

Abstract
Malaria deaths have been decreasing over the last 10–15 years, with global mortality rates having fallen by 47% since 2000. While the World Health Organization (WHO) recommends the use of artemisinin-based combination therapies (ACTs) to combat malaria, the emergence of artemisinin resistant strains underscores the need to develop new antimalarial drugs. Recent in vivo efficacy improvements of the historical antimalarial ICI 56,780 have been reported, however, with the poor solubility and rapid development of resistance, this compound requires further optimization. A series of piperazine-containing 4(1H)-quinolones with greatly enhanced solubility were developed utilizing structure–activity relationship (SAR) and structure–property relationship (SPR) studies. Furthermore, promising compounds were chosen for an in vivo scouting assay to narrow selection for testing in an in vivo Thompson test. Finally, two piperazine-containing 4(1H)-quinolones were curative in the conventional Thompson test and also displayed in vivo activity against the liver stages of the parasite.
Graphical Abstract
∎. INTRODUCTION
Malaria is one of the deadliest public health problems in the world, accounting for nearly half a million casualties annually.1 A protozoan parasitic species, Plasmodium is responsible for transmitting the disease to humans through a mosquito vector. The various developmental stages of the parasite within the host make the design and development of curative antimalarial agents challenging. In addition, the widespread parasite resistance to almost all antimalarial drugs in use emphasizes the pressing need for new drugs with novel chemotypes that are safe and effective against multiple stages of highly resistant parasites.2,3 In the past few years, several research groups reported their optimization efforts in developing antimalarial 4(1H)-pyridone- and 4(1H)-quinolone-based agents, which are structurally related.4–13
One of the major challenges in advancing these 4(1H)-quinolones into antimalarial drugs is the poor aqueous solubility of these scaffolds, which limits the oral bioavail-ability.14 Quinolone ester ICI 56,78015,16 (1, Figure 1) is one of these antimalarial 4(1H)-quinolones displaying very potent activity against blood, liver, and transmission stages of the parasite. 7,16,17 Quinolone ester 1 even produced radical cures (eradicated dormant exoerythrocytic stages of the parasite) in Plasmodium cynomolgi infected rhesus monkeys.15 However, the development of 1 was halted as resistance emerged after only one passage in Plasmodium berghei (Pb) infected mice.16 Nevertheless, recent improvements in preclinical efficacy models and compound property assessment motivated the laboratories of Guy,4,5 Ward and O’Neill,18 and Manetsch and Kyle7,17 to reexamine studies on the antimalarial 4(1H)-quinolone ester from slightly different angles. Previous work completed by our laboratory resulted in a set of 46 compounds with in vitro activities against clinically relevant, multidrug resistant malarial strains W2 and TM90-C2B. Because cytochrome bc1 is known to be the biological target of antimalarial 4(1H)-quinolones, cross resistance with atovaquone is a concern for any new antimalarial chemotype series. Therefore, all compounds were routinely tested against W2, a chloroquine and pyrimethamine resistant strain, and TM90-C2B, a chloroquine-, mefloquine-, pyrimethamine-, and atovaquone-resistant strain, to determine the resistance index (RI) as the calculated ratio of the effective concentrations of W2 and the atovaquone resistant TM90-C2B (RI = EC50(TM90-C2B)/EC50(W2)).
Figure 1.
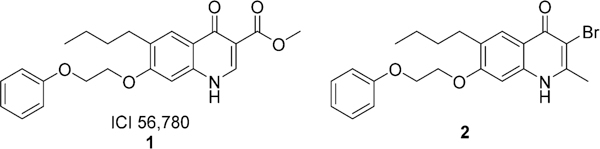
Antimalarial 4(1H)-quinolones.
Lead compound 3-bromo-6-butyl-2-methyl-7-(2-phenoxyethoxy)quinolin-4(1H)-one (2, Figure 1) addressed atovaquone cross resistance concerns over 1 by lowering the resistance index (RI) to 4.7 and exemplified a potent liver stage activity of 2.12 nM.17 It further exemplified the need to not only improve potency but also optimize aqueous solubility. Therefore, the main objective for the next optimization phase focused on the design and synthesis of a series of 6- and 7- substituted 4(1H)-quinolones with enhanced aqueous solubility without compromising blood and liver stage activity. Herein, we report detailed structure–activity relationship and structure–property relationship studies, leading to a set of analogues with improved aqueous solubility and oral bioavailability for which a subset has been further assessed for in vivo efficacy in targeting the blood and liver stages of the parasite.
∎. RESULTS AND DISCUSSION
Design and Synthesis.
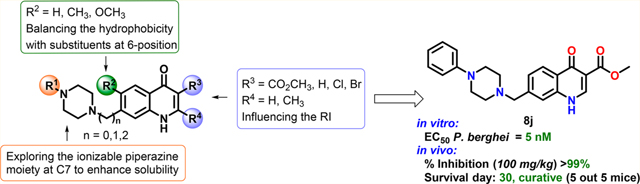
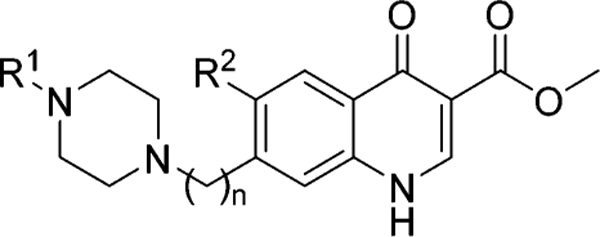
The rationale for the design of the next generation 4(1H)-quinolones was based on insights gained from structure—activity relationship (SAR) studies on 4(1H)-quinolone esters reported by us and others.7,15,18 The 4(1H)-quinolone core, the ester group in the 3-position, and the phenoxyethoxy substituent in the 7-position were identified to be key structural elements rendering this chemotype’s potent antimalarial activity. Substituents in 2- and 6-positions were considered to be of secondary importance, with a negligible influence on the RI and/or the overall hydrophobicity of the 4(1H)-quinolone ester analogues. As a starting point for the next compound design, we decided to proceed with a 4(1H)-quinolone pharmacophore containing a 3-carboxylic acid ester and a 7-piperazinyl group (Figure 2). The piperazinyl moiety was selected for the following reasons: (1) an ionizable piperazine will enhance the aqueous solubility of 4(1H)-quinolones, (2) a straightforward, base-mediated or reductive N-alkylation of a piperazine provides an easy route to access highly functionalized 4(1H)-quinolone ester analogues, and (3) the commercial availability of various N-substituted piperazines allows the straightforward synthesis of a diversified set of piperazinyl-substituted 4(1H)-quinolones. In addition to the design of piperazine-substituted 4(1H)-quinolones, a general synthetic route was chosen by introducing the 4(1H)-quinolone core at the end of the synthesis sequence, reducing the number of cumbersome synthetic and purification steps which are complicated due to solubility limitations typical for 4(1H)-quinolone moieties.
Figure 2.
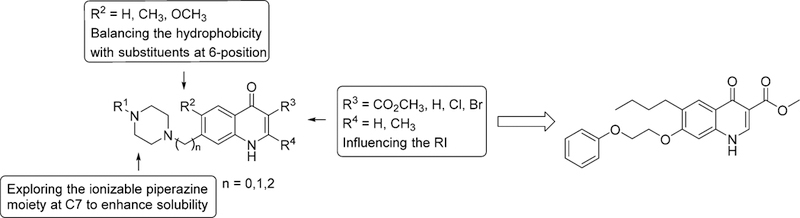
Design of piperazinyl-substituted scaffolds based on 1.
Initially, a set of 7-piperazinyl-4(1H)-quinolone esters with differing alkyl chain lengths between the 4(1H)-quinolone core and the piperazinyl moiety were synthesized. The connectivity ranged from a direct attachment of the piperazine to the 4(1H)-quinolone’s 7-position to a methylene or ethylene chain with each linker requiring a different synthetic path. The nucleophilic aromatic substitution was initially attempted for analogues with the piperazinyl moiety directly attached to the 4(1H)-quinolone core, using substituted nitrobenzenes 3 along with the required substituted piperazine. The substitution reaction was followed by a reduction of the nitro group and a thermal cyclization to yield 4(1H)-quinolones 8a—8b. This synthetic approach, however, was only successful when the 4(1H)-quinolone core was sufficiently electron deficient (Scheme 1).19
Scheme 1.
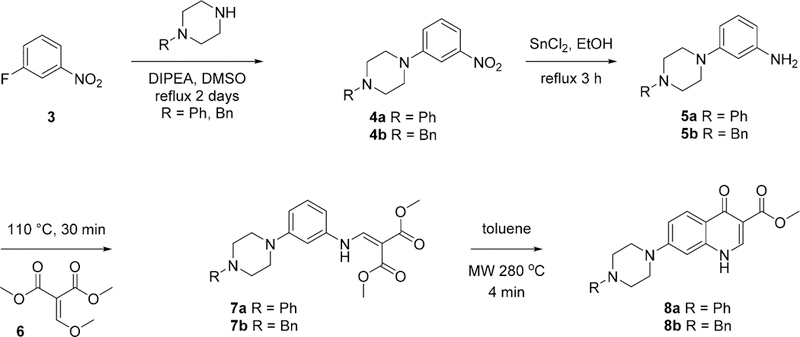
Synthesis of 4(1H)-Quinolones with Direct Attachment of Piperazinyl Moiety
For analogues in which the 4(1H)-quinolone core was not sufficiently electron deficient, a two-step sequence was required to obtain the necessary nitro intermediates 4. First, the corresponding para-substituted aniline or benzylamine 9 was reacted with 2 equiv of 2-chloroethanol to give diols 10. Diols 10 were chlorinated, and their products were reacted with substituted nitroanilines to yield piperazines 4.20 Subsequent reduction with tin(II) chloride gave piperazine-substituted anilines 5, which were further reacted using standard Gould— Jacob sequence of reactions to afford final products 8c—8i (Scheme 2).7,21
Scheme 2.
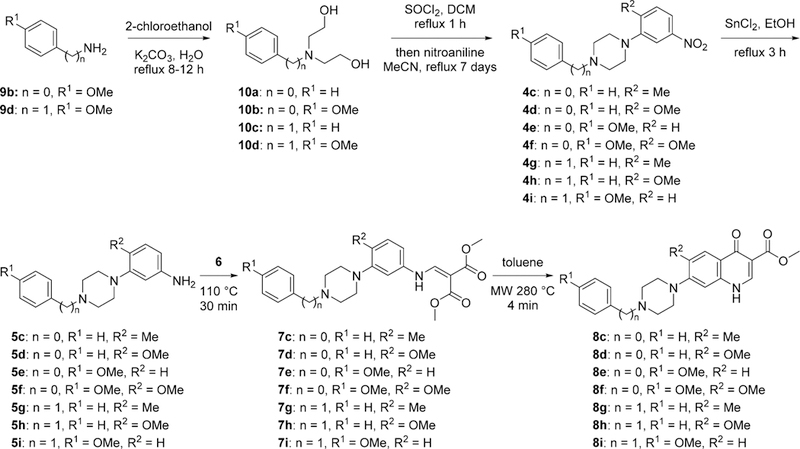
Alternate Synthesis of 4(1H)-Quinolones with Direct Attachment of the Piperazinyl Moiety to the 4(1H)-Quinolone Core
Compounds with a methylene unit between the 4(1H)-quinolone core and the piperazine were synthesized starting with 3-amino-benzyl alcohol 11, which was reacted with dimethyl 2-(methoxymethylene)malonate (6) to yield the corresponding enamines 12. The alcohol was oxidized using Dess—Martin periodinane22 to the corresponding benzaldehyde 13, which was subjected to direct reductive amination conditions to yield substituted piperazines 7j—7ab.23 These piperazinyl-substituted enamines were then cyclized using a microwave reactor to yield 4(1H)-quinolones 8j—8ab (Scheme 3).
Scheme 3.
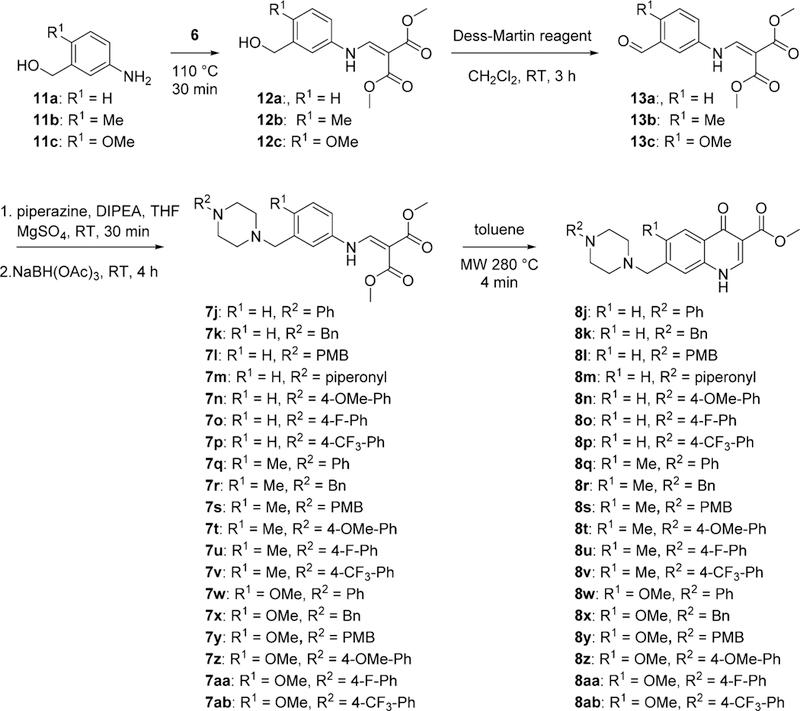
Synthesis of 4(1H)-Quinolones with a Methylene Between the Piperazinyl Moiety to the 4(1H)-Quinolone Core
Compounds with an ethylene between the piperazine and 4(1H)-quinolone core were synthesized through a four-step reaction sequence that was initiated by an alkylation of 3-nitrophenethyl bromide with corresponding piperazines 14 to yield intermediates 4.24 The nitrophenyl intermediates 4 were reduced to anilines 5 using tin(II) chloride, which were subjected to the standard Gould—Jacob reaction sequence to give 4(1H)-quinolone esters 8ac—8ae (Scheme 4).
Scheme 4.
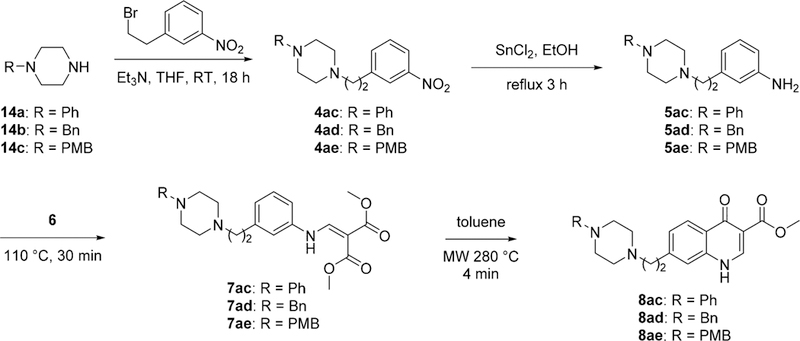
Synthesis of 4(1H)-Quinolones with an Ethylene between the Piperazinyl Moiety to the 4(1H)-Quinolone Core
A similar approach described for 8j—8ab was used for the synthesis of 6-piperizino-4(1H)-quinolone esters, however, 4-amino-benzyl alcohol 11d was used instead. The same four-step reaction sequence involving the enamine formation, a Dess—Martin oxidation, a direct reductive amination, followed by the cyclization was performed to yield 6-piperizino-4(1H)-quinolone esters 8af—8ai (Scheme 5).
Scheme 5.
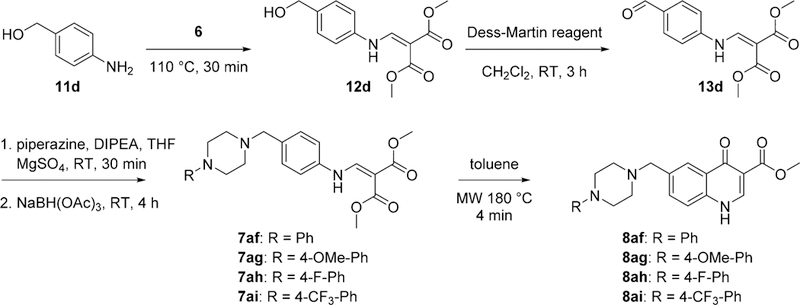
Synthesis of 6-Piperazinyl-4(1H)-Quinolones
Finally, 7-piperazinyl-3-halo-4(1H)-quinolones 8aj—8at were synthesized by alkylation of substituted piperazines 1424 with nitrophenethyl bromides to give nitro intermediates 4, followed by a reduction using tin(II) chloride. The resulting piperazine aniline 5aj was reacted with Meldrum’s acid,25 resulting in enamine 15aj, which was further reacted in a thermal cyclization to give 7-((4-(4-fluorophenyl)piperazin-1-yl)-methyl)quinolin-4(1H)-one 8aj. This was followed by the use of an appropriate N-halo succinimide to obtain the required 3-halo-7-piperazinyl-4(1H)-quinolones 8ak and 8al. The same approach was used for the preparation of 3-halo-2-methyl-4(1H)-quinolones, with the exception being that a Conrad—Limpach cyclization using ethyl acetoacetate was used following the formation of anilines 5aj—5as instead of the abovementioned cyclization with Meldrum’s acid to give compounds 8am, 8ap, and 8as. These 4(1H)-quinolones were reacted with the appropriate N-halo succinimide to procure compounds 8an, 8ao, 8aq, 8ar, and 8at (Scheme 6).
Scheme 6.
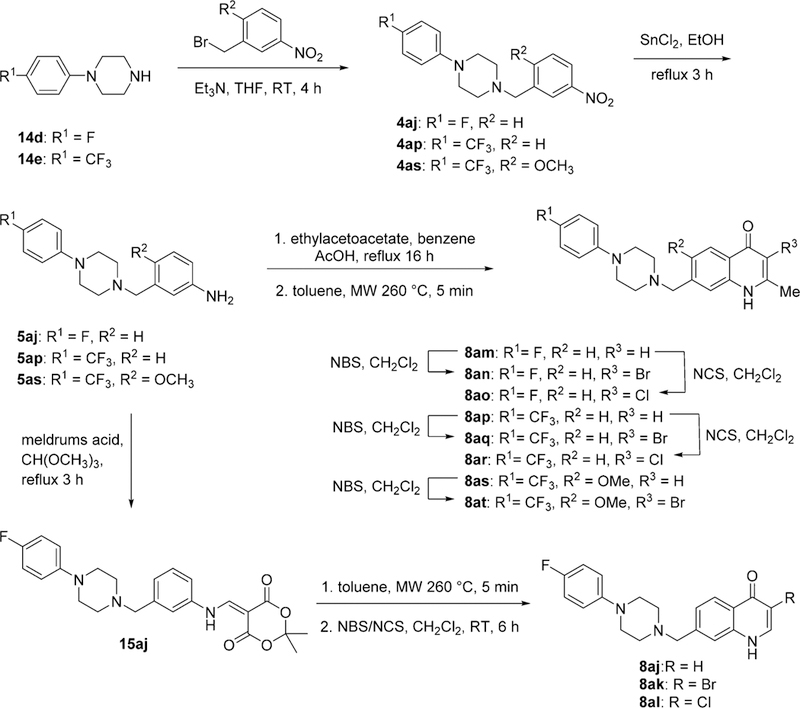
Synthesis of 3-Halo Substituted 4(1H)-Quinolones
Antimalarial Activity and Cytotoxicity.
All compounds were tested against clinically relevant multidrug resistant malarial strains W2 (pyrimethamine- and chloroquine-resist-ant) and TM90-C2B (mefloquine-, chloroquine-, atovaquone-, and pyrimethamine-resistant) as previously reported.6,9,26 Because of the emergence and rapid acquisition of cross-resistance,27 each compound was also evaluated based on its RI (EC50) for TM90-C2B and W2 strains (RI = EC50 TM90-C2B/ EC50 W2). Ideally, the RI of a compound should lie between 0.3 and 3.0 in order to avoid rapidly inducing resistance in the parasite. This range is based upon the natural resistance patterns observed for drugs like chloroquine and mefloquine.28,29 Selected compounds were also tested for in vitro liver stage activity using Pb sporozoites expressing luciferase, harvested from mosquito salivary glands and allowed to infect HEPG2 hepatoma cells in order to assess if the compounds possessed causal prophylactic activity.26 Additionally, each compound was tested for cytotoxicity using mammalian J774 cell lines in a 96-well plate format.6,8,9,26
Structure—Activity Relationships.
The poor aqueous solubility of our 4(1H)-quinolone esters7 motivated us to design and prepare a set of ionizable piperazinyl-substituted analogues with the primary aim being to significantly enhance the aqueous solubility without compromising antimalarial activity. The initial, small set of 6-hydrogen-7-piperazinyl-4(1H)-quinolones containing various linkages between the piperazinyl moiety and the 4(1H)-quinolone’s benzenoid ring was prepared to identify the optimal spacer length (Table 1). In general, compounds with an ethylene between the 4(1H)-quinolone core and the piperazine showed the poorest blood stage activity of the group, with N-phenylpiperazinyl-4(1H)-quinolone 8ac displaying EC50 values of 26 nM for W2 and 1500 nM for TM90-C2B, while benzyl-substituted analogue 8ad was less active, with EC50 values of 120 nM for W2 and >2.0 μM for TM90-C2B. In contrast, p-methoxybenzylpiper-azinyl-4(1H)-quinolone 8ae was the most active for W2, with EC50 values of 12 nM, but the least active for TM90-C2B, showing activities greater than 2.0 μM. Compounds with piperazines directly attached to the 4(1H)-quinolone core were more active than compounds containing an ethylene linker. N-Phenylpiperazinyl-4(1H)-quinolone 8a was the most active member, with EC50 values of 4.5 and 250 nM for W2 and TM90-C2B. In comparison, N-benzylpiperazinyl-4(1H)-quinolone 8b was 4-fold less potent, with and EC50 of 16 nM for W2, along with 860 nM for TM90-C2B, respectively.
Table 1.
Exploration of Various N-Substituted Piperazinyl Moieties on 7-Position of 4(1H)-Quinolone Benzenoid Ring to Enhance the Solubility and Antimalarial Activity
 | |||||||
|---|---|---|---|---|---|---|---|
| compd | R | n | EC50 W2 (nM)a |
EC50 TM90-C2B (nM) |
RIb | EC50Pb (nM) |
EC50 J774 (μM) |
| 8ac |  |
2 | 25 | 1500 | 60 | -c | >20 |
| 8ad |  |
2 | 120 | >2000 | >17 | 160 | >20 |
| 8ae | 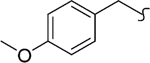 |
2 | 12 | >2000 | >167 | -c | >20 |
| 8a | 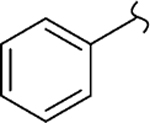 |
0 | 4.5 | 250 | 56 | 74 | >20 |
| 8b | 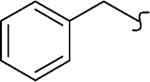 |
0 | 16 | 860 | 53 | 85 | >20 |
| 8i | 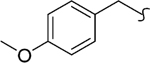 |
0 | 20 | 1300 | 65 | -c | -c |
| 8j | 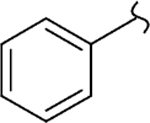 |
1 | 1.3 | 480 | 369 | 4.7 | 3.9 |
| 8k | 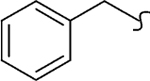 |
1 | 1.4 | 150 | 106 | 44 | 10 |
| 8l | 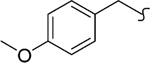 |
1 | 2.5 | 800 | 320 | 83 | 5.1 |
| 8m | 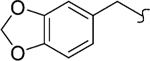 |
1 | 160 | >2000 | >12 | -c | >20 |
Chloroquine (CQ), atovaquone (ATO), and dihydroartemisinin (DHA) are internal controls for each in vitro assay: CQ, 0.42 μM W2, 0.23 μM TM90-C2B, and 47.2 μM J774; ATO, 1.4 nM W2, 18 μM TM90-C2B, and 28 μM J774; DHA, 5.5 nM W2, 5.9 nM TM90-C2B and 1.5 μM J774.
