

Abstract
Intracellular pathogens and danger signals trigger the formation of inflammasomes, which activate inflammatory caspases and induce pyroptosis. The anthrax lethal factor metalloprotease and small-molecule DPP8/9 inhibitors both activate the NLRP1B inflammasome, but the molecular mechanism of NLRP1B activation is unknown. In this study, we used genome-wide CRISPR-Cas9 knockout screens to identify genes required for NLRP1B-mediated pyroptosis. We discovered that lethal factor induces cell death via the N-end rule proteasomal degradation pathway. Lethal factor directly cleaves NLRP1B, inducing the N-end rule–mediated degradation of the NLRP1B N terminus and freeing the NLRP1B C terminus to activate caspase-1. DPP8/9 inhibitors also induce proteasomal degradation of the NLRP1B N terminus but not via the N-end rule pathway. Thus, N-terminal degradation is the common activation mechanism of this innate immune sensor.
Mammals express a diverse array of intra-cellular pattern-recognition receptors (PRRs) that detect cytoplasmic microbial structures and activities (1). Upon recognition of their cognate danger signals, several PRRs form large, multiprotein complexes called inflammasomes, which recruit and activate caspase-1. Caspase-1, in turn, cleaves and activates inflammatory cytokines and gasdermin D (GSDMD), triggering an inflammatory form of cell death called pyroptosis (1–3).
Anthrax lethal factor (LF), the active component of lethal toxin (LT), is a metalloprotease that activates the NLRP1B (nucleotide-binding domain leucine-rich repeat pyrin domain-containing 1B) inflammasome by cleaving NLRP1B after Lys44 (4–6). However, it is unclear how proteolytic cleavage activates NLRP1B. Small-molecule inhibitors of the serine dipeptidases DPP8 and DPP9 (DPP8/9) also activate NLRP1B. The mechanism of DPP8/9 inhibitor-induced NLRP1B activation is unknown, but, unlike LF, it does not involve direct NLRP1B cleavage (7, 8). Proteasome inhibitors block both LF-and DPP8/9 inhibitor-induced pyroptosis (8–10) but do not block pyroptosis mediated by other inflammasomes (10,11). Thus, although LF and DPP8/9 inhibitors activate NLRP1B in different ways, a component of the NLRP1B activation mechanism—the degradation of a key protein—appears to be shared between these two stimuli.
To investigate the mechanism underlying NLRP1B activation, we performed two genome-wide CRISPR-Cas9 screens in RAW 264.7 cells to identify gene knockouts that provide resistance to the DPP8/9 inhibitor Val-boroPro (VbP) or LT (fig. S1) (12). As expected, Nlrp1b, Casp1, and Gsdmd were among the most enriched genes in both the LT and VbP screens (Fig. 1, table S1, and data S1). Nlrp1a, which is not expressed in Balb/c macrophages from which RAW 264.7 cells were derived (13), was likely enriched as a result of off-target knockout of Nlrp1b by the Nlrp1a single guide RNAs. Also as expected, many genes encoding proteins required for LT cell penetrance, including the anthrax toxin receptor Antxr2 (14), the protease furin (15), and members of the vacuolar adenosine triphos-phatase proton pump (16), were enriched in LT-treated samples (Fig. 1, table S2, and data S2). Several genes with no known involvement in inflammasome biology were also identified, including genes encoding members of the protein-folding machinery, the RNA methyltransferase complex, and the INO80 chromatin-remodeling complex (Fig. 1, data S2, and figs. S2 and S3).
Fig. 1. Genome-wide CRISPR-Cas9 screening identifies genes involved in NLRP1B-mediated pyroptosis.
