughout the genome, LRCI are associated with CTCF, enhancers and promoters (Rao et al., 2014; Sanyal et al., 2012). CTCF is a major architectural protein organizing the entire genome through the creation of long-range loops (Rao et al., 2014). CTCF also plays an important role in modulating Ig 3D structure and function (Choi and Feeney, 2014; Guo et al., 2011; Ribeiro de Almeida et al., 2011). There are many CTCF sites throughout the Ig loci, and knockdown of CTCF in pro-B cells results in a decrease in Igh locus contraction (Degner et al., 2011; Loguercio et al., 2018). The LRCI at the Igh locus that lead to its contraction in pro-B cells are also dependent on TF such as Pax5, YY1, and Ikaros (Hesslein et al., 2003; Liu et al., 2007; Reynaud et al., 2008).
Less is known about the regulation of rearrangement and 3D conformation of the Igκ locus, although YY1 reconstitution experiments in YY1−/− mice indicated that YY1 affects Vκ gene usage (Pan et al., 2013). Additionally, PU.1 and E2A have been associated with rearrangement fitness and LRCI (Batista et al., 2017; Lin et al., 2012; Stadhouders et al., 2014). The Igκ locus has three well characterized enhancers (iEκ, 3’Eκ, and Ed), all located at the 3’ end of the locus. Deletion of iEκ greatly reduces Igκ rearrangement, whereas deletion of 3’Eκ has only a modest effect, but together with iEκ deletion, eliminates all rearrangement (Inlay et al., 2002). In contrast, Ed has no effect on rearrangement (Xiang and Garrard, 2008). The loss of CTCF in pre-B cells results in greatly increased usage of the most proximal V genes and increased interactions of iEκ and 3’Eκ to the most proximal portion of the Igκ locus (Ribeiro de Almeida et al., 2011). Deletion of the CTCF-containing elements, Cer and/or Sis, located between the Vκ and Jκ genes, results in increased rearrangements of proximal V genes and reduced distal Vκ rearrangement (Xiang et al., 2013; Xiang et al., 2011). Cer deletion results in a lack of locus contraction, implicating the CTCF sites, and/or some other sites in Cer, in LRCI leading to locus contraction (Xiang et al., 2013).
The Igκ locus predominantly undergoes rearrangement in pre-B cells, although it is reported that ~15% of pro-B cells harbor Igκ rearrangements (Novobrantseva et al., 1999). 4C and Hi-C studies revealed that LRCI within the Igκ locus are developmentally regulated. During the pre-pro-B cell stage, the Igκ locus is devoid of significant LRCI (Lin et al., 2012). In contrast, IL7-cultured pro-B cells and pre-B cells show a complex network of LRCI involving iEκ and 3’Eκ, among other sites (Lin et al., 2012; Stadhouders et al., 2014). During the transition from pro-B cells to pre-B cells, there is a redistribution of the LRCI coming from the enhancers in the regulatory region (Stadhouders et al., 2014). The Igκ locus contraction is reported to occur at the pro-B cell stage as measured by 3D-FISH (Rother et al., 2016; Stadhouders et al., 2014) suggesting that this conformation, and its alteration during development, may play a role in Vκ rearrangement.
In this study, we identified and characterized the most striking hub of LRCI which is located in the middle of the V region of the Igκ locus. We show that this element (E88) has the highest epigenetic marks of an enhancer at the pro-B cell stage, and has B cell-specific enhancer activity (Predeus et al., 2014). E88 has many key TF binding to it, and it strongly interacts with several regions within the Igκ locus including the enhancers at the 3’ end of the locus. Deletion of E88 alters the LRCI pattern of the Igκ locus. Importantly, E88 impacts the frequency of individual Vκ gene rearrangements throughout the Igκ locus in a domain-specific manner that correlates with subTAD boundaries. We propose that the altered Igκ repertoire is due to changes in the 3D conformation of Igκ when E88 is absent. Our data show that E88 plays a critical role in orchestrating the proper folding of the Igκ locus in preparation for Vκ rearrangement and that is required to generate the natural V diversity of the B cell repertoire.
RESULTS
Major hub of LRCI in the Igκ V region has epigenetic marks of an active enhancer in pro-B cells
Previous Hi-C data showed that some regions in the Vκ locus have a high density of LRCI in IL7-cultured pro-B cells (Lin et al., 2012). These LRCI hubs are not randomly distributed but show a network of interactions connecting the iEκ/Jκ/3’Eκ regulatory region to many sites in the large Vκ gene region, and connecting different parts of the Vκ gene region to each other. To systematically analyze LRCI hubs, we used the Hi-C interaction coordinates data to determine the number of interactions in 10 kb bins along the Igκ locus (Figure 1A, top panel). Bins that have ≥35 interactions (>1.6 SD above average) are indicated by the horizontal line. We used ChIP-seq data to determine the TF binding and epigenetic profiles of these LRCI hubs (Figure 1A). All but one of the interaction hubs were located in regions containing bound CTCF and/or the enhancer mark H3K4me1. Some enriched H3K4me1 areas were also high in the epigenetic mark of active enhancers H3K27ac. We classified hubs into 6 categories according to their CTCF binding and epigenetic profile (Figure 1B). Although CTCF is associated with LRCI formation genome wide, we found that many more hubs of LRCI within the Igκ locus were at sites associated with enhancer marks than with bound CTCF. Thus, enhancer-associated epigenetic marks, rather than CTCF occupancy, dominated the conformational landscape of the Igκ locus.
Figure 1. V gene region of the Igκ locus contains multiples hubs of LRCI that correlated with enhancer epigenetic marks.
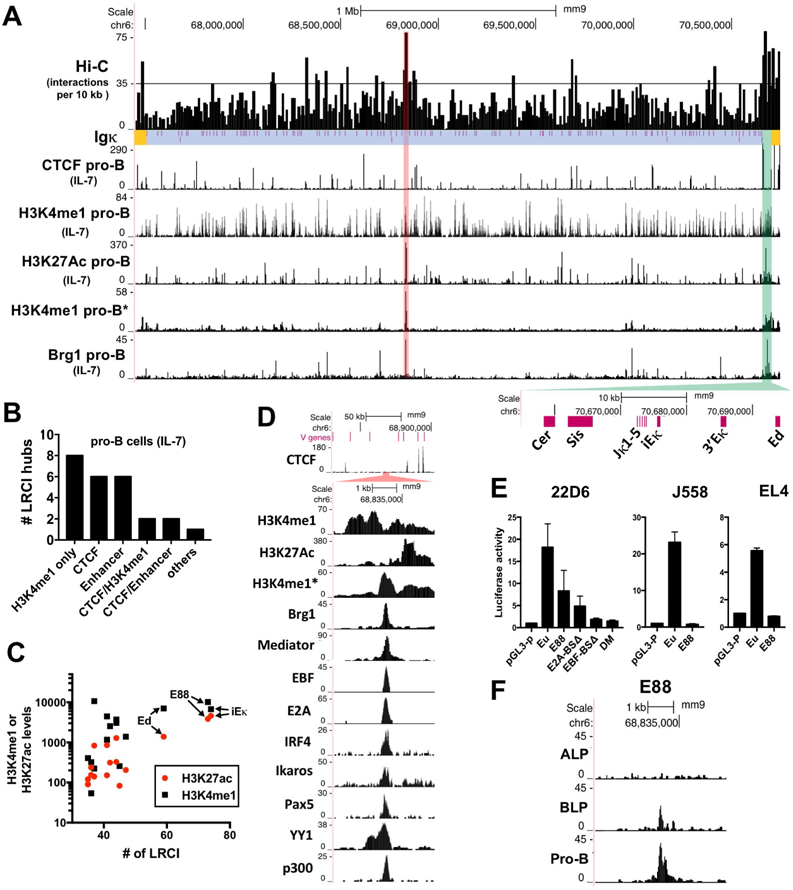
(A) Hi-C from IL7-cultured pro-B cells plotted as number of interactions per 10 kb bins for the V region of the Igκ locus (blue), Igκ regulatory region (green) and flanking regions (yellow). Vκ genes locations are shown (purple lines). Horizontal black line indicates 35 interactions (>1.6 SD above average). Red highlight marks the bin with the highest number of interactions in the V region (E88). Below are shown several ChIP-seq datasets. ChIP-seqs in panels A and D are from IL7-cultured pro-B cells, except for one non-IL7-cultured pro-B set (*) and Mediator that is from a v-Abl transformed pro-B cell line (See Table S2 for GEO numbers). (B) Bar graph showing the number of Hi-C LRCI hubs above horizontal line in panel A that are located at CTCF sites, H3K4me1-only enriched areas and/or putative enhancers (H3K27ac and H3K4me1). (C) Correlation between the number of interactions at all hubs containing H3K4me1 with read numbers of H3K27ac (red) or H3K4me1(black) in the 10 kb bins. (D) ChIP-seq in the E88 region. (E) Luciferase assay of E88, and E88 with the indicated point mutations. Cells assayed are a v-Abl pre-B cell line (22D6), myeloma cell line (J558L) and T cell line (EL4). Eµ is the positive control for a strong enhancer. DM= double EBF and E2A binding site mutations. BS=binding site. Luciferase data is the average of 3 independent biological replicates. (F) ATAC-seq tracks of ALP, BLP, and pro-B cells around E88.
Analysis of the epigenetic marks in all the hubs containing H3K4me1 shows that the levels of the active enhancer mark H3K27ac in those bins have a higher correlation (r=0.9172), compared to the levels H3K4me1 only (r=0.6356), with the number of LRCI (Figure 1C). This analysis also shows that iEκ and a region close to the IGKV1–88 gene (hereinafter all Vκ genes are referred to without IGΚ) are the most prominent hubs of LRCI in the Igκ locus, with high levels H3K4me1 and the highest level of H3K27ac. Putative enhancers and CTCF binding sites were named, and hereinafter referred to, by their closest V gene, e.g. E88, CTCF45, and V genes are numbered consecutively in the locus, starting with Jκ-proximal. Although there are many peaks of H3K4me1 in IL7-cultured pro-B cells as well as pre-B cells, there are very few peaks of H3K4me1 in non-cultured pro-B cells in the Igκ locus (Figure 1A) (Loguercio et al., 2018). Thus, extensive marking of Igκ near most Vκ genes with H3K4me1 happens during the transition from pro-B cells to pre-B cells. E88 (highlighted in red) is located in the most prominent H3K4me1 peak in non-cultured pro-B cells. Further analysis shows that E88 also has high levels of Brg1, Mediator, EBF1, E2A, IRF4, Ikaros, Pax5, YY1, and p300 (Figure 1D), all indicative of active enhancers or B cell regulatory elements (Heinz et al., 2010; Lin et al., 2010). ChromHMM analysis also identified this region as a potential enhancer (Figure S1A) (Predeus et al., 2014). Thus, E88 is associated with all the landmarks of an active enhancer.
E88 is a B cell-specific enhancer
E88 had significant enhancer activity in luciferase assays in an Abelson virus (v-Abl) transformed pre-B cell line (22D6) but not in a myeloma cell line or T cell line (Figure 1E). The activity of E88 harboring mutations in the E2A or/and EBF binding sites (Figure S1B) was partially or substantially attenuated, respectively (Figure 1E). DNase hypersensitive sites (DHS) are areas of open chromatin associated with genetic regulatory elements. DNAse-seq data show that E88 is a DHS in multiple B cell types, but not in other cell types (Vierstra et al., 2014) (Figure S1C). Also, analysis of E88 accessibility by ATAC-seq (Bossen et al., 2015) during B cell development in cells with multi-lineage potential (ALP), B cell biased lymphoid progenitors (BLP), and pro-B cells reveals that E88 accessibility appears only in BLP and pro-B stages (Figure 1F). LRCI between enhancers and their distal cis-regulatory elements form concomitantly as enhancers emerge in a developmental stage-specific manner (de Laat and Duboule, 2013; Dixon et al., 2015). We therefore hypothesized that E88 might be very important in initiating the proper folding of the Igκ locus in pro-B cells by establishing LRCI during locus contraction.
