

Summary:
Autophagy is required in diverse paradigms of lifespan extension, leading to the prevailing notion that autophagy is beneficial for longevity. However, why autophagy is harmful in certain contexts, remains unexplained. Here we show that mitochondrial permeability defines the impact of autophagy on aging. Elevated autophagy unexpectedly shortens lifespan in C. elegans lacking serum/glucocorticoid regulated kinase-1 (sgk-1) because of increased mitochondrial permeability. In sgk-1 mutants, reducing levels of autophagy or mitochondrial permeability transition pore (mPTP) opening restores normal lifespan. Remarkably, low mitochondrial permeability is required across all paradigms examined of autophagy-dependent lifespan extension. Genetically induced mPTP opening blocks autophagy-dependent lifespan extension resulting from caloric restriction or loss of germline stem cells. Mitochondrial permeability similarly transforms autophagy into a destructive force in mammals, as liver-specific Sgk knockout mice demonstrate marked enhancement of hepatocyte autophagy, mPTP opening, and death with ischemia/reperfusion injury. Targeting mitochondrial permeability may maximize benefits of autophagy in aging.
Keywords: mTORC2, SGK, autophagy, mitochondrial permeability, aging
In Brief
The role of autophagy in lifespan extension depends on modulation of mitochondrial permeability via the action of the kinase SGK1.
Graphical Abstract
INTRODUCTION
A hallmark of aging is the accumulation of molecular damage, dysfunctional organelles, and defective enzymes (Lopez-Otin et al., 2013). Autophagy is an ancient, stress response present in all living eukaryotic cells, responsible for clearing intracellular damaged proteins, protein aggregates and dysfunctional organelles. Autophagy is generally thought of as a positive longevity modulatory factor. An unbiased screen for chronological aging factors in yeast identified multiple short-lived mutants with defects in autophagy (Matecic et al., 2010). In organisms such as the nematode C. elegans, autophagy is also critical for increased longevity evident in insulin-signaling mutants, during caloric restriction, and loss of germline stem cells (Hansen et al., 2018). In mice, disruption of the Beclin 1-BCL2 complex increases autophagy and promotes longevity (Fernandez et al., 2018).
In spite of these generally beneficial effects, autophagy may also play a detrimental role in health, promoting diseases such as cancer, diabetes, neurodegeneration, and ischemic injury (Thorburn, 2014). With extended starvation, high levels of autophagy promote death in C. elegans (Kang et al., 2007). These observations imply that cellular factors and context determine whether autophagy is beneficial or detrimental to health.
Mitochondrial oxidative stress and high cellular calcium concentrations trigger opening of the mitochondrial permeability transition pore (mPTP). Opening of the mPTP, which is regulated by the voltage-dependent anion channel (VDAC), the adenine nucleotide translocators (Ant proteins), BAX, PiC, F1FO ATP synthase and cyclophilin D, permits diffusion of molecules of less than 1500 Da to flow freely across the mitochondrial inner membrane (Kwong and Molkentin, 2015). Transient mPTP opening regulates calcium and mitochondrial reactive oxygen species (ROS) signaling, while prolonged mPTP opening results in mitochondrial energetic dysfunction, organelle swelling, rupture and apoptotic or necrotic cell death (Kwong and Molkentin, 2015). Recent evidence suggests that opening of the pore also plays a role in mitochondrial quality control, as mPTP opening depolarizes mitochondria, marking them for autophagic clearance (Gottlieb and Mentzer, 2010). However, direct and mechanistic connections of the mPTP with regulation of autophagy have not been made.
The heteromeric protein kinase mTOR complex 2 (mTORC2) controls cellular metabolism, proliferation, and survival by phosphorylating members of the AGC (PKA/PKG/PKC) family of protein kinases, including serum/glucocorticoid regulated kinase 1 (SGK1) (Garcia-Martinez and Alessi, 2008). SGK1 is also regulated at different levels by growth factors, glucocorticoids, and inflammatory factors and has been implicated in responses to oxidative stress, apoptosis, and DNA damage (Di Cristofano, 2017). In worms, sgk-1 is a major effector of metabolic regulation downstream of mTORC2. rict-1, the C. elegans ortholog of the essential mTORC2 component Rictor, and sgk-1 modulate lifespan in a complex manner, depending on food source and temperature (Mizunuma et al., 2014; Soukas et al., 2009). Molecular determinants of longevity, metabolism and development acting downstream of mTORC2 and sgk-1 remain incompletely understood.
Here we show that in spite of shortened lifespan in rict-1 and sgk-1 mutants under standard laboratory conditions, these animals have increased levels of autophagy. Reducing function of autophagy genes restores normal lifespan in mTORC2 mutants, indicating that elevated autophagy is detrimental rather than beneficial in the context of dysfunctional mTORC2 signaling to SGK-1. We establish that the reason that autophagy shortens lifespan is due to a concomitant increase in mitochondrial permeability in mTORC2 mutants, attributable to increased VDAC1 protein level. We find that even in wild-type C. elegans, increased mitochondrial permeability shortens lifespan. Most importantly, this mechanism generalizes to many long-lived genetic mutants dependent upon autophagy. Under each circumstance tested, induction of mitochondrial permeability completely reverses autophagy-dependent lifespan extension. Finally, we show that hepatic ischemia/reperfusion injury is enhanced in liver-specific Sgk knockout mice due to increased mPTP opening. Thus, enhanced autophagy in the setting of increased mitochondrial permeability is a detrimental combination conserved from nematodes to mammals.
RESULTS
mTORC2 Pathway Members RICT-1 and SGK-1 Negatively Regulate Autophagy
Levels of a SGK-1::GFP reporter decrease in the starved state, suggesting that reduced SGK-1 signaling participates in the metabolic adaptation to starvation (Figures 1A and S1A). Phosphorylation of α catalytic subunit AMP-activated protein kinase Thr172, which is elevated in starvation, is increased in sgk-1 mutants (Figure S1B). sgk-1 mutant worms deliver a reduced brood size over an extended period (Figure S1C), similar to animals that are calorically restricted (Crawford et al., 2007; Wang et al., 2014). These results suggest that a key role of SGK-1 is to suppress adaptive responses to starvation in the fed state.
Figure 1. mTORC2 Regulates Autophagy in Both C. elegans and Mouse Hepatocytes.

(A) SGK-1::GFP detected by fluorescence microscopy, scale bar: 200 μm. (n = 67 worms for control and 69 for starvation, student’s t-test).
(B) C. elegans sgk-1 mutants show increased autolysosomes (arrows) by electron microscopy versus wild type (wt), scale bar: 500 nm. (n = 59 and 87 for wt and sgk-1, respectively student’s t-test).
(C) Increased autophagy levels in seam cells of rict-1 and sgk-1 mutants by GFP::LGG-1 puncta (n = 70, 73 and 71 for wt, rict-1, and sgk-1, respectively, one-way ANOVA).
(D) Western blotting of LGG-1::GFP, LGG-1::GFP-PE and cleaved free GFP protein levels in fed and starved rict-1 and sgk-1 mutants (n = 8 biological replicates, one-way ANOVA).
(E) Fluorescence microscopy of GFP::LGG-1 puncta in the proximal intestinal cells of rict-1 and sgk-1 mutants expressing wildtype LGG-1 or lipidation defective mutant LGG-1(G116A).
(F and G) Quantification of GFP::LGG-1 puncta in the intestine, muscle, hypodermis and pharynx of rict-1 (F) and sgk-1 (G) mutants expressing LGG-1 or LGG-1(G116A). (n > 25 cells analyzed per group, two-way ANOVA).
(H) GFP::LGG-1 and free GFP levels in wild type, rict-1, and sgk-1 mutants under vehicle and chloroquine (CQ, 100 mM for 18h) treatment. (n = 5 biological replicates, two-way ANOVA).
(I) Autophagy in Sgk1 knockout (Sgk1(−/−)) AML12 hepatocytes as indicated by LC3A/B turnover in both full medium and Hank’s balanced salt solution (HBSS) starvation conditions (n = 6 for control and n = 5 HBSS, two-way ANOVA).
(J) LC3A/B conversion in Sgk1(−/−) versus wild type (wt) AML12 cells with or without 50 μM chloroquine (CQ) for 4h (n =10 for vehicle and n = 6 for CQ, two-way ANOVA).
See also Figure S1. *p < 0.05, **p < 0.01. All bars indicate means and SEM.
We hypothesized that mTORC2 and SGK-1 may function as negative regulators of catabolic processes such as autophagy during nutrient sufficiency. In line with this, the number of autophagic vesicles evident by electron microscopy and green fluorescent protein GFP::LGG-1 puncta (C. elegans ATG8 ortholog, which marks autophagosomes) is increased in fed rict-1 and sgk-1 mutant worms (Figures 1B and 1C). Western blotting confirms increased LGG-1 protein levels and LGG-1 processing in rict-1 and sgk-1 mutant worms under both fed and starved conditions (Figure 1D). Numbers of GFP::LGG-1 puncta are increased in intestine, muscle, hypodermis and pharynx in rict-1 mutant worms; in sgk-1 mutant worms, numbers of GFP::LGG-1 puncta are increased in intestine and hypodermis but not in muscle and pharynx (Figures 1E–1G). This suggests that sgk-1 modulates intestinal and hypodermal autophagy downstream of rict-1. We confirmed that GFP::LGG-1-positive puncta represent autophagosomes and not unspecific aggregates by using mutant GFP-tagged LGG-1(G116A) protein that is expected to be defective in lipidation and autophagosome-membrane targeting (Manil-Segalen et al., 2014). In GFP::LGG-1(G116A) reporter animals, no increased puncta were seen in rict-1 mutant and sgk-1 mutant worms (Figures 1E–1G).
Increased numbers of GFP::LGG-1 puncta can be seen either in the setting of increased autophagic flux or, alternatively, due to the inhibition of late stages of autophagy (Zhang et al., 2015). We investigated autophagic flux with chloroquine (CQ) treatment, which increases lysosomal pH and impairs lysosomal function (Klionsky et al., 2016). CQ treatment induces accumulation of GFP::LGG-1 and free GFP protein in rict-1 and sgk-1 mutants versus wild type worms (Figure 1H). This indicates increased autophagic flux in rict-1 and sgk-1 mutants, and that increased GFP::LGG-1 puncta seen in these animals are not due to a block in late steps of autophagy.
To explore whether Sgk also modulates autophagy in mammals, we generated two distinct loss of function models for Sgk1: CRISPR/Cas9 Sgk1 knockout AML12 hepatocytes and liver-specific Sgk1 knockout mice (hereafter referred to as Sgk1Lko) (Fejes-Toth et al., 2008). Autophagy initiation is increased in both Sgk1 knockout AML12 hepatocytes and primary Sgk1Lko hepatocytes, as indicated by both immunoblotting for LC3A/B conversion (LC3A/B I to LC3A/B II) (Figures 1I and S1E) and CYTO-ID staining autophagic vacuoles (Figures S1D and S1F). Finally, CQ treatment in Sgk1 knockout AML12 hepatocytes leads to LC3A/B II accumulation, indicating increased autophagic flux (Figure 1J).
Inhibition of Autophagy Restores Normal Lifespan in Short-Lived mTORC2 Mutants
Given the strong positive association of elevated autophagy and longevity, we evaluated the role of autophagy in C. elegans mTORC2 and sgk-1 mutant lifespan. Surprisingly, RNAi of autophagy components bec-1 (C. elegans Beclin1 homolog) and lgg-1 restored normal lifespan in short-lived sgk-1 and rict-1 mutants (Figures 2A–2D). Confirming that bec-1 RNAi inhibits autophagy, GFP::LGG-1 puncta decrease in sgk-1 mutants after bec-1 RNAi (Figure 2E). RNAi of bec-1 extends lifespan similarly in both sgk-1 mutants and rict-1;sgk-1 double mutants (Figure S2A), suggesting sgk-1 is the main modulator of lifespan downstream of rict-1. RNAi of the autophagy regulator unc-51 (C. elegans ULK1 homolog) also partially restored lifespan, and nearly completely normalized the low brood size of sgk-1 mutant worms (Figures 2F, 2G and S2B). Silencing of autophagy components also accelerates sgk-1 mutant growth (Figure S2C). In contrast, RNAi of these components in wild type worms had no effect on development and modestly shortens lifespan (Figures 2A–2D, and S2D). RNAi of autophagy genes has no effect on fat levels in sgk-1 mutants (Figure S2E).
Figure 2. Inhibition of Autophagy Restores Normal Lifespan in Short-Lived sgk-1 and rict-1 Mutants.

(A-D) In sgk-1 and rict-1 mutants, RNAi of bec-1 (A and C) and lgg-1 (B and D) restores normal lifespan versus wild type (wt) worms treated with empty vector RNAi (ev) (log rank test).
(E) GFP::LGG-1 puncta in the intestine are reduced by bec-1 RNAi in sgk-1 mutants, scale bar: 20 μm. (n = 45 worms for ev and n = 46 for bec-1, student’s t-test).
(F and G) RNAi of unc-51 partially restores normal lifespan in sgk-1 (F) and rict-1 (G) mutants (log rank test).
(H) HLH-30::GFP protein level in sgk-1 mutants and wildtype animals, scale bar: 200 μm. (n =20 worms for wt and n = 15 for sgk-1, student’s t-test).