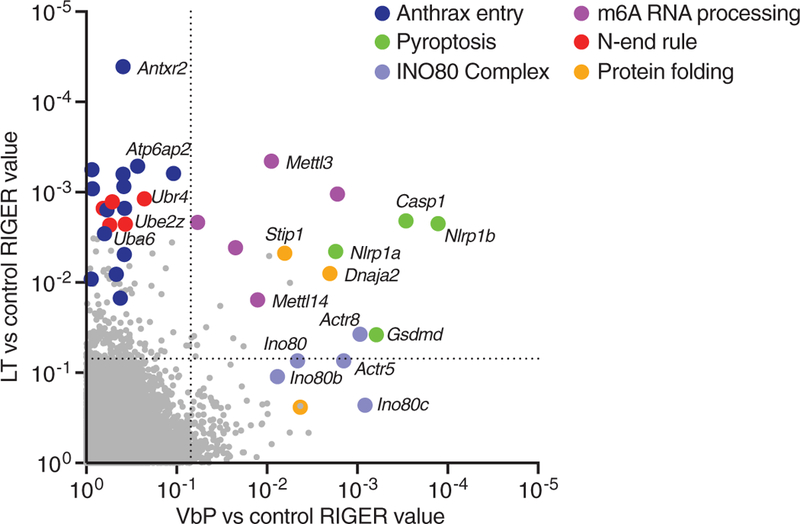
Screens were performed in RAW 264.7 cells (see fig. S1). RIGER (RNAi gene enrichment ranking) values indicating the relative enrichment of genes after treatment with VbP (x axis) or LT (y axis) relative to control. The dotted lines indicate a RIGER p-value of 0.01.
Several genes involved in the N-end rule proteasomal degradation pathway (17–20), Ubr2, Ubr4, Uba6, Ube2z, and Kcmf1, were highly enriched by LT but not by VbP (Fig. 1, table S2, and data S2). The N-end rule pathway recognizes, ubiquitinates, and degrades proteins with destabilizing N-terminal residues (17,18). Wickliffe et al. showed that inhibitors of the N-end rule pathway, bestatin and amino acid derivatives, block LT-mediated cell death (21). They proposed that LF might cleave a key substrate protein to generate a destabilizing N-terminal residue, inducing that protein’s degradation via the N-end rule and triggering cell death. However, such an N-end rule substrate has not been identified, and the direct involvement of N-end rule proteins has not been established. Our screening results suggested that the N-end rule pathway is indeed involved in LT-mediated cytotoxicity. We hypothesized that NLRP1B itself, which was discovered to be directly cleaved by LF after the Wickliffe study (4–6), may be the key LF substrate degraded by the N-end rule pathway.
NLRP1B contains nucleotide-binding (NACHT), leucine-rich repeat (LRR), function-to-find (FIIND), and caspase activation and recruitment (CARD) domains (Fig. 2A). NLRP1B undergoes post-translational autoproteolysis within the FIIND domain, resulting in N-and C-terminal fragments that remain associated in an autoinhibited state (22–24). Autoproteolysis is necessary for inflammasome formation (8, 23, 24), but why it is necessary remains unknown. The N-terminal fragment was nontoxic in human embryonic kidney (HEK) 293T cells stably expressing caspase-1, whereas C-terminal fragments containing the CARD were toxic (Fig. 2, B and C) (24). We predicted that N-end rule degradation of the NLRP1B N terminus after LF cleavage could free the C terminus, as the break in the polypeptide chain would prevent its concomitant destruction, resulting in caspase-1 activation. We reasoned that VbP might induce the degradation of the NLRP1B N terminus via a different mechanism, as VbP did not enrich the N-end rule genes.
Fig. 2. LT and VbP induce proteasome-mediated degradation of NLRP1B.