E88 deletion severely reduces H3K4me1 deposition around E88
To address the role of E88, we deleted a 244 bp region (Figure S1B) containing the TF binding sites in a v-Abl transformed pro-B cell line 445.3-WT using CRISPR-Cas9 editing (445.3-E88∆). v-Abl transformed cell lines are blocked in differentiation between the pro-B cell and pre-B cell stages. Culture of 445.3-WT cells with the Abl kinase inhibitor STI571 removes the block in differentiation and induces robust Igκ rearrangement and germline transcription (GLT) (Muljo and Schlissel, 2003). The 445.3-WT cells are on a Rag1−/− background which maintains an intact germline Igκ locus while creating the E88-deleted clones by avoiding prior V(D)J rearrangement. For the same reasons, the Rag1−/− background is necessary for all ChIP, RNA-seq and 4C experiments. To determine whether E88 controls H3K4me1 deposition around this large area, we performed chromatin immunoprecipitation (ChIP) followed by qPCR at several sites around the E88 deletion (Figure 2A). H3K4me1 levels were profoundly reduced for >7 kb surrounding E88 in 445.3-E88∆ cells, confirming that it represents the primary enhancer element in the region. Levels of H3K4me1 were reduced (~2-fold) at V1–88, 22 kb away, and unaffected in other locations in Igκ.
Figure 2. E88 controls H3K4me1 deposition, GLT, and rearrangement in an inducible pro-B cell line.
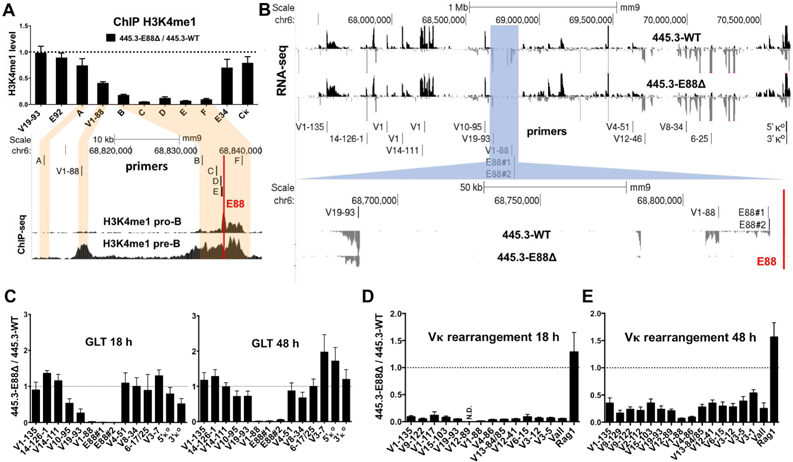
(A, top) H3K4me1 ChIP qPCR analysis was performed in 445.3-WT or 445.3-E88∆ Abl cell lines. Results are expressed as the E88∆ / WT ratio of the relative level of H3K4me1. (A, bottom) H3K4me1 ChIP-seq in RAG−/− pro-B cells and RAG−/−.Igh-Tg+ pre-B cells and the location of primers used for ChIP qPCR shown above. (B, top) RNA-seq data from STI571-stimulated 445.3-WT and 445.3-E88∆ cells. Reads are directionally mapped, with sense strand on top in black and antisense below in gray. (B, highlighted blue and bottom) Region around E88 is shown. Red line indicates the location of E88 in (A, B). (C) GLT was assessed at 18 and 48 hours post STI571 stimulation by qPCR with primers along the Igκ locus (location shown in B). (D, E) Vκ rearrangement was assessed in 445.3-WT or 445.3-E88∆ cells. RNA was harvested at 18 and 48 hours post STI571 stimulation and analyzed by qPCR. Data from Vκ rearrangement and GLT were normalized with GAPDH and expressed as E88∆ / WT ratio. N.D.= not detected in the E88∆ samples. Data is the average of at least 3 independent biological replicates from 3 independent E88∆ clones ± SEM. RNA-seq is the representative of 2 independent experiments.
E88 regulates local V gene-derived and intergenic GLT in inducible cell lines
GLT throughout AgR loci is associated with rearrangement and chromatin accessibility (Yancopoulos and Alt, 1986). Unrearranged V genes and some intergenic regions in AgR loci undergo low levels of transcription and J and C genes display high level transcription, all occurring at the specific developmental stage at which that locus is undergoing recombination (Van Ness et al., 1981; Yancopoulos and Alt, 1985). We assessed GLT in STI571-stimulated 445.3-WT and 445.3-E88∆ cells by RNA-seq and qPCR (Figures 2B and 2C). Three independent clones of E88∆ were assayed for GLT and Vκ rearrangement. We found that GLT around E88 was significantly reduced at 18 and 48 hours after STI571 stimulation (Figure 2C). Both time points show an almost complete elimination of intergenic GLT initiating ~5 kb upstream of E88 (primer sets E88#1 and E88#2) as well as GLT at the V1–88 gene. GLT was modestly affected at other nearby Vκ segments (e.g. V19–93 and V10–95) but not at more distant sites, indicating that the impact of E88 on transcription is largely restricted to a region of ~200 kb.
E88∆ affects global patterns of V gene rearrangement
Deletion of iEκ, and to a lesser extent 3’Eκ, enhancers in the Igκ regulatory region greatly reduces Vκ rearrangement. Since E88 is the first enhancer described within the large V gene-containing region of any AgR loci, the role of this class of enhancers in rearrangement is unknown. Therefore, we evaluated the effects of E88∆ in Vκ rearrangement. As 445.3 cells were on a Rag1−/− background, we transduced 445.3-WT and 445.3-E88∆ cells with a Rag1-expressing retrovirus. Cells were then stimulated with STI571 to induce Vκ rearrangement. We quantified rearrangement from RNA and genomic DNA (gDNA) using qPCR with a panel of individual Vκ gene primers spread throughout the locus and a degenerate “Vall” primer (Schlissel and Baltimore, 1989). We observed profound defects on Vκ rearrangement in 445.3-E88∆ cells at 18 hours post-stimulation when compared to 445.3-WT (Figure 2D). The effects were still significant at 48 hours post-stimulation (Figure 2E). Vκ genes around E88 (V4–86, V1–88, and V12–89) have much more severe defects in rearrangement compared to other Vκ genes further away from the deletion. Rag1 expression was comparable between cell lines (Figures 2D and 2E). Similar results were obtained for assays with gDNA, confirming that the defects were not due to transcriptional differences following rearrangement, but due to direct effects on rearrangement per se (Figure S2A).
To probe early stages of rearrangement, we analyzed 445.3-WT and 445.3-E88∆ cells that were unstimulated, or at 12 and 24 hours post STI571 stimulation (Figure S2B). Using the Vall primer, we determined that rearrangement in unstimulated 445.3-WT cells was 6 times higher than in 445.3-E88∆ cells. After 12 hours of STI571 stimulation, 445.3-WT cells had 25 times more rearrangement than 445.3-E88∆ cells. This indicates that 445.3-WT cells rearranged 5 times faster than 445.3-E88∆ cells (Figure S2C). In summary, this data suggest that E88 has a global effect on the pattern, initiation, and kinetics of Vκ rearrangement.
E88 controls GLT in mice
Since deletion of E88 had a profound effect on the pattern of rearrangement and on GLT in 445.3 cells, we wished to define its role in the natural generation of the Igκ repertoire at different stages of B cell differentiation. Therefore, we generated germline mutations in mice using CRISPR/Cas9 technology. Mice with deletions of interest were bred to homozygosity (Figure 3A) and E88∆ mice were also crossed with Rag−/− mice for GLT and 4C analyses.
Figure 3. E88 regulates GLT and Vκ gene utilization in mice.
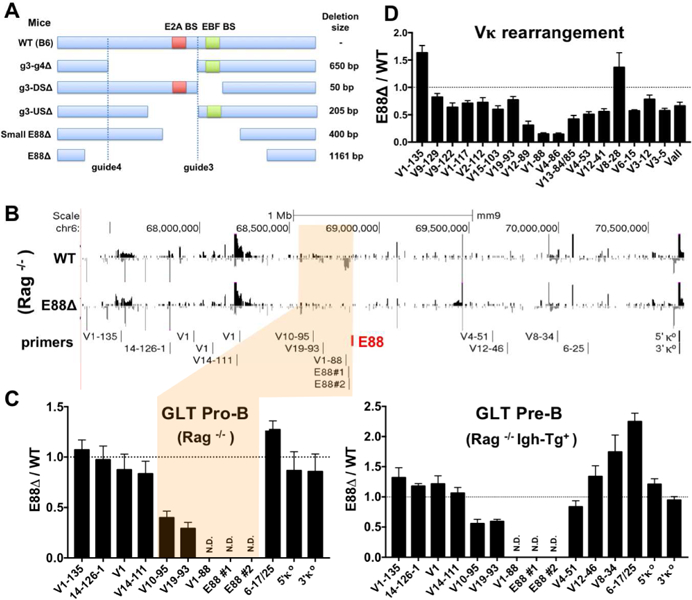
(A) E88 was deleted in mice using CRISPR/Cas9 genome editing. Guide 3 and guide 4 indicate the location of the sgRNAs. Schematic of the selected E88 deletions in mice show the size and location of each of the deletions. Red and green squares show the relative location of the E2A and EBF binding sites respectively. DS=downstream, US=upstream. (B) RNA-seq data from BM-derived CD19+ pro-B cells from WT.Rag−/− and E88∆.Rag−/− mice. Red line indicates the location of E88. (C, left) qPCR with the indicated set of primers was performed on RNA from WT.Rag−/− and E88∆.Rag−/−pro-B cells. E88 surrounding region is shown (highlighted tan) with primers locations shown above. (C, right) GLT in WT.Rag1−/−.Igh-Tg+ or E88∆.Rag1−/−Igh-Tg+ pre-B cells was quantified by qPCR with the indicated primers. (D) BM-derived CD19+ cells were purified from WT and E88∆ mice, and RNA was harvested. Vκ rearrangement was assessed by qPCR for specific individual Vκ genes. Data was normalized to GAPDH and is shown as the ratio of E88∆ / WT. Data is average of at least 3 independent experiments ± SEM. Two to five mice 6–10 weeks of age were used for each experiment. RNA-seq is representative of 2 independent experiments.
To evaluate GLT, we performed RNA-seq and qPCR on RNA from bone marrow (BM)-derived CD19+ pro-B cells from WT.Rag1−/− and E88∆.Rag1−/− mice. As in the 445.3 cell lines, GLT was severely affected near E88 in pro-B cells from E88∆ mice (Figure 3B, highlighted area). Intergenic GLT upstream of E88 was completely ablated as well as transcription of the V1–88 gene (Figures 3B and 3C, left panel). Other nearby GLT, such as V19–93 and V10–95 were reduced, ~4 and ~3 fold respectively. To assess GLT at the pre-B cell stage, we crossed WT.Rag−/− and E88∆.Rag−/− mice with mice that express a transgene encoding a rearranged heavy chain allowing the differentiation from pro-B cells to pre-B cells (WT.Rag−/−Igh-Tg+ and E88∆.Rag1−/−Igh-Tg+). As with the E88∆ Rag1−/− mice, GLT was ablated near E88 and the V1–88 gene in E88∆ pre-B cells (Figure 3C, right panel). Also, GLT from V19–93 and V10–95 were reduced ~2-fold. In contrast to the pro-B cells, some GLT at the pre-B cell stage were modestly increased, e.g., V19–25 (~2 fold) in E88∆ pre-B cells. In summary, deletion of the E88 in mice reduces local GLT (~200 kb) with the most dramatic effects close to E88.
E88 controls Vκ rearrangement patterns in mice
To assess Vκ rearrangement in the mouse lines we generated, BM-derived CD19+ cells, which are composed predominantly of pre-B cells but also contain pro-B, immature, and recirculating B cells, were isolated and analyzed. Vκ rearrangement in all mouse lines have a similar pattern in rearrangement although the extent varies modestly. Genes near E88, such as V4–86 and V1–88, were more severely affected by the deletions, in agreement with the cell line data. The fold reduction for V4–86, V1–88, and V12–89 vary from 6–7-fold for E88∆, small E88∆, and g3-US∆ deletions (Figures 3D, S3A and S3D) to 4–5-fold for g3-g4∆ and g3-DS∆ deletions (Figures S3B and S3C). V genes further away from the deletion tend to have minimal reductions in rearrangement (<2-fold). In contrast, some Vκ genes even further away showed an increase in rearrangement compared to WT (i.e. V1–135). This data demonstrates that deletion of the entire E88 region, or partial deletions removing specific TF binding site-containing areas, affects Igκ rearrangement patterns in vivo in a similar manner suggesting that the full function of E88 depends on multiple cis-regulatory elements.