(I) Two different RNAi targeting hlh-30 both extend lifespan in sgk-1 and rict-1 mutants (log rank test).
See also Figure S2. For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All bars indicate means and SEM.
C. elegans HLH-30, homolog of mammalian transcription factor EB, is a positive transcriptional regulator of autophagy (Lapierre et al., 2013). Levels of HLH-30 are elevated in sgk-1 mutants as indicated by a HLH-30::GFP reporter (Figure 2H). As with bec-1 RNAi, knockdown of hlh-30 significantly extends the lifespan of sgk-1 mutants (Figure 2I).
Because these results are opposite to the positive effect of enhanced autophagy in many long-lived mutants, we surmised that the cellular context in mTORC2 mutants renders autophagy detrimental rather than beneficial. To begin to elucidate this context, we evaluated the spatiotemporal requirements for bec-1. Consistent with our prior observation that mTORC2 regulates metabolism, growth, and reproduction via action in C. elegans intestine (Soukas et al., 2009), intestine-specific, bec-1 RNAi restores normal lifespan and slow development in rict-1 mutant worms (Figures S2F–S2H). Whole life bec-1 knockdown had the greatest quantitative effect on restoration of normal lifespan in sgk-1 mutants, versus larval and adult only knockdowns (Figures S2I and S2J, RNAi efficiency in Figure S2K). Thus, intestinal autophagy in all life stages is detrimental to sgk-1 mutant longevity.
SGK-1 Regulates mPTP Opening
We employed unbiased protein mass spectrometry in order to determine the mechanistic tie between SGK-1 and autophagy. We used GFP tagged SGK-1 transgenic C. elegans in order to co-immunoprecipitate (co-IP) SGK-1 interacting proteins, versus a GFP only transgenic strain. Analysis of proteins that co-IP with SGK-1 indicated nearly the entire complement of protein regulators of the mPTP: VDAC-1, ANT-1.1, ANT-1.3, SPG-7, F-ATP synthase components ATP-5 and F58F12.1, and phosphate carrier protein SLC25A3 homologue F01G4.6 (Kwong and Molkentin, 2015; Shanmughapriya et al., 2015). The mPTP is a critical regulator of mitochondrial homeostasis, and it initiates autophagy under serum starvation or glucagon treatment (Elmore et al., 2001). To confirm that the mPTP and SGK-1 interact, we selected as bona fide SGK interactors only proteins that 1) have two or more unique peptide matches, and 2) are not present in GFP control or have at least 5-fold the number of total peptides and 5-fold the average precursor intensity versus GFP control. Using these criteria, six mPTP proteins interact with SGK-1. These six mPTP proteins are overrepresented in the total set of 244 SGK interactors (from our Mass Spec data), assuming 1150 mitochondrial proteins as the universe (mitoCarta 2.0) (Calvo et al., 2016) (hypergeometric test P value = 0.000113).
We evaluated direct protein-protein interaction between SGK-1 and regulators of the mPTP. GST-pull down suggests that the direct interaction of SGK-1 with the mPTP occurs through binding with VDAC-1 (Figure 3A). Interaction between mammalian SGK1 and VDAC1 was confirmed by endogenous co-IP (Figures 3B and 3C) and reciprocal co-IP of overexpressed SGK1 and VDAC1 (Figure S3A) in 293T cells, and is not affected by PI3K pathway inhibition (Figure S3B). Overall, these data are in line with our and others’ findings that SGK1 localizes to mitochondria (Cordas et al., 2007).
Figure 3. SGK-1 Regulates the mPTP Through Physical Interaction with VDAC-1.

(A) Purified C. elegans Myc-tagged VDAC-1 protein is pulled down by GST-SGK-1 and not GST alone in vitro.
(B) Endogenous VDAC1 is pulled down by native anti-SGK1 co-IP.
(C) Endogenous SGK1 is pulled down by native anti-VDAC1 co-IP.
(D) Calcium retention capacity assay in mouse Sgk1(−/−) and wt AML12 cells (arrows indicate calcium pulses). Equal numbers (1×107) of cells were used and equivalent biomass was confirmed by western blotting for Actin (right panel).
(E) RNAi of ant-1.1 restores normal lifespan in rict-1 and sgk-1 mutants (log rank test).
(F) RNAi of vdac-1 partially restores normal lifespan in sgk-1 mutants (log rank test).
(G) Lifespan extension by RNAi of F01G4.6 in sgk-1 mutants and wt C. elegans (log rank test).
(H) RNAi of spg-7 extends lifespan in sgk-1 mutants (log rank test).
(I) Fragmentation and abnormal cristae in sgk-1 mutants versus wt by electron microscopy (arrows indicate mitochondria), scale bar: 2 μm.
(J) Oxygen consumption is decreased in both rict-1 and sgk-1 mutants versus wt (n = 6, one-way ANOVA).
(K) Western blot for cytochrome C in wt animals and rict-1 and sgk-1 mutants. (n = 4, one-way ANOVA).
See also Figure S3. For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ****p < 0.0001. All bars indicate means and SEM.
Since SGK1 interacts with VDAC, we next asked whether SGK1 modulates mitochondrial permeability by regulating mPTP opening. As assessed with the calcium retention capacity assay, the mPTP opens after significantly fewer calcium pulses in AML12 hepatocytes lacking Sgk1 (Figures 3D and S3C). These data suggest that a normal role of SGK1 is to maintain mitochondrial homeostasis by supporting mPTP closure and mitochondrial calcium buffering capacity.
We next evaluated whether aberrant opening of the mPTP in sgk-1 mutant C. elegans is deleterious to longevity. Indeed, RNAi of the mPTP regulating gene ant-1.1 in rict-1 and sgk-1 mutants completely reverses their shortened lifespan and moderately extends lifespan of wild type worms (Figure 3E). Concordantly, RNAi of vdac-1, F01G4.6, spg-7, atp-5 or treatment with the mPTP inhibitor cyclosporine A also significantly extends the lifespan of sgk-1 mutants (Figures 3F–3H, S3D and S3E). Intestine specific RNAi of ant-1.1 in rict-1 mutants also partially restores normal lifespan, consistent with intestine as a major site of mPTP action in modulation of longevity, as for mTORC2 and autophagy components (Figure S3F).
Given the important role of sgk-1 in regulation of the mPTP, we examined mitochondrial morphology and function in sgk-1 mutants. By transmission electron microscopy, mitochondria in sgk-1 mutants exhibit more fragmentation, more dense morphology, and less apparent cristae (Figure 3I). Oxygen consumption and ATP levels are decreased in mTORC2 mutant worms, as are levels of the electron transport chain protein cytochrome C (Figures 3J, 3K and S3G). Reduced oxidative capacity of sgk-1 mutants is not due to decreased mitochondrial mass, as indicated by quantification of mitochondrial DNA and a mitochondrially localized RFP reporter (Figures S3H and S3I), rather may be due to greater mPTP opening and more fragmented mitochondria. RNAi of electron transport chain genes nuo-1, nuo-6 and cco-4 all significantly increase SGK-1 protein levels (Figures S3J and S3K). Together with the observation that mTORC2-SGK1 regulate mitochondrial function, this suggests that SGK-1 is activated by mitochondrial stress as part of a negative feedback loop maintaining mitochondrial homeostasis.
SGK-1 Negatively Regulates mPTP Function by Decreasing VDAC-1 Protein Levels
Levels of VDAC1 are associated with mPTP opening frequency (Tomasello et al., 2009). As SGK1 and VDAC1 physically interact (Figures 3A–3C), it logically follows that SGK-1 may regulate mPTP function by regulating VDAC1 abundance. In order to measure VDAC-1 protein in sgk-1 mutant worms, we created one integrated and two extrachromosomal transgenic C. elegans lines expressing Flag-tagged VDAC-1 (Figures S4A and S4B). VDAC-1-Flag levels are elevated ~2.5 fold in sgk-1 mutants (Figure 4A), in spite of equivalent mRNA levels (not shown). Native VDAC1 protein level is also higher in Sgk1 knockout primary hepatocytes (Figure 4B). In contrast, ANT protein in sgk-1 mutant C. elegans and ANT1 and VDAC2 protein levels in Sgk1 knockout hepatocytes remain unchanged (Figures S4C and S4D). The proteasome inhibitor MG132 eliminated the difference between wild type and Sgk1 knockout hepatocytes (Figure 4C), indicating that SGK1 regulates proteasomal VDAC1 degradation. Expression of constitutively active SGK1(S422D) increases the level of ubiquitinated VDAC1 to a greater extent than wild type SGK1, an effect eliminated by the PI3K inhibitor wortmannin (Figures 4D and 4E). Finally, overexpression of SGK1(S422D) decreases co-expressed VDAC1 protein levels when compared with SGK1(S422A) (Figure 4F), as the latter lacks the capacity to be activated by mTORC2 (Park et al., 1999).
Figure 4. SGK1 Modulates VDAC1 Protein Ubiquitination and Degradation by Regulating Its Phosphorylation.

(A) Western blot for VDAC-1::Flag protein levels in sgk-1 mutant worms versus wild type (n = 3, student’s t-test).
(B) Western blotting for native VDAC1 protein in primary hepatocytes from Sgk1 liver specific knockout (Sgk1Lko) versus Sgk1flox/flox control mice (control) (n = 3 cell samples per group, each sample from 1 mouse, student’s t-test).
(C) Native VDAC1 level by western blot in control Sgk1flox/flox and Sgk1Lko primary hepatocytes with or without proteasome inhibitor MG132 (5 μM for 16h) (n = 6 cell samples per group, each sample from 1 mouse, student’s t-test).
(D-E) Overexpression of active SGK1-S422D (D) increases VDAC1 ubiquitination by western blotting for HA-Ub and (E) is unaffected by the PI3K inhibitor wortmannin. 293T cells were co-transfected with wildtype SGK1 (SGK1-WT) or active SGK1 (SGK1-S422D) with VDAC1-Myc and HA-ubiquitin for 48h. Results shown are representative of 3 biological replicates.
(F) Overexpression of active SGK1-S422D decreases co-transfected VDAC1 protein level versus overexpression of inactive SGK1-S422A (n = 4, student’s t-test).
(G) In vitro kinase assay shows increased serine phosphorylation of VDAC1 after incubation with SGK1 by western blot (n = 3, student’s t-test).
(H) MS2 spectra identifies VDAC1 Ser104 (S#) as the SGK1 phosphorylation site (n = 2, student’s t-test).
(I) SGK1 phosphorylates serine in wildtype VDAC1 but not VDAC1-S104A in vitro (n = 3, two-way ANOVA).
(J) Ubiquitination of VDAC1 and VDAC1-S104A with overexpression of active SGK1-S422D by western blot. Results shown are representative of 3 biological replicates.
(K) Overexpression of active SGK1-S422D decreases wildtype VDAC1, VDAC1-S101/102A but not VDAC1-S104A protein levels compared with inactive SGK1-S422A overexpression (n = 3, two-way ANOVA).
See also Figure S4. *p < 0.05, **p < 0.01. All bars indicate means and SEM.
We reasoned that direct VDAC1 phosphorylation by SGK1 may prompt VDAC1 degradation. Constitutively active SGK1(S422D) increases serine phosphorylation of mammalian VDAC1 protein in vitro (Figure 4G). Mass spectrometric analysis indicates that Ser104 of VDAC1 is a bona-fide, direct, but non-canonical SGK1 target (Figure 4H). In confirmation, VDAC1(S104A), which cannot be phosphorylated, reduces baseline serine phosphorylation and eliminates SGK1-mediated VDAC1 serine phosphorylation in vitro (Figure 4I). Ubiquitination level of VDAC1(S104A) is decreased versus wild type VDAC1 when co-expressed with activated SGK1 (Figure 4J). Further, VDAC1(S104A) is resistant to SGK1 mediated protein degradation seen with wild type VDAC1 protein (Figure 4K). In contrast, mutation of other serine residues on VDAC1 did not prevent SGK1-mediated reductions in protein level (Figures 4K and S4E). Taken together, these data indicate that Ser104 phosphorylation of VDAC1 by SGK1 increases its ubiquitination and subsequent cellular clearance.
Accumulation of VDAC-1 Protein Induces Autophagy
Opening of the mPTP is associated with activation of autophagy (Elmore et al., 2001), and VDAC1 regulates mitochondrial autophagy (Geisler et al., 2010). Since VDAC-1 is increased in sgk-1 mutants, we analyzed autophagy with RNAi of vdac-1, which inhibits mPTP opening (Tomasello et al., 2009). Knockdown of vdac-1 in sgk-1 mutants decreases numbers of GFP::LGG-1 puncta and cleavage of GFP::LGG-1 to free GFP (Figures 5A and 5B). RNAi of ant-1.1 and treatment with cyclosporine A also reduces cleavage of GFP::LGG-1 to free GFP in sgk-1 mutants (Figures 5C and 5D). Vdac1 overexpression increases LC3A/B I/II conversion in mammalian cells, indicating that the association of mPTP opening with autophagy is conserved (Figure 5E). Taken together, these data demonstrate that aberrant mPTP opening attributable to excess VDAC-1 increases autophagy levels. Finally, double RNAi of bec-1 and ant-1.1 does not additively extend lifespan of sgk-1 mutants, suggesting action in the same genetic pathway (Figure S5A).