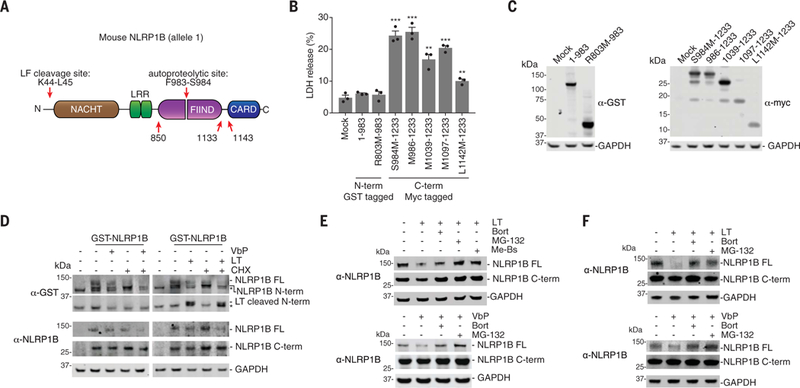
(A) Diagram of NLRP1B. Single-letter abbreviations for the amino acid residues are as follows: F, Phe; K, Lys; L, Leu; M, Met; R, Arg; and S, Ser. (B and C) HEK 293T cells stably expressing mCasp1 (“m” denotes mouse) were transiently transfected with the indicated constructs (2 μg) for 24 hours, before cell viability was evaluated by lactate dehydrogenase (LDH) release (B) and expression was evaluated by immunoblotting (C). Residues that were mutated to create start sites are indicated. Data are means ± SEM of three biological replicates. ***P < 0.001 and **P < 0.01 by two-sided Student’s t test compared with mock. GST, glutathione S-transferase; GAPDH, glyceraldehyde phosphate dehydrogenase. (D) HEK 293T cells stably expressing mCasp1 were transiently transfected with a construct encoding GST-NLRP1B (30 ng). After 24 hours, cycloheximide (CHX, 100 mg/ml; used to block new protein synthesis), LT (1 μg/ml), and VbP (10 μM) were added to the indicated samples, which were then incubated for an additional 6 hours. FL, full-length. Asterisks indicate background bands. (E) HEK 293Tcells stably expressing mCasp1 were transiently transfected with a construct encoding V5-GFP-NLRP1B-FLAG (0.1 μg). After 24 hours, cells were treated with dimethyl sulfoxide (DMSO), bortezomib (Bort, 20 μM), MG-132 (20 μM), or Me-Bs (20 μM) for 30 min before the addition of either LT (1 μg/ml, 6 hours) or VbP (10 μM, 6 hours). Protein levels were evaluated by immunoblotting. (F) RAW 264.7 cells were treated with DMSO, bortezomib (20 μM), or MG-132 (20 μM) for 30 min before the addition of LT (1 μg/ml, 3 hours) or VbP (2 μM, 6 hours). Protein levels were evaluated by immune-blotting. Data are representative of three or more independent experiments.
To determine whether the NLRP1B protein was degraded after LT and VbP treatment, we evaluated NLRP1B protein levels in HEK 293T cells ectopically expressing caspase-1 and NLRP1B, which are sensitive to both LT-and VbP-induced pyroptosis (8). Full-length NLRP1B was lost after treatment of these cells with either LT or VbP (Fig. 2, D and E). Loss of the N-terminal fragment of NLRP1B itself was also observed after VbP treatment (Fig. 2D). However, because LF removes the N-terminal tag and the α-NLRP1B antibody detects the C terminus, the N-terminal fragment could not be directly observed after LF cleavage. Similarly, LT and VbP both induced the loss of endogenous NLRP1B in RAW 264.7 cells (Fig. 2F and fig. S4). Consistent with our model, there was considerably more of the C-terminal fragment remaining relative to the full-length protein in both the HEK 293T and RAW 264.7 cells (Fig. 2, D to F, and fig. S4). Bortezomib and MG-132 rescued NLRP1B protein loss, indicating that NLRP1B was being targeted for proteasome-mediated degradation (Fig. 2, E and F). In humans, DPP8/9 inhibitors activate CARD8, a homolog of NLRP1B that only contains the FIIND-CARD region (25). Similar to NLRP1B, VbP induced the proteasome-mediated degradation of the CARD8 N terminus (fig. S5). Thus, proteasome-mediated N-terminal degradation is a key feature of NLRP1/CARD8 activation.
We generated RAW 264.7 cells lacking the N-end rule proteins UBR2, UBR4, and UBA6 (Fig. 3A), which were identified in the LT screen. UBR2 and UBR4 are N-recognins that directly bind to destabilizing N-terminal residues of N-end rule substrates via UBR box domains (17,18). UBR2 also possesses a RING domain and acts as an E3 ligase to transfer ubiquitin to N-end rule substrates. Unlike UBR2, UBR4 is not an E3 ligase, but the E3 ligase KCMF1, also a hit in the LT screen, is proposed to function with UBR4 to ubiquitinate N-end rule substrates (20). UBA6 is an E1 enzyme that has the ability to charge the E2 enzyme UBE2Z, which functions with N-recognin E3 ligases UBR1-UBR3 (19) and was also enriched by LT. Ubr2, Ubr4, and Uba6 knockout (KO) RAW 264.7 cells were all significantly resistant to LT-induced cell killing (Fig. 3B) and NLRP1B degradation (Fig. 3C and fig. S4) at 3 hours. In contrast, and as expected, knockout of the N-end rule genes did not affect VbP-induced pyroptosis (fig. S6). Although these knockout cells were highly resistant to LT, some LT-induced cell death was nevertheless observed at 3 hours (Fig. 3B). Moreover, Ubr2 KO cells treated with LT for 6 hours showed some NLRP1B degradation (Fig. 3, D and E). We hypothesized that the Ubr2 KO cells were not completely resistant to LT-induced pyroptosis owing to redundancy with other N-recognins, in particular UBR4. Indeed, LT induced little, if any, cell death or NLRP1B degradation in Ubr2/4 double-KO RAW 264.7 cells after 6 hours (Fig. 3, F to H). Moreover, the full-length NLRP1B protein appeared to migrate slightly faster in the LT-treated cells (Fig. 3H), which is consistent with the formation of a highly stable, LT-cleaved NLRP1B protein.