RI = TM90-C2B/W2.
Not determined.
Compounds with a methylene spacer between the piperazine and 4(1H)-quinolone were the most active analogues of this first set of 4(1H)-quinolones. Compounds 8j, 8k, and 8l were similarly potent, with low single-digit nM inhibitory concentrations for W2. However, the same compounds displayed reduced activity against TM90-C2B, producing RI values ranging from 100 to 390 for analogues 8j, 8k, and 8l.
A selection of this first set of piperazinyl-4(1H)-quinolones (Table 1) was tested for in vitro liver stage activity using P. berghei sporozoites expressing luciferase as previously described.19 The best results were obtained with analogues whose piperazinyl moiety was attached to the quinolone’s benzenoid ring via a methylene unit. N-Phenylpiperazinyl-4(1H)-quinolone 8j was the most potent compound, with an EC50 of 4.7 nM for Pb, while its benzyl analogue 8k or its 4-methoxybenyl analogue 8l were approximately 10-fold less potent, with EC50 values of 44–83 nM. All the other analogues 8a, 8b, and 8ad, with the piperazinyl group substituted to the 4(1H)-quinolone’s core directly or via an ethylene, were even less potent.
Follow-up SAR studies focused solely on piperazines directly attached to the 4(1H)-quinolone core or via one methylene unit, as the antimalarial activity of the N-phenyl or N-benzylpiperazinyl-4(1H)-quinolones appeared to be more potent than ethylene-connected analogues. This, in conjunction with previous observations that substituents in the 6-position alter antimalarial activity predominantly against TM90-C2B, led to the design of a small series of 6-methyl- or 6-methoxy-4(1H)-quinolone esters retaining in 7-position an N-phenyl-, N-benzyl-, or 4-methoxybenzyl-substituted piperazine. Piperazinyl-4(1H)-quinolones 8q, 8r, 8w, and 8x with a methylene spacer were approximately 10-fold more potent against W2 than their structurally related analogues 8c, 8g, 8d, and 8h, whose 4(1H)-quinolone core is directly substituted with the piperazine moiety (Table 2). In contrast, the 7-piperazinyl-4(1H)-quinolones 8c, 8g, 8d, and 8h possessed approximately 10-fold more favorable RI values in comparison to the analogues 8q, 8r, 8w, and 8x with a methylene spacer. Furthermore, for both the N-phenylpiperazinyl-or N-benzylpiperazinyl-substituted 4(1H)-quinolones, the 6-methyl substituent appeared to improve antimalarial activity, whereas the opposite effect was true for the 6-methoxy-susbtituted analogues. 6-Methyl-7-phenylpiperazinyl-4(1H)-quinolone 8q was the most potent of the group against W2, with an EC50 value of 0.44 nM and 0.15 μM against TM90-C2B. When the N-phenylpiperazinyl moiety of 8q was exchanged by an N-benzylpiperazine, activity for analogue 8r fell slightly for W2, with an EC50 value of 1.5 nM, and more noticeable for TM90-C2B, with an EC50 value of 0.89 μM. Additional potency losses were observed with compound 8s when the N-phenyl-piperazinyl moiety of 8q was exchanged by a 4-methoxybenzylpiperazine. Exchange of the 6-methyl group of 4(1H)-quinolone 8q by a 6-methoxy substituent in compound 8w dropped the potency approximately 3-fold for both W2 and TM90-C2B.
Table 2.
Effect of Various Substitutions in 6-Position of the 4(1H)-Quinolone’s Benzenoid Ring on Antimalarial Activity
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | n | EC50W2 (nM)a |
EC50 TM90-C2B (nM) |
RIb | EC50Pb (nM) |
EC50 J774 (μM) |
| 8q | 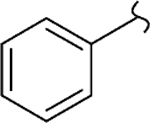 |
−CH3 | 1 | 0.44 | 150 | 345 | 6.9 | 12 |
| 8r | 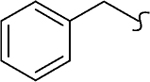 |
−CH3 | 1 | 1.5 | 890 | 614 | 9.5 | >20 |
| 8s | 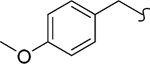 |
−CH3 | 1 | 2.4 | 1000 | 417 | -c | >20 |
| 8w | 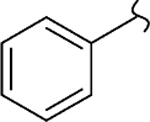 |
−OCH3 | 1 | 1.6 | 490 | 306 | 110 | >20 |
| 8x | 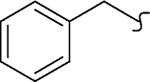 |
−OCH3 | 1 | 5.5 | >2000 | >364 | 9.3 | >20 |
| 8y | 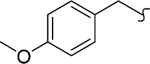 |
−OCH3 | 1 | 10 | >2000 | >200 | 52 | >20 |
| 8c | 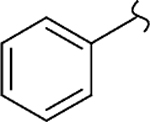 |
−CH3 | 0 | 46 | 1100 | 24 | -c | 17 |
| 8g |  |
−CH3 | 0 | 13 | 980 | 75 | >100 | >20 |
| 8d |  |
−OCH3 | 0 | 37 | 800 | 22 | -c | 17 |
| 8h |  |
−OCH3 | 0 | 64 | 200 | 3 | -c | 12 |
Chloroquine (CQ), atovaquone (ATO), and dihydroartemisinin (DHA) are internal controls for each in vitro assay: CQ, 0.42 μM W2, 0.23 μM TM90-C2B, and 47.2 μM J774; ATO, 1.4 nM W2, 18 μM TM90-C2B, and 28 μM J774; DHA, 5.5 nM W2, 5.9 nM TM90-C2B, and 1.5 μM J774.
RI = TM90-C2B/W2.
Not determined.
The compounds displaying the best W2 activity were also tested for in vitro liver stage activity. Analogues 8q, 8r, and 8x were the most potent ones, with EC50 values of 6.9, 9.5, and 9.3 nM, respectively.
Next, a subseries was designed to determine how steric and/ or electronic effects of the N-phenylpiperazinyl moiety influences the antimalarial activity (Table 3). The para position of the N-phenylpiperazinyl group was substituted with a fluorine, a trifluoromethyl, or a methoxy group, while simultaneously the quinolone’s 6-position was probed with a hydrogen, a methyl, or a methoxy group. The previously observed trend was confirmed as analogues 8n and 8z with the methylene spacer between the piperazine and the 4(1H)-quinolone’s core were more potent against W2 than compounds 8e and 8f with a directly attached piperazine. Furthermore, independently of the 4-substituent of the N-phenylpiperazine moiety, activity data against W2 indicated that the 6-methyl-substitued compounds are slightly more potent than the 6-methoxy analogues, which in comparison to the 6-hydrogen, analogues are equipotent or less potent. Finally, substituting the 4-position of the N-phenylpiperazinyl moiety with the electron donating methoxy group generally produced compounds that were slightly less potent than analogues with an electron withdrawing group.
Table 3.
Steric and Electronic Effects of the Phenylpiperazine Moiety and the 4(1H)-Quinolone 6-Position on Antimalarial Activity
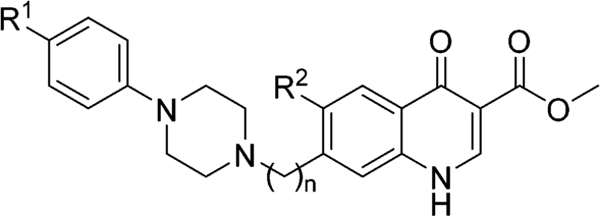 | ||||||||
|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | n | EC50 W2 (nM)a | EC50 TM90-C2B (nM) | RIb | EC50 Pb (nM) | EC50J774 (μM) |
| 8o | −F | −H | 1 | 1.8 | 130 | 74 | 1.1 | 2.0 |
| 8u | −F | −CH3 | 1 | 0.84 | 350 | 417 | 2.6 | 16 |
| 8aa | −F | −OCH3 | 1 | 1.8 | 220 | 122 | 32 | >20 |
| 8e | −OCH3 | −H | 0 | 7.8 | 150 | 20 | c | >20 |
| 8f | −OCH3 | −OCH3 | 0 | 43 | 2000 | 47 | >100 | >20 |
| 8n | −OCH3 | −H | 1 | 3.6 | 220 | 61 | 0.12 | 4.7 |
| 8t | −OCH3 | −CH3 | 1 | 0.57 | 180 | 316 | 4.1 | >20 |
| 8z | −OCH3 | −OCH3 | 1 | 3.4 | 860 | 253 | 10 | 16 |
| 8p | −CF3 | −H | 1 | 0.32 | 120 | 375 | 16 | 11 |
| 8v | −CF3 | −CH3 | 1 | 0.066 | 100 | 1515 | c | >20 |
| 8ab | −CF3 | −OCH3 | 1 | 6.4 | 210 | 33 | 9.6 | >20 |
Chloroquine (CQ), atovaquone (ATO), and dihydroartemisinin (DHA) are internal controls for each in vitro assay: CQ, 0.42 μM W2, 0.23 μM TM90-C2B, and 47.2 μM J774; ATO, 1.4 nM W2, 18 μM TM90-C2B, and 28 μM J774; DHA, 5.5 nM W2, 5.9 nM TM90-C2B, and 1.5 μM J774.
RI = TM90-C2B/W2.
Not determined.
The fluorinated methylene-spaced compounds 8o, 8u, and 8aa showed significant antimalarial activity against all strains. Analogues 8o and 8aa were equipotent against W2, with EC50 values of 1.8 nM, whereas compound 8u displayed subnanomolar potency. The trifluoromethylphenylpiperazinyl-4(1H)-quinolones 8p, 8v, and 8ab were more potent than the fluorophenyl-substituted analogues 8o, 8u, and 8aa, suggesting a strong electron-withdrawing effect on the phenyl group to be beneficial. Compound 8v was the most potent analogue of the entire series, with an EC50 value of 66 pM against W2 and an EC50 value of 100 nM against TM90-C2B.
Compounds in this series also displayed very potent liver stage activity. Of the compounds chosen for testing, p-methoxyphenyl-substituted analogue 8n was the most potent one, with an EC50 value of 120 pM. Fluorophenyl- or methoxyphenyl-substituted 4(1H)-quinolones 8o, 8u, and 8t displayed single-digit nanomolar activity.
We next considered whether a positional change of the piperazine moiety from the 7-position to the 6-position would retain or improve the antimalarial activity and possibly improve the RI value. A set of analogues 8af–8ai was prepared by switching the piperazine moiety from the 7-position to 6-position of the 4(1H)-quinolone’s benzenoid ring and evaluated for their activity (Table 4). These piperazinyl analogues 8af–8ai lost significantly in activity in comparison to their 7-substituted counterparts, with EC50 values ranging from 45 to 160 nM against W2. Against TM90–C2B, these compounds were considered to be nearly inactive. Surprisingly, compounds 8af and 8ai showed moderate activity against Pb, with EC50 values of 85–90 nM. However, the lack of potency against W2, TM90-C2B, and Pb further substantiated our initial hypothesis that for antimalarial activity the piperazine moiety must be attached at the 4(1H)-quinolone’s 7-position.
Table 4.
4(1H)-Quinolones Substituted in 6-Position with the Piperazine Moiety
 | ||||||
|---|---|---|---|---|---|---|
| compd | R | EC50 W2 (nM)a |
EC50 TM90- C2B (nM) |
RIb | EC50
Pb
(nM) |
EC50 J774 (μM) |
| 8af | −H | 150 | >2000 | >13 | 91 | >20 |
| 8ag | −OCH3 | 160 | >2000 | >12 | >100 | >20 |
| 8ah | −F | 110 | >2000 | >18 | c | >20 |
| 8ai | −CF3 | 45 | >2000 | >44 | 85 | >20 |
Chloroquine (CQ), atovaquone (ATO), and dihydroartemisinin (DHA) are internal controls for each in vitro assay: CQ, 0.42 μM W2, 0.23 μM TM90-C2B, and 47.2 μM J774; ATO, 1.4 nM W2, 18 μM TM90-C2B, and 28 μM J774; DHA, 5.5 nM W2, 5.9 nM TM90-C2B, and 1.5 μM J774.
RI = TM90-C2B/W2.
Not determined.
Previously, 3-halo-substitutions 7 were shown to significantly improve the RI values of phenoxyethoxy-4(1H)-quinolones and several piperazinyl-substituted analogues were prepared for this purpose (Table 5). With the exception of compounds 8ar and 8at, all other 3-halo-4(lH)-quinolones 8ak-8aq possessed acceptable RI values smaller than 3. Like the 7-phenoxyethoxy-4(lH)-quinolone analogues, the addition of a 2-methyl substituent gave rise to more potent compounds, with the 3-chloro-substituted 4(lH)-quinolone 8ao having EC50 values of 39 and 52 nM for W2 and TM90-C2B, respectively, producing an RI of 1.4. The 3-bromo 8an was even more potent, with EC50s of 25 nM for both W2 and TM90-C2B with an RI of 1.0. However, there was a 30-fold difference in Pb activities between the 2-unsubstituted and 2-methyl substituted with 8ak displaying subnanomolar activity while 8an had an EC50 of 26 nM. Finally, trifluoromethylphenyl-substituted piperazine variants were synthesized to give 3-chloro 8ar and 3-bromo 8aq. Both compounds showed a significant decrease in activity compared to their fluoro-substituted piperazine counterparts 8ao and 8an.
Table 5.
3-Halo-Substituted 4(1H)-Quinolones
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | R4 | EC50 W2 (nM)a | EC50 TM90-C2B (nM) | RIb | EC50 Pb (nM) | EC50J774 (μM) |
| 8ak | −F | −H | −Br | −H | 810 | >2000 | >2.5 | 0.86 | >20 |
| 8al | −F | −H | −Cl | −H | 1100 | >2000 | >1.8 | c | >20 |
| 8am | −F | −H | −H | −CH3 | 1200 | 1500 | 1.2 | c | >20 |
| 8an | −F | −H | −Br | −CH3 | 25 | 26 | 1.0 | 26 | >20 |
| 8ao | −F | −H | −Cl | −CH3 | 39 | 52 | 1.3 | c | >20 |
| 8aq | −CF3 | −H | −Br | −CH3 | 120 | 95 | 0.8 | c | >20 |
| 8ar | −CF3 | −H | −Cl | −CH3 | 140 | 1500 | 11 | c | >20 |
| 8at | −CF3 | −OCH3 | −Br | −CH3 | 130 | >2000 | >15 | c | >20 |
Chloroquine (CQ), atovaquone (ATO), and dihydroartemisinin (DHA) are internal controls for each in vitro assay: CQ, 0.42 μM W2, 0.23 μM TM90-C2B and 47.2 μM J774; ATO, 1.4 nM W2, 18 μM TM90-C2B and 28 μM J774; DHA, 5.5 nM W2, 5.9 nM TM90-C2B and 1.5 μM J774.
RI = TM90-C2B/W2.
Not determined
Proposed Structural Basis of Antimalarial Activity and Molecular Docking.
Plasmodium cytochrome bcl complex is an effective and validated target in the treatment and prophylaxis of malaria. Many different classes of compounds, including 4(lH)-pyridone30,31 and related 4(lH)-quinolone6–9,26,32 derivatives developed by our and other groups, have been identified as potent inhibitors of bcl mitochondrial electron transport chain component. Nevertheless, only one bcl inhibitor, atovaquone, has been approved for clinical use as a key component of the fixed-dose antimalarial combination therapy, marketed as Malarone, since 2000. Unfortunately, several point mutations at Qo atovaquone binding site of the bcl complex lead to clinically relevant atovaquone-resistant strains of P. falciparum (one of these atovaquone-resistant strains is TM90-C2B that we routinely used for our SAR campaign in this study). Up until 2015, majority of the investigated bcl inhibitors were believed to target Qo site of the protein. Disclosing the structure of bovine heart cytochrome bcl complex with 4(lH)-pyridones GW844520 and GSK932121 revealed clear evidence of these compounds unexpectedly binding to the reductive Qi site.33 These findings explain the ability of 4(lH)-pyridones (and related 4(lH)-quinolones) to overcome parasite Qo-based atovaquone resistance. Additionally, TM90-C2B cross resistance and RI data itself could be used as an indicator of preferential Qo/Qi site binding. The discovery of preferred binding of the 4(lH)-pyridones class of inhibitors to Qi site as well as RI data provided critical structural and biological information that we applied to structure-guided drug design efforts of herein disclosed piperazine substituted 4(1H)-quinolone inhibitors.
To understand the structural basis of observed antimalarial activity, we performed molecular docking studies using coordinates of the bc1GW844520 (Qi site, PDB 4D6T33) and bc1 atovaquone (Qo site, PDB 4PD434). Receptor grids were generated and validated by docking GW844520 and atovaquone into Qi and Qo sites accordingly (Figure 3E,F). The docked models for both inhibitors were in good agreement with the reported crystal structures coordinates. We then selected two representative piperazine substituted 4(1H)-quinolone inhibitors with high RI (8ae, RI > 495, Table 1) suggesting preferential Qo site inhibition and low (8ao, RI = 1.35, pan Qi/ Qo or selective Qi inhibition, Table 5) RI values. The binding poses of both 8ao and 8ae (Figure 3A–D) as well as historic atovaquone, GW844520 (Figure 3E,F) and quinolones ELQ-30026 and 1 (data not shown) in the Qo and Qi sites of bc1 were subsequently analyzed.
Figure 3.
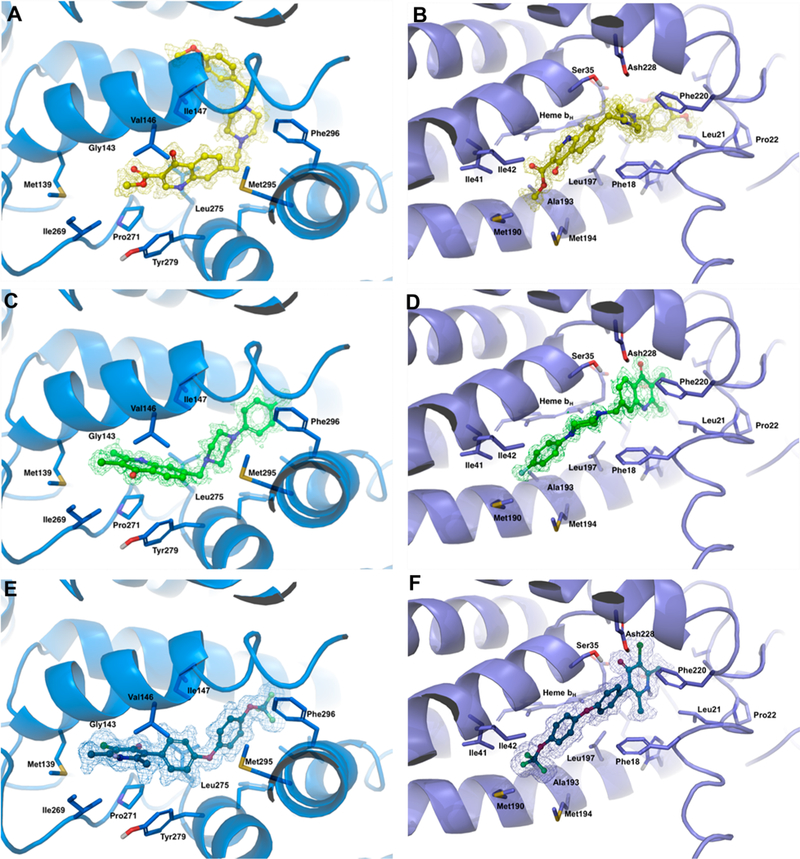
Binding of 8ae (yellow), 8ao (green) and GW844520 (blue) within the hydrophobic Qo and Qi cavities as predicted by Glide docking. The electron density map (Gaussian type, yellow or green, contoured at level 1.0 in Pymol) was calculated after docking of the ligands to the model. Protein residues within 5 Å to Qo site (marine blue) or Qi site (deep blue) are shown in stick representation. (A) Docking pose of 8ae (yellow sticks) within Qo binding site (marine blue). (B) Docking pose of 8ae (yellow sticks) within Qi binding site (deep blue). (C) Docking pose of 8ao (green sticks) within Qo binding site (marine blue). (D) Docking pose of 8ao (green sticks) within Qi binding site (deep blue). (E) Docking pose of GW844520 (blue sticks) within Qo binding site (marine blue). (F) Docking pose of GW844520 (blue sticks) within Qi binding site (deep blue).
On the basis of the phenotypic readout (EC50 W2 = 12 nM, TM90-C2B > 5740 nM) and RI data (RI > 495), we hypothesize that 8ae is a preferential Qo site binder. Indeed, 8ae docked well into Qo binding pocket similarly to atovaquone (docking scores: atovaquone, –9.86; 8ae, –8.77) with 4(1H)-quinolone core occupying space in between Val146 and Leu275 (Figure 3A) and C7 piperazine tale extending outside of the pocket. On the other hand, the orientation of compound 8ae docked into the Qi site of the protein (Figure 3B) was opposed to the orientation of GW844520 (Figure 3F) in the original crystal structure, suggesting diminished mode of action and confirming the loss of activity against TM90-C2B. The 4(1H)-pyridone core of GW844520 sits deep in the Qi site, making favorable π–π interactions with Phe220 and hydrogen bond interactions with Ser35 of loop A (Figure 3F, also see the original report33 for more details). Instead, docking of 8ae into Qi site revealed positioning of the quinolone core in the hydrophobic pocket away from the heme (Figure 3B). Switching the 3-ester group of 8ae to halogen led to compound 8ao. Interestingly, compound 8ao (EC50 W2 = 39 nM, TM90-C2B = 52 nM) docked well in the Qi site of bc1 with quinolone core sitting deep in the binding pocket making similar to GW844520 interactions and piperazine tale extending out of the channel (Figure 3D,F). This mode of action can explain the ability of 8ao bind to Qi site, hence the activity against Qo-based atovaquone resistance TM90-C2B strain. Compounds 8ao and GW844520 also docked well in Qo site (WT) of the bc1 (Figure 3C,E).
Molecular docking studies performed in combination with biological data suggest that 3-halo-substituted 4(lH)-quinolones (Table 5) are Qi site inhibitors showing significantly improved RI values and activity against TM90-C2B strain. At the same time, it is very likely that vast majority of piperazine-containing 4(lH)-quinolone esters (Tables 1–4) are selective inhibitors of Qo site of cytochrome bc1 complex.
Cytotoxicity.
All compounds were tested in vitro for cytotoxicity to J774 mammalian cells using a protocol, which was used in all of our previous published studies with various 4(lH)-quinolone derivatives as a guide and to produce selectivity index data to assist with the SAR optimization of the compounds (Tables 1–5).6–9 Of all the compounds tested, only a few compounds displayed signs of cytotoxicity at concentrations lower than 20 μM. Onset of cytotoxicity was recorded for compounds 8j, 8l, 8n, and 8o with EC50 values of 3.94, 5.13, 4.73, and 2.00 μM, respectively. Nevertheless, these analogues and the majority of the piperazinyl-4(lH)-quinolones can be considered selective chemotypes as they display single-digit nanomolar or subnanomolar activity against W2.
Structure–Property Relationships.
Calculated properties such as molecular weight, polar surface area, and number of H-bond donors and acceptors were within the recommended ranges typically needed for good oral bioavailability, suggesting that the piperazinyl-4(lH)-quinolone design provided excellent spatial leeway for structural modifications to occupy physicochemical space unique for orally bioavailable compounds. Furthermore, to profile the properties of the piperazinyl-4(lH)-quinolones, and to identify potential limitations, aqueous solubility and lipophilicity log D were experimentally determined via LC/MS-based assays as described by the Manetsch laboratory previously6,8,35,36 (see Supporting Information, Tables S1–S5). Encouragingly, the piperazinyl-4(lH)-quinolones were much more soluble (average solubility ≥50 μM) than the previously described phenoxyethoxy-4(lH)-quinolones (average solubility ≤5 μM) or 3-phenyl-susbstituted analogues (average solubility ≤5 μM).17 As expected, the solubility of all compounds was affected by pH, with better solubility under more acidic conditions.