Figure 5. mPTP Opening Regulates Autophagy and Accelerates Aging.

(A and B) RNAi of vdac-1 decreases GFP::LGG-1 and free GFP protein levels (A) and decreases GFP::LGG-1 foci (B) in intestine of sgk-1 mutants, scale bar: 20 μm (n = 3, two-way ANOVA for A; n = 60 for ev and n = 51 for vdac-1, student’s t-test for B).
(C and D) In sgk-1 mutants, RNAi of ant-1.1 (C) or CsA treatment (D) decreases GFP::LGG-1 or free GFP protein levels (n = 3, two-way ANOVA).
(E) Autophagic flux determined by LC3A/B I/II levels detected in 293T cells following overexpression of Vdac1 for 48h with and without 50 μM chloroquine (CQ) for 4h (n = 3, two-way ANOVA).
(F and G) bec-1, lgg-1, unc-51 (F) and hlh-30 (G) mRNA levels in L4 stage wild type, rict-1, sgk-1 mutants and vdac-1 overexpression transgenics (n = 6, two-way ANOVA (F); n = 6, one-way ANOVA (G)).
(H) mPTP opening by calcium retention assay (arrows indicate calcium pluses) in 293T cells expressing Myc-tagged mouse VDAC1 and C. elegans VDAC-1 (A). Equal numbers (2×106) of cells were used and confirmed by blotting for Tubulin (right).
(I) Decreased cytochrome C protein in vdac-1 transgenic C. elegans. (n = 3, student’s t-test).
(J) vdac-1 overexpression shortens lifespan versus wt C. elegans (log rank test).
(K-L) RNAi of bec-1 (K) or ant-1.1 (L) extends lifespan of the vdac-1 overexpression strain (log rank test).
See also Figure S5. For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ****p < 0.0001. All bars indicate means and SEM.
Our earlier result that HLH-30 levels increase in mTORC2 mutants suggests that mTORC2 and VDAC-1 may regulate autophagy at the transcriptional level. mRNA levels of lgg-1, unc-51, and hlh-30, but not bec-1 are significantly increased in rict-1 and sgk-1 mutants as well as in transgenic vdac-1 overexpressing animals (Figures 5F–5G). These data suggest that elevated VDAC-1 levels stimulate autophagy at least partially transcriptionally.
Increased Mitochondrial Permeability Is Detrimental to Longevity and Organismal Health
To explore the impact of increased mitochondrial permeability on aging, we first confirmed that overexpression of both mouse Vdac1 and C. elegans vdac-1 in HEK293 cells facilitates mPTP opening (Figure 5H). Second, remarkably, vdac-1 overexpression alone in C. elegans is sufficient to shorten lifespan and decrease cytochrome C protein versus wild type animals (Figures 5I, 5J, S5B and S5C). As hypothesized based upon our observation that activated SGK-1 reduces VDAC-1 protein levels, sgk-1 overexpression normalizes the shortened lifespan of vdac-1 transgenics (Figure S5D).
Opening of the mPTP is also triggered by paraquat treatment in mammalian cells (Costantini et al., 1995). As expected, rict-1 mutants, sgk-1 mutants, and vdac-1 overexpression worms, which demonstrate facilitated mPTP opening, exhibit decreased survival with paraquat (Figures S5E and S5F).
Our results suggest that autophagy exerts a progeric, rather than a beneficial effect, in the setting of elevated mitochondrial permeability. To formally test this, we inhibited autophagy in our vdac-1 overexpression transgenic worms. Indeed, in vdac-1 overexpressers, RNAi of bec-1 restores normal lifespan, as does RNAi of ant-1.1 (Figures 5K–5L). RNAi of bec-1 or ant-1.1 also attenuates paraquat toxicity in rict-1 and sgk-1 mutants and vdac-1 overexpressers, and even moderately in wild type animals (Figures S5G–S5K). In aggregate, these results demonstrate that the toxic accumulation of VDAC-1 protein in sgk-1 mutants is responsible for three major defects: increased mitochondrial permeability, increased autophagy, and shortened lifespan.
One possibility is that blocking autophagy extends lifespan in sgk-1 mutants by activating the mitochondrial unfolded protein response (via atfs-1) (Lamech and Haynes, 2015) or oxidative stress responses (via FoxO/daf-16 and Nrf2/skn-1) (Blackwell et al., 2015). RNAi of bec-1 extends lifespan in both atfs-1;sgk-1 and daf-16;sgk-1 double mutant worms (Figures S6A and S6B), as does double RNAi of bec-1/skn-1 (Figure S6C). Therefore, the lifespan extending properties of autophagy RNAi in sgk-1 mutants does not require canonical mitochondrial defenses.
Inhibition of Autophagy Restores Lifespan by Enhancing Mitochondrial Function in sgk-1 Mutant Worms
We reasoned that excessive clearance of dysfunctional mitochondria could explain the detrimental effects of autophagy on lifespan in the setting of increased mitochondrial permeability. In line with this possibility, RNAi of bec-1 or vdac-1 improves oxygen consumption in sgk-1 mutants (Figures 6A and S6D–S6F). RNAi of bec-1 also improves ATP levels in sgk-1 mutants (Figure 6B). In addition, RNAi of bec-1, lgg-1 and unc-51 all increase cytochrome C protein levels in sgk-1 mutants (Figure 6C). Taken together, these data confirm that autophagy has a negative impact on mitochondrial functional capacity when mitochondrial permeability is increased.
Figure 6. Low Mitochondrial Permeability Is Required for Autophagy-Dependent Lifespan Extension.

(A and B) Basal and maximal (FCCP) oxygen consumption (A) and ATP level (B) in sgk-1 mutants with empty vector (ev) or bec-1 RNAi. (n = 12, two-way ANOVA; n = 7, student’s t-test).
(C) Cytochrome C western blot in sgk-1 mutants following RNAi of bec-1, lgg-1 or unc-51 (n = 4, one-way ANOVA).
(D) bec-1 RNAi does not extend lifespan in mev-1(kn1);sgk-1 double mutants.
(E) Larval knockdown of drp-1 extends lifespan in sgk-1 mutant worms (log rank test).
(F-I) sgk-1 loss of function or overexpression of vdac-1 eliminate autophagy-dependent lifespan extension in both eat-2(ad465) mutants (F and H) and glp-1(e2141) mutants (G and I) (log rank test).
(J) RNAi of frh-1 and nuo-6 extends lifespan in wild type worms but has no effect on vdac-1 transgenic worms (log rank test).
See also Figure S6. For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ****p < 0.0001. All bars indicate means and SEM.
We next tested whether the reduction in mitochondrial capacity is causally linked to shortened lifespan in sgk-1 mutants. To this end, we examined the impact of superimposition of severe mitochondrial electron transport dysfunction in sgk-1 mutants. In a short-lived mitochondrial complex II mutant mev-1(kn1) background (Ishii et al., 1998), RNAi of bec-1 does not extend the lifespan of sgk-1 mutants (Figure 6D). Thus, when autophagy inhibition can no longer increase overall mitochondrial oxidative capacity, as in the case of the mev-1;sgk-1 double mutant, it can no longer restore normal lifespan.
Our results suggest that enhanced mitochondrial clearance may be detrimental to sgk-1 mutants. Mitochondrial fragmentation has been shown to facilitate mitophagy (Burman et al., 2017). To test whether fragmentation in sgk-1 mutants (Figure 3I) is responsible for increased mitochondrial clearance and decreased lifespan, we performed RNAi of the mitochondrial fission gene drp-1. Knockdown of drp-1 during larval development significantly extends lifespan in sgk-1 mutants (Figure 6E). However, knockdown of mitophagy components pink-1 and pdr-1/PAKN, only partially restore normal lifespan (Figures S6G and S6H). Thus, a mitochondrial clearance pathway dependent upon drp-1 and bec-1 is principally responsible for decreased sgk-1 mutant lifespan.
Enhanced Autophagy Is Not Beneficial for Lifespan Extension with Increased Mitochondrial Permeability
Multiple longevity paradigms mechanistically depend upon autophagy, including long-lived C. elegans mutants lacking germline stem cells (glp-1), calorically restricted eat-2 mutants, and deficiency of daf-2/InsulinR (Hansen et al., 2018). We hypothesize that these paradigms are based upon a foundation of low mitochondrial permeability, as our results suggest that autophagy is transformed into a destructive force whenever mitochondrial permeability is increased. To determine if facilitated mPTP opening is sufficient to mitigate the beneficial effects of increased autophagy, we knocked down a member of mitochondrial complex II, sdhb-1, which induces mitochondrial ROS generation and facilitates mPTP opening (Chouchani et al., 2014; Pujol et al., 2013). Consistent with our hypothesis, RNAi of sdhb-1 eliminated lifespan extension in both eat-2 and glp-1 mutants (Figures S6I and S6J). Second, sgk-1;glp-1 and sgk-1;eat-2 double mutant worms do not have a long-lived phenotype versus wildtype, as loss of sgk-1 similarly facilitates mPTP opening (Figures 6F and 6G). Finally, direct stimulation of mPTP opening by vdac-1 overexpression in both eat-2 and glp-1 mutants completely eliminates lifespan extension seen in these animals (Figures 6H and 6I). Remarkably, daf-2 knockdown in vdac-1 overexpression transgenics still extends lifespan (Figure S6K), because daf-2 deficiency can independently reduce VDAC-1 levels (Figure S6L).
To further extend the generality of these findings, we next examined RNAi of two mitochondrial genes frh-1 or nuo-6, which is known to upregulate autophagy levels, and extend lifespan in an autophagy-dependent manner (Schiavi et al., 2013; Yang and Hekimi, 2010). vdac-1 overexpression completely suppressed the autophagy-dependent lifespan extension by frh-1 or nuo-6 knockdown (Figure 6J). Thus, autophagy only benefits longevity under conditions of low mitochondrial permeability.
Liver Specific Sgk Knockout Mice Are More Sensitive to Hepatic Ischemia/Reperfusion (I/R) Induced Injury
Hepatic I/R injury during liver surgery or liver transplantation negatively influences prognosis. The opening of mPTP in the first few minutes of reperfusion is known to be a critical determinant of I/R injury (Ong et al., 2015). Preventing its opening at this time using mPTP inhibitors such as cyclosporine A has been reported to reduce I/R injury in preclinical and clinical studies (Ong et al., 2015).
Given the importance of mPTP opening in I/R injury, we hypothesized that Sgk1Lko mice may have increased sensitivity to hepatic I/R injury due to increased mitochondrial permeability and autophagy (Figures S1E, S1F, and 3D). Indeed, following hepatic I/R (Figure 7A), serum alanine transaminase (ALT) and serum aspartate transaminase (AST) levels are more than doubled in Sgk1Lko versus control mice (Figures 7B and 7C). Apoptosis (indicated by cleaved Caspase 3) and autophagy (indicated by LC3A/B II) are also higher in Sgk1Lko mice after I/R injury (Figure 7D). Concordantly, Sgk1Lko mice also have elevated hepatic mRNA levels of pro-inflammatory cytokines IL-1β and IL-6 (Figure 7E).
Figure 7. Mice lacking Sgk Are More Sensitive to Liver Ischemia/Reperfusion (I/R) Injury.

(A) Hepatic warm I/R injury model.
(B and C) Serum ALT (B) and AST (C) levels in 12-week-old Sgk1Lko versus control Sgk1flox/flox mice after hepatic I/R injury (n = 10 per group, student’s t-test).
(D) Active, cleaved caspase 3 and LC3A/B II protein levels mice after hepatic I/R injury (n = 5 for control and n = 6 for Sgk1Lko, student’s t-test).
(E) mRNA levels of inflammatory genes IL-1β and IL-6 by QPCR (n = 11 per group, student’s t-test).
(F and G) Serum ALT (F) and AST (G) levels of 24-week-old SgkTKO mice versus Sgk1flox/flox control after hepatic I/R injury with or without cyclosporine A (CsA) pretreatment (n =8 for control and SgkTKO, and n = 7 for SgkTKO+CsA, one-way ANOVA).
(H) Active, cleaved caspase 3 and LC3A/B protein levels in liver after hepatic I/R injury with or without CsA pretreatment (n =7 for control and SgkTKO+CsA, and n = 8 for SgkTKO, two-way ANOVA).
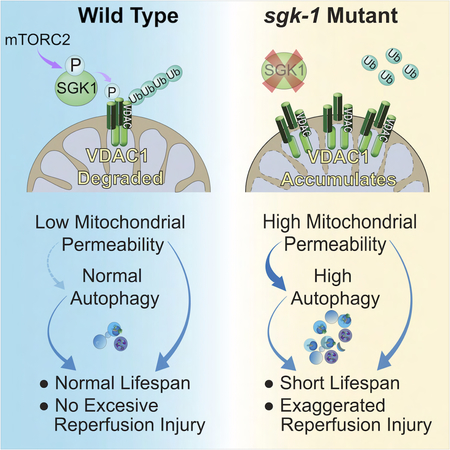
(I) Model of the effect of autophagy on lifespan with facilitated opening of the mPTP (mPTP open) versus normal mitochondrial permeability (mPTP closed) conditions.
*p < 0.05, **p < 0.01. All bars indicate means and SEM.
