

SUMMARY
Interactions between stromal fibroblasts and cancer cells generate signals for cancer progression, therapy resistance, and inflammatory responses. Although endogenous RNAs acting as damage-associated molecular patterns (DAMPs) for pattern recognition receptors (PRRs) may represent one such signal, these RNAs must remain unrecognized under non-pathological conditions. We show that triggering of stromal NOTCH-MYC by breast cancer cells results in a POL3-driven increase in RN7SL1, an endogenous RNA normally shielded by RNA binding proteins SRP9/14. This increase in RN7SL1 alters its stoichiometry with SRP9/14 and generates unshielded RN7SL1 in stromal exosomes. After exosome transfer to immune cells, unshielded RN7SL1 drives an inflammatory response. Upon transfer to breast cancer cells, unshielded RN7SL1 activates the PRR RIG-I to enhance tumor growth, metastasis, and therapy resistance. Corroborated by evidence from patient tumors and blood, these results demonstrate that regulation of RNA unshielding couples stromal activation with deployment of RNA DAMPs that promote aggressive features of cancer.
In Brief
Stromal cells shed exosomes containing an RNA that, in its protein-free form, drives anti-viral signaling in recipient breast cancer cells that ultimately results in tumor growth as well as therapy resistance.
Graphical Abstract
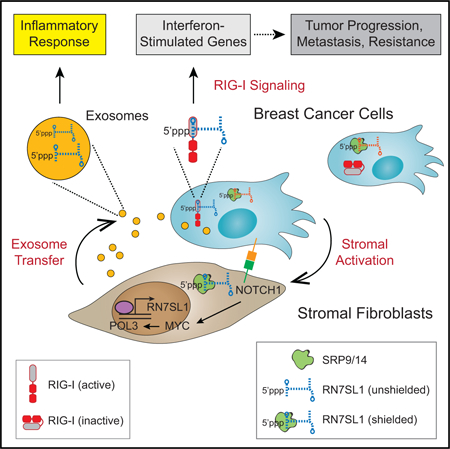
INTRODUCTION
The dynamic interaction between cancer cells and stromal cells of the tumor microenvironment critically regulates important features of cancer (Kalluri, 2016). Reciprocal signaling between these heterotypic cell types can be paracrine or juxtracrine in nature and includes multiple oncogenic and developmental pathways. However, how the interaction between cancer and stromal cells generates and then integrates signals that result in tumor growth, metastasis, therapy resistance, and sterile inflammation are not well understood.
Across many common human cancers, a large proportion of tumors unexpectedly express high levels of interferon-stimulated genes (ISGs) that are typically associated with anti-viral signaling (Weichselbaum et al., 2008). We reported that these ISGs can be induced in a subset of breast cancer cells upon cell-cell contact with stromal fibroblasts (Boelens et al., 2014). This subset of breast cancer cells is denoted as ISG responders (ISG-R) and are predominantly basal/triple-negative breast cancers (TNBC). In contrast, ISG non-responders (ISG-NR) fail to upregulate ISGs and primarily belong to the luminal/ER-positive subtypes. ISG induction results from the transfer of stromal-derived exosomes, which are small extracellular vesicles implicated in a myriad of processes related to cancer progression (Becker et al., 2016). These exosomes contain RNA (exoRNA) that is enriched in non-coding transcripts. Upon transfer to ISG-R breast cancer cells, the exoRNA stimulates the viral RNA pattern recognition receptor (PRR) RIG-I, resulting in STAT1 activation and ISG induction. STAT1 amplifies the NOTCH3 transcriptional response, resulting in expansion of tumor-initiating cells and therapy resistance (Figure 1A). Consistent with these experimental findings, patients with tumors expressing high levels of ISGs are more likely to relapse after chemotherapy or radiation therapy. Similar examples of PRRs recognizing exoRNA in the tumor microenvironment have been reported to influence cancer progression (Liu et al., 2016). However, given that cancer-associated anti-viral signaling is occurring in a sterile microenvironment, this raises questions on the nature of the endogenous RNA that is activating RIG-I and the extent to which it influences the multitude of effects that stromal cells exert on cancer progression and therapy response.
Figure 1. Stromal RNA Is Transferred to Breast Cancer Cells by Exosomes.