The majority of the Vκ rearrangement happens at the pre-B cell stage, but ~15% of pro-B cells are estimated to have Vκ rearrangements (Novobrantseva et al., 1999). Since we postulated that E88 would initially orchestrate LRCI in Igκ at the pro-B cell stage, and since the effects of E88∆ on rearrangement in the 445.3 cells were most dramatic at earlier times after STI571 induction, we hypothesized that pro-B cells from E88∆ mice would have a more profound defect in Igκ rearrangement than pre-B cells. We sorted pro-B cells and small pre-B cells (Figure S4A) and showed that rearrangement at the pro-B cell stage was significantly altered, with Vκ genes near E88 in E88∆ mice more dramatically reduced, 30 to 65-fold, compared to WT (Figure 4A). Other genes along the Vκ locus have also reduced rearrangement that varies between 2 to 8-fold, including Vall. As expected, the pattern and degree of Vκ rearrangement at the small pre-B cell stage resembles rearrangement from unfractionated BM-derived CD19+ cells (Figure 4B). Similar repertoire perturbations were observed in B cell populations from day 17 fetal liver (mostly pro-B, Figure S4B) and spleen (Figure S4C). As we observed in the 445.3 cells, the effect of E88∆ on rearrangement in gDNA and cDNA from pre-B cells is very similar indicating that E88∆ affects V gene rearrangement directly (Figure 4C).
Figure 4. Vκ rearrangement during B cell development and repertoire analysis of E88∆ mice.
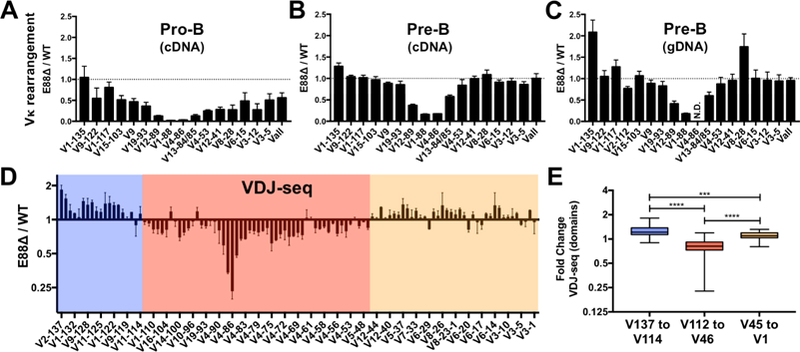
(A-C) BM-derived CD19+ cells from WT and E88∆ mice were purified and sorted for pro-B cells and small pre-B cells. Pro-B cell RNA (A), small pre-B cell RNA (B) and small pre-B cell gDNA (C) were harvested and analyzed for Vκ rearrangement by qPCR. Data was normalized to GAPDH for RNA and to a DNA-loading control (Eµ) for gDNA and is shown as a ratio of E88∆ / WT. N.D.= not detected for WT or E88∆. (D) Small pre-B cell gDNA was analyzed by deep sequencing (VDJ-seq). Data is the average of two experiments and is expressed as the ratio of E88∆ / WT of the percentage of total reads per sample ± SEM. Domains where the majority of the Vκ gene rearrangements are underrepresented (pink box) or overrepresented (blue and tan boxes) in the E88∆ mice are indicated. (E) VDJ-seq results were separated into 3 groups corresponding to the 3 colored regions in panel D and analyzed. T-test was used to calculate significant differences between groups (***p<0.001, and ****p<0.0001).
Vκ rearrangement deep sequencing of E88∆ mice reveals domains of E88 influence
To examine the effects of E88∆ on every Vκ gene in the Igκ repertoire, we performed unbiased amplification of gDNA from cell sorter-purified small pre-B cells of WT and E88∆ mice followed by paired end sequencing (VDJ-Seq) (Bolland et al., 2016). Strikingly, VDJ-seq data showed that changes in the relative usage of all Vκ genes occurred in a domain-specific manner (Figure 4D) in E88∆ mice. We found that almost all Vκ genes in a 1.5 Mb region between V12–46 and V2–112 (pink box) are decreased in rearrangement in E88∆ pre-B cells relative to WT. In contrast, Vκ genes in the two flanking regions V11–114 to V2–137 (blue box) and V3–1 to V5–45 (tan box) have modestly increased relative rearrangement in E88∆ mice, with the genes at the far distal end showing the biggest increase (V1–135 and V2–137). Although the extent of increase or decrease was modest, it was very consistent through these large regions, and we found highly significant differences relative to WT in the fold change within these 3 groups (Figure 4E). As we observed with our qPCR data, the Vκ genes closest to the E88 region (V4–86, V1–88, and V12–89) are the most severely reduced (Figure 4D). Altogether, we find that E88 regulates global patterns of rearrangement, revealing the concept that Vκ rearrangement is regulated in a domain-specific manner.
E88∆ influences individual Vκ rearrangement in a subTAD-specific manner
Topologically associating domains (TADs) are genomic regions that have high levels of chromatin interactions within a specific domain (Dixon et al., 2012; Nora et al., 2012). TAD boundaries are enriched for CTCF and cohesin binding, with CTCF binding sites predominantly in a convergent orientation (Rao et al., 2014). Inside TADs, smaller self-interacting domains have been designated subTADs (Phillips-Cremins et al., 2013). They also often have CTCF at their boundaries and frequently emerge in a cell-type specific manner. Our VDJ-seq data showed that changes in V gene rearrangement upon E88 deletion occurred in a domain-specific manner. To determine whether these changes correlated with potential subTADs in the Igκ locus, we used published IL7-cultured pro-B cell Hi-C data (Lin et al., 2012) to generate a heatmap of the normalized intrachromosomal interaction frequencies (Figure 5A). This heatmap revealed the presence of at least 5 subTADs (A to E) in the Igκ locus. Other overlapping subTADs are also evident, probably as a result of the highly complex CTCF landscape in the Igκ locus. Boundaries of the subTADs correlated with highly enriched binding of CTCF and cohesin subunit Rad21 (Figure 5B). Two subTADs, C and D, overlapped with the location of the Vκ rearrangement domain affected by E88∆ (Figure 5C, pink box) and have CTCF motifs in convergent orientation at their putative boundaries (Figure 5D). CTCF112 and CTCF85 are at the likely boundaries of subTAD C, and CTCF84 and a cluster of CTCF sites (CTCF48, 46 and 45) are at subTAD D boundaries. The domains with increased Vκ rearrangement in E88∆ (blue and tan boxes) overlapped with subTADs A plus B or subTAD E, respectively. We conclude that the effects of the E88∆ on Vκ rearrangement are likely mediated in a subTAD-specific manner.
Figure 5. E88 regulates Vκ rearrangement in a subTAD-specific manner.
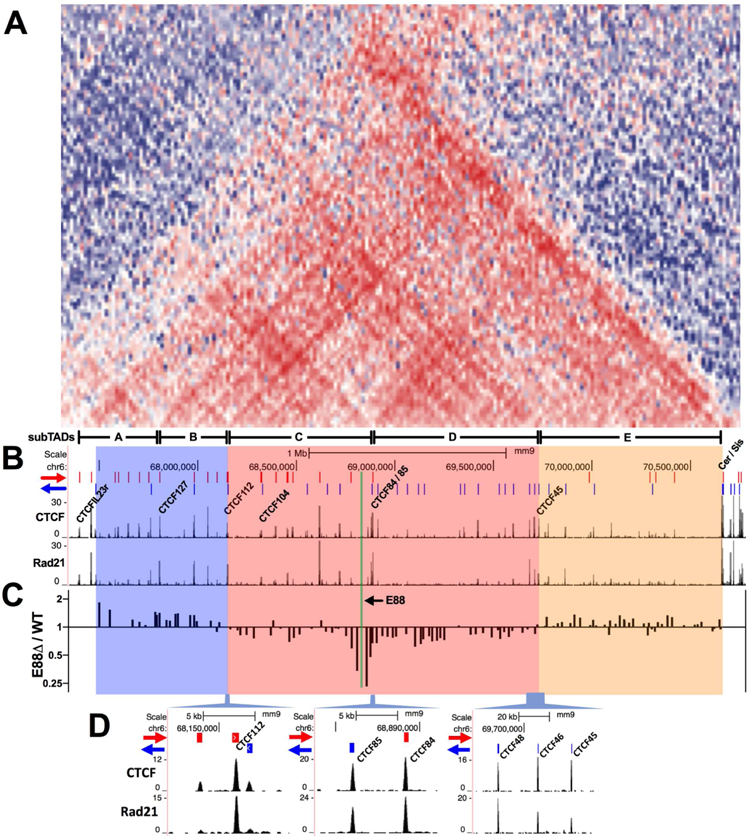
(A) Hi-C interaction frequencies expressed as a heat map over the Igκ locus. Blue areas represent lower than expected while red areas represent higher than expected relative interaction frequencies. Identified subTADs are indicated at the bottom (A to E). (B) ChIP-seq for CTCF and Rad21 in Rag1−/− pre-B cells . Red and blue lines at the top indicate the orientation of the CTCF binding sites (Loguercio et al., 2018). (C) V gene rearrangement frequencies in the Igκ locus expressed as E88∆ / WT ratio in their respective genomic location. (D) Close-up of the CTCF sites located at the putative subTADs boundaries of the area affected by the E88∆.
The 3D conformation of the Igκ locus is altered by E88 deletion in pro-B cells
To assess whether changes in 3D structure of the Igκ locus are caused by the deletion of E88, we performed 4C at the pro-B cell stage from BM-derived CD19+ cells from WT.Rag−/− and E88∆.Rag−/− mice. 4C offers a high-resolution interaction profile from a specific viewpoint and is ideal for complex genetic loci (Stadhouders et al., 2013). Using E88 as the viewpoint, we found that there are two sets of LRCI that are visibly E88 dependent (Figure 6A). The first one is from E88 to the 5’ end of the locus where the most distal Vκ gene is located, a location which also has the characteristics of an enhancer (E137) (Figure S5). The second set of LRCI is to the regulatory region at the 3’ end of the locus. A closer inspection of these latter LRCI shows that E88 interacts with many regulatory elements including the CTCF-containing elements Cer and Sis and all three known enhancers (Figure 6B). There is also an increase in local interactions downstream of E88 in E88∆ compared to WT pro-B cells (Figure 6A, highlighted green). Using iEκ as a viewpoint, we see that the E88 area is a major site of LRCI with iEκ in WT pro-B cells (Figure 6C). This data also shows that these LRCI are mediated by E88 (Figure 6D). Since the interaction of iEκ with E88 is the main prominent interaction in pro-B cells, these data suggest that the E88-dependent LRCI may be responsible for initiating proper folding of the Igκ locus.
Figure 6. LRCI in the Igκ locus are altered by E88 deletion at the pro-B cell stage.