In order to address potential functional redundancy in the Sgk kinase family we generated mice lacking Sgk1, Sgk2, and Sgk3 in liver (SgkTKO). Similar to Sgk1 knockout hepatocytes, SgkTKO hepatocytes demonstrate facilitated mPTP opening in the calcium retention assay (Figure S7A). VDAC1 protein level and the LC3 II/LC3 I ratio are also higher in SgkTKO hepatocytes (Figure S7B). To determine whether mPTP opening under I/R is causal in increased hepatic injury in Sgk deficiency, we pharmacologically inhibited mPTP opening by pretreatment of SgkTKO mice with cyclosporine A. Remarkably, mPTP inhibition with CsA reduced post-injury ALT and AST levels in SgkTKO mice to a level comparable to control mice (Figures 7F and 7G). Concordantly, levels of cleaved Caspase 3 and LC3A/B II protein were restored to control levels in the CsA treated SgkTKO group (Figure 7H).
DISCUSSION
Our results demonstrate a mechanism connecting the nutrient and growth factor responsive kinase SGK1 to health and longevity through its regulation of mitochondrial permeability and autophagy. Work presented here suggests a model whereby the status of the mPTP is a major determinant of the health promoting effects of autophagy (Figure 7I). This represents a significant conceptual advance in our understanding of the way that autophagy modulates health and longevity.
mTORC2 and SGK1 Maintain Mitochondrial Homeostasis
We conclude that mitochondrial dysfunction in the setting of mTORC2 deficiency is related to increased propensity to open the mPTP due to toxic accumulation of VDAC-1 protein. Facilitated mPTP opening in rict-1 and sgk-1 mutant worms, in turn, directly compromises oxidative capacity, and ATP and cytochrome C protein levels. Thus, progeria in C. elegans mTORC2 pathway mutants is a direct consequence of increased mPTP opening and ensuing mitochondrial dysfunction.
mTORC2 is known to regulate mitochondrial function by maintaining mitochondria-associated ER membrane integrity (Betz et al., 2013). Our data demonstrate a previously unappreciated dimension of mitochondrial regulation by mTORC2. SGK1 has been reported to localize to mitochondria under stress, which permits access to physiologically appropriate mitochondrial interacting proteins and substrates, as part of cellular stress responses (O’Keeffe et al., 2013). We conclude that a major way that mTORC2 and SGK1 regulate mitochondrial permeability is through physical association of mTORC2 with mPTP regulatory molecules, modulating the abundance of the outer mitochondrial membrane protein VDAC1 through SGK1.
mTORC2 and SGK-1 Suppress Maladaptive Autophagy
We suggest that increased intestinal autophagic flux in sgk-1 mutant worms has negative consequences for longevity. However, our data leave open the possibility that autophagy is important in additional tissues in modulation of longevity downstream of mTORC2. Autophagic activity is regulated spatiotemporally in multiple tissues during aging (Chang et al., 2017). Spatiotemporal examination of autophagy in rict-1 and sgk-1 mutants may provide greater mechanistic understanding of how autophagy contributes to progeria. Finally, while our data suggest cell autonomous modulation of the mPTP and autophagy by mTORC2, they do not exclude the possibility of non-cell autonomous regulation of autophagy.
Data presented here suggest that a mechanism by which mPTP opening triggers autophagy is through transcriptional induction of the key autophagy transcription factor TFEB/HLH-30. Opening of the mPTP also induces ROS production (Batandier et al., 2004), and two major ROS H2O2 and O2− have been suggested to regulate autophagy (Scherz-Shouval and Elazar, 2011). Further, VDACs specifically interact with Parkin on defective mitochondria, targeting mitochondria for subsequent mitophagy (Geisler et al., 2010). Future work is needed to determine precisely how mPTP opening leads to induction of autophagy.
While mTORC2 regulates autophagy through modulation of the mPTP, our work leaves open the possibility of additional mechanisms. mTORC2-SGK signaling has been shown to activate mTORC1 (Castel et al., 2016), and mTORC1 plays a central role in suppression of autophagy (Saxton and Sabatini, 2017). Therefore, mTORC2 may indirectly reduce autophagy through SGK by activating mTORC1. Consistent with this possibility, our results show decreased mTORC1 activity in SgkTKO hepatocytes (Figure S7C). Irrespective of the mechanism, we conclude that a major role of mTORC2 and SGK1 is to act as autophagy gatekeepers.
mPTP Opening Negatively Modulates Longevity and Health
The work presented here indicates that in the absence of functional mTORC2 or SGK-1, increased opening of the mPTP drives both mitochondrial dysfunction and increased autophagy. We put forward that this combination is particularly devastating, shortening lifespan and increasing susceptibility to I/R injury.
Our results mechanistically connect increased mPTP opening to shortened lifespan and hypersensitivity to oxidative stress. Emerging data indicate that mitochondrial permeability increases in aged animals and humans and in aging-related degenerative diseases, and that diverse manipulations that promote health and extend lifespan decrease mPTP opening (Rottenberg and Hoek, 2017). Caloric restriction protects mitochondria from mPTP opening in rat heart (Hofer et al., 2009). Metformin, an antidiabetic drug with anti-aging properties, inhibits mPTP opening (Bhamra et al., 2008). Thus, maintenance of low mitochondrial permeability may be an obligate step in order to extend lifespan.
Autophagy as a Double-Edged Sword in Ischemia/Reperfusion Injury
The role of autophagy in I/R injury is complex. Beneficial effects of autophagy during I/R have been attributed to ATP generation, improved cellular homeostasis and decreased expression of inflammatory genes (Ma et al., 2015). On the other hand, excessive autophagy contributes to pressure overload-induced cardiac remodeling and heart failure in cardiac I/R injury (Gottlieb and Mentzer, 2010). In a rat hepatic I/R injury model, chloroquine, which inhibits autophagic flux, prevents ischemic liver damage at the early phase, but aggravates liver damage at the late phase in liver I/R injury (Fang et al., 2013).
Based upon our results, we hypothesize that the status of the mPTP is a major governing influence on the effects of autophagy on ischemia-reperfusion injury. Because widespread mPTP opening occurs shortly after reperfusion (Ong et al., 2015), and the ensuing mitochondrial permeability drives autophagy to be detrimental, inhibition of autophagy during early reperfusion reduces injury. At the late phase of reperfusion, the mPTP closes in mitochondria that are able to restore functionality, and autophagy plays a beneficial role by selectively degrading dysfunctional mitochondria and improving cellular homeostasis (Ma et al., 2015). Recent work suggests that accumulation of succinate is responsible for mitochondrial ROS production during I/R injury (Chouchani et al., 2014), sensitizing mitochondria to mPTP opening (Briston et al., 2017). It is tantalizing to speculate that mTORC2 mutants are more sensitive to succinate induced mPTP opening because of accumulation of VDAC1.
Resolving the Paradox: How Inhibition of Autophagy Restores Lifespan in mTORC2 Mutants
Increased autophagy is generally thought to be beneficial for longevity, as almost every manipulation that extends lifespan is dependent on autophagy (Madeo et al., 2015). Enhancing autophagy using genetic methods by activating Beclin increases longevity in mice, and similarly, overexpression of hlh-30 in C. elegans also extends lifespan (Fernandez et al., 2018; Lapierre et al., 2013). However, we conclusively demonstrate that elevated autophagy is detrimental for organismal health when coupled with increased mitochondrial permeability. We surmise that in mTORC2 mutant worms, increased mitochondrial permeability triggers mitochondrial fragmentation and initiates autophagy, resulting in excessive, non-selective mitochondrial clearance and failure to replace those mitochondria with normal, healthy organelles. Thus, inhibition of autophagy in mTORC2 mutants restores normal lifespan by increasing mitochondrial mass and restoring oxidative capacity. This is further supported by the failure of lifespan extension under bec-1 RNAi in sgk-1/mev-1 double mutants, indicating that mitochondria with intact electron transport chain activity are required to restore normal lifespan.
Several lines of evidence support the idea that increased mitochondrial permeability rather than a defect in ATP synthase activity underlies lifespan shortening in mTORC2 mutants. First, RNAi of ATP synthase extends lifespan (Dillin et al., 2002; Lee et al., 2003). Second, knockdown of ant-1.1 or atp-5, which decreases ATP production, further extends lifespan in sgk-1 mutant worms. Finally, RNAi of bec-1 or vdac-1 increases oxygen consumption and ATP level in sgk-1 mutant worms but not in wildtype worms, suggesting the energetic defect in these mutants is due to mPTP dysregulation and ensuing autophagy rather than a primary deficit in energy generation.
The work presented here suggests an over-arching idea that cellular context determines whether autophagy has beneficial or detrimental effects on health. An emerging theme in longevity studies is that in order for a manipulation to extend lifespan, it must suppress mitochondrial permeability. Further, increases in mitochondrial permeability accompany aging and many disease states, and our work indicates that these increases in mitochondrial permeability are causal in aging. Our data also intimates that autophagy induced during I/R injury is harmful, exacerbating damage, if accompanied by increases in mPTP opening. Future work will be needed to determine whether mitochondrial permeability can be favorably manipulated in preclinical and clinical studies in order to promote healthy aging and reduce morbidity associated with diseases such as ischemic stroke, heart attack, ischemic renal and hepatic disease, and with organ transplantation.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alexander A. Soukas (asoukas@mgh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS Nematode Strains
Nematode Strains
N2 Bristol (RRID:WB-STRAIN:N2_(ancestral)) was used as the wild-type strain. The listed mutant strains were used: MGH12 mgIs60[sgk-1::GFP pRF4], MGH48 mgIs43[ges-1p::GFP_PTS1], MGH266 rict-1(mg451) 8x, MGH300 sgk-1(mg455) 8x, MGH55 daf-16(mgDf47) 3x, MGH361 atfs-1(tm4525) 3x, TK22 mev-1(kn1) 5x (RRID:WB-STRAIN:TK22), DA465 eat-2(ad465) (RRID:WB-STRAIN:DA465),CB4037 glp-1(e2141) (RRID:WB-STRAIN:CB4037), MGH358 mev-1(kn1);sgk-1(mg455), MGH359 atfs-1(tm4525);sgk-1(mg455), MGH360 daf-16(mgDf47);sgk-1(mg455), MGH369 sgk-1(mg455), DA2123 adIs2122 [lgg-1p::GFP::lgg-1 + rol-6(su1006)] (RRID:WB-STRAIN:DA2123), MGH371 rict-1(mg451); adIs2122 [lgg-1p::GFP::lgg-1 + rol-6(su1006)], MGH372 sgk-1(mg455);adIs2122 [lgg-1p::GFP::lgg-1 + rol-6(su1006)], MGH392 mgIs60[sgk-1::GFP pRF4]; alxIs28[vdac-1p::VDAC-1::FLAG myo-2p::GFP], RD202 unc-119(ed3) III; ppIs??[unc-119(+) lgg-1p::gfp::lgg-1(G116A)], MAH885 sgk-1(mg455);unc-119(ed3)? III; ppIs?[unc-119(+) lgg-1p::gfp::lgg-1(G116A)] and MAH886 sgk-1(mg455);unc-119(ed3)? III; ppIs?[unc-119(+) lgg-1p::gfp::lgg-1(G116A)]; JIN1821 jinIs[hlh-30p::HLH-30::GFP]. N2, TK22 mev-1(kn1), DA465 eat-2(ad465) and CB4037 glp-1(e2141) strains were obtained from the Caenorhabditis Genetics Center (CGC). MGH361 atfs-1(tm4525) was a gift from Cole Haynes’ lab. JIN1821 jinIs[hlh-30p::HLH-30::GFP] was a gift from Javier Irazoqui. For mitochondrial targeted RFP expression: MGH84 mgIs54[myo-3p::mito::mRFP3] and MGH353 sgk-1(mg455);mgIs54[myo-3p::mito::mRFP3] were used. For vdac-1 overexpression, MGH354 alxEx91[vdac-1p::VDAC-1::FLAG::SL2::GFP] (vdac-1 oe1), MGH355 alxEx92[vdac-1p::VDAC-1::FLAG myo-2p::GFP] (vdac-1 oe2), MGH356 alxIs28[vdac-1p::VDAC-1::FLAG myo-2p::GFP] (vdac-1 oe) 8x and MGH357 glp-1(e2141);alxEx91[vdac-1p::VDAC-1::FLAG::SL2::GFP] were generated for this study. For tissue-specific RNAi, MGH167 sid-1(qt9);alxIs9[vha-6p::SID-1::SL2::GFP], MGH206 rict-1(mg451);sid-1(qt9);alxIs9[vha-6p::SID-1::SL2::GFP] were used. Nematode strains were maintained on nematode growth medium (NGM) plates seeded with E. coli OP50. All experiments were conducted at 20 °C unless otherwise noted.
E. coli Strains
Non-RNAi experiments were all conducted on NGM plates containing E. coli OP50–1 (CGC) as the food source. Cultures of E. coli OP50 were grown in LB broth for 15h at 37 °C without shaking and seeded directly onto NGM plates. Plates were used 3–7 days after seeding.
All RNAi experiments were conducted using E. coli HT115 (DE3) bacteria (Ahringer and Vidal RNAi Library) as the food source. Cultures of E. coli HT115 (DE3) were grown in LB broth for 15h at 37 °C with shaking, then cultures were concentrated to 1/10 volume by centrifugation and seeded directly onto NGM plates containing 5 mM isopropyl-B-D-thiogalactopyranoside and 200 μg/ml carbenicillin. Plates were used 1–5 days after seeding.