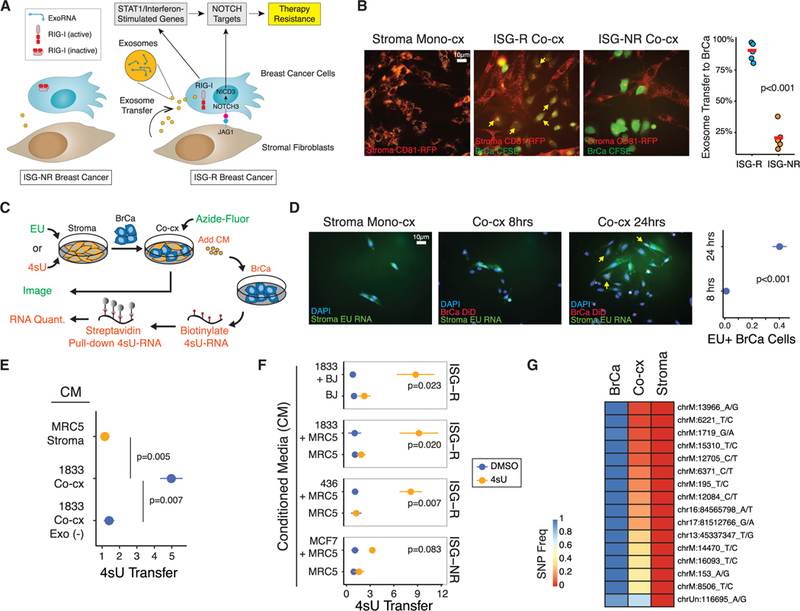
(A) Summary of ISG-R and ISG-NR breast cancer cells and differential exosome transfer and RIG-I activation upon interaction with stromal fibroblasts.
(B) MRC5 fibroblasts expressing a CD81-RFP exosome reporter were co-cultured with CFSE-labeled ISG-R 1833 or ISG-NR MCF7 breast cancer cells. Exosome transfer is quantitated (right) and representative transfer is shown (arrows).
(C) Schema for measuring RNA transfer from stromal to breast cancer cells utilizing the uridine analog EU for fluorescence microscopy (green) or 4sU for streptavidin pull-down (orange).
(D) MRC5 fibroblasts were labeled with EU and co-cultured with DiD lipid-labeled 1833 breast cancer cells. EU-positive 1833 cells (yellow arrows) and quantitation are shown.
(E) Relativetransferof4sU RNA to mono-cultured 1833 breast cancer cells after addition of conditioned media (CM) isolated from 4sU-labeled MRC5 fibroblasts grown in mono-culture (Stroma, orange) or from 1833 ISG-R co-culture (Co-cx, blue). Co-culture CM depleted of exosomes (Co-cx Exo(—) CM) is shown as a control for exosome-dependency (n = 5).
(F) Same as in (E) except CM was isolated from MRC5 or BJ 4sU-labeled fibroblasts grown in mono-culture or co-cultured with the indicated ISG-R or ISG-NR breast cancer cells. Shown is relative 4sU RNA transfer after CM addition to each breast cancer cell mono-culture(n = 3). Transfer is relative to mock4sU labeling using DMSO.
(G) Allelic frequency of exoRNA SNPs from exosomes isolated from 1833 breast cancer (BrCa), MRC5 fibroblasts (Stroma), or from co-culture of both cell types (Co-cx). Analysis is based on SNPs present in exoRNA from breast cancer cells and not present in fibroblasts. Error bars are SEM of biological replicates. See also Figure S1.
There are many properties that RIG-I utilizes to distinguish self from non-self RNA. Typically, RIG-I recognizes cytoplasmic double-stranded RNA that is 5’-triphosphorylated, short (<300 bp) and has a blunt 5’ end (Schlee and Hartmann, 2016). For viral RNAs, polyuridine motifs can favor recognition (Saito et al., 2008), while RNA modifications such as 2’-O-methylation can critically prevent RIG-I binding to 5’ capped cellular RNAs (De-varkar et al., 2016; Schuberth-Wagner et al., 2015). However, much of the RNA features and requirements for optimal RIG-I activation are based on synthetic and/or artificial RNAs in vitro. Emerging evidence indicates that endogenous RNA can function as a damage-associated molecular pattern (DAMP) to activate PRRs under a variety of stress and pathological conditions, such as after chemotherapy (Chiappinelli et al., 2015; Roulois et al., 2015; Sistigu et al., 2014) or radiation (Bernard et al., 2012; Ranoa et al., 2016), or in autoimmunity (Eckard et al., 2014; Hung et al., 2015). How endogenous RNAs can function as DAMPs to activate PRRs while avoiding recognition under non-pathological conditions is not well understood.