The piperazine analogues such as 8ac, 8ad, 8ae, 8j, or 8k with an ethylene or a methylene spacer between the piperazine and the quinolone moiety displayed good aqueous solubility of 80 μM or more at both pH 2.0 and pH 6.5. Only compounds such as 8a with an N-phenylpiperazinyl group directly attached to the 4(lH)-quinolone core had reduced solubility below 20 μM at pH 2.0 and pH 6.5. Replacement of the N-phenylpiperazinyl group in 4(lH)-quinolone 8a by an N-benzylpiperazinyl group in compound 8b reestablished the solubility in the ranges of 60–80 μM. The aqueous solubility was also in good correlation with calculated pKa values of the piperazine nitrogens. Specifically, compounds with an ethylene spacer (8ac, 8ad, 8ae) showed a pKa of approximately 6–7, compounds with a methylene spacer (8j and similar) between the piperazine and the quinolone showed a pKa of approximately 5.0, while compounds with the piperazinyl group directly attached to the 4(lH)-quinolone core (8c and similar) displayed a pKa of 3.8 or less.
Additional solubility dependencies were observed with the various 4(lH)-quinolone compound series. Analogues in which the piperazinyl moiety was moved from the 4(lH)-quinolone’s 7-position to the 6-position were slightly less soluble at pH 6.5 (see Supporting Information, Tables S1–S5). Furthermore, 3-halo-4(lH)-quinolones had marked solubility differences between pH 2.0 and 6.5, displaying significantly higher solubility at low pH. The addition of a 2-methyl group to the 4(lH)-quinolone further lowered solubility, with 3-bromo-2-methyl-4(lH)-quinolone 8an being more than 20 times less soluble at pH 6.5 compared to its 2-hydrogen counterpart 8ak (see Supporting Information, Tables S1–S5). This observation is in agreement with calculated pKa values of 4(lH)-quinolone’s nitrogens of 4.2 for 8ak and 2.8 for 8an, respectively.
In Vivo Efficacy Evaluation of Selected Compounds in an Efficacy Scouting Assay Against Blood Stages of the Parasite.
Of all prepared and tested 4(lH)-quinolones, 29 with potent in vitro activity against both P. falciparum strains were chosen to undergo a scouting assay in Pb-infected mice. The screening involved a single oral 50 mg/kg dose 1 day post infection (PI) and an assessment of parasitemia on days 3 PI and 6 PI (Table 6). The threshold for active compounds was inhibition greater than 50% on days 3 and 6 PI. Compounds 8h, 8m, 8t, and 8ad all showed no inhibition on day 6 PI, whereas 4(lH)-quinolones 8b and 8l were just under the 50% threshold of activity with both showing inhibition in the low 40% ranges. Compounds 8a, 8c, 8d, 8k, 8n, 8q, 8r, 8s, 8x, 8y, and 8ag displayed little to moderate protection on day 6 PI, delaying the parasite’s growth. Lastly, compounds 8o, 8p, 8u, 8v, 8w, 8z, 8aa, 8ab, 8ak, 8al, 8an, and 8ao all showed excellent activities in these scout assays with trifluoromethylpiperazinyl-4(lH)-quinolone esters 8p, 8v, and 8ab and 3-bromo-4(lH)-quinolone 8an having completely inhibited parasite growth even on day 6 PI. These results clearly underscore the significant advantages the piperazinyl-substituted 4(1H)-quinolones have over the previously reported 4(lH)-quinolone esters.17
Table 6.
In Vivo Efficacy Scout Screening
| compd | % inhibition day 3 PIa | % inhibition day 6 PIa | compd | % inhibition day 3 PIa | % inhibition day 6 PIa |
|---|---|---|---|---|---|
| 8a | 38.5 | 27.5 | 8u | 100 | 98.3 |
| 8b | 46.2 | 43.1 | 8v | 100 | 100 |
| 8c | 80.0 | 31.6 | 8w | 100 | 82.4 |
| 8d | 70.0 | 33.0 | 8x | 80.0 | 19.3 |
| 8h | 100 | <1 | 8y | 100 | 24.6 |
| 8k | 100 | 12.3 | 8z | 100 | 70.6 |
| 81 | 69.2 | 41.2 | 8aa | 100 | 56.9 |
| 8m | 40.0 | <1 | 8ab | 100 | 100 |
| 8n | 100 | 39.2 | 8ad | 46.2 | <1 |
| 8o | 100 | 98.0 | 8ag | 84.6 | 31.4 |
| 8p | 100 | 100 | 8ak | 100 | 94.2 |
| 8q | 46.2 | 15.7 | 8al | 100 | 71.9 |
| 8r | 100 | 24.6 | 8an | 100 | 100 |
| 8s | 80.0 | 14.0 | 8ao | 100 | 86.4 |
| 8t | 100 | <1 | atovaquone | 96.3 | 99.8 |
| amodiaquine | 95.5 | 99.9 |
Percent inhibition compared to untreated animals.
In Vivo Efficacy Evaluation of Frontrunner Compounds Against Blood Stages of the Parasite.
Using a modified Thompson test model, frontrunner compounds 8o, 8p, 8u, 8v, 8ab, 8ak, 8an, and 8ao were evaluated in vivo. These frontrunner compounds were selected for further in vivo efficacy testing as they displayed full inhibition on day 3 PI and over 85% inhibition on day 6 PI in the scouting assay. Mice were infected with 1 × 106 P. berghei–GFP parasites, and compounds were dosed orally on days 3, 4, and 5 PI with a dose of 10 mg/kg of compound suspended or dissolved in HEC/Tween or PEG 400. Parasitemia was observed by flow cytometry on days 3, 6, 9, 13, 21, and 30 PI. Compounds with animal survival up to day 30 PI, and parasitemia levels of less than 1% on day 30 PI were considered to be cures. Lastly, animals with more than 40% parasitemia levels were euthanized. For all experiments, atovaquone was used as the positive control.9,26
4(1H)-Quinolone esters 8ak and 8ao and 3-bromo-4(1H)-quinolone 8an all had the same day of death as the untreated control animals. While 4(1H)-quinolone esters 8ak and 8ao displayed a low inhibition on day 6 PI, analogue 8an was much more potent with a 90.9% inhibition on day 6 PI. These results suggest that compound 8an is possibly rapidly cleared after day 6 PI. Compounds 8o, 8p, and 8u had greater 100% inhibition on day 6 PI, nevertheless, all animals succumbed to the parasite by day 21 PI, possibly indicating a longer half-life than 3-bromo-4(1H)-quinolone 8an. The remaining compounds, 8v and 8ab, both produced cures in more than half of the animals, curing 3 of the 5 animals (Table 7).
Table 7.
In Vivo Efficacy Thompson Test
| compd | dose (mg/kg) |
% inhibition day 6 PIa |
av day of death |
no. of cures |
|---|---|---|---|---|
| 8o | 10 | l00 | 2l | 0/5 |
| 8p | 10 | l00 | 2l | 0/5 |
| 8u | 10 | l00 | 2l | 0/5 |
| 8v | 10 | l00 | N/A | 3/5 |
| 8ab | 10 | l00 | N/A | 3/5 |
| 8ak | l0 | 33.0 | l3 | 0/5 |
| 8an | l0 | 90.9 | l3 | 0/5 |
| 8ao | l0 | 11.0 | l3 | 0/5 |
| atovaquone | l0 | l00 | N/A | 5/5 |
Percent inhibition as compared to untreated control animals.
In Vivo Efficacy Evaluation of Frontrunner Compounds Against Liver Stages of the Parasite.
The potent in vitro activity of piperazinyl-substituted 4(1H)-quinolones against liver stages of the parasite prompted a study to determine in vivo these compounds in P. berghei sporozoite infected mice. Five animals per group were dosed as previously reported only 1 h after infection with piperazinyl-substituted 4(1H)-quinolones 8j and 8l. At 44 h PI, day 6 PI, day 9 PI, and day 13 PI, compound efficacy was determined by bioluminescence imaging via injection of D-luciferin. Both compounds were administered orally or subcutaneously at increasing doses of 25, 50, and 100 mg/kg in PEG 400. of the two piperazinyl-substituted 4(1H)-quinolones, 8j performed significantly better than 8l (Figure 4). With the exception of an infection on days 6, 9, 13 PI of a single mouse, which was orally dosed with 50 mg/kg, no luminescence was observed for 8j at any other doses and time points. In comparison, compound 8l displayed full protection at all time points only at an oral dose of 100 mg/kg. Progression of parasitemia was monitored up to 30 days after infection (Table 8). Differences between the two test 4(1H)-quinolones were more obvious following survival cures (Figure 5) as 8j cured two or more out of five animals. In contrast, one of five animals was cured only at a high dose of 100 mg/kg of 8l. These results with 8j and 8l underscore that piperazinyl-substituted 4(1H)-quinolones have potential as single-dose prophylactic and curative agents.
Figure 4.
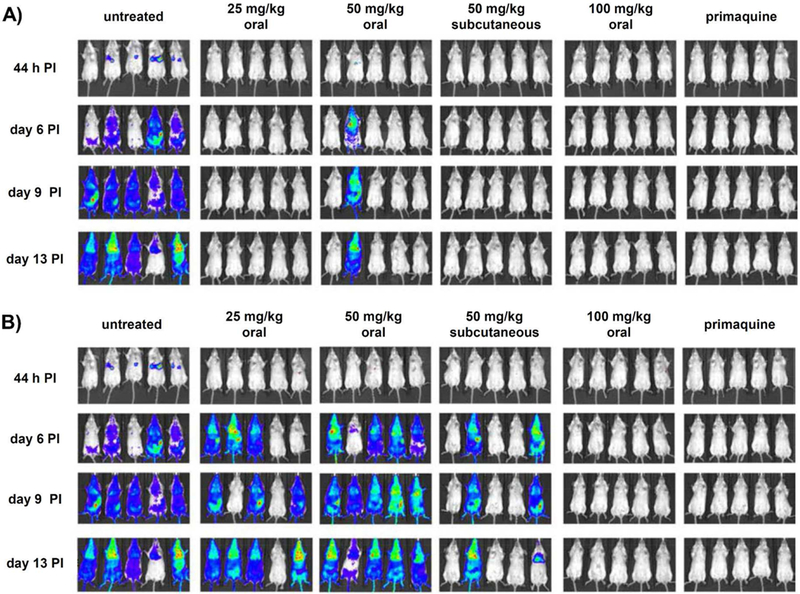
Whole-animal bioluminescence imaging of mice infected with luciferase-transfected P. berghei sporozoites. Mice were treated with different doses of 8j (A), 81 (B), and primaquine (50 mg/kg, oral). Animals (n = 5 per group) received a single dose by gavage 1 h after inoculation with sporozoites. Representative images taken at 44 h, day 6, day 9, and day 13 after infection are shown. At 44 h, bioluminescent signal was detected in control untreated animals, with the highest intensity noted in the area overlaying the liver, consistent with the presence of liver-stage parasites.
Table 8.
In Vivo Efficacy of Compounds 8j and 8l Against Liver Stages of the Parasite
| compd | dose (mg/kg) |
route | % inhibition day 9 PIa |
no. of cures |
|---|---|---|---|---|
| 8j | 25 | oral | 65.3 | 2/5 |
| 8j | 50 | oral | 85.1 | 2/5 |
| 8j | 50 | subcutaneous | >99.0 | 3/5 |
| 8j | 100 | oral | >99.0 | 5/5 |
| 8l | 25 | oral | 49.1 | 0/5 |
| 8l | 50 | oral | 38.8 | 0/5 |
| 8l | 50 | subcutaneous | 20.1 | 0/5 |
| 8l | 100 | oral | 43.9 | 1/5 |
| primaquine | 50 | oral | >99.0 | 2/5 |
Percent inhibition as compared to untreated control animals.
Figure 5.
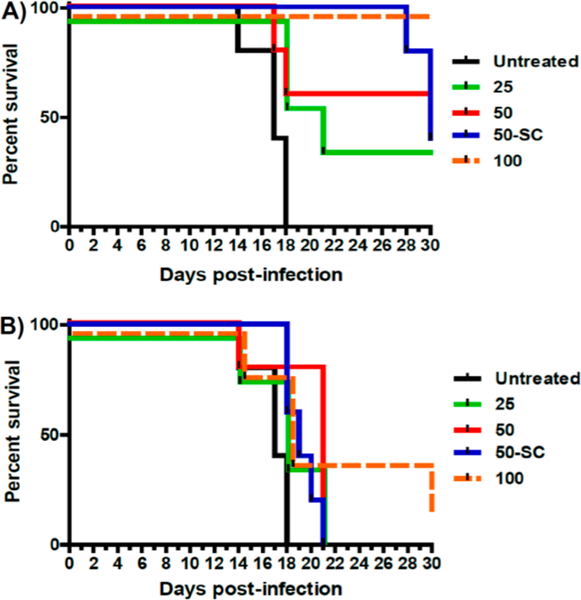
Survival curves for 4(1H)-quinolones 8j (A) and 8l (B) that demonstrate antimalarial activity against liver stages of the parasite. Compounds have been tested oral at 25 mg/kg (25), 50 mg/kg (50), and 100 mg/kg (100) and subcutaneous at 50 mg/kg (50-SC) doses.
∎. CONCLUSIONS
In 1970, Ryley and Peters demonstrated that 4(1H)-quinolone ester 1 possesses causal prophylactic activity (kill growing exoerythrocytic stage parasites) and erythrocytic stage inhibition in avian malaria models but not against malaria parasites in mammals.16 Twenty years after Ryley and Peters’ work was published, Puri and Dutta showed that 1 produced radical cures (eradicate dormant EE parasites) in Plasmodium cynomolgi infected rhesus monkeys.15 Despite the promise of this 4(1H)-quinolone ester, the development of this compound was challenged due to major physicochemical property limitations requiring parenteral dosing. Furthermore, the propensity for rapid acquisition of resistance following exposure to 1 hampered its further development.16
Recent optimization studies led to the synthesis and identification of 4(1H)-quinolones that displayed potent in vitro antimalarial activity against the liver and the blood stages of the parasite. In particular, 4(1H)-quinolones, whose ester group in 3-position was replaced by a halogen, appeared to significantly reduce cross resistance with atovaquone and the best compound of this subseries displayed significantly better in vivo efficacy than compound 1. Nevertheless, as a follow-up, 46 additional piperazinyl-substituted 4(1H)-quinolone esters or 3- halo-4(1H)-quinolones were synthesized with the objective to increase the aqueous solubility while maintaining or improving the antimalarial activity against the blood and liver stages of the parasite. As a secondary objective, compound optimization also focused on equipotency against W2, a chloroquine- and pyrimethamine-resistant strain, and against TM90-C2B, a chloroquine-, mefloquine-, pyrimethamine-, and atovaquone-resistant strain.
Structure—activity relationship studies concentrated primarily on analogues in which (a) a piperazine containing moiety was installed either in 6- or 7-position, (b) the 2-positon was probed with a methyl or a hydrogen, and (c) the 3-position was varied with a methyl ester or a halogen. The best antimalarial activity was observed when the piperazine-containing moiety was installed in 7-position, while the 3-position was equipped with a methyl ester group. Of the 7-substituted 4(1H)-quinolones, analogues with an ethylene spacer between the piperazine and the 4(1H)-quinolone core were approximately 10-fold less potent than counterparts in which the piperazine was directly attached to the 4(1H)-quinolone moiety or via a methylene spacer. Furthermore, N-phenylpiperazinyl-4(1H)-quinolones were slightly better than N-benzylpiperazinyl-substituted counterparts. Antimalarial activity was lost if the piperazine-containing substituent was moved from the 7-position to the 6-position or when the methyl ester in the 3-position was replaced by a halide. Nevertheless, 3-halo-substituted 4(1H)-quinolones possessed significantly improved resistance indices as they displayed equipotent activity against W2 and TM90-C2B. Specifically, analogues 8p, 8q, 8t, 8u, 8v, and 8w were among the most potent 4(1H)-quinolone esters, displaying low single-digit nanomolar or subnanomolar EC50 values against W2, double- or triple-digit nanomolar activity against TM90-C2B, and single- or double-digit nanomolar activity against the Pb liver stages. In contrast, despite 3-bromo-4(1H)-quinolone 8an and other 3-halo-4(1H)-quinolones being slightly less potent against W2, they maintained the potency against TM90-C2B and consequently produced excellent resistance indices smaller than 3. Follow-up docking studies with cytochrome bc1 the molecular target of antimalarial 4(1H)-quinolones or 4(1H)-pyridones, revealed that 3-ester-substituted 4(1H)-quinolones presumably prefer binding to the Qo site of the cytochrome, while 3-halosusbtituted 4(1H)-quinolones bind to the Qi site. This data is in full agreement with the phenotypic data as Y268S mutations in Qo site is directly associated with resistance to atovaquone.
The preferred candidates for in vivo efficacy studies were selected based on log D and aqueous solubility data at two different pHs. The majority of the piperazinyl-substituted 4(1H)-quinolones displayed good aqueous solubility of 40μM or higher. The 29 best compounds were tested for in vivo efficacy, first in a scouting assay, and the eight most promising 4(1H)-quinolones were subsequently assessed for in vivo efficacy in the conventional Thompson test at a lower oral dose. N-Trifluoromethylpiperazinyl-substituted analogues 8v and 8ab were the most efficacious 4(1H)-quinolone esters, reducing parasitemia on day 6 PI completely and curing three out of five animals, whereas 8o, 8p, and 8u were slightly less potent with a survival period of mice to 21 days PI. 3-Bromo-4(1H)-quinolone 8an displayed a 91% inhibition of parasitemia on day 6 PI, nevertheless, mice were sacrificed on day 13 PI due to increased level of parasitemia. Finally, compounds 8j and 81 displaying good activity in vitro against liver stages were also tested for in vivo efficacy against liver stages. The more in vitro potent 4(1H)-quinolone 8j was highly efficacious in vivo, generating cures at oral doses of 25 mg/kg or higher. This in conjunction with the promising in vivo activity against the blood stages renders the piperazinyl-substituted 4(1H)-quinolone compound series potential for further optimization as an orally bioavailable antimalarial compound series.
∎. EXPERIMENTAL SECTION
General.
All reagents and solvents were obtained from Aldrich Chemical Co. and used without further purification. NMR spectra were recorded at ambient temperature on a 400 or 500 MHz Varian NMR spectrometer in the solvent indicated. All 1H NMR experiments are reported in δ units, parts per million (ppm) downfield of TMS, and were measured relative to the signals of chloroform (7.26 ppm) and dimethyl sulfoxide (39.5 ppm) with 1H decoupled observation. Data for 1H NMR are reported as follows: chemicals shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), integration and coupling constant (Hz), whereas 13C NMR analyses were reported in terms of chemical shift. NMR data was analyzed by using MestReNova Software version 6.0.2–5475. The purity of the final compounds was determined to be ≥95% by highperformance liquid chromatography (HPLC) using an Agilent 1100 LC/MSD-VL with electrospray ionization. Low-resolution mass spectra were performed on an Agilent 1100 LC/MSD-VL with electrospray ionization. High-resolution mass spectra (HRMS) were acquired on an Agilent LC/MSD TOF system G3250AA. Analytical thin layer chromatography (TLC) was performed on silica gel 60 F254 precoated plates (0.25 mm) from EMD Chemical Inc., and components were visualized by ultraviolet light (254 nm). EMD silica gel 230—400 (particle size 40—63 μm) mesh was used for all flash column chromatography. Microwave heating was performed in a single-mode Anton Paar Monowave 300, and all microwave-irradiated reactions were conducted in heavy-walled glass vials sealed with Teflon septa.
In Vitro Antimalarial Activity against Blood Stages of P. falciparum (W2 and TM90-C2B).
In vitro antimalarial activity against blood stage parasites W2 and TM90-C2B was determined as previously reported. 6–9,17,37 p. falciparum clone W2 (Indochina) and TM90C2B (Thailand) were grown in continuous culture using RPMI 1640 media containing 10% heat-inactivated type A+ human plasma, sodium bicarbonate (2.4 g/L), HEPES (5.94g/L), and 4% washed human type A+ erythrocytes. Cultures were gassed with a 90% N2, 5% O2, and 5%CO2 mixture followed by incubation at 37 °C. Test compounds in DMSO at 10.0 mM concentration were diluted at least 1:400 and then serially diluted in duplicate over 11 concentrations. P. falciparum cultures with >70% ring stage parasites were diluted to 0.5— 0.7% parasitemia and 1.5% hematocrit in RPMI 1640 media. In 96-well plates, a volume of 90 μL/well of parasitized erythrocytes was added on top of 10 μL/well of the test compound. A separate plate containing chloroquine, dihydroartemisinin, and atovaquone was added to each set of assay plates as control drugs. A Beckman Coulter Biomek 3000 was used to dispense test compounds, control drugs, and parasitized erythrocytes into the microtiter plates. Positive and negative controls were included in each plate. Positive controls consisted of drug-free parasitized erythrocytes, and negative controls consisted of parasitized erythrocytes dosed with a high concentration of chloroquine or dihydroartemisinin that ensured 100% parasite death. Assay plates were placed into a plastic gassing chamber and equilibrated with 90% N2, 5% O2, and5%CO2 mixture then incubated at 37 °C for 48 h, then 3H-hypoxanthine was added and plates incubated another 24 h. After 72 h of incubation, the assay plates were frozen at –80 °C until later processing for parasite growth determinations. Assay plates were removed from –80 °C and allowed to thaw at room temperature. Using a plate harvester, the contents of the plate were collected on filtermats and then CPMs counted in a Topcount liquid scintillation counter. Data analysis was performed using a custom database manager (Dataspects, Inc.). Nonlinear regression analysis was used to calculate EC50.
In Vitro J774 Cytotoxicity Assay.
In vitro J774 cytotoxicity was determined as previously reported.6–9,17,37 Mouse macrophage cell line J774 was cultured in RPMI-1640 media with phenol-red containing L-glutamine then supplemented with 10% fetal bovine serum, penicillin (50 Units/mL), and streptomycin (50 μg/mL). For seeding into 96-well plates, the J774 cells were diluted to 5 × 105 cells/mL. Cells were dispensed into 96-well plates at a volume of 100 μL/well, giving a final concentration of 5 × 104 cells/well. Plates were incubated for 24 h at 37 °C and 5% CO2 to allow the attachment of J774 to the bottom of the plate wells. Test compounds were prepared by diluting to 10 μg/mL or 20 μM, followed by 1:2 serial dilutions over 11 concentrations. After 24 h, the media was removed from the wells and serially diluted test compounds were added to each well. Positive and negative control wells were included on each assay plate. Plates containing cells and test compounds were then incubated for 72 h at 37 °C and 5% CO2. After the incubation period, cell proliferation was assessed using CellTiter 96 Aqueous One Solution Cell Proliferation Assay reagent (Promega). To each well 20 μL of reagent were added followed by incubation for 4 h at 37 °C and 5% CO2. A Spectramax M2e (Molecular Devices) plate reader was used to read absorbance at 490 nM. IC50 values were determined using a custom database manager (Dataspects, Inc.). Nonlinear regression analysis was used to calculate IC50s.
In Vitro Antimalarial Activity against P. berghei Liver Stages.