Mice
All mice were housed in a temperature-controlled room under a 12 hr light–dark cycle and under specific pathogen-free conditions, and provided ad libitum access to water and standard laboratory chow (Prolab Isopro RMH 3000). Sgk1[flox/flox] (RRID: MGI:5317851) mice were as we previously published (Fejes-Toth et al., 2008). Sgk1Lko mice were generated by crossing Albumin-Cre mice (The Jackson Laboratory, RRID: IMSR_JAX: 003574) with Sgk1[flox/flox] mice. Double Sgk2;Sgk3 global knockout mice were generated by C57BL/6 zygotic injection of small guide RNAs targeting Sgk2 and Sgk3 and S.p.Cas9 mRNA at the Genome Modification Facility, Harvard University. Mice were backcrossed four times prior to experimentation. SgkTKO mice were generated by crossing Sgk1Lko mice to Sgk2;Sgk3 global knockout mice. Twelve-week-old male Sgk1Lko and control mice, and 24-week-old male SgkTKO and control mice were used for hepatic ischemia/reperfusion assays. All animal procedures were approved under protocol 2010N000186 by Massachusetts General Hospital Subcommittee on Research Animal Care. Guide RNA sequences used for generating double Sgk2;Sgk3 global knockout mice are as follows (See also Table S2):
Sgk2: CGGAGCCTTCTACGCCGTGA;
Sgk3: CAAGGCACTGGCGATCTCCG.
Cell Lines, Culture Conditions and Transfection
The AML12 (alpha mouse liver 12) cell line was established from hepatocytes from a male mouse (CD1 strain, line MT42) transgenic for human TGF alpha (ATCC, ATCC® CRL-2254™). AML12 hepatocytes were grown in a 1:1 mixture of DMEM and Ham’s F12 medium (Thermo Fisher) with 0.005 mg/ml insulin, 0.005 mg/ml transferrin, 5 ng/ml selenium, 40 ng/ml dexamethasone, 10% fetal bovine serum (FBS, Sigma) and 1% penicillin-streptomycin (PS, Life Technologies), in a 5% CO2 atmosphere at 37°C. Primary cultured mouse hepatocytes were prepared from male C57BL/6, liver-specific Sgk1 knockout mice (genotype Albumin-Cre;Sgk1[flox/flox]) and their wild-type littermate controls (genotype Sgk1[flox/flox]) by collagenase perfusion method as described previously (Li et al., 2010), and were grown in Williams’ Medium E supplemented with 10% FBS and 1% PS. All animal procedures were approved under protocol 2010N000186 by Massachusetts General Hospital Subcommittee on Research Animal Care. HEK293T cells (a gift from Joseph Avruch’s lab), generated from female human embryonic kidney with stably expressing the SV40 T-antigen, were grown in high glucose DMEM (Thermo Fisher) supplemented with 10% FBS and 1% PS in a 5% CO2 atmosphere at 37 °C. For transfection, Lipofectamine 3000 was used a ccording to the instructions of manufacturer (Thermo Fisher).
METHOD DETAILS
Generation of Sgk1 Knockout AML12 Cells
Sgk1 knockout AML12 cells were generated using CRISPR/Cas9 (Cong et al., 2013) with modifications. The px330 vector expressing Cas9 and guide RNA was electro-transfected into AML12 cells together with a plasmid conferring puromycin resistance. Forty-eight hours after transfection, puromycin was added to the cells at 5 μg/ml for 48 hours, then puromycin resistant cells were trypsinized, counted and diluted to 10 cells/ml, then 100 μl of the diluted cell mixture per well was seeded into a 96 well plate, and single-clones were expanded to generate stable cell lines. Sgk1 knockout was confirmed by western blotting. Primers used for sgRNA expression are (See also Table S2): sense, 5’- CACCGTAAGCAGCCGTATGACCGGA-3’, antisense, 5’-AAACTCCGGTCATACGGCTGCTTAC-3’.
Transmission Electron Microscopy (TEM)
C. elegans TEM was performed by the Transmission Electron Microscopy Core at Massachusetts General Hospital (Lapierre et al., 2013). Wild type and sgk-1(mg455) synchronized L4 populations were collected and incubated in fixation buffer (2.5% glutaraldehyde and 1.0% paraformaldehyde in 0.05M sodium cacodylate at pH7.4 plus 3.0% sucrose). During the initiation of fixation, animals were cut in half with a surgical blade in a drop of fixative under a dissecting microscope, fixed overnight at 4°C, rinsed in 0.1M cacodylate, post-fixed in 1.0% osmium tetroxide 0.1M cacodylate buffer, rinsed in buffer and water. Animals were embedded in 2% agarose in phosphate buffered saline (PBS), dehydrated through a graded series of ethanol washes to 100%, then 100% propylene oxide, and finally 1:1 propylene oxide/EPON overnight. Blocks were infiltrated in 100% EPON and then embedded in fresh EPON overnight at 60 °C. Thin sections were cut on a Leica UC6 ultramicrotome and collected on formvar-coated grids. Sections were post-stained with uranyl acetate and lead citrate and viewed using a JEOL 1011 transmission electron microscope at 80 kV with an AMT digital imaging system (Advanced Microscopy Techniques). Autolysosomes were identified as vesicles containing material undergoing degradation in micrographs of a cross-section of the intestine.
Measuring Autophagy Levels in C. elegans and Cells
Autophagy levels were assessed following published guidelines (Klionsky et al., 2016; Zhang et al., 2015). Autophagosomes were scored as follows (Lapierre et al., 2013). For C. elegans starvation, worms were kept in M9 for 24h in a 3cm dish with gentle shaking. For imaging and puncta quantification, animals were mounted on a 2% agarose pad in M9 medium containing 0.1% NaN3 and GFP::LGG-1/Atg8 puncta were counted using a Zeiss Imager Z1 including apotome.2 with a Hamamastsu orca flash 4LT camera and Zen 2.3 software. The total number of GFP::LGG-1/Atg8 puncta was counted in all visible hypodermal seam cells, the striated body-wall muscle, the three to four most proximal intestinal cells or the terminal pharyngeal bulb at 100x magnification. For mammalian cells, autophagosomes were stained using Cyto-ID according to manufacturer instructions (Enzo Life Sciences). Cells were fixed with 4% paraformaldehyde for 20 min and rinsed with PBS twice. Cells were mounted and visualized under a Leica DM6000 microscope with a 63X oil immersion objective.
Chloroquine C. elegans Treatment
For chloroquine treatment, 100 mM chloroquine was added on top of the NGM plates seeded with OP50. About 500 synchronized L4 animals were seeded onto these NGM plates for 18h before harvest.
Western Blotting
Worm and cell lysates were prepared in RIPA buffer (50 mM Tris-HCl pH 7.4,150 mM NaCl, 1% Triton x-100, 1% Sodium deoxycholate, 0.1% SDS,1 mM EDTA with proteinase inhibitors cocktail from Roche) unless otherwise indicated, followed by water bath sonication on maximum energy in a Diagenode Bioruptor XL 4 °C water bath at 30s/30s on and off intervals for a total of 15 min. Lysates were cleared of insoluble material by centrifugation at 21,000g at 4 °C and the supernatant was retained for western blotting. Protein concentration was determined using the Pierce BCA assay (Thermo Fisher). SDS-PAGE was conducted followed by electrophoretic transfer to nitrocellulose membrane at 100 V for 1 hour at 4 °C. Immunoblots were performed according to primary antibody manufacturers’ protocols. For immunodetection of primary antibodies, goat-anti-rabbit-HRP conjugate or goat-anti-mouse-HRP conjugate (GE Healthcare) was used at 1:5,000 in 5% BSA dissolved in TBST, and HRP was detected using West-Pico chemiluminescence substrate (Thermo Pierce). The western blot results shown are representative of at least three independent experiments.
Longevity Assay
Lifespan analysis was conducted at 20°C except where indicated, as previously published (Soukas et al., 2009). Synchronized L1 animals were seeded onto NGM (for mutants and CsA treatment) or RNAi plates and allowed to grow until the L4 stage (for RNAi). On day 0 as indexed in the figure legend, about 100 L4 worms per plate were transferred onto NGM or RNAi plates. All plates were supplemented with 100 μM 5-fluorodeoxyuridine (FUdR) to suppress progeny production. For CsA treatment, about 100 synchronized L1 animals were seeded onto NGM plates containing 5 μM CsA, and at the L4 stage, worms were transferred to NGM plates containing both CsA and FUdR for the remainder of their life. For glp-1 mutant worms, synchronized L1 animals were raised at 25°C (to eliminate germ cells) until young adulthood, then worms were shifted to 20°C for the remainder of life. When compared with other strains using FUdR, strains carrying the glp-1 mutation were also put on plates with FUdR for lifespan analysis. Dead worms were counted every day. Statistical analysis was performed with online OASIS2 resources (Han et al., 2016).
RNA Interference (RNAi)
RNAi clones were isolated from a genome-wide E. coli RNAi library, sequence verified, and fed to animals. RNAi feeding plates (6 cm) were prepared using a standard NGM recipe with 5 mM isopropyl-B-D-thiogalactopyranoside and 200 μg/ml carbenicillin. RNAi clones were grown for 15 hours in Luria Broth (LB) containing 200 μg/ml carbenicillin with shaking at 37°C. The stationary phase culture was then collected, concentrated through centrifugation, the supernatant was discarded and the pellet was resuspended in LB to 10% of the original culture volume; 300 μl of each RNAi clone concentrate was added to RNAi plates and allowed to dry no more than 48 hours prior to adding the worm embryos or animals.
Quantitative RT-PCR
Worms and mouse tissues were flash frozen in liquid nitrogen and kept in −80 °C until RNA preparation. To quantify changes in mRNA abundance in nematode and mammalian cells, total RNA was extracted using RNAzol RT (Molecular Research Center) according to manufacturer instructions. RNA was treated with RNAse free DNAse prior to reverse transcription with the Quantitect reverse transcription kit (Qiagen). Quantitative RT-PCR was conducted in triplicate using a Quantitect SYBR Green PCR reagent (Qiagen) following manufacturer instructions on a Bio-Rad CFX96 Real-Time PCR system (Bio-Rad). For C. elegans, all assays were performed with 1000 animals per sample at late L4 stage or as indicated in the figure legend. For mouse tissue samples, liver samples were homogenized in RNAzol using a TissuleLyser II (Qiagen), centrifuged for 10 min at 12,000g to pellet debris, and the supernatant was collected and subjected to RNA extraction per the manufacturer protocol. Expression levels of tested genes were presented as normalized fold changes to the mRNA abundance of control genes indicated in the figures by the δδCt method.
The primers used for qPCR are as follows (See also Table S2):
act-3, Forward: TGCGACATTGATATCCGTAAGG
act-3, Reverse: GGTGGTTCCTCCGGAAAGAA
nd-1, Forward: AGCGTCATTTATTGGGAAGAAGAC
nd-1, Reverse: AAGCTTGTGCTAATCCCATAAATGT
IL-10, Forward: GCTCTTACTGACTGGCATGAG
IL-10, Reverse: CGCAGCTCTAGGAGCATGTG
IL-1β, Forward: AAATACCTGTGGCCTTGGGC
IL-1β, Reverse: CTTGGGATCCACACTCTCCAG
bec-1, Forward: ACGAGCTTCATTCGCTGGAA
bec-1, Reverse: TTCGTGATGTTGTACGCCGA
lgg-1, Forward: GCCGAAGGAGACAAGATCCG
lgg-1, Reverse: GGTCCTGGTAGAGTTGTCCC
unc-51, Forward: CGCCGGTGGTTCAGCGGATT
unc-51, Reverse: TATCCTGGGTGTCGGCGGGG
hlh-30, Forward: GGATTAAGGAGCTCGGACAA
hlh-30, Reverse: TTTTTGGAGCACTCGGATGT
Brood Size
Ten synchronized animals per strain were transferred daily to fresh RNAi plates containing E. coli HT115 expressing target dsRNAs, and hatched progeny were counted 3 days later. Total brood size was determined by adding progeny produced across all days.
Developmental Timing
Developmental timing analysis was conducted on synchronized L1 animals. Synchronous animals were dropped onto RNAi plates containing empty vector RNAi control or a sequence-verified target RNAi in E. coli HT115 and grown at 20°C. Hermaphrodite animals were scored for their transition into adulthood by appearance of the vulvar slit at the indicated time points. Two to three biological replicates were carried out for each condition.
Nile-Red Staining
Fixative Nile-Red staining was conducted by washing 200–300 young adult animals from synchronized L1 worms obtained by egg prep. The worms were resuspended and washed twice with PBS and then suspended in 40% isopropanol for 5 min. Then the worms were stained in Nile-Red staining solution (3μg/ml Nile-Red in 40% isopropanol) in the dark for at least 2 hours. After staining, worms were washed and put in PBS with 0.01% Triton in the dark for 30 min. Imaging and quantitation was then conducted in fluorescence with a GFP/FITC filter using a Leica DM6000 microscope and Leica MM AF software.