RESULTS
Stromal RNA Is Transferred to Breast Cancer Cells by Exosomes
Increased exosome production and transfer to breast cancer cells only occurs when stromal cells are co-cultured with ISG-R breast cancer cells (ISG-R co-culture) but not when co-cultured with ISG-NR breast cancer cells (ISG-NR co-culture) (Figure 1A). To examine whether the transfer of stromal exosomes also results in transfer of stromal RNA, we first labeled MRC5 stromal fibroblasts with a stably expressed CD81-RFP exosome reporter (Figure 1B). This confirmed a high level of exosome transfer from stromal cells to 1833 ISG-R breast cancer cells, which is a metastatic derivative of MDA-MB-231 (Kang et al., 2003). In contrast, ISG-NR co-cultures show minimal stromal exosome transfer. To examine if RNA from stromal cells accompanies exosome transfer, we metabolically labeled stromal RNA with 5-ethynyl uridine (EU) prior to co-culture with ISG-R 1833 breast cancer cells that were fluorescently marked with lipid dye (Figure 1C). After 24 hours, over 40% of breast cancer cells acquired stromal cell RNA as measured by EU-modification by azide-linked fluorescein (Figure 1D). Moreover, when stromal cell RNA was similarly labeled with 4-thiouridine (4sU) prior to ISG-R co-culture (Figures 1C and S1), application of the exo-some-containing conditioned media (CM) to mono-cultured breast cancer cells also resulted in stromal RNA transfer, as determined by streptavidin pull-down of biotinylated 4sU-labeled stromal RNA (Figure 1E). In contrast, stromal RNA was not transferred when exosomes were depleted from the CM, consistent with our previous findings that the ability of CM to induce ISGs is strictly exosome-dependent (Boelens et al., 2014). Exosome-mediated transfer of stromal RNA was also observed using another ISG-R breast cancer cell line, MDA-MB-436, and using BJ fibroblast cells in co-culture (Figure 1F). Markedly less stromal RNA was transferred by exosomes using CM from co-cultures with the ISG-NR breast cancer cell line MCF7.
To corroborate the transfer of stromal RNA by exosomes, we also performed exoRNA SNP analysis using exosomes from mono-cultures of either ISG-R 1833 breast cancer cells or MRC5 stromal cells and compared SNP allelic frequencies to the exoRNA from co-culture (Figure 1G). Multiple SNPs, primarily from mitochondrial RNA, were discovered to have an allelic frequency of near one in the exoRNA from breast cancer cells but near zero in stromal exoRNA. Examination of exoRNA from coculture revealed that most of these SNPs maintained a frequency closer to zero, consistent with the exoRNA primarily originating from stromal cells. In total, these results suggest that cellular RNAs are transferred from stromal to breast cancer cells in an exosome-dependent manner.
Stromal RNA Polymerase III Generates 5’ppp ExoRNA that Activates RIG-I in Breast Cancer Cells
Classification of non-ribosomal exoRNA transcripts from ISG-R co-cultures reveals an enrichment in non-coding RNAs compared to cellular RNA (Figure 2A). These non-coding RNAs include repeat and transposable elements, small nuclear RNA (snRNA), signal recognition particle RNA (srpRNA), and others, but no viral RNAs were detected. Previously, we demonstrated that upon transfection this exoRNA activates RIG-I to induce ISGs in recipient breast cancer cells, and this activity requires a5’-triphosphate (5’ppp) RNA end. To confirm this notion, we utilized CRISPR/Cas9 to knock out RIG-I in breast cancer cells and re-expressed either wild-type (WT) RIG-I or RIG-I with alanine substitution mutations in key lysine residues (K858 and K861) that make contacts with the 5’ppp motif (RIG-IK858/861A) (Wang et al., 2010) (Figures S2A and S2B). Co-culture-derived exosomes were purified (Figures S2C and S2D) and transfection of the exoRNA failed to induce ISGs in RIG-I KO breast cancer cells (Figure 2B). Re-expression of WT RIG-I rescued this defect whereas RIG-IK858/861A was markedly less effective at restoring activity. In contrast, cellular RNA failed to induce ISGs regardless of RIG-I status. Thus, these results provide evidence that 5’ppp exoRNA from stromal cells activates RIG-I.