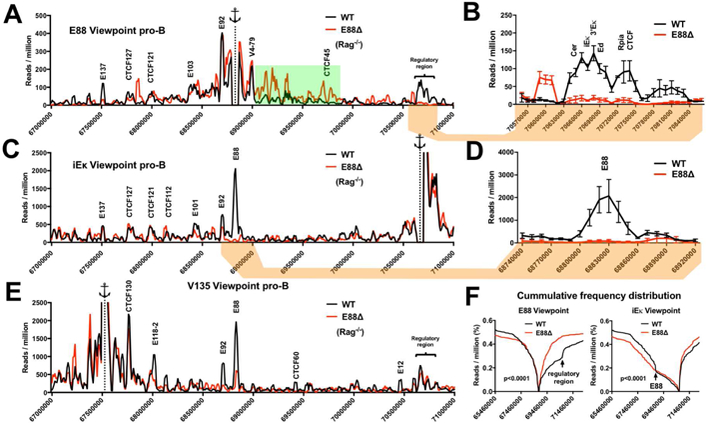
4C libraries from WT.Rag−/− and E88∆.Rag−/− CD19+ pro-B cells were assessed by deep sequencing. (A, C, E) 4C interactions plots for the Igκ locus from E88, iEκ, or E135 viewpoints for WT (black) or E88∆ (red). The interaction frequencies are expressed as reads per million (RPM) in 10 kb windows using a running window of 30 kb for the indicated genomic locations. The anchor represents the viewpoint. Green highlight indicates an area with higher density of specific LRCI in E88∆ pro-B cells. (B, D) Close-up view of indicated areas showing the average of the 3 experiments ± SEM. (F) Cumulative frequency distributions (CFD) from E88 and iEκ viewpoints show the cumulative 4C reads in E88∆ compared to WT pro-B cells (p<0.0001). Mann-Whitney test was used to calculate the statistically significant difference between CFD from WT and E88∆. Data is the average of 3 independent biological samples. Significant differential 4C interactions between the compared cell types are shown in Table S3.
To appreciate changes in interaction frequency more clearly, we used cumulative frequency distribution (CFD) to determine the number of 4C reads upstream or downstream of the viewpoint. CFD from the E88 viewpoint shows a substantial increase in interactions downstream of E88 in the E88∆ pro-B cells compared to WT pro-B cells (Figure 6F, left). CFD from the iEκ viewpoint shows an increase of interactions downstream of iEκ in E88∆.Rag−/− pro-B cells (Figure 6F, right). Additionally, a flattening shift in the trend of the upstream interactions beginning at the E88 location is seen in E88∆ cells (Figure 6F, right, arrow). This suggests that E88 is a crucial element to focus the interactions from iEκ to the V gene region of the locus.
Since E88 and iEκ both appear to interact with the distal part of the locus (Figures 6A and 6B), we used a region close to the V1–135 gene as viewpoint (called E135 since it had epigenetic enhancer marks) (Figure S5). We found that this region strongly interacts with E88 and iEκ (Figure 6E). E135 interacts with iEκ in both WT and E88∆ cells (Figure 6E) indicating that this E135-iEκ interaction is not E88 dependent. These data demonstrate that iEκ, E88, and the distal end of the locus are in close physical proximity at the pro-B cell stage and that E88 mediates the LRCI of the E88 region with the distal and proximal regions of the Igκ locus.
Dynamics of LRCI between pro-B and pre-B cells shape the 3D structure of the Igκ locus
The majority of Vκ gene rearrangement occurs at the pre-B cell stage. This requires a mechanism that allows all V genes across the Igκ locus to have a comparable opportunity to come in proximity to the Jκ genes. Accordingly, during the transition from WT pro-B cells to WT pre-B cells, there is a redistribution of interactions coming from elements in the regulatory region adjacent to the Jκ genes (Stadhouders et al., 2014). Using E88 as a viewpoint, we see that the developmentally-associated, pro-B to pre-B stage, E88 interaction patterns are significantly different in the range and magnitude (Figure 7A). Interactions from the E88 viewpoint towards putative enhancers E101 and E103 (Figure S5, for epigenetic marks) show distinct pre-B cell-specific peaks. Importantly, interactions from the E88 viewpoint going downstream of V4–79 to CTCF45, mostly to subTAD D, are greatly increased in the pre-B cell stage (Figure 7A, green highlight). We also noticed that LRCI from the E88 viewpoint had a change in the relative magnitude at specific locations (area between red dotted lines, Figure 7A). This change occurred between the CTCF45 and CTCF104 binding sites. CTCF45 is one of the putative boundaries of subTAD D and of the E88 zone of influence for rearrangement (Figures 5), suggesting that distribution and amount of certain E88 interactions can be limited by CTCF binding and are developmentally regulated.
Figure 7. LRCI are developmentally regulated and correlate with alterations in V gene rearrangement frequency.
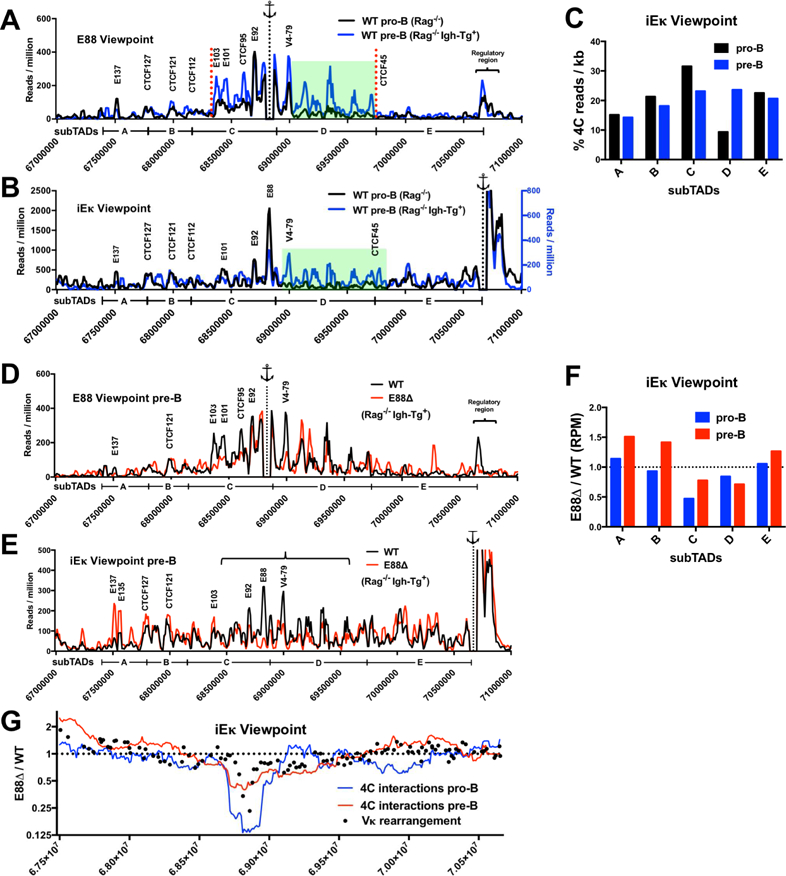
(A, B) 4C interactions plots from E88 (A) and iEκ (B) viewpoints are shown for WT.Rag−/− pro-B (black lines) and WT.Rag−/− Igh-Tg+ pre-B (blue lines, right axis in panel B) cells. Green highlights show the interactions that are significantly increased in pre-B cells compared to pro-B cells. (C) 4C reads from the iEκ viewpoint in the indicated subTADs expressed as a percentage of RPM per kb for the Igκ locus. (D, E) 4C interaction plots in pre-B cells from WT.Rag−/−.Igh-Tg+ (black line) and E88∆.Rag−/−.Igh-Tg+ (red line) from E88 and iEκ viewpoints are shown. 4C interaction frequencies are expressed as RPM in 10 kb windows using a running window of 30 kb. (F) Ratio of E88∆ / WT normalized 4C reads for the indicated subTADs in pro-B and pre-B cells from the iEκ viewpoint. (G) Running window (300 kb) analysis of the normalized 4C reads is shown for pro-B cells (blue line) and pre-B cells (red line), and expressed as the ratio of the E88∆ / WT. This data is overlapped with the VDJ-seq rearrangement frequency data (black dots) from mice expressed as the ratio of E88∆ / WT. Data is the average of 3 (pro-B) or 2 (pre-B) independent biological samples. Significant differential 4C interactions between the compared cell types are shown in Table S3.
Notably, the LRCI from the iEκ viewpoint into the V region show a much more uniform distribution along the entire locus in WT pre-B cells compared to WT pro-B cells (Figure 7B). The E88 interaction peak is not as prominent compared to other peaks as it was in pro-B cells. Additionally, as from the E88 viewpoint, interactions from iEκ to the region between V12–38 and V33–84 are more frequent compared to those seen in pro-B cells indicating that these interactions are largely pre-B cell specific (Figure 7B, green highlight). This is noticeably evident in Figure 7C, where the % of 4C interactions from CTCF45 to CTCF84, subTAD D, is increased ~2.5-fold in WT pre-B cells compared to WT pro-B cells. Thus, 4C data show that interactions from iEκ in WT pro-B cells have a bias for the distal half part of the locus and that some regions in the middle of the locus downstream of E88 are virtually not interacting with the regulatory region. In contrast, WT pre-B cells show a more uniform distribution of interactions from iEκ. In summary, this 4C data shows that the Igκ locus has very different type of contracted structure at the WT pro-B compared to WT pre-B cell stage suggesting that these changes may play a role in Vκ rearrangement.
E88∆ changes the LRCI pattern locus wide and shapes the Igκ repertoire
To evaluate LRCI changes caused by E88 deletion in the Igκ locus at the pre-B cell stage, we performed 4C from E88 and iEκ viewpoints with pre-B cells from WT.Rag−/−.Igh-Tg+ and E88∆.Rag−/−.Igh-Tg+ mice. The LRCI from E88 to the 3’ regulatory region and to the distal end of the locus (near E137) are impaired in the E88∆ pre-B cells, similar to the observations made at the pro-B cell stage (Figure 7D). Other interactions from the E88 viewpoint also show significant reduction in the E88∆ pre-B cells. These reduced interactions were to some CTCF sites but were predominantly to other putative enhancer elements, most notably at E103, E101, and other H3K4me1 and H3K27Ac enriched areas such as the one near V4–79 (Figure 7D).
Because the location of the iEκ viewpoint is just 0.3–1.6 kb from the J genes, LRCI from this viewpoint can be considered as an approximate measure of the LRCI frequency with Jκ genes, hence pertinent for the assessment of rearrangement frequency. As expected, LRCI from the iEκ viewpoint to the E88 area are essentially absent in the E88∆ pre-B cells (Figure 7E). Other than this, the pattern of LRCI from iEκ to the Vκ region is moderately similar between WT and E88∆ pre-B cells. Significantly, LRCI from iEκ to the distal portion of the locus near E137 and E135 are more prominent in E88∆ pre-B cells. In contrast, LRCI peaks from the iEκ viewpoint to a region around E88 and further downstream are lower (Figure 7E, bracket). To quantitate these changes, we evaluated the 4C data by calculating the total number of normalized reads within specific subTADs. As expected, subTAD C, which includes E88, is much lower in LRCI with iEκ in both E88∆ pro-B and pre-B cells, although less so in pre-B cells (Figure 7F). SubTAD D also shows a reduction in the number of reads. In contrast, subTADs A, B and E have an increase in the number of reads in E88∆ pre-B cells. These changes in interaction frequency of the 5 subTADs between WT and E88 from the iEκ viewpoint reflect the rearrangement pattern differences between WT and E88∆ mice.
To have a more detailed picture of how LRCI between iEκ and the entire V region affected rearrangements, we used a running window analysis of the 4C reads in pro-B cells and pre-B cells and overlapped this with rearrangement frequencies of individual V genes in pre-B cells (Figure 7G). There is a strong correspondence between the levels of iEκ to V region LRCI and Vκ rearrangement frequencies indicating that E88 regulates the ability of iEκ to interact with V genes throughout the locus, with the interaction data in pre-B cells showing a similar trend to the pre-B VDJ-seq repertoire data. We conclude that increased or decreased rearrangement correlates with increased or decreased interactions from iEκ to the Vκ genes, respectively.