Fig. 3. The N-end rule pathway mediates LT-induced NLRP1B degradation.
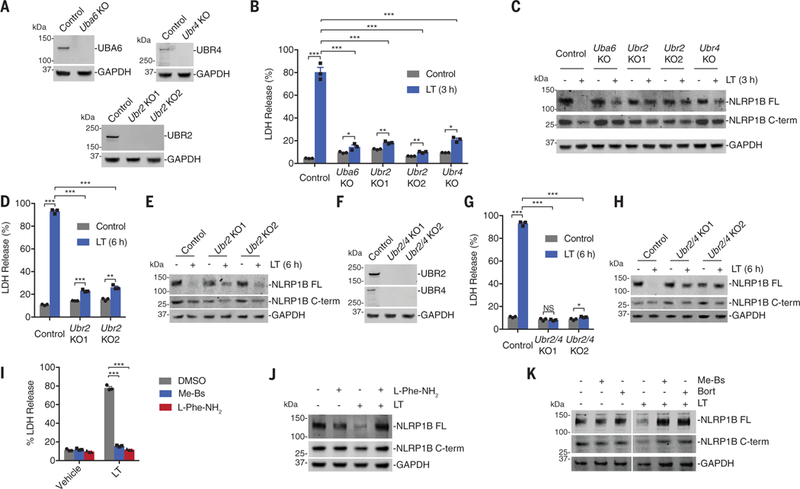
(A) Knockout of Uba6, Ubr2, and Ubr4 in RAW 264.7 cells was confirmed by immunoblotting. (B to E) RAW 264.7 cells with the indicated genotypes were treated with LT (1 μg/ml) for 3 hours [(B) and (C)] or 6 hours [(D) and (E)] before supernatants were assessed for LDH release [(B) and (D)] and lysates were evaluated by immunoblotting [(C) and (E)]. (F) Knockout of both Ubr2 and Ubr4 in RAW 264.7 cells was confirmed by immunoblotting. (G and H) RAW 264.7 cells with the indicated genotypes were treated with LT (1 μg/ml) for 6 hours before supernatants were assessed for LDH release (G) and lysates were evaluated by immunoblotting (H). In (B), (D), and (G), data are means ± SEM of three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001, and NS (not significant) by two-sided Student’s t test. The same samples were used as controls in (D) and (G). (I) RAW 264.7 cells were treated with Me-Bs (10 μM) or L-Phe-NH2 (1 μM) for 30 min before the addition of LT (1 μg/ml, 6 hours). Supernatants were then evaluated for LDH release. Data are means ± SEM of three biological replicates. ***P < 0.001 by two-sided Student’s t test. (J and K) RAW 264.7 cells were treated with Me-Bs (10 μM), L-Phe-NH2 (1 μM), or bortezomib (1 μM) for 30 min before the addition of LT (1 μg/ml, 3 hours). Lysates were then evaluated by immunoblotting. Data are representative of three or more independent experiments.