In vitro antimalarial activity against liver stage parasites was determined as previously reported.2,37 HepG2 cells (75000 per well) were seeded into collagen-coated, black 96-well plates with optically clear bottoms (Beckton Dickson, Franklin Lakes, NJ) for viewing on the IVIS spectrum system (Caliper Life Sciences, Hanover, MD) and white 96-well plates for analysis with the TopCount microplate luminometer (Packard, Meriden, CT). Cells were maintained at 37 °C in 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 1.0% penicillin-streptomycin (Sigma), and 1.0% L-glutamine. Mosquito salivary glands were dissected as described above, and 5000 sporozoites were added per well. Plates were incubated at 37 °C for 3 h and then washed three times with PBS. Serial dilutions of test compounds were prepared as previously described, added to parasite-infected HepG2 cells in triplicate, and incubated at 37 °C for 44 h. Following the incubation, cells were washed once with PBS and then lysed with 10 μL of cell culture lysis reagent (Promega Luciferase Assay system kit; Promega, Madison, WI). Immediately after cell lysis, 100 μL of luciferase assay substrate was added and then the parasite lysates were analyzed.
In Vivo Blood Stage Antimalarial Activity (Thompson Test, P. berghei).
In vivo antimalarial activity against blood stage parasites was determined as previously reported. 9,17,37 We used a “modified Thompson model”, in which infections were established by an intraperitoneal inoculation of 2 × 106 P. berghei (GFP)-infected red blood cells. By day 3, the first day of drug treatment, parasitemia typically reached 1% (i.e., 1% of the RBC will be parasitized). Test compounds were administered per os once-daily (qd) for 3 days. Atovaquone was included as a standard drug for comparison. Infected mice, compound treated on days 3–5 post parasite inoculum, were followed for 30 days total. Parasitemia was determined from blood collected from the tail vein on days 3, 6, 9, 12, 15, 18, 21, 24, 27, and 30; parasitemia was quantified by microscopic examination of Giemsa-stained blood smears. We also monitored whether (and when) recrudescence occurred. End points for the efficacy analysis were the ED50 and ED90 (dose of drug that suppressed 50% and 90% of parasitemia on day 6, respectively), the no recrudescence dose (ie., no parasitemia following initial clearance of parasitemia), and percent cure at 30 days. This study was conducted in compliance with the Guide for the Care and Use of Laboratory Animals of the National Research Council for the National Academies. The protocol was approved by the University of South Florida Institutional Animal Care and Use Committee.
In Vivo Antimalarial Activity against Liver Stages (P. berghei).
In vivo antimalarial activity against liver stage parasites was determined as previously reported.2,37 There were five mice per treatment group. Experimental groups were treated with 50 or 25 mg/ kg sc or administered at 100 or 50 mg/kg po. Mice were treated once at 2 h after infection. Untreated, noninfected, and infected mice (infection controls) as well as compound-treated and atovaquone-treated mice (drug controls) were also included. In vivo imaging was performed 44 h post infection to assess liver stage development and on days 6, 9, and 13 PE to monitor blood stage development using methods described previously.38 Briefly, the luciferase activities of whole animals were assessed using IVIS Spectrum (Caliper Life Sciences, Hanover, MD). Prior to analysis, the abdomens of all mice were shaved. Animals (n = 5) were injected ip with D-luciferin (100 mg/kg), anesthetized with isoflurane, and imaged 5 min postinjection. While animals were continuously exposed to isoflurane, images were acquired with a 23 cm field of view (FOV), medium binning factor, and an exposure time of 20 to 120 s. Images were analyzed using the Living Image 3.0 software (Caliper Life Sciences, Hanover, MD). This study was conducted in compliance with the Guide for the Care and Use of Laboratory Animals of the National Research Council for the National Academies. The protocol was approved by the University of South Florida Institutional Animal Care and Use Committee.
Molecular Modeling.
The original co-crystal structures of bc1 atovaquone (Qo site, PDB 4PD434) and bc1GW844520 (Qi site, PDB 4D6T33) were refined using the Protein Preparation Wizard39 implemented in the Maestro 11.2 (Schrodinger Release 2017–2) interface, and invalid atom types were corrected using this same wizard. The protein structure was imported into workspace and preprocessed to assign bond orders, add hydrogen atoms, create disulfide bonds, and to delete water molecules beyond 5 Å from hetero groups. Additionally, the protein structure was refined via automated H-bond assignment and restrained minimization with OPLS 2005 force field by converging heavy atoms to 0.5 Å RMSD. Ligand structures were sketched in ChemDraw and prepared with LigPrep in the Maestro interface. A receptor grid was generated from the refined structures using default values while centered on the position of the ligand (atovaquone or GW844520) that occupied either Qo and Qi sites of bc1. Selected inhibitors were docked into the grid using Glide 7.440–42 in standard precision (SP) mode, without any constraints, by sampling of the conformational and positional degrees of freedom of the ligand. The binding poses of inhibitors in the Qo and Qi sites of bc1 were further refined using minimization of the binding pocket around the docked ligands and subsequently analyzed.
General Procedure A.
A mixture of piperazine (1 equiv), 3-nitrobenzyl bromide/3-nitrophenethyl bromide (1.1 equiv), and Et3N (1.5 equiv) in anhydrous THF was stirred for overnight at RT. The reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by flash chromatography (90:20 → 70:30, hexanes/ EtOAc) to afford the title compounds.
General Procedure B.
A mixture of nitro compound (1 equiv) and SnCl2 (3 equiv) in absolute ethanol was refluxed for 3 h. The reaction was neutralized with 4N KOH solution and extracted with EtOAc. The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by flash chromatography using 100% EtOAc.
General Procedure C.
A neat mixture of aniline/aminobenzyl alcohol (1 equiv) and dimethyl 2-(methoxymethylene)malonate (1.05 equiv) was heated at 110 °C for 30 min. The reaction mixture was allowed to cool to RT while precipitation arose. Diethyl ether was added to the mixture to improve the precipitation. The solid was filtered off and washed with diethyl ether, dried under vacuum, and used directly for the further transformations.
General Procedure D.
To a solution of alcohol (1 equiv) in anhydrous CH2Cl2 was added the Dess–Martin periodinane (1.5 equiv) at RT. The resulting mixture was stirred for 3 h at RT. The mixture was treated with aqueous NaHCO3 solution and filtered through a sintered funnel while washing with CH2Cl2. The organic phase was separated, dried over Na2SO4, filtered, and concentrated to give the aldehyde in an almost pure form which was used directly for further transformations.
General Procedure E.
To a mixture of aldehyde (1 equiv) and piperazine/piperazine hydrochloride (1.2 equiv) in anhydrous THF were added anhydrous MgSO4 (2 equiv) followed by N,N-diisopropylethylamine (2.5 equiv) at RT, and the resulting solution was stirred vigorously for 30 min. To this was then added sodium triacetoxyborohydride (2 equiv). The reaction was stirred for an additional 4 h at RT, quenched with saturated NaHCO3 solution, and extracted with EtOAc. The combined organic fractions were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude was purified by flash chromatography gradient elution (80:20 → 30:70, hexanes/EtOAc) to afford the title compounds.
General Procedure F.
The enamine in toluene was subjected to microwave heating at 280 °C for 4 min. The reaction mixture was allowed to cool to RT while precipitation rose. Diethyl ether was added to the mixture to improve the precipitation. The solid was filtered off and washed with diethyl ether. The solid containing the unreacted enamine and quinolone regio-isomers was then refluxed in methanol (in most of the cases unless it is mentioned otherwise) for 1 h and filtered hot to give the title quinolones in purest form.
General Procedure G.
To a solution of quinolone (1 equiv) in anhydrous CH2Cl2 was added freshly recrystallized NBS/NCS (1.2 equiv) at RT, and the resulting mixture was stirred overnight. The reaction was concentrated, and the crude was purified by either recrystallization or HPLC.
General Procedure H.
To a stirred solution of diol 10 (12.5 mmol) in anhydrous CH2Cl2 (50 mL) was added thionyl chloride (2.5 mL, 34.7 mmol) dropwise. The mixture was heated at reflux for 1 h. The reaction was concentrated, and the residue was diluted with MeCN (200 mL). To this was then added KI (100 mg, 0.62 mmol) followed by nitroaniline (13 mmol), and the resulting mixture was refluxed for 7 days. The reaction was concentrated, and the crude product was used for the next reaction without further purification.
1-(3-nitrophenyl)-4-phenylpiperazine 4a.
To a solution of 1-fluoro-3-nitrobenzene (2 g, 14.2 mmol) in DMSO (28.5 mL) was added 1-phenylpiperazine (6.5 mL, 42.5 mmol) and DIPEA (9.9 mL, 56.8 mmol). The reaction was refluxed for 2 days. DI water was added, then extracted with EtOAa 3 times. The organic layer was dried over sodium sulfate, filtered, and concentrated under reduced pressure followed by separation by flash column chromatography to give 4a as a yellow solid in 50% yield. 1H NMR (399 MHz, (CD3XCO) δ 7.77 (t, J = 2.3 Hz, 1H), 7.63 (ddd, J = 7.9, 2.1, 1.0 Hz, 1H3, 7.50 (t, J = 8.1 Hz, 1H), 7.44 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H), 7.29–7.23 (m, 2H), 7.05–7.00 (m, 2H), 6.84 (tt, J = 7.4, 1.0 Hz, 1h), 3.49 (dd, J = 6.3, 3.9 Hz, 4H), 3.37 (dd, J = 6.3, 3.9 Hz, 4H). 13C NMR (100 MHz, (CD3)2CO) δ 153.1, 152.4, 150.5, 131.0, 130.1, 122.4, 120.7, 117.2, 114.2, 110.1, 49.9, 49.2.
1-Benzyl-4-(3-nitrophenyl)piperazine 4b.
To a solution of 1-fluoro-3-nitrobenzene (2 g, 14.2 mmol) in DMSO (28.5 mL) were added 1-benzylpiperazine (7.4 mL, 42.5 mmol) and DIPEA (9.9 mL, mmol). The reaction was refluxed for 2 days. DI water was added, then extracted with EtOAc 3 times. The organic layer was dried over sodium sulfate, filtered, and concentrated under reduced pressure followed by separation by flash column chromatography to give 4a as a yellow solid in 65% yield. 1H NMR (399 MHz, (CD3)2CO) δ 7.69 (t, J = 2.3 Hz, 1H), 7.59 (ddd, J = 8.0, 2.1, 0.8 Hz, 1h3, 7.45 (t, J = 8.2 Hz, 1H), 7.40–7.31 (m, 5H), 7.29–7.23 (m, 1H), 3.57 (s, 2H), 3.35–3.31 (m, 4H), 2.62–2.58 (m, 4H). 13C NMR (100 MHz, (CD3)2CO) δ 152.4, 149.6, 138.7, 130.1, 129.1, 128.4, 127.2, 121.2, 112.9, 109.0, 62.7, 52.9, 48.3.
1-(3-Nitrophenethyl)-4-phenylpiperazine 4ac.
4ac was obtained as a pale-yellow semisolid (600 mg, 62% yield) by alkylation of 1-phenylpiperazine (500 mg, 3.08 mmol) with 3-nitrophenethyl bromide (780 mg, 3.4 mmol) following general procedure A. 1H NMR (400 MHz, CDCl3): δ 8.16–8.04 (m, 2H), 7.57 (d, J = 7.6 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 7.33–7.23 (m, 2h), 7.00–6.82 (m, 3H), 3.31–3.17 (m, 4H), 2.96 (t, J = 7.8 Hz, 2H), 2.73–2.69 (m, 6h). 13C NMR (101 MHz, CDCl3): δ 151.2, 148.3, 142.3, 135.0, 129.2, 129.1 (2C), 123.6, 121.3, 119.8, 116.1 (2C), 59.5, 53.2 (2C), 49.1 (2C), 33.1. HRMS (ESI-TOF) calcd for C18H22N3O2 [M + H]+ 312.1712, found 312.1719.
1-Benzyl-4-(3-nitrophenethyl)piperazine 4ad.
4ad was obtained as a an orange-yellow oil (800 mg, 63% yield) by alkylation of 1-benzylpiperazine (690 mg, 4.0 mmol) with 3-nitrophenethyl bromide (1.0 g, 4.3 mmol) following general procedure A. 1H NMR (400 MHz, CDCl3): δ 8.06 (t, J = 2.0 Hz, 1h), 8.03 (ddd, J = 8.1, 2.4, 1.1 Hz, 1H), 7.54–7.50 (m, 1H), 7.41 (t, J = 7.9 Hz, 1H), 7.32–7.20 (m, 5H), 3.51, (s, 2H), 2.91–2.85 (m, 2H), 2.65–2.59 (m, 2H), 2.59–2.45 (m, 8H). 13C NMR (101 MHz, CDCl3): δ 148.3, 142.5, 138.0, 135.0, 129.2, (2C), 129.1, 128.2 (2C), 127.0, 123.6, 121.2, 63.0, 59.6, 53.1 (2C), 53.0 (2C), 33.1. HRMS (eSI-TOF) calcd for C19H24N3O2 [M + H]+ 326.1869, found 326.1869.
1-(4-Methoxybenzyl)-4-(3-nitrophenethyl)piperazine 4ae.
4ae was obtained as an orange-yellow oil (1.3 g, 59% yield) by alkylation of 1-(4-methoxybenzyl)piperazine (1.3 g, 6.3 mmol) with 3-nitrophenethyl bromide (1.5 g, 6.9 mmol) following general procedure A. 1H NMR (400 MHz, CDCl3): δ 8.07–7.95 (m, 2H), 7.48 (t, J = 8.3, 1H), 7.37 (dd, J = 16.5, 8.6, 1H), 7.23–7.15 (m, 2H), 6.86–6.78 (m, 2h), 3.74 (s, 3H), 3.42 (s, 2H), 2.87–2.80 (m, 2H), 2.59 (dd, J = 9.0, 6.7, 2H), 2.54–2.40 (m, 8H). 13C NMR (101 MHz, CDCl3): δ 158.7, 148.2, 142.5, 135.0, 130.3 (2C), 130.0, 129.1, 123.5, 121.1, 113.5 (2C), 62.4, 59.5, 55.2, 53.1 (2C), 52.9 (2c), 33.1. HRMS (ESI-TOF) calcd for C20H25N3O3 [M + H]+ 356.1974, found 356.1981.
1-(4-Fluorophenyl)-4-(3-nitrobenzyl)piperazine 4aj.
4aj was obtained as a white solid (8.5 g, 97% yield) by alkylation of 1-(4-(trifloromethyl)phenyl)piperazine (5.0 g, 27.7 mmol) with 3-nitro-benzyl bromide (6.6 g, 30.5 mmol) following general procedure A. 1H NMR (400 MHz, CDCl3): δ 8.21 (t, J = 2.0 Hz, 1H), 8.09 (ddd, J = 8.2, 2.4, 1.1 Hz, 1H), 7.68 (dt, J = 7.7, 1.3 Hz, 1H), 7.47 (t, J = 7.9 Hz, 1H), 6.95–6.89 (m, 2H), 6.87–6.81 (m, 2H), 3.63 (s, 2H), 3.16–3.05 (m, 4H), 2.65–2.56 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 158.2, 155.8, 148.3, 147.8, 140.5, 134.9, 129.1, 123.6, 122.1, 117.7, 115.5, 115.3, 61.8, 53.0 (2C), 50.0 (2C). HRMS (ESI-TOF) calcd for C17H19FN3O2 [M + H]+ 316.1461, found 316.1466.
1-(3-Nitrobenzyl)-4-(4-(trifluoromethyl)phenyl)piperazine (4ao).
4ao was obtained as a pale-yellow solid (3.7 g, 90% yield) by alkylation of 1-(4-(trifluoromethyl)phenyl)piperazine (3 g, 13.0 mmol) with 3-nitrobenzyl bromide (2.2 g, 14.3 mmol) following general procedure A. 1H NMR (399 MHz, CDCl3) δ 8.25 (s, 1H), 8.12, (d, J = 8.2 Hz, 1H), 7.72 (d, J = 7.6 Hz, 1H), 7.51 (d, J = 7.9 Hz, 1H), 7.47 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 3.65 (s, 2H), 3.34–3.26 (m, 4H), 2.69–2.55 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 153.3, 148.5, 140.5, 135.1, 129.3, 126.4 (dd, J = 7.4, 3.7 Hz), 124.9 (d, J = 270.5 Hz), 123.7, 122.4, 120.4 (d, J = 32.7 Hz), 114.6, 62.0, 52.8, 48.0.
1-(2-Methoxy-5-nitrobenzyl)-4-(4-(trifluoromethyl)phenyl)-piperazine (4as).
4as was obtained as a white solid (1.4 g, 90% yield) by alkylation of 1-(4-(trifluoromethyl)phenyl)piperazine (850 mg, 3.7 mmol) with 2-(bromomethyl)-1-methoxy-4-nitrobenzene (1 g, 4.1 mmol) following general procedure A. 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J = 2.8 Hz, 1H), 8.19 (dd, J = 9.0, 2.8 Hz, 1H), 7.49 (d, J = 8.8 Hz, 2H), 6.95 (t, J =8.1 Hz, 3H), 3.97 (s, 3H), 3.65 (s, 2H), 3.38–3.29 (m, 4H), 2.73–2.65 (m, 4h). 13C NMR (126 MHz, CDCl3) δ 162.7, 153.4, 141.5, 127.7, 126.4 (q, J = 3.9 Hz), 125.6, 124.9 (d, J = 270.9 Hz), 124.6, 120.4 (d, J = 32.7 Hz), 114.6, 110.1, 56.23, 55.5, 53.0, 48.1.
3-(4-Phenylpiperazin-1-yl)aniline 5a.
A nitro reduction reaction of 4a (178 mg, 0.63 mmol) following general procedure B resulted in 5a, isolated as a crude orange solid which was used in the next step without further purification.
3-(4-Benzylpiperazin-1-yl)aniline 5b.
A nitro reduction reaction of 4b (658 mg, 2.21 mmol) following general procedure B resulted in 5b, isolated as a crude orange solid which was used in the next step without further purification.
3-(2-(4-Phenylpiperazin-1-yl)ethyl)aniline 5ac.
A nitro reduction reaction of 4ac (500 mg, 1.6 mmol) following general procedure B resulted in 5ac as an orange-yellow semisolid (450 mg, 99% yield). 1H NMR (400 MHz, CDCl3): S 7.30–7.24 (m, 2H), 7.08 (t, J = 7.7 Hz, 1H), 6.97–6.92 (m, 2H), 6.88–6.84 (m, 1H), 6.63 (dt, J = 7.6, 1.2 Hz, 1H), 6.56–6.52 (m, 2H), 3.60 (bs, 2H), 3.26–3.22 (m, 4h), 2.79–2.74 (m, 2H), 2.71–2.62 (m, 6H). 13C NMR (101 MHz, CDCl3): δ 151.3, 146.4, 141.4, 129.3, 129.1 (2C), 119.7, 119.0, 116.0, 115.4 (2C), 112.9, 60.4, 53.2 (2C), 49.1 (2C), 33.6. HRMS (ESI-TOF) calcd for C18H24N32 [M + H]+ 282.1970, found 282.1965.
3-(2-(4-Benzylpiperazin-1-yl)ethyl)aniline 5ad.
A nitro reduction reaction of 4ad (700 mg, 2.1 mmol) following general procedure B resulted in 5ad as a yellow oil (550 mg, 86% yield). 1H NMR (400 MHz, CDCl3): S 7.32–7.29 (m, 4H), 7.28–7.20 (m, 1H), 7.04 (td, J = 7.3, 1.4 Hz, 1H), 6.58 (dt, J = 7.3, 1.3 Hz, 1H), 6.50 (dd, J = 7.1, 1.1 Hz, 2H), 3.91 (bs, 2H), 3.52 (s, 2H), 2.76–2.66 (m, 2H), 2.57 (ddd, J = 23.4, 12.1, 4.9 Hz, 10H). 13C NMR (101 MHz, CDCl3): δ 146.4, 141.3, 137.8, 129.2, 129.2 (2C), 128.1 (2C), 127.0, 118.9, 115.4, 112.9, 62.9, 60.2, 52.9 (2C), 52.7 (2C), 33.3.
3-(2-(4-(4-Methoxybenzyl)piperazin-1-yl)ethyl)aniline 5ae.
A nitro reduction reaction of 4ae (1.0 g, 3.0 mmol) following general procedure B resulted in 5ae as an orange-yellow semisolid (820 mg, 90% yield). 1H NMR (400 MHz, CDCl3): δ 7.20 (t, J = 8.4 Hz, 2H), 7.03 (t, J = 7.7 Hz, 1H), 6.83 (dd, J = 8.4, 5.8 Hz, 2H), 6.57 (d, J = 7.6 Hz, 1H), 6.49 (d, J = 7.0 Hz, 2H), 3.77 (s, 3H), 3.43 (s, 2H), 2.69 (dd, J = 10.6, 5.7 Hz, 2H), 2.64–2.39 (m, 10H). 13C NMR (101 MHz, CDCl3): S 158.6, 146.4, 141.4, 130.3 (2C), 129.9, 129.2, 118.8, 115.3, 113.5 (2C), 112.8, 62.3, 60.3, 55.1, 53.0 (2C), 52.8 (2C), 33.5. HRMS (ESI-TOF) calcd for C20H28N3O [M + H]+ 326.2232, found 326.2225.
3-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)aniline 5aj.
A nitro reduction reaction of 4aj (8.4 g, 26.6 mmol) following general procedure B resulted in 5aj as a pale-yellow solid (7.3 g, 96% yield). 1H NMR (400 MHz, methanol-d4): δ 7.04 (t, J = 7.7 Hz, 1H), 6.92–6.82 (m, 4H), 6.68 (t, J = 2.0 Hz, 1H), 6.65 (dt, J = 7.5, 1.3 Hz, 1H), 6.60 (ddd, J = 8.0, 2.3, 1.0 Hz, 1H), 3.40 (s, 2h), 3.06–3.01 (m, 4h), 2.58–2.52 (m, 4H). 13C NMR (101 MHz, methanol-d4) δ 159.5, 157.1, 148.9, 148.2, 138.7, 129.9, 120.4, 118.9, 117.4, 116.2, 116.0, 115.4, 63.9, 53.8 (2C), 50.7 (2C). HRMS (eSI-TOF) calcd for C17H21FN3 [M + H]+ 286.1720, found 286.1730.
3-((4-(4-(Trifluoromethyl)phenyl)piperazin-1-yl)methyl)aniline 5ap.
A nitro reduction reaction of 4ap (2.8 g, 7.55 mmol) following method B resulted in 5ap as a pale-orange solid (2.4 g, 94% yield). 1H NMR (399 MHz, CDCl3) δ 7.48 (d, J = 8.7 Hz, 2H), 7.13 (t, J = 7.7 Hz, 1H), 6.92 (d, J = 8.7 Hz, 2H), 6.79–6.70 (m, 2h), 6.66–6.57 (m, 1H), 3.66 (bs, 2H), 3.49 (s, 2H), 3.35–3.22 (m, 4h), 2.70–2.56 (m, 4h). 13C NMR (100 MHz, CDCl3) δ 153.4, 146.6, 139.1, 129.3, 126.4 (q, J = 3.6 Hz), 124.9 (d, J = 270.6 Hz), 120.3 (d, J = 32.5 Hz), 119.6, 115.8, 114.5, 114.2, 63.1, 52.9, 48.0.
4-Methoxy-3-((4-(4-(trifluoromethyl)phenyl)piperazin-1-yl)-methyl)aniline 5as.
A nitro reduction reaction of 4as (1.5 g, 3.79 mmol) following method B resulted in 5as, isolated as a crude orange solid which was used in the next step without further purification.
Dimethyl 2-(((3-((4-Phenylpiperazin-1-yl)methyl)phenyl)amino)-methylene)malonate 7j.
A direct reductive amination (DRA) reaction between aldehyde 13a (1.0 g, 3.8 mmol) and 1-phenyl-piperazine (740 mg, 4.6 mmol) following general procedure E afforded 7j as a brown yellow semisolid (1.1 g, 72% yield). 1H NMR (400 MHz, CDCl3): δ 11.02 (d, J = 13.6 Hz, 1H), 8.52 (dd, J = 13.7, 2.1 Hz, 1H), 7.31–(5.96 (m, 6H), 6.90–6.75 (m, 3H), 3.79 (d, J = 2.5 Hz, 3h), 3.73 (d, J = 2.2 Hz, 3H), 3.51 (d, J = 2.8 Hz, 2H), 3.16 (t, J = 4.9 Hz, 4H), 2.57 (t, J = 5.0 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 169.0, 165.6, 151.9, 150.9, 139.8, 138.9, 129.4, 128.8 (2C), 125.5, 119.4, 117.6, 115.7 (2C), 115.6, 92.6, 62.3, 52.8 (2C), 51.3, 51.2, 48.7 (2C). HRMS (ESI-TOF) calcd for C23H27N3O4 [M + H]+ 410.2080, found 410.2105.