SGK-1-GFP Co-IP for Mass Spectrometry
Transgenic C. elegans strains that express SGK-1::GFP (MGH12) or GFP only (MGH48) were used for Co-IP. Ten 15cm dishes of mixed age worms were used to get enough protein for mass spectrometry. Briefly, worm lysates were made using RIPA buffer (10mM Tris-HCl pH 7.2, 5mM EDTA, 150mM NaCl, 0.1% SDS, 1.0% Triton X-100, 1% Deoxycholate with Protease inhibitors). SGK-1::GFP was pulled down using an anti-GFP antibody (ThermoFisher) and eluted with 2X Laemmli Buffer without DTT. Samples were loaded onto a 10% SDS-PAGE gel and stained using Coomassie Brilliant Blue. Bands corresponding to broad ranges of molecular weight were cut and sent to The Taplin Biological Mass Spectrometry Facility at Harvard for in gel trypsin digestion and subsequent mass spectrometric peptide/protein identification.
Oxygen Consumption
Synchronized, young adult worms grown on either NGM OP50 or RNAi plates were washed repeatedly with M9 media to remove bacteria. Oxygen consumption of 2500 worms was then measured immediately in 1.5 mL of M9 using a Strathkelvin 928 6-channel Clark electrode respirometer with stirring. Independent measurements were conducted and averaged per strain, per RNAi and normalized to total protein content determined using a BCA assay (Pierce).
ATP Measurement
ATP measurement was carried out according to a modified protocol based on the published literature (Palikaras and Tavernarakis, 2016). 100 synchronized young adult worms were collected in M9 buffer and washed three times. Worm pellets were resuspended with 50ml M9 and boiled for 15 min to release ATP and destroy ATPase activity and then spun at 4° at 12,000 × g for 10 m in, and the supernatant was diluted 10 times for ATP measurement. ATP content was measured with an ATP detection kit according to the manufacturer’s instructions (ThermoFisher, A22066).
Creation of the VDAC-1::FLAG C. elegans Transgenic Lines
For VDAC-1 expression, the entire genomic sequence of the vdac-1 locus (1367 bp), including introns and exons, plus 2418 bp of promoter were amplified and cloned into a modified fire vector driving VDAC-1 with a C-terminal Flag epitope tag and GFP expression from the same promoter separated by the gpd-2/3 splice leader sequence (pAS21). The following cloning primers were used: forward (See also Table S2): 5’-ATCGTCGACTCCAGTTGTGGCTTAGAGACTTC −3’ and reverse: 5’-CCCGGGCTACTTGTCGTCATCGTCTTTGTAGTCGTTGGATGGATCGAATTCGAGTC −3’. The VDAC-1 expression construct was injected at 100 ng/ml into the gonad of wild type adult animals.
VDAC1 In Vitro Pull-Down Assay and Ubiquitination Assay
C.elegans vdac-1 cDNA was amplified using following primers (See also Table S2): vdac-1 forward, 5’- CATGCGGCCGCATGGCCCCACCAACCTTCGCTG −3’, vdac-1 reverse, 5’- CGAGATATCGTTGGATGGATCGAATTCGAGT −3’, Amplified fragments were inserted into pcDNA3.1-Myc-His vectors with NotI and EcoRV double digestion.
C.elegans sgk-1 was cloned into the pEBG vector to express a GST-SGK-1 fusion protein using these primers:
forward, 5’- CGCACTAGTGTGAGGAAAGATGAGGTGACAT −3’,
reverse, 5’- ATAGCGGCCGCTCAGACCAAAACGCGATTGGTG −3’.
Mammalian Sgk1 and Vdac1 were cloned into pCMV5-Flag and pcDNA3.1-Myc-His vectors respectively using the following primers:
Sgk1 forward, 5’- ATTTGCGGCCGCTGGAAGATGGTAAACAAAGACA −3’,
Sgk1 reverse, 5’- TAGGATCCTCAGAGGAAGGAATCCACAGGA −3’,
Vdac1 forward, 5’- CATGCGGCCGCATGGCCGTGCCTCCCACATACG −3’,
Vdac1 reverse, 5’- CGAGATATCTGCTTGAAATTCCAGTCCTAGG −3’.
To make the constitutively active SGK1, Ser422 was changed to Asp by site directed mutagenesis, a change previously shown to increase kinase activity (Park et al., 1999).
For GST-pulldown assays, GST-SGK-1 protein was purified from HEK293T cells transfected with expression vectors using the Pierce GST Spin Purification Kit (Thermo Scientific, Prod# 16106). Five micrograms of purified GST and GST-SGK-1 proteins were re-immobilized with pre-equilibrated 20 μl GST beads in lysis buffer. VDAC-1-Myc-His protein was purified from HEK293T cells following transfection of the indicated expression vectors using the Pierce HisPur Ni-NTA Purification Kit (Thermo Scientific, Prod# 88227). Protein concentration was determined with the Pierce BCA Protein Assay Kit (Thermo Scientific) before the GST-pulldown assay. Five micrograms of His purified protein were incubated with the immobilized beads at 4°C for 1.5 hours. After washing the beads three times with lysis buffer, samples were analyzed by western blot using the anti-Myc antibody.
For the Co-IP assay with overexpressed protein, GST-SGK-1 and VDAC-1-Myc-His were co-transfected into HEK293T cells. Forty-eight hours after transfection, cells were lysed using Co-IP buffer (0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, 5% glycerol; pH 7.4), GST-SGK-1 protein was pulled down using the Pierce GST Spin Purification Kit, and samples were analyzed by western blot using anti-Myc antibody. Similarly, for mammalian SGK1 and VDAC1, Flag-SGK1 and VDAC1-Myc-His were co-transfected into HEK293T cells for 48h, cells were then lysed using Co-IP buffer, and lysate was incubated with M2-Flag magnetic beads from SIGMA following its Co-IP protocol. Western blot was used for detection of VDAC1-Myc. For the Co-IP assay of mammalian endogenous protein, 107 HEK293T cells were lysed using Co-IP buffer, SGK1 or VDAC1 protein was pulled down using SGK1 antibody (Cell signaling Technology) and protein A magnetic beads (New England Biolabs) or VDAC1 antibody (Santa Cruz) and protein G magnetic beads (New England Biolabs) following the manufacturer’s protocol.
For detection of VDAC1 ubiquitination, Flag-SGK1, VDAC1-Myc-His and HA-Ub plasmids were co-transfected into HEK293T cells for 48h, then cell lysates were made using RIPA buffer, and magnetic Protein A beads (New England Biolabs) were used for Myc IP following the manufacturer’s protocol. Anti-HA antibody was used to detect the ubiquitination of VDAC1 protein.
mtDNA Copy Number
The mtDNA copy number was measured by quantitative PCR. Worms at the indicated developmental stage were singled in worm lysis buffer for PCR. Primers for NADH dehydrogenase subunit 1 (nd1) and actin-3 (act-3) were used in determination of mtDNA copy number. Primers used were as follows (See also Table S2): act-3 forward primer: 5′- TGCGACATTGATATCCGTAAGG −3′, and reverse primer 5′-GGTGGTTCCTCCGGAAAGAA −3′; nd1 forward primer 5′-AGCGTCATTTATTGGGAAGAAGAC −3′, and reverse primer 5′-AAGCTTGTGCTAATCCCATAAATGT −3′. PCR conditions were 2 min at 50°C, 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 60 s at 60°C. Quantitative PCR was conducted in triplicate using Quantitect SYBR Green PCR reagent (QIAGEN) following manufacturer instructions on a CFX96 Real-Time PCR system (Bio-Rad). The CT values of nd1 were normalized to nuclear genomic DNA (act-3). Quantitative PCR was performed for each initial template copy number at least four times and the results were reproducible.
Mitochondrial Ca2+ Uptake
Cells were permeabilized in a buffer containing 125 mM KCl, 2 mM K2HPO4, 1 mM MgCl2, 20 mM Hepes pH 7.2, 0.005% digitonin, 5 mM glutamate and malate, and a cell-impermeable Ca2+ indicator (0.5 μM Calcium Green-5N). The fluorescence of the Ca2+ indicator dye was monitored in real time using a PerkinElmer LS55 fluorimeter using a cuvette with stirring at room temperature. Pulses of CaCl2 were given sequentially (5 μl of 3 mM CaCl2 was added to 500 μl assay buffer). At least three replicates were performed for each experiment, and representative traces are shown.
Paraquat Treatment
For paraquat treatment, 8 mM paraquat was added on top of the NGM (for mutants) or RNAi agar plates. About 100 synchronized L4 animals were seeded onto the NGM or RNAi plates seeded with OP50 or HT115 expressing dsRNA for RNAi respectively, and dead worms were counted every day. Statistical analysis was performed with online OASIS2 resources.
Identification of the VDAC1 Phosphorylation Site by Mass Spectrometry
Proteins were separated by SDS-PAGE, gel bands were cut and destained in 50 mM ammonium bicarbonate:acetonitrile (50:50) until clear. Protein were in-gel reduced with dithiothreitol (DTT), alkylated with iodoacetamide, and digested with trypsin. Peptides were extracted and subjected to C18 solid phase extraction. The generated peptides were subjected to microcapillary liquid chromatography followed by tandem mass spectrometry (LC-MS2) on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). Full MS spectra were acquired using the Orbitrap analyzer, and MS2 spectra were acquired in a data denpdent mode after CID fragmentation using the linear ion trap. MS2 spectra were assigned using a SEQUEST proteomics analysis platform requiring cysteine residues to be carbamidomethylated (mass increment of 57.02146 Da), while methionine residues were allowed to be oxidized (15.99492 Da) and serine, threonine and tyrosine residues were allowed to be phosphorylated (79.96633 Da). The database used in the search was the UniProt database (02/04/2014) of human protein sequences including known contaminants such as trypsin. Based on the target-decoy database search strategy and employing linear discriminant analysis and posterior error histogram sorting, peptide and protein assignments were filtered to false discovery rate (FDR) of ˂ 1 %. The localization of phosphorylation sites was determined using the modscore algorithm (Schwartz and Gygi, 2005). For quantification, the MS1 signal for the phosphorylated peptide and unphosphorylated peptide was integrated using Qual Browser software (Buisson et al., 2017). Integration was done for VDAC1 protein incubated with or without SGK1 S422D in duplicate and statistical significance confirmed by students t-test.
Hepatic Ischemia/Reperfusion Model
The hepatic warm I/R model was performed as described below. Under ketamine (100 mg/kg body weight, administered intraperitoneally [IP]) and xylazine (10 mg/kg IP) anesthesia, the liver was exposed, and the left and median lobes were clamped with a microvascular clamp for 45 minutes. Ischemia was confirmed by visualizing the blanching of the ischemic lobes. The clamp was removed, and reperfusion confirmed on the basis of immediate color change before the abdomen was closed with continuous silk suture. Absence of ischemic color change or lack of reperfusion resulted in exclusion from further analysis. Temperature was maintained at 37°C by a warming pad. Six hours after reperfusion, mice were sacrificed by exsanguination after being anesthetized with ketamine and xylazine. For Cyclosporine A pretreatment, a dose of 10 mg/kg CsA were injected IP twice at 12h and 1h before ischemia.
QUANTIFICATION AND STATISTICAL ANALYSIS
All western blotting quantifications were conducted in ImageJ. Statistical analyses were performed using Prism (GraphPad Software). The statistical differences between control and experimental groups were determined by two-tailed students t-test (two groups), one-way ANOVA (more than two groups), or two-way ANOVA (two independent experimental variables), with corrected P values < 0.05 considered significant. Asterisks denote corresponding statistical significance *p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001. The statistical tests performed and definition of “n” numbers in this study are indicated in the figure legends. For Figures 1A, 1B, 2H, S1C, S2B–S2F and S3K, “n” means the number of worms. For Figures 7B–7H, “n” means the number of mice. For Figures 1C, 1F, 1G, 2E and 5B, “n” means the number of cells that were analyzed. For Figures 1D, 1H, 1I, 1J, 3J, 3K, 4A–4C, 4G–4K, 5A, 5C–5G, 5I, 6A–6C, S1A, S1B, S1E, S2K, S3G, S3H, S3J, S6D–S6F, S6L, S7B and S7C, “n” means biological replicates. For C. elegans western blotting, oxygen consumption, and ATP determination, each sample within each biological replicate corresponds to a sample pooled from 100–1000 animals. For cell culture western blotting, each sample within each biological replicate corresponds to one well from a tissue culture plate. For primary hepatocyte experiments, each biological replicate corresponds to hepatocytes isolated from one mouse per genotype, divided evenly between experimental conditions. For mouse liver western blotting (Figures 7D, 7H) each sample corresponds to protein extract from one mouse. The log rank test was used to determine significance in lifespan analyses using online OASIS2. For details of survival analyses and statistics, see Table S1.
Supplementary Material
(A) SGK-1 protein level is decreased under starvation. SGK-1::GFP fusion protein is detected by western blotting for GFP. (n = 3, student's t-test)
(B) Phosphorylation of C. elegans aAMPK(Thr172) in sgk-1 mutant and wildtype animals, as assessed by western blot. (n = 3, student's t-test)
(C) Brood size of individual sgk-1 mutant and wildtype C. elegans as determined daily from day 1 of adulthood for five days. (n = 10 per genotype, two-way ANOVA)
(D) Imaging in situ using the fluorescent autophagosome indicator Cyto-ID indicates elevated autophagy levels in AML12 Sgk1(−/−) cells. Scale bar: 20 μm.