Figure 2. Stromal POL3-Derived ExoRNA Activates Breast Cancer RIG-I in a 5’-Triphosphate-Dependent Manner.
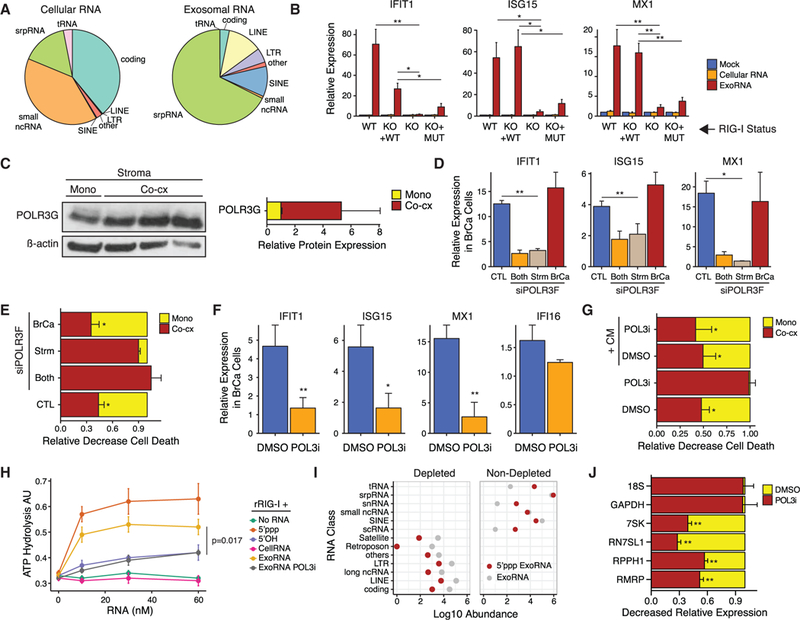
(A) Distribution of RNA classes found in cellular RNA and exosome RNA by RNA-seq after co-culture of1833 ISG-R breast cancer cells with MRC5 stromal cells. Ribosomal RNA counts were removed.
(B) ISG expression after transfection of co-culture exoRNA or co-culture cellular RNA into 1833 cells with wild-type RIG-I (WT), RIG-I knockout (KO), or RIG-I KO cells restored with either wild-type RIG-I (KO + WT) or RIG-IK858/861A (KO + MUT) (n = 5). Baseline was established by mock transfection (see legend).
(C) Immunoblot for POLR3G and ß-actin in sorted MRC5 fibroblasts after mono- or ISG-R co-culture (left) along with quantification of replicates (right).
(D and E) Expression of ISGs in sorted 1833cells(D) or(E) RT-mediated cell death in 1833 cells after co-culture with MRC5cells(CTL) or after siRNA knockdown of POLR3F in 1833 (BrCa), MRC5 (Strm), or both cell types (Both). Gene expression is relativeto1833 cells in mono-culture, and cell death was assessed4days after 10 Gy RT (n = 3).
(F) ISG expression in 1833 cells after addition of conditioned media (CM) from ISG-R co-cultures treated with DMSO or POL3 inhibitor (POL3i). Values are relative to 1833 cells in mono-culture (n = 3).
(G) RT-mediated cell death of 1833 cells in mono-culture (Mono) or co-culture with MRC5 cells (Co-cx). Cells were grown in the presence of DMSO or POL3i and with (+CM) or without CM from ISG-R co-culture (n = 3).
(H) ATP hydrolysis assay for recombinant RIG-I activation by increasing amount sof the indicated RNA. ExoRNA and cellular RNA are from 1833 ISG-R co-culture treated with or without POL3i (n = 3). 5’OH is a negative control and 5’ppp is a positive control.
(I) Abundance (Log10) of RNA classes from exoRNA sequencing compared to 5’ppp-seq. RNA classes depleted in 5’ppp-seq by ~10-fold or greater are shown on the left (n = 4).
(J) Relative levels of POL3 transcripts in exosomes harvested from ISG-R co-culture treated with DMSO or POL3i (n = 3). Error bars are SEM of biological replicates and *p < 0.05, **p < 0.01. See also Figure S2.