Dimethyl 2-(((3-((4-Benzylpiperazin-1-yl)methyl)phenyl)amino)-methylene)malonate 7k.
A direct reductive amination (DRA) reaction between aldehyde 13a (500 mg, 1.9 mmol) and 1-benzylpiperazine (400 mg, 2.3 mmol) following general procedure E afforded 7k as a yellow oil (600 mg, 75% yield). 1H NMR (400 MHz, CDCl3): δ 11.00 (d, J = 13.8 Hz, 1H), 8.52 (dd, J = 13.8, 1.2 Hz, 1H), 7.30–7.24 (m, 5H), 7.20 (ddt, J = 5.9, 4.8, 2.4 Hz, 1H), 7.11 (t, J =1.8 Hz, 1H), 7.06 (dt, J = 7.6, 1.2 Hz, 1H), 7.02–6.98 (m, 1H), 3.83 (d, J = 1.4 Hz, 3H), 3.76 (d, J = 1.3 Hz, 3H), 3.49 (d, J = 1.2 Hz, 2H), 3.46 (s, 2H), 2.55–2.36 (m, 8H). 13C NMR (101 MHz, CDCl3): δ 169.2, 165.9, 152.2, 140.5, 139.0, 137.9, 129.5, 129.1 (2C), 128.1 (2C), 126.9, 125.7, 117.8, 115.6, 92.6, 62.9, 62.5, 53.0 (2C), 52.9 (2C), 51.5, 51.4. HRMS (ESI-TOF) calcd for C24H30N3O4 [M + H] + 424.2236, found 424.2250.
Dimethyl 2-(((3-((4-(4-Methoxybenzyl)piperazin-1-yl)methyl)-phenyl)amino)methylene)malonate 7l.
A DRA reaction between aldehyde 13k (1.0 g, 3.8 mmol) and 1-(4-methoxybenzyl) piperazine (940 mg, 4.6 mmol) following general procedure E afforded 7l as a yellow oil (1.2 g, 70% yield). 1H NMR (400 MHz, CDCl3): δ 10.99 (d, J = 13.8 Hz, 1H), 8.51 (d, J = 13.8 Hz, 1H), 7.27–7.23 (m, 1H), 7.19–7.15 (m, 2H), 7.10 (t, J = 1.9 Hz, 1H), 7.05 (dt, J = 7.6, 1.2 Hz, 1H), 6.99 (ddd, J = 8.1, 2.5, 1.0 Hz, 1H), 6.82–6.78 (m, 2H), 3.81 (s, 3H), 3.74 (s, 3H), 3.74 (s, 3H), 3.45 (s, 2H), 3.41 (s, 2H), 2.43 (s, 8h). 13C NMR (101 MHz, CDCl3): δ 169.2, 165.9, 158.6, 152.1, 140.5, 139.0, 130.3 (2C), 129.8, 129.5, 125.7, 117.7, 115.6, 113.4 (2C), 92.6, 62.5, 62.3, 55.1, 53.0 (2C), 52.7 (2C), 51.5, 51.3. HRMS (ESI-TOF) calcd for C25H32N3O5 [M + H]+ 454.2342, found 454.2356.
Dimethyl 2-(((3-((4-(Benzo[d][1,3]dioxol-5-ylmethyl)piperazin-1-yl)methyl)phenyl)amino)methylene)malonate 7m.
A DRA reaction between aldehyde 13a (500 mg, 1.9 mmol) and 1-piperonylpiperazine (500 mg, 2.3 mmol) following general procedure E afforded 7m as a yellow oil (600 mg, 67% yield). 1H NMR (400 MHz, CDCl3): S 11.01 (d, J = 13.7 Hz, 1H), 8.53 (d, J = 13.9 Hz, 1H), 7.32–7.23 (m, 1H), 7.12 (t, J = 1.9 Hz, 1H), 7.07 (dt, J = 7.7, 1.1 Hz, 1H), 7.01 (ddd, J = 8.0, 2.5, 0.9 Hz, 1H), 6.82 (d, J = 1.1 Hz, 1H), 6.75–6.68 (m, 2H), 5.90 (d, J = 0.6 Hz, 2H), 3.83 (d, J = 0.6 Hz, 4H), 3.76 (d, J = 0.5 Hz, 3H), 3.48 (s, 2H), 3.41 (s, 2H), 2.46 (s, 8H). 13C NMR (101 MHz, CDCl3): δ 169.3, 166.0, 152.2, 147.5, 146.5, 140.5, 139.1, 131.7, 129.6, 125.8, 122.2, 117.8, 115.7, 109.5, 107.8, 100.8, 92.7, 62.6, 62.6, 53.0 (2C), 52.8 (2C), 51.5, 51.4. HRMS (ESI-TOF) calcd for C25H30N3O6 [M + H]+ 468.2135, found 468.2142.
Dimethyl 2-(((3-((4-(4-Methoxyphenyl)piperazin-1 -yl)methyl)-phenyl)amino)methylene)malonate 7n.
A DRA reaction between aldehyde 13a (500 mg, 1.9 mmol) and 1-(4-methoxyphenyl)-piperazine hydrochloride (604 mg, 2.3 mmol) following general procedure E afforded 7n as a pale-yellow semisolid (565 mg, 68% yield). 1H NMR (400 MHz, CDCl3): δ 11.03 (d, J = 13.8 Hz, 1H), 8.54 (d, J = 13.8 Hz, 1H), 7.30 (t, J = 7.8 Hz, 1H), 7.16 (t, J = 1.9 Hz, 1H), 7.11 (dt, J =7.6, 1.2 Hz, 1h), 7.05–7.01 (m, 1H), 6.89–6.84 (m, 2h), 6.82–6.78 (m, 2H), 3.83 (s, 3H), 3.76 (s, 3H), 3.73 (s, 3H), 3.53 (s, 2H), 3.10–3.05 (m, 4H), 2.59 (dd, J = 6.0, 3.8 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 169.3, 166.0, 153.7, 152.2, 145.6, 140.4, 139.1, 129.6, 125.8, 118.2 (2C), 117.9, 115.8, 114.3 (2C), 92.7, 62.6, 55.5,53.2 (2C), 51.5, 51.4, 50.5 (2C). HRMS (ESI-TOF) calcd for C24H30N3O5 [M + H]+ 440.2185, found 440.2196.
Dimethyl 2-(((3-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-phenyl)amino)methylene)malonate 7o.
A direct reductive amination (DRA) reaction between aldehyde 13a (500 mg, 1.9 mmol) and 1-(4-florophenyl)piperazine (410 mg, 2.3 mmol) following general procedure E afforded 7o as a pale-yellow semisolid (580 mg, 72%). 1H NMR (400 MHz, CDCl3): δ 11.04 (d, J = 13.6 Hz, 1H), 8.54 (dd, J = 13.9, 3.6 Hz, 1H), 7.30 (td, J = 7.8, 3.5 Hz, 1H), 7.16 (s, 1H), 7.12 (d, J = 7.5 Hz, 1H), 7.06–7.01 (m, 1H), 6.92 (td, J = 8.6, 3.5 Hz, 2H), 6.84 (dt, J = 9.0, 4.3 Hz, 2H), 3.83 (d, J = 3.2 Hz, 3H), 3.76 (d, J = 3.2 Hz, 3H), 3.53 (bs, 2H), 3.11–3.08 (m, Hz, 4H), 2.60–2.58 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 169.2, 165.9, 158.2, 155.8, 152.1, 147.8 140.3, 139.1, 129.6, 125.6, 117.7, 117.6, 115.7, 115.4, 115.2, 92.7, 62.4, 53.0 (2C), 51.4, 51.3, 50.0 (2C). HRMS (ESI-TOF) calcd for C24H30N3O5 [M + H]+ 428.1986, found 428.1990.
Dimethyl 2-(((3-((4-(4-(Trifluoromethyl)phenyl)piperazin-1-yl)-methyl)phenyl)amino)methylene)malonate 7p.
A direct reductive amination (DRA) reaction between aldehyde 13a (500 mg, 1.9 mmol) and 1-(4-trifloromethylphenyl)piperazine (840 mg, 2.3 mmol) following general procedure E afforded 7p as a pale-yellow semisolid (980 mg, 68% yield). 1H NMR (400 MHz, CDCl3): δ 11.03 (d, J =13.8 Hz, 1H), 8.53 (dd, J = 13.8, 1.0 Hz, 1H), 7.41 (d, J =8.7 Hz, 2H), 7.28 (t, J = 7.8 Hz, 1H), 7.14 (t, J =1.8 Hz, 1H), 7.09 (dd, J = 7.7, 1.3 Hz, 1H), 7.04–7.00 (m, 1H), 6.85 (d, J = 8.6 Hz, 2h), 3.81 (s, 3H), 3.74 (s, 3H), 3.50 (s, 2H), 3.23 (t, J = 5.1 Hz, 4H), 2.54 (dd, J = 6.1, 3.9 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 169.1, 165.8, 153.1, 152.0 140.1, 139.1, 129.6, 126.2, 126.2, 126.1, 126.1, 125.6, 117.6, 115.7, 114.3 (2C), 92.7, 62.4, 52.6 (2C), 51.4, 51.3, 47.7 (2C). HRMS (ESI-TOF) calcd for C24H27F3N3O4 [M + H]+ 478.1954, found 478.1970.
Dimethyl 2-(((4-Methyl-3-((4-phenylpiperazin-1-yl)methyl)-phenyl)amino)methylene)malonate 7q.
A DRA reaction between aldehyde 13b (600 mg, 2.2 mmol) and 1-phenylpiperazine (430 mg, 2.6 mmol) following general procedure E afforded 7q as a white semisolid (700 mg, 77%). 1H NMR (400 MHz, CDCl3): δ 11.02 (1H), 8.52 (d, J = 13.9 Hz, 1H), 7.23–7.20 (m, 2H), 7.15–7.12 (m, 2H), 6.95 (d, J = 8.2 Hz, 2H), 6.91–6.88 (dd, J = 8.8, 1.1 Hz, 2H), 6.84–6.80 (m, 1H), 3.83 (s, 3H), 3.75 (s, 3H), 3.47 (s, 2H), 3.19–3.13 (m, 4H), 2.62–2.55 (m, 4h), 2.32 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 165.9, 152.2, 151.1, 138.0, 136.7, 134.1, 131.3, 128.9 (2C), 119.4, 118.5, 115.8 (2C), 115.3, 92.0, 60.2, 53.0 (2C), 51.3, 51.2, 49.0 (2C), 18.6. HRMS (ESI-TOF) calcd for C24H30N3O4 [M + H]+ 424.2236, found 424.2257.
Dimethyl 2-(((3-((4-Benzylpiperazin-1-yl)methyl)-4-methylphenyl)amino)methylene)malonate 7r.
A DRA reaction between aldehyde 13b (650 mg, 2.3 mmol) and 1-benzylpiperazine (500 mg, 2.8 mmol) following general procedure E afforded 7r as a pale-yellow semisolid (630 mg, 63%). 1H NMR (400 MHz, CDO3): δ 10.98 (d, J = 13.8 Hz, 1H), 8.50 (dd, J = 13.9, 0.9 Hz, 1H), 7.32–7.20 (m, 5H), 7.13–7.07 (m, 2H), 6.92 (dd, J = 8.1, 2.5 Hz, 1H), 3.83 (s, 3H), 3.76 (s, 3H), 3.50 (s, 2H), 3.41 (s, 2H), 2.47 (m, 8H), 2.28 (s, 3h). 13C NMR (101 MHz, CDCl3): δ 169.3, 166.0, 152.3, 138.4, 137.9, 136.7, 134.1, 131.3, 129.1 (2C), 128.1 (2C), 126.9, 118.5, 115.2, 92.0, 62.9, 60.1, 53.1 (2C), 53.0 (2C), 51.4, 51.3, 18.5. HRMS (ESI-TOF) calcd for C25H32N3O4 [M + H]+ 438.2393, found 438.2394.
Dimethyl 2-(((3-((4-(4-Methoxybenzyl)piperazin-1-yl)methyl)-4-methylphenyl)amino)methylene)malonate 7s.
A DRA reaction between aldehyde 13b (200 mg, 1.4 mmol) and 1-(4-methoxybenzyl)-piperazine (350 mg, 1.7 mmol) following general procedure E afforded 7s as a pale-yellow semisolid (430 mg, 70%). 1H NMR (400 MHz, CDCl3): δ 10.95 (d, J = 13.9 Hz, 1h), 8.46 (d, J = 13.9 Hz, 1H), 7.19–7.12 (m, 2H), 7.07–7.01 (m, 2H), 6.87 (dd, J = 8.1, 2.5 Hz, 1H), 6.79–6.75 (m, 2H), 3.78 (s, 3H), 3.71 (s, 3H), 3.70 (s, 3H), 3.38 (s, 2H), 3.36 (s, 2H), 2.40 (s, 8H), 2.23 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 169.1, 165.8, 158.4, 152.1, 138.3, 136.6, 133.9, 131.1, 130.0 (2C), 129.8, 118.3, 115.0, 113.3 (2C), 91.9, 62.1, 59.9, 54.9, 53.0 (2C), 52.7 (2C), 51.2, 51.1, 18.4.
Dimethyl 2-(((3-((4-(4-Methoxyphenyl)piperazin-1-yl)methyl)-4-methylphenyl)amino)methylene)malonate 7t.
A DRA reaction between aldehyde 13b (600 mg, 2.16 mmol) and 1-(4-methoxyphenyl)piperazine hydrochloride (690 mg, 2.6 mmol) following general procedure E afforded 7t as a pale-yellow semisolid (685 mg, 70%). 1H NMR (400 MHz, CDCl3): δ 11.00 (d, J = 13.9 Hz, 1H), 8.52 (d, J = 13.9 Hz, 1H), 7.15–7.11 (m, 2H), 6.95 (dd, J = 8.2, 2.5 Hz, 1H), 6.90–6.85 (m, 2H), 6.83–6.78 (m, 2H), 3.83 (s, 3H), 3.75 (s, 3H), 3.73 (s, 3H), 3.48 (s, 2H), 3.09–3.05 (m, 4H), 2.60 (dd, J = 6.1, 3.7 Hz, 4h), 2.32 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 169.4, 166.1, 153.7, 152.4, 145.7, 138.3, 136.8, 134.3, 131.5, 118.7, (2C), 115.4, 114.4 (2C), 92.2, 60.3, 55.5, 53.3 (2C), 51.5, 51.4, 50.7 (2C), 18.7. HRMS (ESI-TOF) calcd for C25H32N3O5 [M + H]+ 454.2342, found 454.2351.
Dimethyl 2-(((3-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-4-methylphenyl)amino)methylene)malonate 7u.
A DRA reaction between aldehyde 13b (600 mg, 2.16 mmol) and 1-(4-florophenyl)-piperazine (470 mg, 2.6 mmol) following general procedure E afforded 7u as a yellow semisolid (800 mg, 84% yield). 1H NMR (400 MHz, CDCl3): δ 11.00 (d, J = 13.9 Hz, 1H), 8.49–8.42 (m, 1H), 7.46–7.39 (m, 2H), 7.22 (d, J = 2.9 Hz, 1H), 7.00 (dd, J = 8.8, 2.9 Hz, 1H), 6.89–6.82 (m, 3H), 3.81 (s, 3H), 3.79 (s, 3H), 3.73 (s, 3H), 3.57 (s, 2H), 3.27 (t, J = 5.0 Hz, 4H), 2.61 (t, J = 5.0 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 169.4, 166.1, 158.3, 155.9, 152.4, 148.0, 138.1, 136.9, 134.3, 131.5, 118.7, 117.8, 117.7, 115.6, 115.5, 115.3, 92.2, 60.3, 53.2 (2C), 51.5, 51.4, 50.2 (2C), 18.7. HRMS (ESI-TOF) calcd for C24H29FN3O4 [M + H]+ 442.2142, found 442.2157.
Dimethyl 2-(((4-Methyl-3-((4-(4-(trifluoromethyl)phenyl)-piperazin-1-yl)methyl)phenyl)amino)methylene)malonate 7v.
A DRA reaction between aldehyde 13b (1.0 g, 3.6 mmol) and 1-(4-trifloromethylphenyl)piperazine (1.0 g, 4.4 mmol) following general procedure E afforded 7v as a pale-yellow semisolid (1.17 g, 66% yield). 1H NMR (400 MHz, CDCl3): δ 10.98 (d, J = 13.8 Hz, 1H), 8.47 (d, J = 13.9 Hz, 1H), 7.37 (d, J = 8.6 Hz, 2H), 7.09–7.05 (m, 2H), 6.89 (dd, J = 8.2, 2.5 Hz, 1H), 6.80 (d, J = 8.6 Hz, 2H), 3.76 (s, 3H), 3.70 (s, 3H), 3.40 (s, 2H), 3.17 (dd, J = 6.2, 3.7 Hz, 4H), 2.51 (t, J = 5.0 Hz, 4H), 2.26 (s, 3h). 13C NMR (101 MHz, CDCl3): δ 169.0, 165.7, 153.0, 151.9, 137.7, 136.6, 134.0, 131.2, 126.0, 126.0, 125.9, 125.9, 118.3, 115.2, 114.0 (2C), 92.0, 59.9, 52.5 (2C), 51.1, 51.0, 47.6 (2C), 18.3. HRMS (ESI-TOF) calcd for C25H29F3N3O4 [M + H] + 492.2110, found 492.2115.
Dimethyl 2-(((4-Methoxy-3-((4-phenylpiperazin-1-yl)methyl)-phenyl)amino)methylene)malonate 7w.
A DRA reaction between aldehyde 13c (300 mg, 1.0 mmol) and 1-phenylpiperazine (200 mg, 1.22 mmol) following general procedure E afforded 7w as a pale-yellow semisolid (305 mg, 68% yield). 1H NMR (400 MHz, CDCl3): δ 11. 00 (d, J = 13.9 Hz, 1H), 8.46 (d, J = 14.0 Hz, 1H), 7.27–7.18 (m, 3H), 7.01 (dd, J = 8.7, 2.9 Hz, 1H), 6.94–6.88 (m, 2H), 6.85–6.78 (m, 2H), 3.82 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.58 (s, 2H), 3.22–3.19 (m, 4H), 2.66–2.62 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 169.5, 166.1, 155.4, 152.9, 151.3, 132.4, 129.0 (2C), 128.0, 119.8, 119.5, 116.7, 116.0 (2C), 111.4, 91.65, 55.8, 55.6, 53.1 (2C), 51.4, 51.3, 49.1 (2C). HRMS (ESI-TOF) calcd for C24H30N3O5 [M + H]+ 440.2185, found 440.2199.
Dimethyl 2-(((3-((4-Benzylpiperazin-1-yl)methyl)-4-methoxyphenyl)amino)methylene)malonate 7x.
A DRA reaction between aldehyde 13c (500 mg, 1.7 mmol) and 1-benzylpiperazine (360 mg, 2.0 mmol) following general procedure E afforded 7x as a pale-yellow semisolid (500 mg, 65% yield). 1H NMR (400 MHz, CDCl3): δ 10.97 (d, J = 14.0 Hz, 1h), 8.44 (d, J = 13.9 Hz, 1H), 7.30–7.26 (m, J = 2.6 Hz, 4H), 7.23–7.18 (m, 2H), 6.97 (dd, J = 8.7, 2.9 Hz, 1H), 6.80 (d, J = 8.7 Hz, 1H), 3.83 (s, 3H), 3.77 (s, 3H), 3.75 (s, 3H), 3.52 (s, 2H), 3.49 (s, 2H), 2.50 (m, 8H). 13C NMR (101 MHz, CDCl3): δ 169.5, 166.1, 155.4, 152.9, 138.0, 132.3, 129.2 (2C), 128.2, 128.1 (2C), 126.9, 119.8, 116.6, 111.3, 91.6, 63.0, 55.7, 55.5, 53.1 (2C), 53.0 (2C), 51.4, 51.3. HRMS (ESI-TOF) calcd for C25H32N3O5 [M + H]+ 454.2342, found 454.2365.
Dimethyl 2-(((4-Methoxy-3-((4-(4-methoxyphenyl)piperazin-1-yl)methyl)phenyl)amino)methylene)malonate 7y.
A DRA reaction between aldehyde 13c (350 mg, 1.2 mmol) and 1-(4-methoxybenzyl)-piperazine (295 mg, 1.4 mmol) following general procedure E afforded 7y as a pale-yellow semisolid (350 mg, 61%). 1H NMR (400 MHz, CDCl3) δ 11.02–10.91 (m, 1H), 8.44 (d, J = 14.0 Hz, 1H), 7.25–7.17 (m, 3H), 6.98 (dd, J = 8.7, 2.9 Hz, 1H), 6.85–6.77 (m, 3H), 3.83 (s, 3H), 3.77 (s, 3H), 3.76 (s, 3H), 3.75 (s, 3H), 3.54 (s, 2H), 3.46 (s, 2h), 2.52 (s, 8h). 13C NMR (101 MHz, CDCl3) δ 169.5, 166.2, 158.8, 155.4, 153.0 (2C), 132.4 (2C), 130.5 (2C), 119.9, 116.8, 113.6 (2C), 111.4, 91.6, 62.3, 55.7, 55.4, 55.2, 55.2, 52.9, 52.8, 51.5, 51.4, 51.4. HRMS (ESI-TOF) calcd for C26H34N3O6 [M + H]+ 484.2448, found 484.2357.
Dimethyl 2-(((4-Methoxy-3-((4-(4-methoxyphenyl)piperazin-1-yl)methyl)phenyl)amino)methylene)malonate 7z.
A DRA reaction between aldehyde 13c (200 mg, 0.7 mmol) and 1-(4-methoxyphenyl)-piperazine hydrochloride (220 mg, 0.82 mmol) following general procedure E afforded 7z as a pale-yellow semisolid (250 mg, 78%). 1H NMR (400 MHz, CDCl3): S 11.00 (d, J = 13.9 Hz, 1H), 8.47 (d, J = 13.9 Hz, 1H), 7.28–7.24 (m, 1H), 7.02 (dd, J = 8.7, 2.9 Hz, 1H), 6.92–6.77 (m, 5H), 3.83 (s, 4H), 3.82 (s, 3H), 3.75 (s, 3H), 3.74 (s, 3H), 3.61 (s, 2H), 3.14–3.08 (m, 4H), 2.67 (t, J = 5.0 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 169.5, 166.2, 155.5, 153.8, 153.0 (2C), 145.7, 132.4, 119.9, 118.2 (2C), 116.8, 114.4 (2C), 111.5, 91.7, 55.8, 55.6, 55.5, 53.2, 51.5, 51.4, 50.6. HRMS (ESI-TOF) calcd for C25H32N3O6 [M + H]+ 470.2291, found 470.2309.
Dimethyl 2-(((3-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-4-methoxyphenyl)amino)methylene)malonate 7aa.