(E) Immuno-blotting of LC3A/B turnover in control (littermates without the Cre transgene, Sgk1flox/flox) and Sgk1Lko primary hepatocytes under full medium conditions. (n = 4 replicates from 2 mice per group, student's t-test)
(F) The fluorescent autophagosome indicator Cyto-ID demonstrates elevated autophagy levels in Sgk1Lko primary hepatocytes both under full medium and HBSS starvation conditions, scale bar: 20 μm.
*p < 0.05, **p < 0.01. All bars indicate means and SEM.
(A) RNAi of bec-1 restores normal lifespan in both sgk-1 and rict-1;sgk-1 double mutants (log rank test).
(B) RNAi of unc-51 increases brood size in sgk-1 mutants (n = 5 per group, student's t-test).
(C and D) RNAi of bec-1, lgg-1, and unc-51 accelerates growth rate in sgk-1 mutants. Developmental stage was evaluated 61 hours after seeding of synchronized sgk-1 L1 worms (C) or 48 hours after seeding of synchronized wild type L1 worms (D). YA: young adult; L4: larval stage 4 (n > 50 per genotype, per replicate for 3 replicates, two-way ANOVA).
(E) RNAi of bec-1, lgg-1, and unc-51 does not significantly change fat mass in sgk-1 mutants. Fixative Nile red staining was used to assess fat levels. (n = 69, 108, 70 and 61 animals for ev bec-1, lgg-1, and unc-51 RNAi, respectively).
(F) Intestine specific RNAi of bec-1, lgg-1, and unc-51 accelerates growth rate in rict-1 mutants. Developmental stage was checked 62 hours after seeding of synchronized intestine specific RNAi rict-1 L1 worms (n > 60 per genotype, per replicate for 3 replicates, two-way ANOVA).
(G and H) Intestine-specific RNAi of bec-1 partially rescues shortened lifespan of rict-1 mutant worms (G) but slightly shortens lifespan of wild type worms (H) (log rank test).
(I) RNAi of bec-1 beginning at the L1 stage of larval development partially restores normal lifespan in sgk-1 mutants compared to whole life (wl) RNAi (log rank test).
(J) RNAi of bec-1 beginning in adulthood is insufficient to restore normal lifespan in sgk-1 mutants (log rank test).
(K) Comparison of bec-1 RNAi efficiency in sgk-1 mutant worms only during larval stages (larval) and started from young adulthood for three days (adult) (n = 4, two-way ANOVA).
For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ****p < 0.0001. All bars indicate means and SEM.
(A) Upon co-transfection of mouse Flag-SGK1 and VDAC1-Myc-His in 293T cells, VDAC1-Myc-His co-immunoprecipitates with Flag-SGK1.
(B) Interaction between VDAC1 and SGK1 is not regulated by wortmannin treatment.
(C) The mPTP opens earlier in mouse Sgk1(−/−) AML12 cells vs. wild type cells under a calcium challenge. Two million cells were used for this experiment, versus ten million in Figure 3D.
(D) RNAi of atp-5 extends lifespan in sgk-1 mutants (log rank test).
(E) mPTP inhibitor Cyclosporine A (CsA) treatment partially restores normal lifespan in sgk-1 mutants. No obvious effects are seen in wild type animals (log rank test).
(F) Intestine specific RNAi of ant-1.1, similar to bec-1, partially restores normal lifespan in short-lived rict-1 mutant worms (log rank test).
(G) ATP level is decreased in both rict-1 and sgk-1 mutants compared with wt animals (n = 4, one-way ANOVA).
(H) Mitochondrial DNA copies in wild type N2, rict-1 and sgk-1 mutants as measured by qPCR. nd-1 was used as mitochondrial DNA marker normalized to nuclear DNA act-3 (n = 7).
(I) Representative image of mitochondrially localized RFP in muscle of wild type N2 and sgk-1 mutants, scale bar: 200 μm.
(J) Increased SGK-1 protein levels under mitochondrial stress induced by RNAi to electron transport chain genes, as indicated by western blotting of SGK-1::GFP. Cell lysates were made using SDS lysis buffer (n = 3, one-way ANOVA).
(K) Representative image of the SGK-1::GFP reporter under mitochondrial stress (n = 45, 23, 29 and 25 animals for ev, nuo-1, nuo-6 and cco-4 RNAi, respectively, one-way ANOVA), scale bar: 200 μm.
For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ****p < 0.0001. All bars indicate means and SEM.
(A) VDAC-1::Flag fusion protein detected by western blot using Flag antibody in C. elegans overexpressing VDAC-1 (vdac-1 oe).
(B) Extrachromosomal expression of VDAC-1::Flag confirmed by western blotting in the two vdac-1 overexpression lines.
(C) ANT protein level measured by western blot in wildtype, rict-1 and sgk-1 mutant C. elegans.
(D) ANT1 and VDAC2 protein measured by western blot in wildtype and Sgk1Lko primary hepatocytes.
(E) Western blotting of myc-tagged VDAC1 protein with the indicated mutations of candidate Ser and Thr phosphorylation sites with constitutively active SGK1 (S422D) and inactive SGK1 (S422A) co-overexpression.
(A) Double RNAi of bec-1 and ant-1.1 does not have an additive effect on lifespan extension in sgk-1 worms (log rank test).
(B and C) Shortened lifespan is also observed in two additional extrachromosomal vdac-1 overexpression lines compared with wild type C. elegans (log rank test).
(D) Overexpression of sgk-1 restores normal lifespan of vdac-1 overexpression C. elegans (log rank test).
(E and F) rict-1, sgk-1 mutants (E) and vdac-1 overexpression C. elegans (F) are more sensitive to paraquat (8mM) treatment compared with wild type control (log rank test).
(G-J) Increased resistance to paraquat (8mM) is seen following RNAi of bec-1 (G and I) or ant-1.1 (H and J) in wt animals, sgk-1 and rict-1 mutants (log rank test).
(K) RNAi of bec-1 increases paraquat (8mM) resistance in vdac-1 overexpression C. elegans (log rank test).
For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(A and B) RNAi of bec-1 can still restore normal lifespan in atfs-1(tm4525);sgk-1 (A) and daf-16(mgDf47);sgk-1 (B) double mutants (log rank test).
(C) RNAi of skn-1 does not affect the lifespan extension by bec-1 RNAi in sgk-1 mutants (log rank test).
(D) bec-1 RNAi decreases basal and maximal (FCCP) oxygen consumption in wildtype C. elegans. (n = 12, two-way ANOVA).
(E and F) RNAi of vdac-1 increases basal oxygen consumption in sgk-1 mutants (E) but has no significant effect on wild type C. elegans (F) (n = 4, student's t-test).
(G and H) RNAi of mitophagy related genes pink-1 (G) or pdr-1 (H) only very modestly extends lifespan of sgk-1 mutants (log rank test).
(I and J) RNAi of mitochondrial complex II gene sdhb-1 eliminates autophagy-dependent lifespan extension in both eat-2(ad465) mutants (I) and glp-1(e2141) mutants (J). For glp-1(e2141) lifespan, experiments were conducted at 25°C (log rank test).
(K) RNAi of daf-2 still extends lifespan of vdac-1 overexpression worms (log rank test).
(L) RNAi of daf-2 decreases VDAC-1 protein level. (n = 3, student's t-test) For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All bars indicate means and SEM.
(A) The mPTP opens earlier in mouse SgkTKO primary hepatocytes versus control cells when challenged with multiple calcium pulses (arrows). Equal numbers (2×106) of cells were used and equivalent biomass was confirmed by western blotting for Actin (right panel).
(B) Increased VDAC1 protein level and LC3 II/LC3 I conversion in SgkTKO primary hepatocytes. (n = 4 replicates for VDAC1 protein level and n = 6 for LC3 II/LC3 I conversion analysis from 3 mice per group, student's t-test).
(C) Decreased phosphorylation levels of p70S6K Thr389 and S6 Ser240/244 in SgkTKO primary hepatocytes. (n = 3, two-way ANOVA). *p < 0.05, **p < 0.01. All bars indicate means and SEM.
Table S1. Tabular and Survival Data and Biological Replicates. Related to Figure 2, Figure 3, Figure 5, Figure 6, Figure S2, Figure S3, Figure S5 and Figure S6.
Table S2. Oligonucleotides used in this study. Related to STAR METHODS.
Highlights.
SGK1 regulates autophagy in both C.elegans and mammalian cells
Elevated autophagy and mPTP opening shorten lifespan in sgk-1/mTORC2 mutant worms
SGK-1 phosphorylates mPTP component VDAC1 on Ser104, promoting its degradation
Loss of SGK function exaggerates mPTP dependent hepatic ischemia/reperfusion injury
ACKNOWLEDGEMENTS
We thank Joseph Avruch, Noriko Oshiro-Rapley, Ning Dai, and Laura Regue for discussions and reagents, Johnathan Whetstine for the HA-Ub plasmid, and Diane Capen at the Program in Membrane Biology at MGH for EM. Thanks to the NORC of Harvard (P30DK040561) and the Boston Area DERC (P30DK057521) for core services. Some strains were provided by the CGC, funded by the NIH Office of Research Infrastructure Programs (P40OD010440), and the C. elegans Knockout Consortium. This work was funded by NIH grants K08DK087941 and R01DK101522 (to AAS), R01AG038664 (to MH), and R21AG058038 (to CK), the Ellison Medical Foundation New Scholar in Aging Award (to AAS), the Howard M. Goodman Fellowship (to AAS), the Charles H. Hood Foundation Child Health Research Award (to AAS), the Weissman Family MGH Research Scholar Award (to AAS), a Glenn Award for Research in Biological Mechanisms of Aging (to AAS), the Julie Martin Award from The Ellison Medical Foundation/AFAR (to MH), and an MGH ECOR Postdoctoral Fellowship (to BZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures and one table and can be found with this article online.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Batandier C, Leverve X, and Fontaine E (2004). Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. The Journal of biological chemistry 279, 17197–17204. [DOI] [PubMed] [Google Scholar]
- Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, and Hall MN (2013). Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A 110, 12526–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, Mocanu MM, and Yellon DM (2008). Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol 103, 274–284. [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Steinbaugh MJ, Hourihan JM, Ewald CY, and Isik M (2015). SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic Biol Med 88, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briston T, Roberts M, Lewis S, Powney B,J,MS, Szabadkai G, and Duchen MR (2017). Mitochondrial permeability transition pore: sensitivity to opening and mechanistic dependence on substrate availability. Sci Rep 7, 10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, Hardy EJ, Dellaire G, Haas W, Xia B, et al. (2017). Coupling of Homologous Recombination and the Checkpoint by ATR. Molecular cell 65, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA, et al. (2017). Mitochondrial fission facilitates the selective mitophagy of protein aggregates. The Journal of cell biology 216, 3231–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, et al. (2016). PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer Cell 30, 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Kumsta C, Hellman AB, Adams LM, and Hansen M (2017). Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordas E, Naray-Fejes-Toth A, and Fejes-Toth G (2007). Subcellular location of serum- and glucocorticoid-induced kinase-1 in renal and mammary epithelial cells. Am J Physiol Cell Physiol 292, C1971–1981. [DOI] [PubMed] [Google Scholar]
- Costantini P, Petronilli V, Colonna R, and Bernardi P (1995). On the effects of paraquat on isolated mitochondria. Evidence that paraquat causes opening of the cyclosporin A-sensitive permeability transition pore synergistically with nitric oxide. Toxicology 99, 77–88. [DOI] [PubMed] [Google Scholar]
- Crawford D, Libina N, and Kenyon C (2007). Caenorhabditis elegans integrates food and reproductive signals in lifespan determination. Aging cell 6, 715–721. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A (2017). SGK1: The Dark Side of PI3K Signaling. Curr Top Dev Biol 123, 49–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, and Kenyon C (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. [DOI] [PubMed] [Google Scholar]
- Elmore SP, Qian T, Grissom SF, and Lemasters JJ (2001). The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 15, 2286–2287. [DOI] [PubMed] [Google Scholar]
- Fang H, Liu A, Dahmen U, and Dirsch O (2013). Dual role of chloroquine in liver ischemia reperfusion injury: reduction of liver damage in early phase, but aggravation in late phase. Cell Death Dis 4, e694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejes-Toth G, Frindt G, Naray-Fejes-Toth A, and Palmer LG (2008). Epithelial Na+ channel activation and processing in mice lacking SGK1. American journal of physiology Renal physiology 294, F1298–1305. [DOI] [PubMed] [Google Scholar]
- Fernandez AF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, He C, Ting T, Liu Y, Chiang WC, et al. (2018). Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez JM, and Alessi DR (2008). mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). The Biochemical journal 416, 375–385. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, and Springer W (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature cell biology 12, 119–131. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA, and Mentzer RM (2010). Autophagy during cardiac stress: joys and frustrations of autophagy. Annual review of physiology 72, 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Lee D, Lee H, Kim D, Son HG, Yang JS, Lee SV, and Kim S (2016). OASIS 2: online application for survival analysis 2 with features for the analysis of maximal lifespan and healthspan in aging research. Oncotarget 7, 56147–56152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Rubinsztein DC, and Walker DW (2018). Autophagy as a promoter of longevity: insights from model organisms. Nature reviews Molecular cell biology 19, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer T, Servais S, Seo AY, Marzetti E, Hiona A, Upadhyay SJ, Wohlgemuth SE, and Leeuwenburgh C (2009). Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mech Ageing Dev 130, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, and Suzuki K (1998). A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394, 694–697. [DOI] [PubMed] [Google Scholar]