In the absence of viral infection, the main source of endogenous 5’ppp RNA is from RNA polymerase III (POL3) transcription (White, 2011). Therefore, we sought to examine if stromal POL3 generates the exoRNA that is transferred to breast cancer cells to activate anti-viral signaling. Indeed, the POL3 subunit POLR3G was upregulated in stromal cells after ISG-R co-culture (Figures 2C and S2E). Knockdown of POL3 using a small interfering RNA (siRNA) to the POLR3F subunit (Figure S2F) revealed that inhibiting POL3 in stromal cells, but notin breast cancer cells alone, significantly blunted breast cancer ISG induction (Figure 2D). Interrogation of functional consequences revealed that the ability of stromal cells to protect breast cancer cells after radiation was impaired with stromal POL3 knockdown, but unchanged after breast cancer POL3 knockdown (Figures 2E and S2G). Consistent with these findings, treatment with a POL3 small-molecule inhibitor (Wu et al., 2003) also blunted stroma-mediated resistance and ISG induction in breast cancer cells after co-culture (Figures S2H and S2I). To confirm that exosomes are responsible for the effects resulting from inhibiting stromal POL3, we isolated exosome-containing CM from ISG-R co-culture treated with or without the POL3 inhibitor. CM isolated from co-culture both induced ISGs when added to mono-cultured breast cancer cells (Figure 2F) and re-established stroma-mediated radiation resistance that was abrogated by POL3 inhibition (Figures 2G and S2J). In contrast, CM from ISG-R co-culture treated with POL3 inhibitor failed to induce ISGs, but expression of unrelated genes such as IFI16 was not affected (Figure 2F). To test if exoRNA can directly activate RIG-I in a POL3-dependent manner, we examined the ability of exoRNA to stimulate ATP hydrolysis of recombinant RIG-I. Indeed, addition of exoRNA from ISG-R co-culture, but not exoRNA from POL3 inhibitor-treated co-culture or equimolar amounts of cellular RNA, stimulates RIG-I ATP helicase activity (Figure 2H). Thus, these results suggest that stromal POL3 generates exoRNA that directly activates breast cancer RIG-I and promotes stroma-mediated protection against DNA damage.
To characterize the exoRNA generated by stromal POL3, we developed an approach to identify 5’ppp RNA by sequencing. For this, we utilized a set of enzymatic reactions to sequentially modify the 5’ end of RNA prior to library construction to deplete RNA lacking a 5’ppp modification (5’ppp-seq). Many coding and non-coding RNAs were depleted by ~10-fold or greater, consistent with the absence of a 5’ppp (Figure 2I, left). Examination of RNA classes that maintained or increased abundance revealed many exoRNA transcripts known to be under POL3 regulation, including tRNAs, srpRNA, Y RNA/snRNAs, and ALU/SINE RNAs (Figure 2I, right). As expected, inhibiting POL3 resulted in a decrease in the abundance of several of these 5’ppp RNA in exo-somes (Figure 2J). Thus, multiple 5’ppp exoRNAs regulated by stromal POL3 are present in exosomes and represent candidate RIG-I ligands transferred to breast cancer cells.
RN7SL1 from Stromal Cells Is Transferred to Breast Cancer Cells by Exosomes to Activate RIG-I
As part of a strategy to identify a specific 5’ppp exoRNA from stromal cells that activates breast cancer RIG-I, we examined differences in 5’ppp exoRNA abundance that correlate with the ability of exosomes to induce anti-viral signaling. Specifically, we took advantage of the observation that exosomes and exoRNA from ISG-R co-culture is more effective than exosomes and exoRNA from stromal cell mono-culture at inducing breast cancer ISGs (Figures 3A and S3A). Because 5’ppp-seq may not be quantitative, we first performed RNA sequencing (RNA-seq) from exosomes (exoRNA-seq) isolated from ISG-R co-culture versus stromal mono-culture. Using these data, we specifically examined transcripts that were also identified by 5’ppp-seq. This revealed that most 5’ppp exoRNA does not or only modestly varies in abundance in exosomes from co-culture compared to stromal mono-culture (Figure 3B). In contrast, RN7SL1, an srpRNA, and RN7SL1 pseudo-genes stood out as abundant transcripts that markedly increase in exosomes from co-culture compared to stromal mono-culture (Figures 3B, 3C, and S3B; Table S1). Therefore, to further investigate a potential role for RN7SL1 exoRNA, we metabolically labeled stromal cell RNA with 4sU and assayed for transfer to breast cancer cells. This demonstrated that transfer of RN7SL1 is enhanced after ISG-R co-culture with multiple different stromal cells but not after ISG-NR co-culture (Figure 3D). Tandem pull-down by first immu-noprecipitating FLAG-tagged breast cancer RIG-I and then isolating biotinylated 4sU-labeled stromal RNA with streptavidin beads (Figures 3E, top, and 3F) revealed that stromal RN7SL1 specifically bound to WT RIG-I compared to RIG-IK858/861A (Figure 3G). In contrast, other 5’ppp stromal RNA found in exosomes such as RMRP and RNA without 5’ppp (i.e., capped mRNAs) showed markedly less transfer and binding. Selective stromal RN7SL1 transfer and RIG-I binding was also observed using CM from co-culture of 4sU-labeled stromal cells with ISG-R breast cancer cells (Figures 3E, bottom, and 3G), consistent with exosome-mediated transfer. Thus, stromal RN7SL1 is transferred to ISG-R breast cancer cells by exosomes and binds to RIG-I.