A DRA reaction between aldehyde 13c (500 mg, 1.7 mmol) and 1-(4-florophenyl)-piperazine (220 mg, 2.0 mmol) following general procedure E afforded 7aa as a pale-yellow semisolid (550 mg, 71%). 1H NMR (400 MHz, CDCl3): δ 11.03–10.94 (m, 1H), 8.44 (dd, J = 13.9, 12.3 Hz, 1H), 7.25–7.14 (m, 1H), 7.03–6.97 (m, 1H), 6.96–6.89 (m, 2H), 6.87–6.82 (m, 3H), 3.82 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.59 (s, 2H), 3.15–3.10 (m, 4H), 2.68–2.61 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 169.5, 166.2, 158.3, 155.9, 155.4, 154.6, 152.9, 152.8, 148.0, 132.5, 132.4, 131.0, 127.8, 119.9, 117.8, 117.7, 117.6, 117.5, 116.8, 115.5, 115.3, 111.5, 111.0, 91.7, 60.9, 55.8, 55.6, 55.5, 53.1, 51.5, 51.4, 51.3, 50.1. HRMS (ESI-TOF) calcd for C24H28FN3O5 [M + H]+ 458.2091, found 458.2104.
Dimethyl 2-(((4-Methoxy-3-((4-(4-(trifluoromethyl)phenyl)-piperazin-1-yl)methyl)phenyl)amino)methylene)malonate 7ab.
A DRA reaction between aldehyde 13c (1.0 g, 3.4 mmol) and 1-(4-trifloromethylphenyl)piperazine (950 mg, 4.1 mmol) following general procedure E afforded 7ab as a pale-yellow semisolid (1.2 g, 70% yield). 1H NMR (400 MHz, CDCl3): δ 11.00 (d, J = 13.9 Hz, 1H), 8.48–8.42 (m, 1H), 7.46–7.40 (m, 2H), 7.22 (d, J = 2.9 Hz, 1H), 7.00 (dd, J = 8.8, 2.9 Hz, 1H), 6.89–6.81 (s, 3H), 3.81 (m, 3h), 3.79 (s, 3H), 3.73 (S, 3H), 3.57 (s, 2H), 3.27 (t, J = 5.0 Hz, 4H), 2.61 (t, J = 5.0 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 169.5, 166.2, 155.4, 153.3, 152.9, 132.4, 127.9, 126.3, 126.3, 126.3, 126.2, 119.8, 116.8, 114.4 (2C), 111.5, 91.7, 55.8, 55.6, 52.8 (2C), 51.4, 51.4, 47.9 (2C). HRMS (ESI-TOF) calcd for C25H29F3N3O5 [m + H]+ 508.2059, found 508.2065.
Dimethyl 2-(((4-((4-Phenylpiperazin-1-yl)methyl)phenyl)amino)-methylene)malonate 7af.
A DRA reaction of aldehyde 13d (500 mg, 1.89 mmol) with 1-phenylpiperazine hydrochloride (370 mg, 2.3 mmol) following general procedure E furnished 7af as a pale-yellow semisolid (520 mg, 67% yield). 1H NMR (400 MHz, CDCl3): δ 11.04 (d, J = 13.9 Hz, 1H), 8.52 (d, J = 13.7 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.23 (t, J = 7.7 Hz, 2h), 7.09 (d, J = 8.0 Hz, 2H), 6.90 (d, J = 8.1 Hz, 2H), 6.82 (t, J = 7.3 Hz, 1H), 3.84 (s, 3H), 3.76 (s, 3h), 3.52 (s, 2H), 3.17 (t, J = 4.8 Hz, 4h), 2.58 (t, J = 4.8 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 169.3, 165.9, 152.2, 151.2, 138.1, 135.1, 130.5 (2C), 129.0 (2C), 119.6, 117.1 (2C), 116.0 (2C), 92.7, 62.2, 53.0 (2C), 51.5, 51.4, 49.1 (2C). HRMS (ESI-TOF) calcd for C24H30N3O5 [M + H]+ 440.2185, found 440.2173.
Dimethyl 2-(((4-((4-(4-Methoxyphenyl)piperazin-1-yl)methyl)-phenyl)amino)methylene)malonate 7ag.
A DRA reaction of aldehyde 13d (850 mg, 3.2 mmol) with 1-(4-methoxyphenyl)-piperazine hydrochloride (1.0 g, 3.86 mmol) following general procedure E furnished 7ag as a pale-yellow solid (1.0 g, 71% yield). 1H NMR (400 MHz, CDCl3): δ 11.01 (d, J = 13.8 Hz, 1H), 8.49 (dd, J = 13.7, 1.1 Hz, 1H), 7.31 (d, J = 8.0 Hz, 2H), 7.07 (d, J = 8.1 Hz, 2H), 6.84 (d, J = 9.1 Hz, 2H), 6.80–6.76 (m, 2H), 3.81 (s, 3H), 3.73 (s, 3H), 3.70 (s, 3H), 3.49 (s, 2H), 3.04 (t, J = 4.8 Hz, 4H), 2.55 (t, J = 4.8 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 169.2, 165.8, 153.6, 152.1, 45.5, 138.0, 135.0, 130.4 (2C), 118.0 (2C), 117.1, 117.0, 114.2 (2C), 92.5, 62.1, 55.3, 53.0 (2C), 51.4, 51.3, 50.4 (2C). HRMS (ESI-TOF) calcd for C24H30N3O5 [M + H] + 440.2185, found 440.2173.
Dimethyl 2-(((4-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-phenyl)amino)methylene)malonate 7ah.
A DRA reaction of aldehyde 13d (850 mg, 3.2 mmol) with 1-(4-florophenyl)piperazine (695 mg, 3.86 mmol) following general procedure E furnished 7ah as a pale-white solid (900 mg, 66% yield). 1H NMR (400 MHz, CDCl3): δ 11.01 (d, J = 13.7 Hz, 1H), 8.49 (d, J = 13.8 Hz, 1H), 7.33–7.29 (m, 2H), 7.07 (dd, J = 9.0, 2.5 Hz, 2H), 6.92–6.87 (m, 2H), 6.84–6.79 (m, 2H), 3.81 (s, 3H), 3.73 (s, 3H), 3.49 (s, 2H), 3.06 (t, J = 5.0 Hz, 4H), 2.54 (dd, J = 6.1, 3.9 Hz, 4h). 13C NMR (101 MHz, CDCl3): δ 169.2, 165.8, 158.1, 155.7, 152.1, 147.8, 147.8, 138.0, 135.0, 130.4, 117.6, 117.5, 117.0, 115.4, 115.2, 92.6, 62.1, 52.9 (2C), 51.4, 51.3, 50.0 (2C). HRMS (ESI-TOF) calcd for C23H27FN3O4 [M + H] + 428.1986, found 428.1993.
Dimethyl 2-(((4-((4-(4-(Trifluoromethyl)phenyl)piperazin-1-yl)-methyl)phenyl)amino)methylene)malonate 7ai.
A DRA reaction of aldehyde 13d (500 mg, 1.9 mmol) with 1-(4-trifloromethylphenyl)-piperazine (523 mg, 2.3 mmol) following general procedure E furnished 7ai as a pale-yellow solid (570 mg, 63% yield). 1H NMR (400 MHz, CDCl3): δ 11.03 (d, J = 13.7 Hz, 1H), 8.52 (dd, J = 13.7, 1.1 Hz, 1H), 7.43 (d, J = 1.2 Hz, 2H), 7.34 (d, J = 7.4 Hz, 2H), 7.12–7.08 (m, 2H), 6.90–6.86 (m, 2H), 3.83 (s, 3H), 3.76 (s, 3H), 3.51 (s, 2H), 3.27–3.23 (m, 4H), 2.56 (dd, J = 6.1, 4.1 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 169.4, 165.9, 153.3, 152.2, 138.2, 134.9, 130.5 (2C), 126.4, 126.3, 126.3, 126.3, 117.2 (2C), 114.4 (2C), 92.8, 62.2, 52.7 (2C), 51.6, 51.4, 47.9 (2C). HRMS (ESI-TOf) calcd for C24H27F3N3O4 [M + H]+ 478.1954, found 478.1966.
Methyl 4-Oxo-7-(4-phenylpiperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate 8a.
8a was synthesized from amine 5a following general procedures C and F in 12% yield over two steps. 1H NMR (500 MHz, DMSO-d6) δ 11.95 (s, 1H), 8.44 (s, 1H), 7.97 (d, J = 9.1 Hz, 1H), 7.25 (t, J = 7.8 Hz, 2H), 7.17 (dd, J = 9.1, 2.3 Hz, 1H), 7.01 (d, J = 8.1 Hz, 2h), 6.88 (d, J = 2.4 Hz, 1H), 6.82 (t, J = 7.4 Hz, 1h), 3.71 (s, 3H), 3.46 (t, J = 5.0 Hz, 4H), 3.30 (d, J = 5.2 Hz, 4H). HRMS (ESI-TOF) calcd for C21H22N3O3 [M + H]+ 364.1661, found 364.1653.
Methyl 7-(4-Benzylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8a.
8a was synthesized from amine 5b following general procedures C and F in 16% yield over two steps. 1H NMR (500 MHz, DMSO-d6) δ 11.92 (s, 1H), 8.41 (s, 1H), 7.93 (d, J = 9.1 Hz, 1H), 7.34 (d, J = 4.8 Hz, 4H), 7.30–7.24 (m, 1H), 7.09 (dd, J = 9.2, 2.4 Hz, 1H), 6.79 (d, J = 2.4 Hz, 1H), 3.70 (s, 3H), 3.53 (s, 2H), 3.30 (t, J = 5.1 Hz, 4H), 2.53 (d, J = 5.4 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ 172.8, 165.5, 153.4, 144.7, 140.7, 137.9, 128.9, 128.2, 127.0, 126.7, 119.1, 113.7, 109.0, 100.0, 62.0, 52.2, 51.0, 47.0. HRMS (ESI-TOf) calcd for C22H24N3O3 [M + h]+ 378.1818, found 378.1816.
Methyl 6-Methyl-4-oxo-7-(4-phenylpiperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate 8c.
8c was synthesized from commercially available 10a following general procedure H, followed by general procedures B, C, and F in 5% overall yield. 1H NMR (400 MHz, DMSO-d6) δ 12.12 (s, 1H), 8.46 (d, J = 5.9 Hz, 1H), 7.90 (s, 1H), 7.21 (d, J = 7.8 Hz, 2H), 7.11 (s, 1H), 6.98 (d, J = 8.2 Hz, 2H), 6.79 (t, J = 7.2 Hz, 1H), 3.69 (s, 3H), 3.07 (dd, J = 6.2, 3.7 Hz, 4H), 2.51 (t, J = 5.0 Hz, 4H), 2.35 (s, 3H). HRMS (ESI-TOF) calcd for C22H24N3O3 [M + h]+ 378.1818, found 378.1827.
Methyl 6-Methoxy-4-oxo-7-(4-phenylpiperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate 8d.
8d was synthesized from commercially available 10a following general procedure H, followed by general procedures B, C, and F in 4% overall yield. 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H), 8.47 (s, 1H), 7.53 (s, 1H), 7.26–7.22 (m, 2H), 7.06 (s, 1H), 7.00 (d, J = 8.0 Hz, 2H), 6.81 (t, J = 7.2 Hz, 1H), 3.90 (s, 3H), 3.72 (s, 3H), 3.31–3.25 (m, 8H). HRMS (ESI-TOf) calcd for C22H23N3NaO3 [M + Na]+ 416.1586, found 416.1584.
Methyl 7-(4-(4-Methoxyphenyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8e.
8e was synthesized from 10b following general procedure H, followed by general procedures B, C, and F in 6% overall yield. 1H NMR (500 MHz, DMSO-d6) 5 11.93 (s, 1H), 8.43 (d, J = 6.0 Hz, 1H), 7.97 (d, J = 9.0 Hz, 1H), 7.16 (dd, J = 9.1, 2.3 Hz, 1H), 6.96 (d, J = 8.8 Hz, 2H), 6.90–6.82 (m, 3H), 3.70 (d, J = 9.7 Hz, 6h), 3.44 (t, J =5.1 Hz, 4H), 3.17 (t, J =5.1 Hz, 4H). HRMS (ESI-TOF) calcd for C22H24N3O4 [M + H]+ 394.1767, found 394.1761.
Methyl 6-Methoxy-7-(4-(4-methoxyphenyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8f.
8f was synthesized from 10b following general procedure H, followed by general procedures B, C, and F in 4% overall yield. 1H NMR (400 MHz, DMSO-d6) δ 12.41 (s, 1H), 8.65 (s, 1H), 8.01 (s, 1H), 6.96 (d, J = 8.0 Hz, 2H), 7.11 (s, 1H), 7.08 (d, J = 8.2 Hz, 2H), 3.84 (s, 3H), 3.78 (s, 3H), 3.61 (s, 3H), 3.48–3.38 (m, 8H). HRMS (ESI-TOf) calcd for C23H26N3O5 [M + H]+ 424.1872, found 424.1877.
Methyl 7-(4-Benzylpiperazin-1-yl)-6-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate 8g.
8g was synthesized from commercially available 10c following general procedure H, followed by general procedures B, C, and F in 6% overall yield. 1H NMR (500 MHz, DMSO-d6) δ 11.85 (s, 1H), 8.37 (s, 1H), 7.88 (s, 1H), 7.7 (d, J = 4.2 Hz, 4H), 7.27–7.21 (m, 1H), 6.99 (s, 1H), 3.70 (s, 3H), 3.62 (s, 2H), 3.42 (t, J = 5.1 Hz, 4H), 3.08 (s, 3H) 2.66 (d, J = 5.4 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ 172.8, 165.5, 155.3, 144.5, 138.4, 138.0, 129.0, 128.2, 127.5, 127.0, 122.3, 109.1, 106.7, 103.2, 62.1, 52.8, 51.0, 50.9, 18.0. HRMS (ESI-TOF) calcd for C23H26N3O3 [M + H]+ 392.1974, found 392.1975.
Methyl 7-(4-Benzylpiperazin-1-yl)-6-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate 8h.
8h was synthesized from commercially available 10c following general procedure H, followed by general procedures B, C, and F in 8% overall yield. 1H NMR (500 MHz, DMSO-d6) δ 12.12 (s, 1H), 8.11 (s, 1H), 7.46 (s, 1H), 7.21 (d, J = 3.9 Hz, 4H), 7.19–7.11 (m, 1H), 6.89 (s, 1H), 3.58 (s, 3H), 3.51 (s, 2H), 3.37 (t, J = 5.1 Hz, 4H), 3.01 (s, 3H) 2.64 (d, J = 5.4 Hz, 4H). HRMS (ESI-TOF) calcd for C23H26N3O4 [M + H]+ 408.1923, found 408.1918.
Methyl 7-(4-(4-Methoxybenzyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8i.
8i was synthesized from 10d following general procedure H, followed by general procedures B, C, and F 10% overall yield. 1H NMR (400 MHz, DMSO-d6) δ 11.90 (s, 1H), 8.41 (s, 1H), 7.93 (d, J = 9.2 Hz, 1H), 7.26–7.22 (m, 2H), 7.08 (dd, J = 9.2, 2.3 Hz, 1H), 6.91–6.88 (m, 2H), 6.78 (d, J = 2.4 Hz, 1H), 3.72 (d, J = 14.9 Hz, 10H), 3.46 (s, 2H), 3.28 (d, J = 6.8 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ 172.8, 165.5, 158.4, 153.3, 144.7, 140.7, 130.2, 126.7, 119.1, 113.7, 113.6, 109.0, 99.9, 61.3, 55.0, 52.1, 51.0, 47.0. HRMS (ESI-TOF) calcd for C23H26N3O4 [M + H]+ 408.1923, found 408.1911.
Methyl 4-Oxo-7-((4-phenylpiperazin-1-yl)methyl)-1,4-dihydroquinoline-3-carboxylate 8j.
8j was synthesized from 7j following general procedure F as a pale-yellow solid in 32% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.58 (s, 1H), 8.57 (d, J = 6.4, 1H), 8.22 (d, J = 7.9, 1H), 7.69 (s, 1H), 7.51 (d, J = 8.0, 1H), 7.22 (t, J = 7.9, 2h), 6.94 (d, J = 8.1, 2h), 6.82 (t, J = 7.2, 1H), 3.78 (s, 2h), 3.72 (s, 3h), 3.53 (m, 4H), 3.21 (s, 4H). 13C NMR (101 MHz, DMSO-d6): 5 173.7, 165.9, 151.4, 145.7, 143.8, 139.8, 129.3 (2C), 126.8, 126.1, 125.8, 119.3, 118.7, 115.8 (2C), 109.8, 61.8, 53.1 (2C), 51.5, 48.7 (2C). HRMS (ESI-TOF) calcd for C22H24N3O3 [M + h]+ 378.1818, found 378.1815.
Methyl 7-((4-Benzylpiperazin-1-yl)methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8k.
8k was synthesized from 7k following general procedure F as a pale-yellow solid in 26% yield. 1H NMR (400 MHz, methanol-d4): δ 8.67 (s, 1H), 7.95 (d, J = 8.3, 1H), 7.61 (s, 1H), 7.47–7.34 (m, 6H), 4.41 (s, 2H), 4.30 (s, 2H), 3.83 (s, 3H), 3.61 (s, 4H), 3.49 (s, 4H). 13C NMR (101 MHz, methanol-d4): δ 175.3, 165.6, 145.8 (2C), 138.8, 137.6, 130.9 (2C), 129.8, 128.8 (2C), 127.1, 126.4, 120.6, 117.7, 108.5, 60.0, 59.7, 50.9, 50.0 (2C), 48.9 (2C). HRMS (ESI-TOF) calcd for C23H26N3O3 [M + h]+ 392.1974, found 392.1968.
Methyl 7-((4-(4-Methoxybenzyl)piperazin-1-yl)methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8l.
8l was synthesized from 7l following general procedure F as a light-brown solid in 21% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.27 (s, 1H), 8.52 (s, 1H), 8.08 (d, J = 8.0, 1H), 7.52 (s, 1H), 7.32 (d, J = 8.1, 1H), 7.17 (d, J = 7.0, 2H), 6.85 (d, J = 6.8, 2H), 3.74 (s, 2H), 3.57 (s, 3H), 3.36 (m, 4H), 2.38 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 173.8, 165.6, 159.5, 145.4 139.4, 132.0, 131.7, 126.8, 126.1, 125.8, 118.7, 114.8, 114.1, 109.8, 79.6, 79.3, 78.9, 61.2, 55.4 (2C), 51.7 (2C), 51.4 (2C). HRMS (ESI-TOF) calcd for C24H28N3 [M + H] + 422.2080, found 422.2071.
Methyl 7-((4-(Benzo[d][1,3]dioxol-5-ylmethyl)piperazin-1-yl)-methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8m.
8m was synthesized from 7m following general procedure F as a pale-yellow solid in 15% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.33 (bs, 1H), 8.51 (s, 1H), 8.10 (s, 1H), 7.52 (s, 1H), 7.35 (s, 1H), 6.98–6.89 (m, 3H), 6.00 (s, 2H), 3.92 (m, 4H), 3.70 (s, 3H), 2.82 (m, 8H). 13C NMR (126 MHz, DMSO-d6) δ 173.24, 165.39, 147.16, 146.10, 145.01, 143.62, 139.05, 131.94, 126.27, 125.63, 125.27, 121.90, 117.89, 109.45, 109.02 107.80, 100.74, 61.71, 61.39, 52.72 (2C), 52.42 (2C), 51.09. HRMS (ESI-TOF) calcd for C24H26N3O5 [M + H] + 436.1872, found 436.1853.
Methyl 7-((4-(4-Methoxyphenyl)piperazin-1 -yl)methyl)-4-oxo-1,4--dihydroquinoline-3-carboxylate 8n.
8n was synthesized from 7n following general procedure F as a pale-yellow solid in 20% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.31 (s, 1H), 8.55 (s, 1H), 8.12 (d, J = 8.2 Hz, 1H), 7.60 (s, 1H), 7.38 (d, J = 8.3 Hz, 1H), 6.88 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 9.1 Hz, 2H), 3.74 (s, 3H), 3.68 (s, 3H), 3.65 (s, 2h), 3.03 (t, J = 4.6 Hz, 4h), 2.58–2.53 (m, 4H). 13C NMR (101 MHz, DMSO-d6): 5 173.7, 165.9, 153.3, 145.8, 145.5, 143.9, 139.5, 126.8, 126.1, 125.8, 118.5, 117.8 (2C), 114.7 (2C), 109.9, 61.9, 55.6, 53.2 (2C), 51.6, 50.1 (2C). HRMS (ESI-TOF) calcd for C23H26N3O4 [M + H]+ 408.1923, found 408.1902.
Methyl 7-((4-(4-Fluorophenyl)piperazin-1 -yl)methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8o.
8o was synthesized from 7o following general procedure F as a pale-yellow solid in 20% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.29 (s, 1H), 8.53 (s, 1H), 8.10 (d, J = 8.3 Hz, 1H), 7.57 (d, J = 1.4 Hz, 1H), 7.35 (dd, J = 8.2, 1.5 Hz, 1H), 7.04–6.98 (m, 2H), 6.93–6.89 (m, 2H), 3.72 (s, 3H), 3.63 (s, 2h), 3.07 (t, J = 4.9 Hz, 4H), 2.53 (t, J = 4.9 Hz, 4H). 13C NMR (101 MHz, DMSO-d6): δ 173.7, 165.8, 157.6, 155.3, 148.3, 145.5, 143.8, 139.5, 126.8, 126.1, 125.8, 118.5, 117.6, 117.5, 115.8, 115.6, 109.9, 61.8, 53.1 (2C), 51.5, 49.5 (2C). HRMS (ESI-TOF) calcd for C22H22FN3NaO3 [M + Na]+ 418.1543, found 418.1539.
Methyl 4-Oxo-7-((4-(4-(trifluoromethyl)phenyl)piperazin-1-yl)-methyl)-1,4-dihydroquinoline-3-carboxylate 8p.
8p was synthesized from 7p following general procedure F as a pale-yellow solid in 35% yield. 1H NMR (400 MHz, DMSO-d6): 5 12.30 (s, 1H), 8.54 (s, 1H), 8.12 (d, J = 8.2 Hz, 1H), 7.58 (s, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.37 (dd, J = 8.2, 1.5 Hz, 1H), 7.04 (d, J = 8.6 Hz, 2H), 3.73 (s, 3H), 3.65 (s, 2H), 3.32–3.27 (m, 4H), 2.54 (t, J = 5.0 Hz, 4H). HRMS (ESI-TOF) calcd for C23H23F3N3O3 [M + H]+ 446.1692, found 446.1670.
Methyl 6-Methyl-4-oxo-7-((4-phenylpiperazin-1-yl)methyl)-1,4-dihydroquinoline-3-carboxylate 8q.
8q was synthesized from 7q following general procedure F as a pale-yellow solid in 30% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.22 (s, 1H), 8.48 (s, 1H), 7.89 (s, 1H), 7.58 (s, 1H), 7.16 (t, J = 7.8 Hz, 2H), 6.89 (d, J = 8.1 Hz, 2H), 6.73 (t, J = 7.3 Hz, 1H), 3.69 (s, 3H), 3.56 (s, 2H), 3.11 (m, 4h), 2.55 (t, J = 5.0 Hz, 4H), 2.37 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 173.6, 165.9, 151.4, 145.0, 137.6, 134.3, 129.4 (2C), 126.5 (2C), 119.3, 118.8 (2C), 115.8 (2C), 109.6, 59.7, 53.3 (2C), 51.5, 48.8 (2C), 19.2. HRMS (ESI-TOF) calcd for C23H26N3O3 [M + H] + 392.1974, found 392.1981.
Methyl 7-((4-Benzylpiperazin-1-yl)methyl)-6-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate 8r.
8r was synthesized from 7r following general procedure F as a pale-yellow solid in 23% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.20 (s, 1H), 8.47 (s, 1H), 7.89 (d, J = 1.0 Hz, 1H), 7.52 (s, 1H), 7.33–7.17 (m, 5H), 3.72 (s, 3H), 3.51, (s, 2H), 3.46 (s, 2H), 2.45–2.40 (m, 8H), 2.37 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 173.07, 165.53, 144.62, 142.18, 138.07, 137.18, 133.88, 128.86 (2C), 128.16 (2c), 126.92, 126.01, 125.94, 118.24, 109.14, 62.07, 59.35, 52.91 (2C), 52.67 (2C), 51.05, 18.68. HRMS (ESI-TOF) calcd for C24H28N3O3 [M + H]+ 406.2131, found 406.2122.