- Kang C, You YJ, and Avery L (2007). Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes & development 21, 2161–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, and Molkentin JD (2015). Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell metabolism 21, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamech LT, and Haynes CM (2015). The unpredictability of prolonged activation of stress response pathways. The Journal of cell biology 209, 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE, et al. (2013). The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nature communications 4, 2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, and Ruvkun G (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33, 40–48. [DOI] [PubMed] [Google Scholar]
- Li WC, Ralphs KL, and Tosh D (2010). Isolation and culture of adult mouse hepatocytes. Methods in molecular biology 633, 185–196. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Wang Y, Chen Y, and Cao F (2015). The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim Biophys Acta 1852, 271–276. [DOI] [PubMed] [Google Scholar]
- Madeo F, Zimmermann A, Maiuri MC, and Kroemer G (2015). Essential role for autophagy in life span extension. The Journal of clinical investigation 125, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manil-Segalen M, Lefebvre C, Jenzer C, Trichet M, Boulogne C, Satiat-Jeunemaitre B, and Legouis R (2014). The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Developmental cell 28, 43–55. [DOI] [PubMed] [Google Scholar]
- Matecic M, Smith DL, Pan X, Maqani N, Bekiranov S, Boeke JD, and Smith JS (2010). A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet 6, e1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizunuma M, Neumann-Haefelin E, Moroz N, Li Y, and Blackwell TK (2014). mTORC2-SGK-1 acts in two environmentally responsive pathways with opposing effects on longevity. Aging cell 13, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keeffe BA, Cilia S, Maiyar AC, Vaysberg M, and Firestone GL (2013). The serum- and glucocorticoid-induced protein kinase-1 (Sgk-1) mitochondria connection: identification of the IF-1 inhibitor of the F(1)F(0)-ATPase as a mitochondria-specific binding target and the stress-induced mitochondrial localization of endogenous Sgk-1. Biochimie 95, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SB, Samangouei P, Kalkhoran SB, and Hausenloy DJ (2015). The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. Journal of molecular and cellular cardiology 78, 23–34. [DOI] [PubMed] [Google Scholar]
- Palikaras K, and Tavernarakis N (2016). Intracellular Assessment of ATP Levels in Caenorhabditis elegans. Bio Protoc 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, and Hemmings BA (1999). Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J 18, 3024–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol C, Bratic-Hench I, Sumakovic M, Hench J, Mourier A, Baumann L, Pavlenko V, and Trifunovic A (2013). Succinate dehydrogenase upregulation destabilize complex I and limits the lifespan of gas-1 mutant. PloS one 8, e59493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg H, and Hoek JB (2017). The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging cell 16, 943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, and Elazar Z (2011). Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36, 30–38. [DOI] [PubMed] [Google Scholar]
- Schiavi A, Torgovnick A, Kell A, Megalou E, Castelein N, Guccini I, Marzocchella L, Gelino S, Hansen M, Malisan F, et al. (2013). Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Experimental gerontology 48, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz D, and Gygi SP (2005). An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol 23, 1391–1398. [DOI] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, Hines KJ, Smith DJ, Eguchi A, Vallem S, et al. (2015). SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Molecular cell 60, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukas AA, Kane EA, Carr CE, Melo JA, and Ruvkun G (2009). Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes & development 23, 496–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn A (2014). Autophagy and its effects: making sense of double-edged swords. PLoS biology 12, e1001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina S, Thoraval D, Crouzet M, Ichas F, De Pinto V, et al. (2009). Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell research 19, 1363–1376. [DOI] [PubMed] [Google Scholar]
- Wang MC, Oakley HD, Carr CE, Sowa JN, and Ruvkun G (2014). Gene pathways that delay Caenorhabditis elegans reproductive senescence. PLoS genetics 10, e1004752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, and Hekimi S (2010). Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging cell 9, 433–447. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chang JT, Guo B, Hansen M, Jia K, Kovacs AL, Kumsta C, Lapierre LR, Legouis R, Lin L, et al. (2015). Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 11, 9–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) SGK-1 protein level is decreased under starvation. SGK-1::GFP fusion protein is detected by western blotting for GFP. (n = 3, student's t-test)
(B) Phosphorylation of C. elegans aAMPK(Thr172) in sgk-1 mutant and wildtype animals, as assessed by western blot. (n = 3, student's t-test)
(C) Brood size of individual sgk-1 mutant and wildtype C. elegans as determined daily from day 1 of adulthood for five days. (n = 10 per genotype, two-way ANOVA)
(D) Imaging in situ using the fluorescent autophagosome indicator Cyto-ID indicates elevated autophagy levels in AML12 Sgk1(−/−) cells. Scale bar: 20 μm.
(E) Immuno-blotting of LC3A/B turnover in control (littermates without the Cre transgene, Sgk1flox/flox) and Sgk1Lko primary hepatocytes under full medium conditions. (n = 4 replicates from 2 mice per group, student's t-test)
(F) The fluorescent autophagosome indicator Cyto-ID demonstrates elevated autophagy levels in Sgk1Lko primary hepatocytes both under full medium and HBSS starvation conditions, scale bar: 20 μm.
*p < 0.05, **p < 0.01. All bars indicate means and SEM.
(A) RNAi of bec-1 restores normal lifespan in both sgk-1 and rict-1;sgk-1 double mutants (log rank test).
(B) RNAi of unc-51 increases brood size in sgk-1 mutants (n = 5 per group, student's t-test).
(C and D) RNAi of bec-1, lgg-1, and unc-51 accelerates growth rate in sgk-1 mutants. Developmental stage was evaluated 61 hours after seeding of synchronized sgk-1 L1 worms (C) or 48 hours after seeding of synchronized wild type L1 worms (D). YA: young adult; L4: larval stage 4 (n > 50 per genotype, per replicate for 3 replicates, two-way ANOVA).
(E) RNAi of bec-1, lgg-1, and unc-51 does not significantly change fat mass in sgk-1 mutants. Fixative Nile red staining was used to assess fat levels. (n = 69, 108, 70 and 61 animals for ev bec-1, lgg-1, and unc-51 RNAi, respectively).
(F) Intestine specific RNAi of bec-1, lgg-1, and unc-51 accelerates growth rate in rict-1 mutants. Developmental stage was checked 62 hours after seeding of synchronized intestine specific RNAi rict-1 L1 worms (n > 60 per genotype, per replicate for 3 replicates, two-way ANOVA).
(G and H) Intestine-specific RNAi of bec-1 partially rescues shortened lifespan of rict-1 mutant worms (G) but slightly shortens lifespan of wild type worms (H) (log rank test).
(I) RNAi of bec-1 beginning at the L1 stage of larval development partially restores normal lifespan in sgk-1 mutants compared to whole life (wl) RNAi (log rank test).
(J) RNAi of bec-1 beginning in adulthood is insufficient to restore normal lifespan in sgk-1 mutants (log rank test).
(K) Comparison of bec-1 RNAi efficiency in sgk-1 mutant worms only during larval stages (larval) and started from young adulthood for three days (adult) (n = 4, two-way ANOVA).
For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ****p < 0.0001. All bars indicate means and SEM.
(A) Upon co-transfection of mouse Flag-SGK1 and VDAC1-Myc-His in 293T cells, VDAC1-Myc-His co-immunoprecipitates with Flag-SGK1.
(B) Interaction between VDAC1 and SGK1 is not regulated by wortmannin treatment.
(C) The mPTP opens earlier in mouse Sgk1(−/−) AML12 cells vs. wild type cells under a calcium challenge. Two million cells were used for this experiment, versus ten million in Figure 3D.
(D) RNAi of atp-5 extends lifespan in sgk-1 mutants (log rank test).
(E) mPTP inhibitor Cyclosporine A (CsA) treatment partially restores normal lifespan in sgk-1 mutants. No obvious effects are seen in wild type animals (log rank test).
(F) Intestine specific RNAi of ant-1.1, similar to bec-1, partially restores normal lifespan in short-lived rict-1 mutant worms (log rank test).
(G) ATP level is decreased in both rict-1 and sgk-1 mutants compared with wt animals (n = 4, one-way ANOVA).
(H) Mitochondrial DNA copies in wild type N2, rict-1 and sgk-1 mutants as measured by qPCR. nd-1 was used as mitochondrial DNA marker normalized to nuclear DNA act-3 (n = 7).
(I) Representative image of mitochondrially localized RFP in muscle of wild type N2 and sgk-1 mutants, scale bar: 200 μm.
(J) Increased SGK-1 protein levels under mitochondrial stress induced by RNAi to electron transport chain genes, as indicated by western blotting of SGK-1::GFP. Cell lysates were made using SDS lysis buffer (n = 3, one-way ANOVA).
(K) Representative image of the SGK-1::GFP reporter under mitochondrial stress (n = 45, 23, 29 and 25 animals for ev, nuo-1, nuo-6 and cco-4 RNAi, respectively, one-way ANOVA), scale bar: 200 μm.
For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ****p < 0.0001. All bars indicate means and SEM.
(A) VDAC-1::Flag fusion protein detected by western blot using Flag antibody in C. elegans overexpressing VDAC-1 (vdac-1 oe).
(B) Extrachromosomal expression of VDAC-1::Flag confirmed by western blotting in the two vdac-1 overexpression lines.
(C) ANT protein level measured by western blot in wildtype, rict-1 and sgk-1 mutant C. elegans.
(D) ANT1 and VDAC2 protein measured by western blot in wildtype and Sgk1Lko primary hepatocytes.
(E) Western blotting of myc-tagged VDAC1 protein with the indicated mutations of candidate Ser and Thr phosphorylation sites with constitutively active SGK1 (S422D) and inactive SGK1 (S422A) co-overexpression.
(A) Double RNAi of bec-1 and ant-1.1 does not have an additive effect on lifespan extension in sgk-1 worms (log rank test).
(B and C) Shortened lifespan is also observed in two additional extrachromosomal vdac-1 overexpression lines compared with wild type C. elegans (log rank test).
(D) Overexpression of sgk-1 restores normal lifespan of vdac-1 overexpression C. elegans (log rank test).
(E and F) rict-1, sgk-1 mutants (E) and vdac-1 overexpression C. elegans (F) are more sensitive to paraquat (8mM) treatment compared with wild type control (log rank test).
(G-J) Increased resistance to paraquat (8mM) is seen following RNAi of bec-1 (G and I) or ant-1.1 (H and J) in wt animals, sgk-1 and rict-1 mutants (log rank test).
(K) RNAi of bec-1 increases paraquat (8mM) resistance in vdac-1 overexpression C. elegans (log rank test).
For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(A and B) RNAi of bec-1 can still restore normal lifespan in atfs-1(tm4525);sgk-1 (A) and daf-16(mgDf47);sgk-1 (B) double mutants (log rank test).
(C) RNAi of skn-1 does not affect the lifespan extension by bec-1 RNAi in sgk-1 mutants (log rank test).
(D) bec-1 RNAi decreases basal and maximal (FCCP) oxygen consumption in wildtype C. elegans. (n = 12, two-way ANOVA).
(E and F) RNAi of vdac-1 increases basal oxygen consumption in sgk-1 mutants (E) but has no significant effect on wild type C. elegans (F) (n = 4, student's t-test).
(G and H) RNAi of mitophagy related genes pink-1 (G) or pdr-1 (H) only very modestly extends lifespan of sgk-1 mutants (log rank test).
(I and J) RNAi of mitochondrial complex II gene sdhb-1 eliminates autophagy-dependent lifespan extension in both eat-2(ad465) mutants (I) and glp-1(e2141) mutants (J). For glp-1(e2141) lifespan, experiments were conducted at 25°C (log rank test).
(K) RNAi of daf-2 still extends lifespan of vdac-1 overexpression worms (log rank test).
(L) RNAi of daf-2 decreases VDAC-1 protein level. (n = 3, student's t-test) For tabular survival data and biological replicates see also Table S1. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All bars indicate means and SEM.
(A) The mPTP opens earlier in mouse SgkTKO primary hepatocytes versus control cells when challenged with multiple calcium pulses (arrows). Equal numbers (2×106) of cells were used and equivalent biomass was confirmed by western blotting for Actin (right panel).
(B) Increased VDAC1 protein level and LC3 II/LC3 I conversion in SgkTKO primary hepatocytes. (n = 4 replicates for VDAC1 protein level and n = 6 for LC3 II/LC3 I conversion analysis from 3 mice per group, student's t-test).
(C) Decreased phosphorylation levels of p70S6K Thr389 and S6 Ser240/244 in SgkTKO primary hepatocytes. (n = 3, two-way ANOVA). *p < 0.05, **p < 0.01. All bars indicate means and SEM.
Table S1. Tabular and Survival Data and Biological Replicates. Related to Figure 2, Figure 3, Figure 5, Figure 6, Figure S2, Figure S3, Figure S5 and Figure S6.
Table S2. Oligonucleotides used in this study. Related to STAR METHODS.