Discussion
Changes in 3D structure of the AgR loci during the process of locus contraction are essential for the generation of a diverse repertoire by bringing all the V genes into proximity with their target J genes. Our finding that E88 is the most prominent hub of LRCI within the Igκ locus and is a major interaction site with the regulatory region in pro-B cells, a stage that precedes the majority of Igκ rearrangements, suggests that it plays a critical role in initiating or coordinating these conformation changes. Since the generation of a contracted conformation mediated by LRCI would provide proximity for all Vκ genes to the J genes, we predicted that disruption of this 3D structure at an early stage would affect normal V gene usage. In agreement with our hypothesis, we found that E88 deletion dramatically affected both the initial rearrangement rate and relative usage of individual Vκ genes. These observations shed light on how the Igκ locus topology and relative location of the V genes in the compacted locus affects the likelihood of individual V genes to be utilized in the repertoire during B cell development. Strikingly, VDJ-seq data revealed that the rearrangement of V genes in a wide domain of ~1.5 Mb are globally decreased, with an abrupt increase in Vκ segment usage on both sides of this domain in E88∆ relative to WT pre-B cells. All these domains correlated with the location of subTADs suggesting that E88 regulates Vκ rearrangement in a subTAD-dependent manner. It has not been previously reported that an enhancer in the middle of any AgR locus controls rearrangement frequencies of a large number of V genes. However, such enhancer-mediated subTAD-specific regulation might occur in other AgR loci since subTADs and enhancers have been predicted to exist in other AgR loci (Medvedovic et al., 2013; Montefiori et al., 2016; Predeus et al., 2014). This type of domain-specific regulation of rearrangement of a large cluster of V genes is a previously undescribed mechanism, and suggests that the 3D looping structure of the V gene region greatly influences the V gene rearrangement pattern.
Rag activity is largely confined within convergent CTCF binding sites that form chromosomal loop domains (Hu et al., 2015). Hu et al. propose a tracking mechanism for Rag to find its DNA targets but how Rag is able to access all the CTCF-bound domains in AgR loci is unknown. Our data support a model in which enhancer-mediated LRCI bring distinct CTCF-bound domains to the recombination center at J genes to allow Rag binding and subsequent scanning through the domain. Thus, formation of multiple CTCF-bound subTADs may regulate entire groups of V genes via enhancers within the subTADs by bringing that whole loop of V genes closer to the J genes. Deletion of the element, as we did here with E88, may result in the juxtaposition of a different set of domains to the J genes, giving them the recombinational advantage. Two recent papers demonstrated that in the absence of the CTCF-binding element IGCR1, the Igh enhancer (Eµ) associates with the next available CTCF site, creating a loop whose base is now adjacent to Vh81X instead of to IGCR1, and this altered loop highly promotes rearrangement of 81X (Jain et al., 2018; Qiu et al., 2018). However, deletion of the CTCF site adjacent to 81X greatly impairs 81X rearrangement, and instead results in increased utilization of the Vh gene adjacent to the next upstream CTCF site. Together, these observations illustrate that removal of regulatory elements, such as CTCF sites or enhancers, change long-range interaction patterns, and subsequently alter individual V gene usage.
Conformational analysis showed that E88 is a major site of interaction with iEκ in pro-B cells. E88 deletion reduced the frequency of iEκ LRCI to a large portion of the V gene part of the locus, mainly subTADs C and D suggesting that E88 anchors these subTADs to the regulatory region. Accordingly, since the LRCI from the E88 area to the iEκ are eliminated as a consequence of the E88 deletion, subTADs C and D are impaired in the ability to come to close proximity to the recombination center at the J genes. This has direct consequences for V gene rearrangement since spatial confinement is a major factor that determines the likelihood of V genes finding and interacting with (D-)J elements (Lucas et al., 2014). iEκ is still able to interact with subTADs C and D through other sites within these subTADs, but with a lower frequency.
The strength and prominence of the E88 interactions suggest that it plays a significant role in initiation and maintenance of Igκ locus 3D structure. The timing of E88 emerging as an enhancer suggests that it is required early at the pro-B cell stage. Developmentally-associated enhancers experience a step-wise activation involving TF binding, recruitment of chromatin remodeling complexes, and subsequent changes in chromatin modifications. H3K4me1 is a chromatin modification that marks the initial stage of enhancer activation (Calo and Wysocka, 2013; Mercer et al., 2011; Rada-Iglesias et al., 2011). H3K4me1 deposition around E88 is severely reduced in E88∆ cells suggesting that the normal progression of enhancer activation was interrupted. Enhancer-associated nascent H3K4me1 deposition is correlated with LRCI dynamics including proper chromatin interactions between enhancers and promoters (Dixon et al., 2015; Yan et al., 2018). Since E88 has the highest H3K4me1 enrichment of the V region in pro-B cells, it seems reasonable that regional loss of this modification in E88∆ cells affects initiation and proper conformation of the 3D structure of Igκ before rearrangement begins. Later, in pre-B cells, H3K4me1 levels increase locus-wide including around most functional Vκ genes, and correspondingly, E88 interactions with iEκ are not as prominent, but rather iEκ establishes LRCI more broadly across the Igκ locus.
E88 mainly affected transcription over a relatively small area yet it controls the level of rearrangement in a much broader region. In this way, rearrangement and GLT are functionally separated. Enhancers regulate transcription through LRCI with their cognate promoters (Levine et al., 2014). Instead of using LRCI to regulate transcription, E88-dependent LRCI may serve to connect the neighboring Vκ genes in subTADs C and D with the Jκ segments. Thus, E88 may be viewed as a structural element more than a transcriptional regulator. E88-dependent LRCI were primarily to enhancers in the regulatory region and to enhancer-like regions in the V gene area such as E137, E103, and E101. Also, since CTCF knock-out in pre-B cells does not significantly affect the LRCI from iEκ or 3E’κ to the V region except for the Jκ-proximal V segments (Ribeiro de Almeida et al., 2011), we hypothesize that enhancers, and more specifically enhancer-dependent LRCI, play a significant role in the 3D structure of the Igκ locus (Beagrie et al., 2017).
E88 LRCI extend across multiple subTAD boundaries. CTCF limits the interactions of iEκ and 3’Eκ enhancers to the proximal V gene region suggesting that the regulatory region is partially isolated in a subTAD (Ribeiro de Almeida et al., 2011). Accordingly, most of the interactions emanating from E88 are limited to an area demarcated by convergent CTCF binding sites. In pro-B cells, E88 shows major LRCI to the regulatory region at the 3’ end of the locus. This suggests that E88 can drive and sustain interactions beyond subTAD boundaries towards other enhancers. This enhancer-mediated framework is fundamental at the pro-B cell stage for the natural progression of Vκ gene rearrangement at the pre-B cell stage. Enhancer-regulated LRCI allow the locus to have a poised contracted conformation in pro-B cells. As pro-B to pre-B cell differentiation proceeds, the pro-B to pre-B cell 3D structure transitions, allowing an “equal opportunity” starting point for all the subTADs present in the large 3.1 Mb V region to engage with the recombination center at the J genes.
STAR Methods
Cells.
The Abelson virus transformed pro-B cell lines 445.3 and 22D6 were maintained in RPMI media supplemented with 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine, 1% Penicillin/Streptomycin and 0.05 mM β-mercaptoethanol. 445.3 cells are RAG1-deficient, and were kindly provided by Dr. Amy Kenter (Kumar et al., 2013) . 293T, J558 and EL4 cells were maintained in DMEM supplemented with 10% FBS, 1% Penicillin/Streptomycin, and 2 mM L-glutamine.
Hi-C data analysis.
Previously published Hi-C data from IL7-cultured pro-B cells was used to calculate the number of reads per 10 kb bins (Lin et al., 2012). Hi-C data has two chromosomal coordinates, one for each end of the detected interaction. We sorted and counted all the reads at each specific 10 kb bin, regardless of the end of the interaction they were part of, within the Igκ locus. Interaction frequency heat map was generated as described in (Lin et al., 2012)
Luciferase Assay.
E88 enhancer region for cloning was generated by amplifying genomic DNA from WT cells (see primers in (Table S1)). The 1088 bp PCR-generated DNA was cloned downstream of the firefly luciferase gene in the pGL3 vector (Promega), along with a V gene promoter, using the indicated restriction sites (see primer list). 22D6, J558 and EL4 were transfected at a ratio of 200 ng of experimental plasmid and 20 ng of control plasmid (pTK-renilla) per 2×105 cells using Neon Transfection System (Thermo Fisher Scientific) (Solution R, Program 004). Cells were plated in 24-well plates (500 µl per well) and incubated for the indicated times at 37° C and 5% CO2. Cells were harvested, washed with PBS, lysed using passive lysis buffer (15 min), and luciferase activity was measured. Fold induction = (Firefly luciferase experimental vector/Renilla luciferase cotransfected with experimental vector)/(Firefly luciferase control vector/Renilla luciferase cotransfected with control).
CRISPR-Cas9 mediated mutagenesis.
For the generation of the 244 bp E88 deletion in the 445.3 cells, we used the SBI PrecisionX ™ HR Target ing Vector (HR110PA-1) which allows the creation of a custom mutation using the CRISPR-cas9 system. The HR110PA-1 template vector contains a selectable marker cassette and custom-made flanking regions (homology arms) to the targeted deletion (HR110PA-1-E88). Homology arms were made by PCR from 445.3-WT gDNA with the indicated primers. Guides for E88 region were designed using http://crispr.mit.edu website and were cloned into the pX335-U6-Chimeric_BB-CBh-hSpCas9n vector (Cong et al., 2013). We used pX335-U6-Chimeric_BB-CBh-hSpCas9n which is a version of the Cas9 enzyme that creates single stranded nicks in the target DNA. The use of nickases is recommended to reduce off-target effects. HR110PA-1-E88 and pX335-U6 containing sgRNAs specific for E88 were co-transfected into 445.3 cells using a Neon electroporator and were incubated for 48 hours before addition of puromycin for 72 hours to select for cells which had taken up the vectors. Red fluorescent protein (RFP)+ cells were single cell-sorted, and clones were tested by PCR to confirm bi-allelic deletion of the targeted region. Clones with bi-allelic deletion were transformed with a retrovirus expressing GFP and Cre recombinase to excise the RFP expression cassette. Five days after transformation, GFP+ cells were sorted in bulk. Excision of the RFP cassette was confirmed by PCR.
Assessment of CRISPR/Cas9 guide RNA for targeted deletion of the E88 region in mice was tested in 293T cells using a pCAG-EGxxFP plasmid (Figure S2D) (see full protocol in (Mashiko et al., 2013)). The pCAG-EGxxFP vector was used to clone an 800 bp region containing the E88 enhancer (eGx-E88-xFP) in the multiple cloning site inside of the GFP gene of this vector. The pX330-U6-Chimeric_BB-CBh-hSpCas9 vector (Cong et al., 2013) was used to express the E88 specific gRNAs (pX330-E88g3 and pX330-E88g4).Transfections were done using Lipofectamine 2000 with 1 µg of the GFP vector and 1 µg of the pX330 vector following the supplier recommendations. Cells were assessed 48 hours later for GFP expression.
ChIP/qPCR.
ChIP was performed as previously described (Degner et al., 2011). In brief, 445.3 Rag−/− and 445.3-E88∆.Rag−/− cells were cross-linked with 1% formaldehyde for 10 min at room temperature. Lysates were prepared and were sonicated using a Diagenode Biorupter. Chromatin complexes were precleared with salmon sperm DNA-protein A agarose beads. Chromatin complexes were immunoprecipitated with antibodies against H3K4me1 (Abcam Cat# ab8895). After elution, complexes and input DNA were reverse crosslinked at 65° C overnight. DNA was purified using the Qiagen DNA purification kit. ChIP was followed by a quantitative PCR. Data is presented as fold enrichment over input and relative to WT controls.
Mice.
C57BL/6 wild-type and mutant mice were maintained in our breeding facility in accordance with protocols approved by The Scripps Research Institute Institutional Animal Care and Use Committee. Both male and female mice were used. Ages of mice were 6–10 weeks, except for the fetal livers from embryos at day 17 of gestation. Human heavy chain (Igh-Tg+) transgenic mice (Nussenzweig et al., 1987) were kindly provided by Dr. Cornelis Murre (UCSD).
Generation of the E88∆ mice by CRISPR/Cas9.
To make germline mutations in mice, we used gRNAs specific for the E88 region (E88g3 and E88g4). Pronuclear injection of Cas9 mRNA and sgRNAs E88g3 and E88g4 (Figure 3A) was done in mouse zygotes in our Mouse Genetics Core facility (see full protocol in (Mashiko et al., 2013)).
Fluorescence Activated Cell Sorting (FACS).
Total BM cells were harvested from groups of 4–5 mice 6 to 10 weeks old. BM-derived CD19+ cells were isolated using anti-CD19 conjugated MACS beads (Miltenyi, Auburn CA). CD19+ cells were pre-incubated with CD16/32 Fc Block for 5 min. Subsequently, CD19+ cells were stained with antibodies for CD19 (BV421), CD93 (PE/Cy7), IgM Fab fragment (AF647), CD2 (PE), CD43 (BV510). Sorting scheme is ilustrated in Figure S4A.