Methyl 7-((4-(4-Methoxybenzyl)piperazin-1-yl)methyl)-6-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate 8s.
8s was synthesized from 7s following general procedure F as a pale-yellow solid in 20% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.27 (s, 1H), 8.51 (s, 1H), 7.91 (s, 1H), 7.56 (s, 1H), 7.20 (d, J = 7.8 Hz, 2H), 6.88 (d, J = 8.0 Hz, 2H), 3.74 (s, 6H), 3.52 (s, 2h), 3.41 (s, 2H), 2.45 (bs, 8H), 2.38 (s, 3H). 13C NMR (101 MHz, DMSO-d6) 5 173.6, 165.9, 158.7, 145.0 (2C), 142.6, 137.6, 134.2, 130.6 (2C), 126.5, 126.5, 118.7, 114.0 (2C), 109.6, 61.9, 59.8, 55.4 (2C), 53.3, 53.0 (2C), 51.5, 19.1. HRMS (ESI-TOF) calcd for C25H29N3NaO4 [M + Na]+ 458.2056, found 458.2056.
Methyl 7-((4-(4-Methoxyphenyl)piperazin-1-yl)methyl)-6-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate 8t.
8t was synthesized from 7t following general procedure F as a pale-yellow solid in 22% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.26 (s, 1H), 8.55–8.45 (m, 1H), 7.93 (d, J = 4.8 Hz, 1H), 7.59 (d, J = 4.8 Hz, 1H), 6.87–6.84 (m, 2H), 6.81–6.78 (m, 2h), 3.73 (s, 3h), 3.67 (s, 3H), 3.66 (s, 2H), 3.04 (bs, 4H), 2.66 (bs, 4H), 2.41 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 173.2, 165.4, 153.0, 144.6 (2C), 137.1 (2C), 133.9, 126.2, 117.4 (2C), 114.2 (2C), 114.2 (2c), 109.1, 79.2, 78.9, 78.5, 55.1, 52.8 (2C), 51.0, 49.6 (2C), 18.8. HRMS (ESI-TOF) calcd for C24H28N3O4 [M + H]+ 422.2080, found 422.2076.
Methyl 7-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-6-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate 8u.
8u was synthesized from 7u following general procedure F as a pale-yellow solid in 30% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.23 (s, 1H), 8.50 (s, 1H), 7.92 (s, 1H), 7.59 (s, 1H), 7.03 (t, J = 8.7 Hz, 2H), 6.93 (dd, J = 9.2, 4.6 Hz, 2h), 3.72 (s, 3H), 3.59 (s, 2H), 3.10 (t, J = 4.8 Hz, 4H), 2.58 (t, J = 4.7 Hz, 4H), 2.41 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.12, 165.48, 157.15, 154.81, 147.91, 144.57, 141.99, 137.19, 133.87, 126.08, 118.38, 117.10, 117.02, 115.36, 115.14, 109.19, 59.30, 52.81 (2C), 51.05, 49.13 (2C), 18.71. HRMS (ESI-TOF) calcd for C23H25FN3O3 [M + H]+ 410.1880, found 410.1865.
Methyl 6-Methyl-4-oxo-7-((4-(4-(trifluoromethyl)phenyl)-piperazin-1-yl)methyl)-1,4-dihydroquinoline-3-carboxylate 8v.
8v was synthesized from 7v following general procedure F as a pale-yellow solid in 35% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.25 (s, 1H), 8.51 (s, 1H), 7.92 (s, 1H), 7.60 (s, 1H), 7.49 (d, J =8.5 Hz, 2H), 7.05 (d, J = 8.6 Hz, 2H), 3.72 (s, 3H), 3.59 (s, 2H), 3.29 (m, 4h), 2.62–2.54 (m, 4H), 2.40 (s, 3H). HRMS (ESI-TOF) calcd for C24H25F3N3O3 [M + H]+ 460.1848, found 460.1854.
Methyl 6-Methoxy-4-oxo-7-((4-phenylpiperazin-1-yl)methyl)-1,4-dihydroquinoline-3-carboxylate 8w.
8w was synthesized from 7w following general procedure F as a pale-yellow solid in 16% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.29 (s, 1H), 8.52–8.42 (m, 1H), 7.72 (d, J = 14.8 Hz, 1H), 7.59–7.51 (m, 1H), 7.25–7.12 (m, 2h), 6.93 (d, J = 8.3 Hz, 2H), 6.76 (t, J = 7.2 Hz, 1H), 3.87 (s, 3h), 3.70 (m, 3H), 3.61 (d, J =13.9 Hz, 2H), 3.21–3.12 (m, 4H), 2.61 (t, J = 5.0 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ 172.79, 165.56, 154.83, 151.04, 143.70, 133.30, 133.24, 128.95 (2C), 127.12, 119.10, 118.85, 115.34 (2C), 108.33, 104.08, 55.65, 55.22, 52.93 (2C), 51.06, 48.36 (2C). HRMS (ESI-TOF) calcd for C23H26N3O4 [M + H]+ 408.1923, found 408.1903.
Methyl 7-((4-Benzylpiperazin-1-yl)methyl)-6-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate 8x.
8x was synthesized from 7x following general procedure F as a pale-yellow solid in 15% yield. 1H NMR (500 MHz, DMSO-d6): 12.30 (bs, 1H), δ 8.48 (s, 1H), 7.67 (s, 1H), 7.53 (s, 1H), 7.33–7.23 (m, 5H), 3.85 (s, 3H), 3.72 (s, 3H), 3.55 (s, 2H), 3.47 (s, 2H), 2.44 (m, 8h). HRMS (ESI-TOf) calcd for C24H28N3O4 [M + h]+ 422.2080, found 422.2093.
Methyl 6-Methoxy-7-((4-(4-methoxybenzyl)piperazin-1-yl)-methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8y.
8y was synthesized from 7y following general procedure F as a pale-yellow solid in 15% yield. 1H NMR (500 MHz, DMSO-d6) δ 12.30 (s, 1H), 8.49 (s, 1H), 7.68 (s, 1H), 7.54 (s, 1H), 7.20 (d, J = 8.1 Hz, 2H), 6.88 (d, J = 8.1 Hz, 2H), 3.87 (s, 3H), 3.74 (s, 3H), 3.72 (s, 3H), 3.56 (s, 2H), 3.42 (s, 2h), 2.44 (bs, 8H). 13C NMR (126 MHz, DMSO-d6) δ 172.6, 165.5 158.3, 154.6, 143.5, 133.3, 133.2, 130.1 (2C), 129.9, 126.9, 118.7, 113.5 (2C), 108.3, 104.0, 61.5, 55.6, 55.2, 55.0, 53.0 (2C), 52.5 (2C), 51.0. HRMS (ESI-TOF) calcd for C25H30N3O5 [M + H]+ 452.2185, found 452.2175.
Methyl 6-Methoxy-7-((4-(4-methoxyphenyl)piperazin-1-yl)-methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8z.
8z was synthesized from 7z following general procedure F as a pale-yellow solid in 20% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.29 (s, 1H), 8.48 (s, 1H), 7.72 (s, 1H), 7.55 (s, 1H), 6.91–6.85 (m, 2H), 6.80 (d, J = 9.0 Hz, 2H), 3.87 (s, 3H), 3.71 (s, 3H), 3.66 (s, 3H), 3.62 (s, 2H), 3.05 (t, J = 4.7 Hz, 4H), 2.60 (t, J = 4.8 Hz, 4H). 13C NMR (101 MHz, DMSO-d6): δ 173.2, 165.9, 155.2, 153.3, 145.8, 144.1, 133.7 (2C), 127.5, 119.5, 117.7 (2C), 114.7 (2C), 108.8, 104.5, 56.1, 55.6, 53.4 (2C), 51.5, 50.2 (2C). HRMS (ESI-TOF) calcd for C24H28N3O5 [M + H]+ 438.2029, found 438.2009.
Methyl 7-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-6-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate 8aa.
8aa was synthesized from 7aa following general procedure F as a pale-yellow solid in 28% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.29 (s, 1H), 8.49 (s, 1H), 7.73 (s, 1H), 7.56 (s, 1H), 7.03 (t, J = 8.7 Hz, 2H), 6.94 (dd, J = 9.2, 4.7 Hz, 2h), 3.88 (s, 3h), 3.72 (s, 3H), 3.63 (s, 2H), 3.12 (t, J = 4.6 Hz, 4H), 2.61 (t, J = 4.8 Hz, 4H). 13C NMR (101 MHz, DMSO-d6): δ 173.2 166.0, 157.6, 155.2, 148.4, 144.1, 133.7, 133.6, 127.6, 119.5, 1117.5, 115.8, 115.6, 108.8, 104.5, 56.1, 55.6, 53.3 (2C), 51.5, 49.6 (2C). HRMS (ESI-TOF) calcd for C23H25FN3O4 [M + H]+ 426.1829; found 426.1817.
Methyl 6-Methoxy-4-oxo-7-((4-(4-(trifluoromethyl)phenyl)-piperazin-1-yl)methyl)-1,4-dihydroquinoline-3-carboxylate 8ab.
8ab was synthesized from 7ab following general procedure F as a pale-yellow solid in 30% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.29 (s, 1H), 8.49 (s, 1H), 7.73 (s, 1H), 7.56 (s, 1H), 7.49 (d, J = 8.5 Hz, 2H), 7.06 (d, J = 8.6 Hz, 2H), 3.88 (s, 3h), 3.72 (s, 3H), 3.63 (s, 2H), 3.32 (m, 4H), 2.61 (t, J = 4.8 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 172.78, 165.40, 154.77 (2C), 153.10, 143.78, 143.61, 133.17 (2C), 126.08 (2C), 126.03 (2C), 114.07 (2C), 108.26, 104.07, 55.56, 55.02, 52.45 (2C), 50.92, 46.98 (2C). HRMS (ESI-TOF) calcd for C24H25F3N3O4 [M + H]+ 476.1797, found 476.1796.
Methyl 4-Oxo-7-(2-(4-phenylpiperazin-1-yl)ethyl)-1,4-dihydroquinoline-3-carboxylate 8ac.
8ac was synthesized from 5ac following general procedures C and F as a pale-brown solid in 23% yield over two steps. 1H NMR (400 MHz, DMSO-d6): δ 12.59 (bs, 1H), 8.55 (s, 1H), 8.25 (m, 1H), 7.49 (s, 1h), 7.35 (d, J= 10.5, 1H), 7.26 (t, J = 7.9, 2h), 7.01 (d, J = 8.1, 2h), 6.86 (t, J = 7.3, 1H), 3.85 (m, 2h), 3.73 (s, 3h), 3.51–3.43 (m, 4H), 3.24–3.16 (m, 4H), 3.02 (m, 2H). HRMS (ESI-TOF) calcd for C23H26N3O3 [M + H]+ 392.1974, found 392.1955
Methyl 7-(2-(4-Benzylpiperazin-1-yl)ethyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8ad.
8ad was synthesized from 5ad following general procedures C and F as a brown solid in 15% yield over two steps. 1H NMR (400 MHz, DMSO-d6): δ 12.22 (s, 1H), 8.50 (s, 1H), 8.04 (s, 1H), 7.41 (s, 1H), 7.28 (m, 6H), 3.72 (s, 3H), 3.44 (s, 2h), 3.31 (s, 2H), 2.84 (m, 2H), 2.48–2.37 (m, 8H). HRMS (ESI-TOF) calcd for C24H28N3O3 [M + H]+ 406.2131, found 406.2123.
Methyl 7-(2-(4-(4-Methoxybenzyl)piperazin-1-yl)ethyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8ae.
8ae was synthesized from 5ae following general procedures C and F as a brown solid in 11% yield over two steps. 1H NMR (400 MHz, DMSO-d6) δ 12.24 (bs, 1H), 8.52 (s, 1H), 8.05 (d, J = 7.9 Hz, 1H), 7.42 (s, 1H), 7.29 (s, 1H), 7.19 (d, J = 7.9 Hz, 2H), 6.87 (d, J = 8.1 Hz, 2H), 3.77 (m, 6H), 3.46–3.36 (m, 4H), 2.85 (t, J = 7.4 Hz, 2H), 2.60–2.37 (m, 8H). HRMS (ESI-TOF) calcd for C25H30N3O4 [M + H]+ 436.2236, found 436.2235.
Methyl 4-Oxo-6-((4-phenylpiperazin-1-yl)methyl)-1,4-dihydroquinoline-3-carboxylate 8af.
8af was synthesized following general procedure F as a pale-yellow solid in 42% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.36 (s, 1H), 8.55 (s, 1H), 8.09 (s, 1H), 7.67 (s, 1H), 7.61 (s, 1H), 7.19 (s, 2H), 6.91 (d, J = 7.2 Hz, 2H), 6.76 (s, 1h), 3.74 (s, 3H), 3.64 (s, 2H), 3.13 (m, 4H), 2.50 (m, 4H). HRMS (eSI-TOF) calcd for C22H24N3O3 [M + H]+ 378.1818, found 378.1803.
Methyl 6-((4-(4-Methoxyphenyl)piperazin-1-yl)methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8ag.
8ag was synthesized following general procedure F as a pale-yellow solid in 36% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.34 (s, 1H), 8.53 (s, 1H), 8.07 (s, 1H), 7.68–7.63 (m, 1H), 7.57 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 9.1 Hz, 2H), 6.78 (d, J = 9.1 Hz, 2H), 3.72 (s, 3H), 3.65 (s, 3H), 3.61 (s, 2H), 2.99 (m, 4H), 2.49 (m, 4H). HRMS (ESI-TOF) calcd for C23H26N3O4 [M + H]+ 408.1923, found 408.1935.
Methyl 6-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate 8ah.
8ah was synthesized following general procedure F as a pale-yellow solid in 45% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.36 (s, 1H), 8.54 (s, 1H), 8.08 (s, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.01 (t, J = 8.3 Hz, 2H), 6.92 (d, J = 4.1 Hz, 2H), 3.73 (s, 3H), 3.62 (s, 2h), 3.06 (s, 4H), 2.50 (s, 4H). HRMS (ESI-TOF) calcd for C22H23FN3O3 [M + H]+ 396.1723, found 396.1714.
Methyl 4-Oxo-6-((4-(4-(trifluoromethyl)phenyl)piperazin-1-yl)-methyl)-1,4-dihydroquinoline-3-carboxylate 8ai.
8ai was synthesized following general procedure F as a pale-yellow solid in 45% yield. 1H NMR (400 MHz, DMSO-d6): 8 12.29 (s, 1H), 8.50 (s, 1H), 8.06 (s, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 8.5 Hz, 1H), 7.42 (d, J = 8.6 Hz, 2H), 6.97 (d, J = 8.7 Hz, 2H), 3.69 (d, J = 10.0 Hz, 3H), 3.60 (s, 2H), 3.23 (m, 4H), 2.49 (m, 4H). HRMS (ESI-TOF) calcd for C23H23F3N3O3 [M + H]+ 446.1692, found 446.1685.
3-Bromo-7-((4-(4-fluorophenyl)piperazin-1-yl)methyl)quinolin-4(1H)-one 8ak.
The enamine intermediate 15aj (270 mg, 0.61 mmol) in toluene was subjected to microwave heating at 260 °C for 5 min. The reaction was allowed to cool to RT and concentrated under reduced pressure. The resultant crude was an inseparable mixture of quinolone regio-isomers. To a stirred solution of quinolone isomers in anhydrous CH2Cl2 was added freshly recrystallized NBS (127.0 mg, 0.71 mmol) at RT, and the resulting mixture was stirred overnight. The reaction mixture was concentrated under reduced pressure and the residue was purified by preparative HPLC to give the desired 8ak as a pale-yellow solid (40 mg, 16% over two steps). 1H NMR (400 MHz, DMSO-d6): 8 12.50 (d, J = 6.4 Hz, 1H), 8.53 (d, J = 6.3 Hz, 1H), 8.24 (d, J = 8.3 Hz, 1H), 7.73 (s, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.13–7.06 (m, 2H), 7.02–6.97 (m, 2H), 3.74 (d, J = 13.3 Hz, 2h), 3.44 (d, J = 12.2 Hz, 3H), 3.25 (d, J =11.1 Hz, 3h), 2.97 (d, J = 12.6 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 171.16, 155.72, 146.31, 140.70, 139.13, 133.29, 126.48, 126.21, 12–4.56, 121.79, 117.98, 117.92, 115.65, 115.48, 104.77, 58.11, 50.69 (2C), 46.23 (2C). HRMS (ESI-TOF) calcd for C20H20BrFN3O [M + H]+ 416.0774, found 416.0778.
3-Chloro-7-((4-(4-fluorophenyl)piperazin-1-yl)methyl)quinolin-4(1H)-one 8al.
The enamine intermediate 15aj (270 mg, 0.61 mmol) in toluene was subjected to microwave heating at 260 °C for 5 min. The reaction was allowed to cool to RT and concentrated under reduced pressure. The resultant crude was an inseparable mixture of quinolone regio-isomers. To a stirred solution of quinolone isomers in anhydrous CH2Cl2 was added freshly recrystallized NCS (94.0 mg, 0.71 mmol) at RT, and the resulting mixture was stirred overnight. The reaction mixture was concentrated under reduced pressure, and the residue was purified by preparative HPLC to give the desired 8al as a pale-yellow solid (34 mg, 15% over two steps). 1H NMR (400 MHz, DMSO-d6): δ 12.71 (s, 1H), 8.39 (d, J = 4.8 Hz, 1H), 8.20 (d, J = 8.3 Hz, 1H), 7.86 (d, J = 1.5 Hz, 1H), 7.65 (dd, J = 8.4, 1.5 Hz, 1H), 7.08 (dd, J = 9.9, 7.8 Hz, 2H), 7.01–6.94 (m, 2H), 3.70 (d, J = 12.5 Hz, 2h), 3.37 (d, J = 11.5 Hz, 2H), 3.26–3.03 (m, 6H). 13C NMR (101 MHz, methanol-d6 and 3 drops of CDO3) δ 173.02, 160.83, 158.42, 145.88, 140.41, 139.95, 133.89, 128.07, 127.54, 125.78, 123.04, 120.55, 120.47, 116.84, 116.61, 110.91, 60.34, 52.47 (2C), 48.94 (2C). HRMS (ESI-TOF) calcd for C20H20ClFN3O [M + H]+ 372.1279, found 372.1279.
7-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-2-methylquinolin-4(1H)-one 8am.
A mixture of aniline 5aj (2.0 g, 7.01 mmol) and ethyl acetoacetate (1.34 mL, 10.5 mmol) in benzene (20 mL) and AcOH (cat.) was heated to reflux in an oven-dried round-bottom flask equipped with Dean—Stark trap and a reflux condenser until no water separates out (usually overnight). The solvents were removed under reduced pressure, and the resulting crude intermediate after thorough drying under vacuo was used in next step without further purification. The enamine intermediate in toluene was subjected to microwave heating at 260 °C for 5 min. The reaction mixture was allowed to cool to RT. The crude mixture containing quinolone regio-isomers was then refluxed in methanol for 1 h and filtered to give the unwanted isomer as a white solid. The methanolic solution (filtrate) after evaporation gave 950 mg (39% yield) of light-brown amorphous 8am in pure form. 1H NMR (400 MHz, methanol-d6): δ 8.14 (t, J = 5.5 Hz, 1H), 7.54 (s, 1H), 7.38 (dd, J = 8.4, 1.4 Hz, 1H), 6.94 (d, J = 2.2 Hz, 2H), 6.93 (d, J = 2.4 Hz, 2H), 6.17 (s, 1h), 3.69 (s, 2h), 3.13–3.09 (m, 4H), 2.67–2.62 (m, 4H), 2.44 (s, 3H). HRMS (ESI-TOF) calcd for C21H23FN3O [M + H] + 352.1825, found 352.1829.
3-Bromo-7-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-2-methylquinolin-4(1H)-one 8an.
To a stirred solution of quinolone 8am (50.0 mg, 0.14 mmol) in anhydrous CH2Cl2 was added freshly recrystallized NBS (30.0 mg, 0.17 mmol) at RT, and the resulting mixture was stirred overnight. The reaction mixture was filtered, and the solid was washed with EtOAc followed by MeOH to give the purest 8an as an off-white solid (25 mg, 42% yield). 1H NMR (400 MHz, DMSO-d6): δ 12.07 (s, 1H), 8.04 (d, J = 8.3 Hz, 1H), 7.54 (s, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.03 (t, J = 8.7 Hz, 2H), 6.93 (dd, J = 9.3, 4.6 Hz, 2H), 3.65 (s, 2H), 3.10–3.08 (m, 4H), 2.58–2.55 (m, 4H), 2.54 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 171.20, 158.04, 156.16, 149.85, 146.83, 138.97, 126.62, 126.46, 123.53, 118.42, 118.36, 116.09, 115.92, 109.99, 106.97, 58.70, 51.19 (2C), 46.83 (2C), 21.86. HRMS (ESI-TOF) calcd for C21H22BrFN3O [M + H]+ 430.0930, found 430.0938.
3-Chloro-7-((4-(4-fluorophenyl)piperazin-1-yl)methyl)-2-methylquinolin-4(lH)-one 8ao.
To a stirred solution of quinolone 8am (50.0 mg, 0.14 mmol) in anhydrous CH2Cl2 was added freshly recrystallized NCS (23.0 mg, 0.17 mmol) at RT, and the resulting mixture was stirred overnight. The reaction mixture was concentrated under reduced pressure, and the residue was purified by preparative HPLC to give the purest 8ao as a white solid (30 mg, 55% yield). 1H NMR (400 MHz, DMSO-d6): δ 12.03 (s, 1H), 8.05 (d, J = 8.2 Hz, 1H), 7.54 (s, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.03 (t, J = 8.9 Hz, 2H), 6.97–6.85 (m, 2H), 3.65 (s, 2H), 3.09 (t, J = 4.7 Hz, 4H), 2.55 (t, J = 4.6 Hz, 4H), 2.50 (s, 3h). HRMS (ESI-TOf) calcd for C21H22OFN3O [M + h] + 386.1435, found 386.1434.
3-Bromo-2-methyl-7-((4-(4-(trifluoromethyl)phenyl)piperazin-1-yl)methyl)quinolin-4(1H)-one 8aq.
A mixture of aniline 5ap (380 mg, 1.12 mmol), ethyl acetoacetate (280 μL, 2.98 mmol), and acetic acid (cat.) were stirred overnight in benzene (2 mL) at 100 °C in an oven-dried round-bottom equipped with a Dean–Stark trap and reflux condenser. Solvents were removed under reduced pressure, and the resulting crude intermediate was transferred with toluene (2 mL) into a microwave vessel and subjected to 260 °C in the microwave for 5 min. The resulting crude was a mixture of two quinolone isomers. The quinolone mixture (100 mg, 0.249 mmol) was brominated using NBS (53 mg, 0.299 mmol) according to the general method G. The reaction mixture was filtered, and the solid was recrystallized in methanol to give 8aq as a pale-brown solid (6 mg, 5% yield). 1H NMR (399 MHz, DMSO) δ 12.10 (s, 1H), 8.05 (d, J = 8.2 Hz, 1H), 7.54 (s, 1H), 7.49 (d, J = 8.6 Hz, 2H), 7.32 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 8.7 Hz, 2H), 3.66 (s, 2H), 3.30 (s, 4H), 2.55 (s, 7H).
3-Chloro-2-methyl-7-((4-(4-(trifluoromethyl)phenyl)piperazin-1-yl)methyl)quinolin-4(1H)-one 8ar.