Amino acid derivatives (e.g., L-Phe-NH2) inhibit N-end rule degradation by competing with N-end rule substrates for binding to N-recognins (26). Bestatin methyl ester (Me-Bs), a nonspecific aminopeptidase inhibitor, inhibits N-end rule degradation by preventing aminopeptidases from revealing destabilizing residues (21). Me-Bs and L-Phe-NH2 blocked LT-induced cell death and NLRP1B degradation in RAW 264.7 cells (Fig. 3, I to K), and Me-Bs blocked LT-induced NLRP1B degradation in the HEK assay (Fig. 2E). Consistent with aminopeptidase trimming of the LT-generated N terminus, mutant NLRP1B L45M, which itself should not be strongly bound by N-recognins (17, 18), still triggered pyroptosis (fig. S7). Neither Me-Bs nor L-Phe-NH2 blocked VbP-induced cell death or NLRP1B degradation (fig. S8). Me-Bs actually potentiated VbP-induced RAW 264.7 cell death (fig. S8A), NLRP1B degradation (fig. S8B), in vivo cytokine induction (fig. S8C), and human THP-1 pyroptosis (fig. S9). How Me-Bs synergizes with VbP is unknown, but Me-Bs did not stabilize the NLRP1B C-terminal fragment from N-end rule degradation (fig. S10).
To determine whether the LF-generated NLRP1 N terminus was sufficient to induce degradation, we transfected HEK 293T cells with constructs encoding the intact (M1-L60) or the LF-cleaved (L45-L60) NLRP1B N terminus fused to green fluorescent protein (GFP). In the latter construct, the ubiquitin-fusion strategy was used to release leucine as the N-terminal residue (27). Consistent with our model, NLRP1BL45-L60-GFP, but not NLRP1BM1-L60-GFP, was stabilized by genetic (Fig. 4A and fig. S11) and chemical inhibition (fig. S12) of the N-end rule pathway, pheno-copying the stability of full-length NLRP1B. At higher levels of transfection, ubiquitination of NLRP1BL45-L60-GFP was observed (Fig. 4B). This ubiquitination was indeed mediated by the N-end rule, as the bands were increased by cotransfection with UBR2 (Fig. 4B), decreased by chemical inhibitors of the N-end rule (fig. S13A), and barely detectable in UBR2 and UBA6 KO HEK 293T cells (Fig. 4B and fig. S13, B and C).
Fig. 4. The cleaved NLRP1B N terminus is sufficient to induce protein degradation.
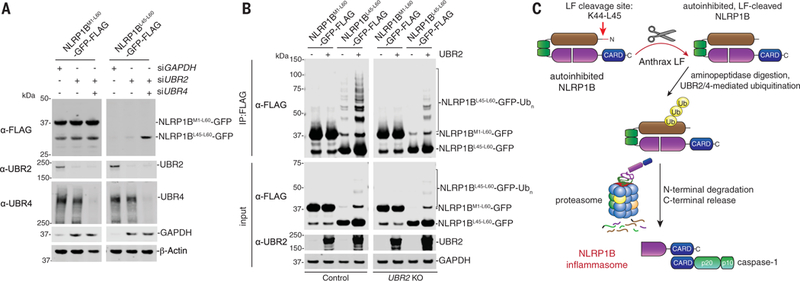
(A) HEK 293T cells were transfected with the indicated small interfering RNAs, incubated for 24 hours, then transfected with NLRP1BM1−60- or NLRP1BL45−60-GFP-FLAG fusion constructs (0.05 μg) for an additional 24 hours. Lysates were then evaluated by immuno-blotting. (B) WT or UBR2 KO HEK 293T cells were transfected with the indicated NLRP1BM1−60- or NLRP1BL45−60-GFP-FLAG fusion constructs (0.5 μg) and UBR2 (1.5 μg) and incubated for 24 hours. Lysates were harvested, immunoprecipitated with anti-FLAG agarose beads, and evaluated by immunoblotting. Data are representative of three or more independent experiments. (C) Proposed model of LT-induced NLRP1B inflammasome activation.
Thus, N-terminal degradation is the unifying mechanism of NLRP1B inflammasome activation. There are at least two distinct degradation pathways, one direct (Fig. 4C) and one indirect (fig. S14). In the direct mechanism, LF protease cleaves NLRP1B and generates a destabilized neo–N terminus that is recognized by the N-end rule pathway. As it is unlikely that anthrax LF evolved to trigger pyroptosis, we speculate that NLRP1B may serve as a booby trap for LF and possibly other pathogen-encoded activities. Indeed, Vance and co-workers found that the Shigella flexneri E3 ligase IpaH7.8 directly ubiquitinates and degrades NLRP1B (28). In the indirect mechanism, DPP8/9 inhibition stimulates an endogenous proteasomal degradation pathway to activate NLRP1B (fig. S14). The biological purpose and molecular details of this pathway remain to be determined. Future studies should leverage these insights to further clarify how NLRP1B senses specific pathogens and how this inflammasome can be modulated for therapeutic benefit.