RNA and gDNA analysis.
RNA extraction was performed from 5–10×106 cells disrupted with QIAshredder (Cat. #79654, Qiagen) and subsequently processed with the RNeasy™ Plus Mini Kit according to manufacturer instructions. First strand cDNA synthesis was done with 1 µg of total RNA with the QuantiTect® Reverse Transcription. DNA was isolated from approximately 5–10×106 cells using the DNeasy Blood and Tissue Kit. Quantitative real-time PCR was performed with the Bio-rad CFX Connect™ Real-Time PCR Detecti on System using SYBR Green Master Mix for cDNA, or TaqMan™ Fast Universal PCR Master Mix for gDNA. Data was normalized by the expression level of GAPDH (RNA) or Eu (gDNA) for each sample.
Circular chromatin conformation capture (4C).
Preparation and analysis of the 4C libraries was adapted from protocols in Stadhouders, et al 2014 and van de Werken, et al, 2012. In brief, WT.Rag−/−, E88∆.Rag−/−, WT.Rag1−/−Igh-Tg+ or E88∆.Rag1−/−Igh-Tg+ BM-derived CD19+ cells were isolated by column separation using anti-CD19 conjugated magnetic beads and crosslinked with 2% formaldehyde for Rag−/− samples and 1% formaldehyde for Rag1−/−Igh-Tg+ samples. BglII and NlaIII were used as primary and secondary restriction enzymes respectively. Inverted PCR was done with Expand™ Long Template PC R System from multiple viewpoints and the resulting products were sequenced using an Illumina NextSeq 500 sequencer with 75 bp single-end reads. The reads were trimmed using cutadapt v1.14 for 3’ and 5’ adapters after removing low-quality ends (Q cutoff = 20) (Martin, 2011). Reads with length of at least 30 bases were retained. Trimmed reads were then aligned to mouse (Mus musculus) genome (UCSC version mm9) using Bowtie2 v2.2.9 (Langmead and Salzberg, 2012). The bedgraph files reporting the summaries of genome coverage by chromosome regions were obtained from the alignments (bam files) using bedtools v2.17.0 (Quinlan and Hall, 2010). Data from bedgraph files was normalized for the size of the library and expressed in reads/million. Reads were binned in 10 kb windows and smoothed with a 30 kb running window for display.
Retrovirus preparation and transduction.
To produce the Rag1 expressing retrovirus, 293T cells at 80% confluence were transfected with the pMSCV-IRES-Bsr-Rag1, which contains full-length WT mouse Rag1 gene and a blasticidin selectable marker, and pEco plasmids, which expresses the ecotropic envelope protein under the control of the CMV immediate-early promoter, using Mirus transfection reagent following the manufacturer’s recommendations. Supernatant was harvested at 48 and 72 hours post transfection. For in vitro rearrangement experiments, 445.3 cell lines were transduced with pMSCV-IRES-Bsr-Rag1 retrovirus. After 48 hours, cells were treated with blasticidin (20 µg/ml) for seven days to generate the lines with constant Rag1 expression. The cells were stimulated with STI571 after the 7 days of blasticidin culture, or after thawing the freshly transduced and short term blasticidin treated cells, in order to avoid the potential for any background rearrangements to occur during excessive culture time.
gDNA Library Preparation for Igκ Chain Repertoire Sequencing (VDJ-seq)
gDNA was isolated from sorted small pre-B cells (Figure S4A). gDNA was processed following the V(D)J-Seq protocol as described, omitting the negative depletion step and using different Jκ primers (Bolland et al., 2016; Matheson et al., 2017). Custom Igκ light chain primers were used for library preparation (Table S1) (EK, SL, and AJF, submitted). Library barcoding was performed using NEBNext Multiplex Oligos for Illumina (E7600S). Samples were paired-end 2×300 sequenced on an Illumina MiSeq System (San Diego, CA) at our Next Generation Sequencing Core.
Quantification and Statistical Analysis
The Standard Deviation (SD) of the number of interactions per 10 kb bin was calculated using excel for the 331 bins from Chr6:67400000 to Chr6:70710000 (Figure 1A). The correlation coefficient (r) was calculated using Prism 7 (GraphPad Software) (Figure 1E).T-test was used to calculate significant differences between the fold change (E88∆ / WT) of the % each gene of the total rearrangements between groups (Figure 4E) using Prism 7 (GraphPad Software). Mann-Whitney test was used to calculate the statistically significant difference between CFD from WT and E88∆ using Prism 7 (GraphPad Software) upstream or downstream of the viewpoint. SEM is shown for each set of the qPCR or luciferase experiments.
For analysis of 4C interactions significant difference between conditions, mapped reads were binned at 2 kb resolution. To smooth the data, the average signal of the running window of 30 kb centered at each bin was calculated. The resulting raw count tables were inputted to DESeq2 (Love et al., 2014) for detecting differential contacting bins across samples. False discovery rate (FDR) adjusted p-value of < 0.1 (significant) or < 0.01 (highly significant) was used as the cutoff (Table S3)
GEO accession numbers.
All ChIP-seq are referenced in Table S2 with their GEO accession numbers.
Primers.
See Table S1 for sequences and details.
Supplementary Material
Table S1. Primers, probes, and oligonucleotides (related to Figures 1, 2, 3, 4, 6, 7, S1, S2, and S3)
Figure S1, (related to figures 1 and 2). E88 ChromHMM analysis, sequence and DHS specificity. (A) ChromHMM assessment of the E88 region done by (Predeus et al., 2014) in pro-B cells. Chromatin states marks: (10), H3K4me1; (19), PU.1; (23), H3K4me2; (34), Pax5; (37), E2A; (1), Mediator and H3K4me1; (9), H3K4me2 and H3K4me1; (16), H3K36me3 and H3K4me1; (17), H3ac and H3K4me1; (6), H3ac, H3K4me2 and H3K4me1; (3), DHS, E2A, H3K4me1, PU.1 and Mediator; (4), DHS, H3K4me3, H3K4me2, H3K4me1, H3ac, PU.1, Mediator, E2A, Pax5 and P300; (5), DHS, H3K4me2, H3K4me1, PU.1 and Mediator; (7), DHS, H3K4me3, H3K4me2, H3K4me1 and H3ac; (35) and (36), empty states. Indicated are the deletions made in 445.3 cells and mice (largest). (B) E88 sequence deleted in 445.3 cells with putative E2A and EBF binding sites highlighted in yellow and blue respectively. (C) DNase-seq data around the E88 area in multiple cells and tissues (Vierstra et al., 2014). An area around the GAPDH gene was used as DHS control for B cell specificity. UCSC Genome Browser views show the mapped read coverage of DNase-seq.
Figure S2, (related to figures 2 and 3). Effects of E88∆ in Vκ rearrangement pattern and early kinetics in 445.3 cell lines. (A) Quantification of Vκ rearrangement on gDNA by qPCR (TaqMan) with specific V gene primers in 445.3-WT or 445.3-E88∆ cells at 48 hours after STI571 stimulation. Data is normalized with a loading gDNA control (Eu) and is expressed as the ratio of E88∆ / WT. (B) Quantification of Vκ rearrangement on RNA by qPCR in 445.3-WT or 445.3-E88∆ cells at 0, 12, and 24 hours after STI571 stimulation with the Vall primer, which gives an estimated measure of the total rearrangement. Data is expressed relative to GAPDH. (C) Relative rate of total rearrangement (Vall) shown as the ratio of WT / E88∆ normalized to t=0 at the indicated time points. Data in A, B and C is representative of at least three independent experiments ± SEM. N.D.= not detected for WT or E88∆. (D) Assessment of sgRNAs specificity and efficiency. pX330-E88g3 and pX330-E88g4 plasmids were tested for efficiency of targeting of the E88 region using the eGx-E88-xFP reporter plasmid (Mashiko et al., 2013). GFP expression indicates that the sgRNA-guided CAS9 endonuclease targets the DNA inserted in the multiple cloning site (MCS) in the middle of the GFP gene. Indicated plasmids were cotransfected in 239T cells and assessed for GFP expression 48 hours post-transfection. (Top left) eGx-E88-xFP plasmid cotransfected with a pX330 plasmid expressing a gRNA not specific for the E88 region. The eGx-control.DNA-xFP plasmid, containing a control DNA fragment that is not targeted by E88g3 or E88g4, cotransfected with pX330-E88g3 (top center) or the pX330-E88g4 (left bottom) plasmids. The eGx-E88-xFP plasmid cotransfected with pX330-E88g3 (top right) or pX330-E88g4 (middle bottom) plasmids. Control cells that were not transfected (right bottom). Pictures are from one of the two experiments performed.
Figure S3, (related to figure 3). E88 enhancer regulates Vκ gene utilization in mice. E88 was deleted in mice using CRISPR/Cas9 editing system. Schematic of the different sized E88 deletions in mice is shown in Figure 3A. DS=downstream, US=upstream. (A-D) BM-derived CD19+ cells were purified, and RNA was harvested. Vκ rearrangement was assessed by qPCR for specific individual Vκ genes for all the mouse lines. Data was normalized with GAPDH and expressed as E88∆ / WT ratio ± SEM. Two to five mice 6–10 weeks of age were used for each experiment. Data was collected from at least three independent biological samples.
Figure S4, (related to figure 4). Sorting scheme for pro-B and small pre-B cells and Vκ rearrangement in fetal liver cells and spleen. CD19+ cells were isolated from BM-cells from WT and E88∆ mice using CD19-conjugated MACS beads. (A) CD19+ cells were stained with antibodies against CD19, CD93, CD2, CD43 and IgM. Sorted pro-B cells (CD19+ CD93+, IgM−, CD2−, CD43+) and small pre-B cells (CD19+ CD93+, IgM−, CD2+, CD43-) were used to isolate RNA or gDNA for qPCR analysis or deep sequencing. Pre-B cells were separated as large or small based on the forward scatter (FSC). (B, C) Vκ rearrangement in fetal liver and spleen CD19+ cells. Isolated CD19+ cells from fetal liver of embryos at day 17 of gestation or spleens from 6–10 week-old mice were used to extract RNA. Quantification of Vκ gene rearrangement was done by qPCR with specific primers for the indicated Vκ genes. Data is shown as the ratio of E88∆ / WT and was normalized to GAPDH expression. Data is representative of at least three independent experiments ± SEM.
Figure S5, (related to figures 6 and 7). Chromatin profile and transcription factor binding for other identified enhancer-like regions. UCSC Genome Browser presentation of ChIP-seq datasets for the indicated putative enhancer regions. H3K4me1 (non-IL7-cultured RAG−/− pro-B cells and IL7-cultured RAG−/− pro-B cells), Mediator (v-Abl transformed pro-B cell line), and the following ChIP-seq using IL7-cultured RAG−/− pro-B cells: H3K27ac, Brg1, EBF, E2A, IRF4, PU.1, and PAX5. E92 has ~89% sequence similarity with E88 over ~ 650 kb. Note that the scales are different for the ChIP-seqs tracks for the different enhancers.