A mixture of aniline 5ap (380 mg, 1.12 mmol), ethyl acetoacetate (280 μL, 2.98 mmol), and acetic acid (cat.) were stirred overnight in benzene (2 mL) at 100 °C in an oven-dried round-bottom equipped with a Dean–Stark trap and reflux condenser. Solvents were removed under reduced pressure, and the resulting crude intermediate was transferred with toluene (2 mL) into a microwave vessel and subjected to 260 °C in the microwave for 5 min. The resulting crude was a mixture of two quinolone isomers. The quinolone mixture (100 mg, 0.249 mmol) was chlorinated using NCS (40 mg, 0.299 mmol) according to the general method G. The reaction mixture was filtered, and the solid was recrystallized in methanol to give 8ar as a pale-brown solid (19 mg, 18% yield). 1H NMR (399 MHz, DMSO) δ 12.06 (s, 1H), 8.06 (d, J = 8.1 Hz, 1H), 7.54 (s, 1H), 7.49 (d, J = 8.6 Hz, 2h), 7.31 (d, J = 8.4 Hz, 1h), 7.05 (d, J = 8.6 Hz, 2H), 3.66 (s, 2h), 3.30 (s, 4H), 2.55 (s, 4H), 2.51 (s, 3H). 13C NMR (100 MHz, DMSO) δ 170.5, 153.2, 142.6, 138.6, 126.2, 125.2, 122.6, 119.1, 117.1, 115.5, 114.2, 113.7, 61.4, 52.4, 47.1, 18.7.
3-Bromo-6-methoxy-2-methyl-7-((4-(4-(trifluoromethyl)phenyl)-piperazin-1-yl)methyl)quinolin-4(1H)-one 8at.
A mixture of aniline 5as (760 mg, 1.37 mmol), ethyl acetoacetate (520 μL, 2.74 mmol), and acetic acid (cat.) were stirred overnight in benzene (3.5 mL) at 100 °C in an oven-dried round-bottom equipped with a Dean–Stark trap and reflux condenser. Solvents were removed under reduced pressure, and the resulting crude intermediate was transferred with toluene (3.5 mL) into a microwave vessel and subjected to 260 °C in the microwave for 5 min. The resulting crude was a mixture of two quinolone isomers. The quinolone mixture (500 mg, 1.16 mmol) was brominated using NBS (250 mg, 1.39 mmol) according to the general method G. The reaction mixture was filtered, and the solid was recrystallized in methanol to give 8at as a yellow solid (74 mg, 12% yield). 1H NMR (399 MHz, CDCl3 and 3 drops of TFA-d) δ 11.12 (s, 1H), 8.60 (s, 1h), 7.76 (d, J = 8.6 Hz, 2H), 7.73 (s, 1H), 7.59 (d, J = 8.7 Hz, 2H), 4.80 (s, 2h), 4.10 (s, 3h), 4.01 (s, 8H), 3.03 (s, 3H). 13C NMR (100 MHz, CDCl3 and 3 drops TFA-d) δ 162.71, 157.95, 155.49, 132.61, 132.35, 128.12 (d, J = 2.9 Hz), 126.5, 121.0, 120.1, 118.8, 116.0, 113.1, 110.3, 105.1, 102.5, 57.2, 55.2, 50.7, 50.1, 22.2.
2,2′-((4-Methoxyphenyl)azanediyl)bis(ethan-1-ol) 10b.
A mixture of p-anisole (36 mmol) and 2-chloroethan-1-ol (90 mmol) were added to a round-bottom flask along with DMF (72 mL) and K2CO3 (72 mmol) and refluxed overnight. The reaction was then concentrated and purified using flash chromatography (hexanes/ethyl acetate) to give the title compound in 85% yield as an off-white solid. 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 8.3 Hz, 2H), 6.82–6.77 (m, 2H), 3.62 (dd, J = 9.4, 7.9 Hz, 4H), 3.89 (s, 3H), 2.64–2.58 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 260.63, 232.07, 215.63 210.11, 164.62, 161.32, 157.38, 157.08. 249.2, 239.6, 218.4, 214.9, 178.3, 151.8, 149.6.
2,2’-((4-Methoxybenzyl)azanediyl)bis(ethan-1-ol) 10d.
A mixture of (4-methoxyphenyl)methanamine (36 mmol) and 2-chloroethan-1-ol (90 mmol) were added to a round-bottom flask along with DMF (70 mL) and K2CO3 (72 mmol) and refluxed overnight. The reaction was then concentrated and purified using flash chromatography (hexanes/ethyl acetate) to give the title compound in 91% yield as an off-white solid. 1H NMR (500 MHz, CDCl3) δ 7.20 (d, J = 8.7 Hz, 2H), 6.86–6.81 (m, 2H), 3.76 (s, 2h), 3.62 (dd, J = 10.3, 8.6 Hz, 4H), 3.47 (s, 3H), 2.70–2.60 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 260.6, 232.1, 215.6, 210.1, 164.6, 161.3, 157.4, 157.1.
Dimethyl 2-(((3-(Hydroxymethyl)phenyl)amino)methylene)-malonate 12a.
12a was prepared from 3-aminobenzyl alcohol (3 g, 24.4 mmol) according to general procedure C (pale-yellow solid, 6.4 g, quantitative yield). 1H NMR (400 MHz, CDCl3): δ 10.93 (d, J = 13.8 Hz, 1H), 8.44 (d, J = 13.8 Hz, 1H), 7.26 (t, J = 7.8 Hz, 1H), 7.11–7.04 (m, 2H), 6.97–6.93 (m, 1H), 4.63 (s, 2H), 3.77 (s, 3H), 3.70 (s, 3H), 2.83 (bs, 1H). 13C NMR (101 MHz, CDCl3): δ 169.1, 166.0, 151.9, 143.2, 139.1, 129.7, 123.2, 116.0, 115.2, 92.6, 64.3, 51.5, 51.4. HRMS (ESI-TOF) calcd for C13H16NO5 [M + H]+ 266.1028, found 266.1021.
Dimethyl 2-(((3-(Hydroxymethyl)-4-methylphenyl)amino)-methylene)malonate 12b.
12b was prepared from 5-amino-2-methoxybenzyl alcohol (3.0 g, 21.9 mmol) according to general procedure C (pale-white solid, 5.5 g, 95% yield). 1H NMR (400 MHz, CDCl3): δ 10.94 (d, J = 13.9 Hz, 1H), 8.44 (dd, J = 13.9, 0.7 Hz, 1H), 7.15 (d, J = 2.5 Hz, 1H), 7.07 (d, J = 8.1 Hz, 1H), 6.88 (dd, J = 8.0, 2.6 Hz, 1H), 4.62 (s, 2H), 3.76 (s, 3H), 3.71 (s, 3H), 2.21 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 169.2, 166.1, 152.1, 140.8, 137.0, 132.2, 131.2, 115.7, 115.6, 92.0, 62.4, 51.4, 51.3, 17.9. HRMS (ESI-TOf) calcd for C14H18NO5 [M + H]+ 280.1185, found 280.1182.
Dimethyl 2-(((3-(Hydroxymethyl)-4-methoxyphenyl)amino)-methylene)malonate 12c.
12c was prepared from 5-amino-2-methoxybenzyl alcohol (3.0 g, 19.6 mmol) according to general procedure C (violet-red semisolid, 5.3 g, 92% yield). 1H NMR (400 MHz, CDCl3) δ 10.96 (d, J = 13.8 Hz, 1H), 8.40 (d, J = 13.9 Hz, 1H), 7.13 (d, J = 2.9 Hz, 1H), 7.01–6.95 (m, 1H), 6.83–6.78 (m, 1h), 4.65 (s, 2H), 3.81 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 169.4, 166.1, 154.6, 152.8, 132.4, 130.9, 117.6, 117.5, 111.0, 91.8, 61.0, 55.6, 51.4, 51.3. HRMS (ESI-TOF) calcd for C14H18NO6 [M + H]+ 296.1134, found 296.1128.
Dimethyl 2-(((4-(Hydroxymethyl)phenyl)amino)methylene)-malonate 12d.
12d was prepared from 4-aminobenzyl alcohol (3 g, 24.4 mmol) according to general procedure C (pale-yellow solid, 6.4 g, quantitative yield). 1H NMR (400 MHz, CDCl3): δ 10.94 (d, J = 13.8 Hz, 1H), 8.43 (ddd, J = 13.8, 8.2, 5.5 Hz, 1H), 7.38–7.28 (m, 2H), 7.06–7.02 (m, 2H), 4.60 (s, 2H), 3.79 s, 3H), 3.71 (m, 3H). 13C NMR (101 MHz, CDCl3): δ 169.2, 166.0, 152.0, 138.1, 138.1, 128.3 (2C), 117.1 (2C), 92.6, 64.2, 51.5, 51.4. HRMS (ESI-TOF) calcd for C13H16NO5 [M + H]+ 266.1028, found 266.1032.
5-(((3-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)phenyl)amino)-methylene)-2,2-dimethyl-1,3-dioxane-4,6-dione 15aj.
Trimethyl orthoformate (3.7 g, 35.0 mmol) and Meldrum’s acid (0.3 g, 2.10 mmol) were refluxed for 3 h under N2 and then cooled to RT. The aniline 5aj (0.5 g, 1.75 mmol) was then added to the reaction, and the resulting mixture was refluxed for another 1 h. The reaction mixture was cooled to RT, ether was added to allow precipitation, and the excess solvents were decanted (the addition of ether and decanting can be repeated couple of times to get rid of excess CH(OCH3)3 and Meldrum’s acid). The resulting orange yellow solid was dried under vacuum to give an NMR-pure enamine 15aj in 84% yield (650 mg) that was used directly in next cyclization step without further purification. 1H NMR (400 MHz, CDCl3): δ 11.19 (m, 1H), 8.64 (d, J = 14.4 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.28 (s, 1H), 7.21 (d, J = 7.6 Hz, 1H), 7.12 (dd, J = 7.9, 1.8 Hz, 1H), 6.95–6.87 (m, 2H), 6.86–6.80 (m, 2H), 3.56 (d, J = 12.8 Hz, 2H), 3.14–3.04 (m, 4H), 2.63–2.55 (m, 4h), 1.72 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 165.38, 163.43, 152.43 (2C), 147.81, 140.78, 137.83, 129.83, 127.34, 118.23, 117.76, 117.69, 116.72 (2C), 115.45, 115.23, 105.02, 62.32, 53.04 (2C), 49.97 (2C), 26.90 (2C). HRMS (ESI-TOF) calcd for C24H27FN3O4 [M + H]+ 440.1986, found 440.1996.
Supplementary Material
∎ ACKNOWLEDGMENTS
We thank Susan A. Charman and Karen L. White (Monash Institute of Pharmaceutical Sciences, Parkville, Australia) for valuable discussions and for proof reading the manuscript. We also thank the Genshaft Family Doctoral Fellowship from the University of South Florida for financial support of J.R.M. and C.L.L. We thank the Medicines for Malaria Venture (MMV 11/ 0022) and the National Institutes of Health (R01 GM097118) for financial support.
∎. ABBREVIATIONS USED
- Ac
acetyl
- ACT
artemisinin combination therapy
- DMF
N,N-dimethylformamide
- EC50
half-maximal effective concentration
- ED50
half-maximal effective dose
- HPLC
high performance liquid chromatography
- LBI
liver blood index
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- ND
not determined
- Pb
Plasmodium berghei
- PI
post infection
- RI
resistance index
- RT
room temperature
- SAR
structure—activity relationship
- SPR
structure—property relationship
- WHO
World Health Organization
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.7b00738.
Additional solubility and log D data for selected compounds (PDF)
Molecular formula strings (CSV)
PDB coordinates of 8ae bound to Qo and Qi sites of bc1 (PDB)
PDB coordinates of 8ac bound to Qo and Qi sites of bc1 (PDB)
The authors declare no competing financial interest.
∎ REFERENCES
- (1).World Malaria Report 2016; World Health Organization: Geneva, 2016.
- (2).Lacrue AN; Saenz FE; Cross RM; Udenze KO; Monastyrskyi A; Stein S; Mutka TS; Manetsch R; Kyle DE 4(1H)-Quinolones with liver stage activity against Plasmodium berghei. Antimicrob. Agents Chemother. 2013, 57, 417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Teixeira C; Vale N; Perez B; Gomes A; Gomes JR; Gomes P “Recycling” classical drugs for malaria. Chem. Rev. 2014, 114, 11164–220. [DOI] [PubMed] [Google Scholar]
- (4).Zhang Y; Guiguemde WA; Sigal M; Zhu F; Connelly MC; Nwaka S; Guy RK Synthesis and structure-activity relationships of antimalarial 4-oxo-3-carboxyl quinolones. Bioorg. Med. Chem. 2010, 18, 2756–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhang Y; Clark JA; Connelly MC; Zhu F; Min J; Guiguemde WA; Pradhan A; Iyer L; Furimsky A; Gow J; Parman T; El Mazouni F; Phillips MA; Kyle DE; Mirsalis J; Guy RK Lead optimization of 3-carboxyl-4(1H)-quinolones to deliver orally bioavailable antimalarials. J. Med. Chem. 2012, 55, 4205–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cross RM; Monastyrskyi A; Mutka TS; Burrows JN; Kyle DE; Manetsch R Endochin optimization: structure-activity and structure-property relationship studies of 3-substituted 2-methyl-4(1H)-quinolones with antimalarial activity. J. Med. Chem. 2010, 53, 7076–94. [DOI] [PubMed] [Google Scholar]
- (7).Cross RM; Namelikonda NK; Mutka TS; Luong L; Kyle DE; Manetsch R Synthesis, antimalarial activity, and structure-activity relationship of 7-(2-phenoxyethoxy)-4(1H)-quinolones. J. Med. Chem. 2011, 54, 8321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cross RM; Maignan JR; Mutka TS; Luong L; Sargent J; Kyle DE; Manetsch R Optimization of 1,2,3,4-tetrahydroacridin-9(10H)-ones as antimalarials utilizing structure-activity and structure-property relationships. J. Med. Chem. 2011, 54, 4399–426. [DOI] [PubMed] [Google Scholar]
- (9).Cross RM; Flanigan DL; Monastyrskyi A; LaCrue AN; Saenz FE; Maignan JR; Mutka TS; White KL; Shackleford DM; Bathurst I; Fronczek FR; Wojtas L; Guida WC; Charman SA; Burrows JN; Kyle DE; Manetsch R Orally Bioavailable 6-Chloro-7-methoxy-4(1H)-quinolones Efficacious against Multiple Stages of Plasmodium. J. Med. Chem. 2014, 57, 8860–8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Saenz FE; LaCrue AN; Cross RM; Maignan JR; Udenze K; Manetsch R; Kyle DE 4-(1H)-Quinolones and 1,2,3,4-tetrahydroacridin-9(10H)-ones prevent the transmission of Plasmodium falciparum to Anopheles freeborni. Antimicrob. Agents Chemother. 2013, 57, 6187–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bueno JM; Manzano P; Garcia MC; Chicharro J; Puente M; Lorenzo M; Garcia A; Ferrer S; Gomez RM; Fraile MT; Lavandera JL; Fiandor JM; Vidal J; Herreros E; Gargallo-Viola D Potent antimalarial 4-pyridones with improved physico-chemical properties. Bioorg. Med. Chem. Lett. 2011, 21, 5214–5218. [DOI] [PubMed] [Google Scholar]
- (12).Winter RW; Kelly JX; Smilkstein MJ; Dodean R; Hinrichs D; Riscoe MK Antimalarial quinolones: synthesis, potency, and mechanistic studies. Exp. Parasitol. 2008, 118, 487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Winter R; Kelly JX; Smilkstein MJ; Hinrichs D; Koop DR; Riscoe MK Optimization of endochin-like quinolones for antimalarial activity. Exp. Parasitol. 2011, 127, 545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Monastyrskyi A; Kyle DE; Manetsch R 4(1H)-Pyridone and 4(1H)-Quinolone Derivatives as Antimalarials with Erythrocytic, Exoerythrocytic, and Transmission Blocking Activities. Curr. Top. Med. Chem. 2014, 14, 1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Puri SK; Dutta GP Quinoline esters as potential antimalarial drugs: effect on relapses of Plasmodium cynomolgi infections in monkeys. Trans. R. Soc. Trop. Med. Hyg. 1990, 84, 759–60. [DOI] [PubMed] [Google Scholar]
- (16).Ryley JF; Peters W The antimalarial activity of some quinolone esters. Ann. Trop. Med. Parasitol. 1970, 64, 209–22. [DOI] [PubMed] [Google Scholar]
- (17).Maignan JR; Lichorowic CL; Giarrusso J; Blake LD; Casandra D; Mutka TS; LaCrue AN; Burrows JN; Willis PA; Kyle DE; Manetsch R ICI 56,780 Optimization: Structure-Activity Relationship Studies of 7-(2-Phenoxyethoxy)-4(lH)-quinolones with Antimalarial Activity. J. Med. Chem. 2016, 59, 6943–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cowley R; Leung S; Fisher N; Al-Helal M; Berry NG; Lawrenson AS; Sharma R; Shone AE; Ward SA; Biagini GA; O’Neill PM The development of quinolone esters as novel antimalarial agents targeting the Plasmodium falciparum bc(l) protein complex. MedChemComm 2012, 3, 39–44. [Google Scholar]
- (19).Reddy MV; Akula B; Cosenza SC; Athuluridivakar S; Mallireddigari MR; Pallela VR; Billa VK; Subbaiah DR; Bharathi EV; Vasquez-Del Carpio R.; Padgaonkar A; Baker SJ; Reddy EP Discovery of 8-cyclopentyl-2-[4-(4-methyl-piperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrid o[2,3-d]pyrimidine-6-car-bonitrile (7x) as a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and AMPK-related kinase 5 (ARK5). J. Med. Chem. 2014, 57, 578–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liu KG; Robichaud AJ A general and convenient synthesis of N-aryl piperazines. Tetrahedron Lett. 2005, 46, 7921–7922. [Google Scholar]
- (21).Gould RG; Jacobs WA The Synthesis of Certain Substituted Quinolines and 5,6-Benzoquinolines. J. Am. Chem. Soc. 1939, 61, 2890–2895. [Google Scholar]
- (22).Dess DB; Martin JC Readily Accessible 12-I-5 Oxidant for the Conversion of Primary and Secondary Alcohols to Aldehydes and Ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar]
- (23).Kuhhorn J; Hubner H; Gmeiner P Bivalent dopamine D2 receptor ligands: synthesis and binding properties. J. Med. Chem. 2011, 54, 4896–903. [DOI] [PubMed] [Google Scholar]
- (24).Rossi C; Porcelloni M; D’Andrea P; Fincham CI; Ettorre A; Mauro S; Squarcia A; Bigioni M; Parlani M; Nardelli F; Binaschi M; Maggi CA; Fattori D Alkyl piperidine and piperazine hydroxamic acids as HDAC inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2305–8. [DOI] [PubMed] [Google Scholar]
- (25).Ghosh B; Antonio T; Reith ME; Dutta AK Discovery of 4-(4-(2-((5-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)-amino)ethyl)piperaz in-1-yl)quinolin-8-ol and its analogues as highly potent dopamine D2/D3 agonists and as iron chelator: in vivo activity indicates potential application in symptomatic and neuroprotective therapy for Parkinson’s disease. J. Med. Chem. 2010, 53, 2114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nilsen A; LaCrue AN; White KL; Forquer IP; Cross RM; Marfurt J; Mather MW; Delves MJ; Shackleford DM; Saenz FE; Morrisey JM; Steuten J; Mutka T; Li Y; Wirjanata G; Ryan E; Duffy S; Kelly JX; Sebayang BF; Zeeman AM; Noviyanti R; Sinden RE; Kocken CH; Price RN; Avery VM; Angulo-Barturen I; Jimenez-Diaz MB; Ferrer S; Herreros E; Sanz LM; Gamo FJ; Bathurst I; Burrows JN; Siegl P; Guy RK; Winter RW; Vaidya AB; Charman SA; Kyle DE; Manetsch R; Riscoe MK Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med. 2013, 5, 177rara37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Plowe CV Malaria: Resistance nailed. Nature 2014, 505, 30–31. [DOI] [PubMed] [Google Scholar]
- (28).Looareesuwan S; Viravan C; Webster HK; Kyle DE; Hutchinson DB; Canfield CJ Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am. J. Trop. Med. Hyg. 1996, 54, 62–66. [DOI] [PubMed] [Google Scholar]
- (29).Milhous WK; Gerena L; Kyle DE; Oduola AM In vitro strategies for circumventing antimalarial drug resistance. Prog. Clin. Biol. Res. 1989, 313, 61–72. [PubMed] [Google Scholar]
- (30).Yeates CL; Batchelor JF; Capon EC; Cheesman NJ; Fry M; Hudson AT; Pudney M; Trimming H; Woolven J; Bueno JM; Chicharro J; Fernandez E; Fiandor JM; Gargallo-Viola D; Gomez de las Heras F; Herreros E; Leon ML Synthesis and structure-activity relationships of 4-pyridones as potential antimalarials. J. Med. Chem. 2008, 51, 2845–52. [DOI] [PubMed] [Google Scholar]
- (31).Bueno JM; Herreros E; Angulo-Barturen I; Ferrer S; Fiandor JM; Gamo FJ; Gargallo-Viola D; Derimanov G Exploration of 4(lH)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochrome bcl. Future Med. Chem. 2012, 4, 2311–2323. [DOI] [PubMed] [Google Scholar]
- (32).Biagini GA; Fisher N; Shone AE; Mubaraki MA; Srivastava A; Hill A; Antoine T; Warman AJ; Davies J; Pidathala C; Amewu RK; Leung SC; Sharma R; Gibbons P; Hong DW; Pacorel B; Lawrenson AS; Charoensutthivarakul S; Taylor L; Berger O; Mbekeani A; Stocks PA; Nixon GL; Chadwick J; Hemingway J; Delves MJ; Sinden RE; Zeeman AM; Kocken CH; Berry NG; O’Neill PM; Ward SA Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Capper MJ; O’Neill PM; Fisher N; Strange RW; Moss D; Ward SA; Berry NG; Lawrenson AS; Hasnain SS; Biagini GA; Antonyuk SV Antimalarial 4(lH)-pyridones bind to the Qi site of cytochrome bcl. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 755–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Birth D; Kao WC; Hunte C Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat. Commun. 2014, 5, 4029. [DOI] [PubMed] [Google Scholar]
- (35).Van Horn KS; Burda WN; Fleeman R; Shaw LN; Manetsch R Antibacterial activity of a series of N2,N4-disubstituted quinazoline-2,4-diamines. J. Med. Chem. 2014, 57, 3075–93. [DOI] [PubMed] [Google Scholar]
- (36).Van Horn KS; Zhu X; Pandharkar T; Yang S; Vesely B; Vanaerschot M; Dujardin JC; Rijal S; Kyle DE; Wang MZ; Werbovetz KA; Manetsch R Antileishmanial activity of a series of N(2),N(4)-disubstituted quinazoline-2,4-diamines. J. Med. Chem. 2014, 57, 5141–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Nilsen A; LaCrue AN; White KL; Forquer IP; Cross RM; Marfurt J; Mather MW; Delves MJ; Shackleford DM; Saenz FE; Morrisey JM; Steuten J; Mutka T; Li Y; Wirjanata G; Ryan E; Duffy S; Kelly JX; Sebayang BF; Zeeman AM; Noviyanti R; Sinden RE; Kocken CHM; Price RN; Avery VM; Angulo-Barturen I; Jimenez-Diaz MB; Ferrer S; Herreros E; Sanz LM; Gamo FJ; Bathurst I; Burrows JN; Siegl P; Guy RK; Winter RW; Vaidya AB; Charman SA; Kyle DE; Manetsch R; Riscoe MK Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med. 2013, 5, 177ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mwakingwe A; Ting LM; Hochman S; Chen J; Sinnis P; Kim K Noninvasive real-time monitoring of liver-stage development of bioluminescent Plasmodium parasites. J. Infect. Dis. 2009, 200, 1470–8. [DOI] [PubMed] [Google Scholar]
- (39).Madhavi Sastry G.; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- (40).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–49. [DOI] [PubMed] [Google Scholar]
- (41).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–96. [DOI] [PubMed] [Google Scholar]
- (42).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.