Supplementary Material
ACKNOWLEDGMENTS
We thank W. Bachovchin, W. Wu, and J. Lai for VbP and 8j; R. Vance for the α-NLRP1B antibody (2A12); R. Garippa for assistance with the genome-wide screens; and J. Hebert for advising ubiquitination assays.
Funding: This work was supported by the Josie Robertson Foundation (D.A.B.); a Stand Up to Cancer-Innovative Research Grant (SU2C-AACR-IRG11–17 to D.A.B.; Stand Up to Cancer is a program of the Entertainment Industry Foundation; research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C); the Pew Charitable Trusts (D.A.B. is a Pew-Stewart Scholar in Cancer Research); the NIH (R01 AI137168 to D.A.B., T32 GM007739-Andersen to A.R.G., T32 GM115327-Tan to E.L.O. and D.C.J., and the MSKCC Core Grant P30 CA008748); an Alfred P. Sloan Foundation Research Fellowship (D.A.B.); the American Cancer Society (Postdoctoral Fellowship PF-17–224-01 - CCG to C.Y.T.); and Gabrielle’s Angel Foundation (D.A.B.).
Footnotes
Competing interests: The authors declare no competing interests. D.A.B. and D.P.B. recently filed a provisional patent regarding methods and compositions for regulating the NLRP1/CARD8 inflammasome.
Data and materials availability: All data are available in the main text or the supplementary materials.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Broz P, Dixit VM, Nat. Rev. Immunol. 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Kayagaki N et al. , Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Shi J et al. , Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Levinsohn JL et al. , PLOS Pathog. 8, e1002638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hellmich KA et al. , PLOS ONE 7, e49741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chavarria-Smith J, Vance RE, PLOS Pathog. 9, e1003452 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okondo MC et al. , Nat. Chem. Biol. 13, 46–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okondo MC et al. , Cell Chem. Biol. 25, 262–267.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang G, Leppla SH, Infect. Immun. 67, 3055–3060 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Squires RC, Muehlbauer SM, Brojatsch J, J. Biol. Chem. 282, 34260–34267 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Fink SL, Bergsbaken T, Cookson BT, Proc. Natl. Acad. Sci. U.S.A. 105, 4312–4317 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shalem O et al. , Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sastalla I et al. , BMC Genomics 14, 188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scobie HM, Rainey GJ, Bradley KA, Young JA, Proc. Natl. Acad. Sci. U.S.A. 100, 5170–5174 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klimpel KR, Molloy SS, Thomas G, Leppla SH, Proc. Natl. Acad. Sci. U.S.A. 89, 10277–10281 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menard A, Altendorf K, Breves D, Mock M, Montecucco C, FEBS Lett. 386, 161–164 (1996). [DOI] [PubMed] [Google Scholar]
- 17.Varshavsky A, Protein Sci. 20, 1298–1345 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sriram SM, Kim BY, Kwon YT, Nat. Rev. Mol. Cell Biol. 12, 735–747 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Lee PC, Sowa ME, Gygi SP, Harper JW, Mol. Cell 43, 392–405 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong JH et al. , Mol. Cell. Proteomics 14, 674–685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickliffe KE, Leppla SH, Moayeri M, Cell. Microbiol. 10, 1352–1362 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Osualdo A et al. , PLOS ONE 6, e27396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finger JN et al. , J. Biol. Chem. 287, 25030–25037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frew BC, Joag VR, Mogridge J, PLOS Pathog. 8, e1002659 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson DC et al. , Nat. Med. 24, 1151–1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker RT, Varshavsky A, Proc. Natl. Acad. Sci. U.S.A. 88, 1090–1094 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachmair A, Finley D, Varshavsky A, Science 234, 179–186 (1986). [DOI] [PubMed] [Google Scholar]
- 28.Sandstrom A et al. , Science 364, eaau1330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.