Table S2. ChIP-seq and RNA-seq datasets (related to Figures 1, 2, 3, 4, and S4)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD16/32 Fc Block | Biolegend | Cat# 562440 |
| CD19 | BD biosciences | Cat# 562440 |
| CD93 | Biolegend | Cat# 136506 |
| IgM Fab fragment | Jackson ImmunoResearch | Cat# 115–607-020 |
| CD2 | Biolegend | Cat# 100108 |
| CD43 | BD biosciences | Cat# 563206 |
| H3K4me1 | abcam | Ab8895 |
| anti-CD19 conjugated MACS beads | Miltenyi Biotech | 130–052-201 |
| Bacterial and Virus Strains | ||
| Rag1-expressing retrovirus | This paper | |
| Cre-expressing retrovirus | This paper | |
| Biological Samples | ||
| B6 CD19+ cells (bone marrow) | This paper | |
| E88Δ CD19+ cells (bone marrow) | This paper | |
| small E88Δ CD19+ cells (bone marrow) | This paper | |
| g3-g4Δ CD19+ cells (bone marrow) | This paper | |
| g3-USΔ CD19+ cells (bone marrow) | This paper | |
| g3-DSΔ CD19+ cells (bone marrow) | This paper | |
| B6.Rag1−/− CD19+ cells (bone marrow) | This paper | |
| E88Δ.Rag1−/− CD19+ cells (bone marrow) | This paper | |
| B6.Rag−/− Igh-Tg+ CD19+ cells (bone marrow) | This paper | |
| E88Δ.Rag−/−Igh-Tg+ CD19+ cells (bone marrow) | This paper | |
| B6 CD19+ cells (spleen) | This paper | |
| E88Δ CD19+ cells (spleen) | This paper | |
| B6 CD19+ cells (liver E17) | This paper | |
| E88Δ CD19+ cells (liver E17) | This paper | |
| B6 pro-B cells (bone marrow) see sorting scheme Figure S4A | This paper | |
| E88Δ pro-B cells (bone marrow) see sorting scheme Figure S4A | This paper | |
| B6 small pre-B cells (bone marrow) see sorting scheme Figure S4A | This paper | |
| E88Δ small pre-B cells (bone marrow) see sorting scheme Figure S4 | This paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| STI571 (Gleevec) | Toronto Research Chemicals | Cat#407000 |
| Formaldehyde 37% | Fisher Scientific | BP531–25 |
| RPMI Medium 1640 | Gibco | 11875–093 |
| BglII (Restriction Endonuclease) | NEB | R0144S |
| Blasticidin | Lifetech Invitrogen | R21001 |
| NlaIII (Restriction Endonuclease) | NEB | R0125S |
| Critical Commercial Assays | ||
| DNeasy Blood & Tissue Kit | QIAGEN | Cat#69504 |
| RNeasy Plus Mini Kit | QIAGEN | Cat#74134 |
| QIAshredder | QIAGEN | Cat#79654 |
| QuantiTect® Reverse Transcription | QIAGEN | Cat#205313 |
| TaqMan™ Fast Universal PCR Master Mix (2X) | Applied Biosystems | Cat# 4352042 |
| SYBR Green Master Mix | Bio-rad | Cat#1725270 |
| Expand™ Long Template PCR System | Roche | Cat#11681842001 |
| SYBR Green Master Mix | Biotool | Cat#B21203 |
| Deposited Data | ||
| See Table S2 | ||
| Experimental Models: Cell Lines | ||
| Abelson virus transformed pro-B cell lines 445.3 (Rag1−/−) | (Kumar et al., 2013) | |
| Abelson virus transformed pro-B cell lines 22D6 | (Yancopoulos and Alt, 1985) | |
| 445.3-E88Δ.Rag−/− | This paper | |
| J558 | ATCC ® | TIB-6 |
| EL4 | ATCC ® | TIB-39 |
| 293T | ATCC® | CRL-3216™ |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Jackson Labs | |
| C57BL/6 E88Δ | This paper | |
| C57BL/6 small E88Δ | This paper | |
| C57BL/6 g3-g4Δ | This paper | |
| C57BL/6 g3-USΔ | This paper | |
| C57BL/6 g3-DSΔ | This paper | |
| B6.Rag1−/− | Jackson Labs | |
| E88Δ.Rag1−/− | This paper | |
| B6.Rag−/−Igh-Tg+ | (Nussenzweig et al., 1987) | |
| E88Δ.Rag−/−Igh-Tg+ | This paper | |
| Oligonucleotides | ||
| See Table S1 for sequences and details | ||
| Recombinant DNA | ||
| pCAG-EGxxFP | (Mashiko et al., 2013) | |
| pX330-U6-Chimeric_BB-CBh-hSpCas | (Cong et al., 2013) | Addgene Plasmid #42230 |
| pX335-U6-Chimeric_BB-CBh-hSpCas9n | (Cong et al., 2013) | Addgene Plasmid #42335 |
| pMSCV-IRES-Bsr- Rag1 | Dr. David Schatz (Yale Universisy) | |
| MSCV-IRES-GFP | Plasmid #20672 | Addgene Plasmid #20672 |
| pEco | CloneTech | 631530 |
| MCS1-EF1α-RFP-T2A-Puro-pA-MCS2 | System Biosciences | HR110PA-1 |
| pGL3 vector | Promega | E1751 |
| Software and Algorithms | ||
| Cutadapt v1.14 | (Martin, 2011) | |
| Bowtie2 v2.2.9 | (Langmead and Salzberg, 2012) | |
| Bedtools v2.17.0 | (Quinlan and Hall, 2010) | |
| UCSC Genome Browser software | The University of California, Santa Cruz | |
| DESeq2 | (Love et al., 2014) | |
| Other | ||
An enhancer, E88, in the middle of the Igκ locus is a major hub of interactions
Deletion of E88 alters Vκ gene rearrangement frequencies throughout the locus
Structure of the Igκ locus in pro-B cells influences Vκ rearrangement in pre-B cells
V gene rearrangement is likely mediated in a subTAD specific manner
Barajas-Mora et al. demonstrate that an enhancer – E88— in the V gene region of the Igκ locus regulates long-range chromatin interactions enabling different V gene-containing subTADs to come in proximity to the J genes to generate a diverse repertoire. Consequently, E88 deletion perturbs the resulting B-cell repertoire.
Acknowledgements
We would like to thank Yinan Liu, Jang Cho, Erika Robinson, Jiadai Ma, and Parsa Parirokh for excellent technical assistance, Greg Martin and Dr. Sergey Kupriyanov in the Mouse Genetics Core for the pronuclear injections, The Center for Computational Biology for deep sequencing and Padmaja Natarajan and Dr. Shuli Kang for initial processing of sequencing results, Dr. Christopher Benner (UCSD) for the analysis and generation of the Hi-C heat map, Dr. David G. Schatz (Yale University) for permission to use the Rag1 pMSCV-IRES-Bsr retrovirus, Dr. Amy Kenter (University of Illinois College of Medicine) for the 445.3 cell line and for comments on the manuscript. This research was supported by NIH grants R56 AI119092, R21 AI113033, R21 AI137867 and R03 AI115486 to AJF. The Hi-C data was supported by NIH grants AI082850, AI00880 and AI09599 to CM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Aoki-Ota M, Torkamani A, Ota T, Schork N, and Nemazee D (2012). Skewed primary Igkappa repertoire and V-J joining in C57BL/6 mice: implications for recombination accessibility and receptor editing. J Immunol 188, 2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista CR, Li SK, Xu LS, Solomon LA, and DeKoter RP (2017). PU.1 Regulates Ig Light Chain Transcription and Rearrangement in Pre-B Cells during B Cell Development. J Immunol 198, 1565–1574. [DOI] [PubMed] [Google Scholar]
- Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas LM, Branco MR, et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolland DJ, Koohy H, Wood AL, Matheson LS, Krueger F, Stubbington MJ, Baizan-Edge A, Chovanec P, Stubbs BA, Tabbada K, et al. (2016). Two Mutually Exclusive Local Chromatin States Drive Efficient V(D)J Recombination. Cell Rep 15, 2475–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossen C, Murre CS, Chang AN, Mansson R, Rodewald HR, and Murre C (2015). The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat Immunol 16, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, and Wysocka J (2013). Modification of enhancer chromatin: what, how, and why? Mol Cell 49, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi NM, and Feeney AJ (2014). CTCF and ncRNA Regulate the Three-Dimensional Structure of Antigen Receptor Loci to Facilitate V(D)J Recombination. Front Immunol 5, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi NM, Loguercio S, Verma-Gaur J, Degner SC, Torkamani A, Su AI, Oltz EM, Artyomov M, and Feeney AJ (2013). Deep sequencing of the murine IgH repertoire reveals complex regulation of nonrandom V gene rearrangement frequencies. J Immunol 191, 2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat W, and Duboule D (2013). Topology of mammalian developmental enhancers and their regulatory landscapes. Nature 502, 499–506. [DOI] [PubMed] [Google Scholar]
- Degner SC, Verma-Gaur J, Wong TP, Bossen C, Iverson GM, Torkamani A, Vettermann C, Lin YC, Ju Z, Schulz D, et al. (2011). CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc Natl Acad Sci U S A 108, 9566–9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxa M, Skok J, Souabni A, Salvagiotto G, Roldan E, and Busslinger M (2004). Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. Genes Dev 18, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan S, Majumder K, Predeus A, Huang Y, Koues OI, Verma-Gaur J, Loguercio S, Su AI, Feeney AJ, Artyomov MN, et al. (2013). Unifying model for molecular determinants of the preselection Vbeta repertoire. Proc Natl Acad Sci U S A 110, E3206–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Yoon HS, Franklin A, Jain S, Ebert A, Cheng HL, Hansen E, Despo O, Bossen C, Vettermann C, et al. (2011). CTCF-binding elements mediate control of V(D)J recombination. Nature 477, 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesslein DG, Pflugh DL, Chowdhury D, Bothwell AL, Sen R, and Schatz DG (2003). Pax5 is required for recombination of transcribed, acetylated, 5’ IgH V gene segments. Genes Dev 17, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Zhang Y, Zhao L, Frock RL, Du Z, Meyers RM, Meng FL, Schatz DG, and Alt FW (2015). Chromosomal Loop Domains Direct the Recombination of Antigen Receptor Genes. Cell 163, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inlay M, Alt FW, Baltimore D, and Xu Y (2002). Essential roles of the kappa light chain intronic enhancer and 3’ enhancer in kappa rearrangement and demethylation. Nat Immunol 3, 463–468. [DOI] [PubMed] [Google Scholar]
- Jain S, Ba Z, Zhang Y, Dai HQ, and Alt FW (2018). CTCF-Binding Elements Mediate Accessibility of RAG Substrates During Chromatin Scanning. Cell 174, 102–116 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhunjhunwala S, van Zelm MC, Peak MM, Cutchin S, Riblet R, van Dongen JJ, Grosveld FG, Knoch TA, and Murre C (2008). The 3D structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions. Cell 133, 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Wuerffel R, Achour I, Lajoie B, Sen R, Dekker J, Feeney AJ, and Kenter AL (2013). Flexible ordering of antibody class switch and V(D)J joining during B-cell ontogeny. Genes Dev 27, 2439–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Cattoglio C, and Tjian R (2014). Looping back to leap forward: transcription enters a new era. Cell 157, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Benner C, Mansson R, Heinz S, Miyazaki K, Miyazaki M, Chandra V, Bossen C, Glass CK, and Murre C (2012). Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat Immunol 13, 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. (2010). A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Schmidt-Supprian M, Shi Y, Hobeika E, Barteneva N, Jumaa H, Pelanda R, Reth M, Skok J, Rajewsky K, et al. (2007). Yin Yang 1 is a critical regulator of B-cell development. Genes Dev 21, 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loguercio S, Barajas-Mora EM, Shih HY, Krangel MS, and Feeney AJ (2018). Variable Extent of Lineage-Specificity and Developmental Stage-Specificity of Cohesin and CCCTC-Binding Factor Binding Within the Immunoglobulin and T Cell Receptor Loci. Front Immunol 9, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JS, Zhang Y, Dudko OK, and Murre C (2014). 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell 158, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. [Google Scholar]
- Mashiko D, Fujihara Y, Satouh Y, Miyata H, Isotani A, and Ikawa M (2013). Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci Rep 3, 3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson LS, Bolland DJ, Chovanec P, Krueger F, Andrews S, Koohy H, and Corcoran AE (2017). Local Chromatin Features Including PU.1 and IKAROS Binding and H3K4 Methylation Shape the Repertoire of Immunoglobulin Kappa Genes Chosen for V(D)J Recombination. Front Immunol 8, 1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedovic J, Ebert A, Tagoh H, Tamir IM, Schwickert TA, Novatchkova M, Sun Q, Huis In ‘t Veld PJ, Guo C, Yoon HS, et al. (2013). Flexible long-range loops in the VH gene region of the Igh locus facilitate the generation of a diverse antibody repertoire. Immunity 39, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer EM, Lin YC, Benner C, Jhunjhunwala S, Dutkowski J, Flores M, Sigvardsson M, Ideker T, Glass CK, and Murre C (2011). Multilineage priming of enhancer repertoires precedes commitment to the B and myeloid cell lineages in hematopoietic progenitors. Immunity 35, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori L, Wuerffel R, Roqueiro D, Lajoie B, Guo C, Gerasimova T, De S, Wood W, Becker KG, Dekker J, et al. (2016). Extremely Long-Range Chromatin Loops Link Topological Domains to Facilitate a Diverse Antibody Repertoire. Cell Rep 14, 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muljo SA, and Schlissel MS (2003). A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines. Nat Immunol 4, 31–37. [DOI] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novobrantseva TI, Martin VM, Pelanda R, Muller W, Rajewsky K, and Ehlich A (1999). Rearrangement and expression of immunoglobulin light chain genes can precede heavy chain expression during normal B cell development in mice. J Exp Med 189, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig MC, Shaw AC, Sinn E, Danner DB, Holmes KL, Morse HC 3rd, and Leder P (1987). Allelic exclusion in transgenic mice that express the membrane form of immunoglobulin mu. Science 236, 816–819. [DOI] [PubMed] [Google Scholar]
- Pan X, Papasani M, Hao Y, Calamito M, Wei F, Quinn Iii WJ, Basu A, Wang J, Hodawadekar S, Zaprazna K, et al. (2013). YY1 controls Igkappa repertoire and B-cell development, and localizes with condensin on the Igkappa locus. EMBO J 32, 1168–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Predeus AV, Gopalakrishnan S, Huang Y, Tang J, Feeney AJ, Oltz EM, and Artyomov MN (2014). Targeted chromatin profiling reveals novel enhancers in Ig H and Ig L chain Loci. J Immunol 192, 1064–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Kumari G, Gerasimova T, Du H, Labaran L, Singh A, De S, Wood WH 3rd, Becker KG, Zhou W, et al. (2018). Sequential Enhancer Sequestration Dysregulates Recombination Center Formation at the IgH Locus. Mol Cell 70, 21–33 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud D, Demarco IA, Reddy KL, Schjerven H, Bertolino E, Chen Z, Smale ST, Winandy S, and Singh H (2008). Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol 9, 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro de Almeida C, Stadhouders R, de Bruijn MJ, Bergen IM, Thongjuea S, Lenhard B, van Ijcken W, Grosveld F, Galjart N, Soler E, et al. (2011). The DNA-binding protein CTCF limits proximal Vkappa recombination and restricts kappa enhancer interactions to the immunoglobulin kappa light chain locus. Immunity 35, 501–513. [DOI] [PubMed] [Google Scholar]
- Roldan E, Fuxa M, Chong W, Martinez D, Novatchkova M, Busslinger M, and Skok JA (2005). Locus ‘decontraction’ and centromeric recruitment contribute to allelic exclusion of the immunoglobulin heavy-chain gene. Nat Immunol 6, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother MB, Palstra RJ, Jhunjhunwala S, van Kester KA, van IWF, Hendriks RW, van Dongen JJ, Murre C, and van Zelm MC (2016). Nuclear positioning rather than contraction controls ordered rearrangements of immunoglobulin loci. Nucleic Acids Res 44, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, and Dekker J (2012). The long-range interaction landscape of gene promoters. Nature 489, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayegh CE, Jhunjhunwala S, Riblet R, and Murre C (2005). Visualization of looping involving the immunoglobulin heavy-chain locus in developing B cells. Genes Dev 19, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlissel MS, and Baltimore D (1989). Activation of immunoglobulin kappa gene rearrangement correlates with induction of germline kappa gene transcription. Cell 58, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, de Bruijn MJ, Rother MB, Yuvaraj S, Ribeiro de Almeida C, Kolovos P, Van Zelm MC, van Ijcken W, Grosveld F, Soler E, et al. (2014). Pre-B cell receptor signaling induces immunoglobulin kappa locus accessibility by functional redistribution of enhancer-mediated chromatin interactions. PLoS Biol 12, e1001791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Kolovos P, Brouwer R, Zuin J, van den Heuvel A, Kockx C, Palstra RJ, Wendt KS, Grosveld F, van Ijcken W, et al. (2013). Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat Protoc 8, 509–524. [DOI] [PubMed] [Google Scholar]
- Van Ness BG, Weigert M, Coleclough C, Mather EL, Kelley DE, and Perry RP (1981). Transcription of the unrearranged mouse C kappa locus: sequence of the initiation region and comparison of activity with a rearranged V kappa-C kappa gene. Cell 27, 593–602. [DOI] [PubMed] [Google Scholar]
- Vierstra J, Rynes E, Sandstrom R, Zhang M, Canfield T, Hansen RS, Stehling-Sun S, Sabo PJ, Byron R, Humbert R, et al. (2014). Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science 346, 1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GS, Martinez A, Montalbano A, Tang A, Mauhar A, Ogwaro KM, Merz D, Chevillard C, Riblet R, and Feeney AJ (2001). Unequal VH gene rearrangement frequency within the large VH7183 gene family is not due to recombination signal sequence variation, and mapping of the genes shows a bias of rearrangement based on chromosomal location. J Immunol 167, 257–263. [DOI] [PubMed] [Google Scholar]
- Xiang Y, and Garrard WT (2008). The Downstream Transcriptional Enhancer, Ed, positively regulates mouse Ig kappa gene expression and somatic hypermutation. J Immunol 180, 6725–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Park SK, and Garrard WT (2013). Vkappa gene repertoire and locus contraction are specified by critical DNase I hypersensitive sites within the Vkappa-Jkappa intervening region. J Immunol 190, 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Zhou X, Hewitt SL, Skok JA, and Garrard WT (2011). A multifunctional element in the mouse Igkappa locus that specifies repertoire and Ig loci subnuclear location. J Immunol 186, 5356–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Chen SA, Local A, Liu T, Qiu Y, Dorighi KM, Preissl S, Rivera CM, Wang C, Ye Z, et al. (2018). Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res 28, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos GD, and Alt FW (1985). Developmentally controlled and tissue-specific expression of unrearranged VH gene segments. Cell 40, 271–281. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, and Alt FW (1986). Regulation of the assembly and expression of variable-region genes. Annu Rev Immunol 4, 339–368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers, probes, and oligonucleotides (related to Figures 1, 2, 3, 4, 6, 7, S1, S2, and S3)
Figure S1, (related to figures 1 and 2). E88 ChromHMM analysis, sequence and DHS specificity. (A) ChromHMM assessment of the E88 region done by (Predeus et al., 2014) in pro-B cells. Chromatin states marks: (10), H3K4me1; (19), PU.1; (23), H3K4me2; (34), Pax5; (37), E2A; (1), Mediator and H3K4me1; (9), H3K4me2 and H3K4me1; (16), H3K36me3 and H3K4me1; (17), H3ac and H3K4me1; (6), H3ac, H3K4me2 and H3K4me1; (3), DHS, E2A, H3K4me1, PU.1 and Mediator; (4), DHS, H3K4me3, H3K4me2, H3K4me1, H3ac, PU.1, Mediator, E2A, Pax5 and P300; (5), DHS, H3K4me2, H3K4me1, PU.1 and Mediator; (7), DHS, H3K4me3, H3K4me2, H3K4me1 and H3ac; (35) and (36), empty states. Indicated are the deletions made in 445.3 cells and mice (largest). (B) E88 sequence deleted in 445.3 cells with putative E2A and EBF binding sites highlighted in yellow and blue respectively. (C) DNase-seq data around the E88 area in multiple cells and tissues (Vierstra et al., 2014). An area around the GAPDH gene was used as DHS control for B cell specificity. UCSC Genome Browser views show the mapped read coverage of DNase-seq.
Figure S2, (related to figures 2 and 3). Effects of E88∆ in Vκ rearrangement pattern and early kinetics in 445.3 cell lines. (A) Quantification of Vκ rearrangement on gDNA by qPCR (TaqMan) with specific V gene primers in 445.3-WT or 445.3-E88∆ cells at 48 hours after STI571 stimulation. Data is normalized with a loading gDNA control (Eu) and is expressed as the ratio of E88∆ / WT. (B) Quantification of Vκ rearrangement on RNA by qPCR in 445.3-WT or 445.3-E88∆ cells at 0, 12, and 24 hours after STI571 stimulation with the Vall primer, which gives an estimated measure of the total rearrangement. Data is expressed relative to GAPDH. (C) Relative rate of total rearrangement (Vall) shown as the ratio of WT / E88∆ normalized to t=0 at the indicated time points. Data in A, B and C is representative of at least three independent experiments ± SEM. N.D.= not detected for WT or E88∆. (D) Assessment of sgRNAs specificity and efficiency. pX330-E88g3 and pX330-E88g4 plasmids were tested for efficiency of targeting of the E88 region using the eGx-E88-xFP reporter plasmid (Mashiko et al., 2013). GFP expression indicates that the sgRNA-guided CAS9 endonuclease targets the DNA inserted in the multiple cloning site (MCS) in the middle of the GFP gene. Indicated plasmids were cotransfected in 239T cells and assessed for GFP expression 48 hours post-transfection. (Top left) eGx-E88-xFP plasmid cotransfected with a pX330 plasmid expressing a gRNA not specific for the E88 region. The eGx-control.DNA-xFP plasmid, containing a control DNA fragment that is not targeted by E88g3 or E88g4, cotransfected with pX330-E88g3 (top center) or the pX330-E88g4 (left bottom) plasmids. The eGx-E88-xFP plasmid cotransfected with pX330-E88g3 (top right) or pX330-E88g4 (middle bottom) plasmids. Control cells that were not transfected (right bottom). Pictures are from one of the two experiments performed.
Figure S3, (related to figure 3). E88 enhancer regulates Vκ gene utilization in mice. E88 was deleted in mice using CRISPR/Cas9 editing system. Schematic of the different sized E88 deletions in mice is shown in Figure 3A. DS=downstream, US=upstream. (A-D) BM-derived CD19+ cells were purified, and RNA was harvested. Vκ rearrangement was assessed by qPCR for specific individual Vκ genes for all the mouse lines. Data was normalized with GAPDH and expressed as E88∆ / WT ratio ± SEM. Two to five mice 6–10 weeks of age were used for each experiment. Data was collected from at least three independent biological samples.
Figure S4, (related to figure 4). Sorting scheme for pro-B and small pre-B cells and Vκ rearrangement in fetal liver cells and spleen. CD19+ cells were isolated from BM-cells from WT and E88∆ mice using CD19-conjugated MACS beads. (A) CD19+ cells were stained with antibodies against CD19, CD93, CD2, CD43 and IgM. Sorted pro-B cells (CD19+ CD93+, IgM−, CD2−, CD43+) and small pre-B cells (CD19+ CD93+, IgM−, CD2+, CD43-) were used to isolate RNA or gDNA for qPCR analysis or deep sequencing. Pre-B cells were separated as large or small based on the forward scatter (FSC). (B, C) Vκ rearrangement in fetal liver and spleen CD19+ cells. Isolated CD19+ cells from fetal liver of embryos at day 17 of gestation or spleens from 6–10 week-old mice were used to extract RNA. Quantification of Vκ gene rearrangement was done by qPCR with specific primers for the indicated Vκ genes. Data is shown as the ratio of E88∆ / WT and was normalized to GAPDH expression. Data is representative of at least three independent experiments ± SEM.
Figure S5, (related to figures 6 and 7). Chromatin profile and transcription factor binding for other identified enhancer-like regions. UCSC Genome Browser presentation of ChIP-seq datasets for the indicated putative enhancer regions. H3K4me1 (non-IL7-cultured RAG−/− pro-B cells and IL7-cultured RAG−/− pro-B cells), Mediator (v-Abl transformed pro-B cell line), and the following ChIP-seq using IL7-cultured RAG−/− pro-B cells: H3K27ac, Brg1, EBF, E2A, IRF4, PU.1, and PAX5. E92 has ~89% sequence similarity with E88 over ~ 650 kb. Note that the scales are different for the ChIP-seqs tracks for the different enhancers.
Table S2. ChIP-seq and RNA-seq datasets (related to Figures 1, 2, 3, 4, and S4)