Figure 3. 5’ppp Stromal RN7SL1 Is Transferred to Breast Cancer Cells by Exosomes to Activate RIG-I.
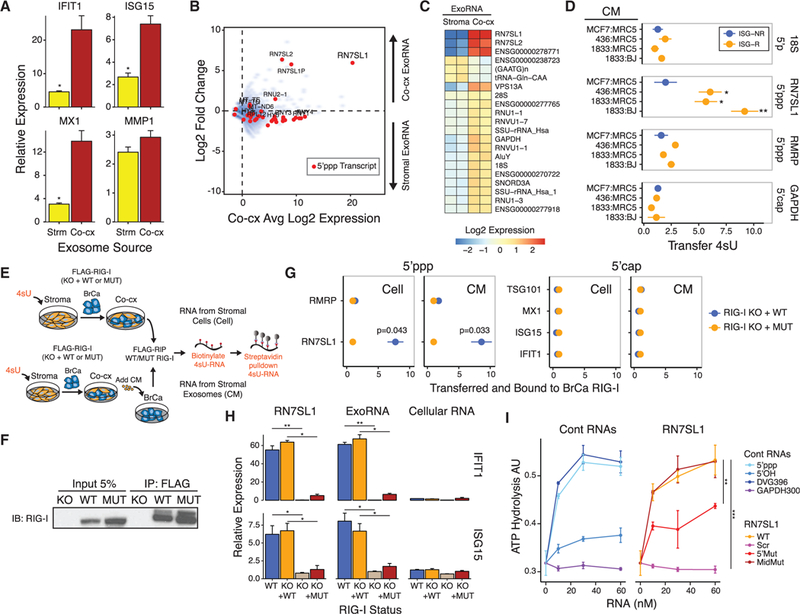
(A) ISG expression in 1833 breast cancer cells after addition of exosomes from MRC5 stromal cell mono-culture(Strm) or from co-culture of1833 and MRC5 cells (Co-cx) (n = 3). Values are relative to mock control.
(B) ExoRNA and 5’ppp exoRNA enriched in ISG-R co-culture exosomes. Shown is average expression (Log2) by exoRNA-seq in co-culture versus fold change from co-culture compared to MRC5 stromal cell mono-culture (n = 2). Transcripts identified by 5’ppp-seq are shown in red. ExoRNA was rRNA-depleted.
(C) Differentially expressed exoRNA from MRC5 mono-culture (Stroma) compared to co-culture of 1833 and MRC5 cells (Co-cx) (n = 2).
(D) Relative transfer of indicated stromal 4sU RNA (right margin) to breast cancer cells after addition of conditioned media (CM) from 4sU-labeled MRC5 or BJ fibroblasts co-cultured with either ISG-R (orange) or ISG-NR (blue) breast cancer cells (n = 3).
(E) Schema to measure 4sU-labeled stromal RNA bound to breast cancer RIG-I after co-culture (Cell, top schema) or after addition of co-culture conditioned media (CM, bottom schema).
(F) Representative immunoprecipitation of FLAG-RIG-I.
(G) Quantitation of indicated 4sU-labeled stromal RNA transferred and bound to wild-type RIG-I (blue) or RIG-IK858/861A (orange) reconstituted in RIG-I KO 1833 cells after co-culture (Cell) or addition of co-culture CM (CM) (n = 3).
(H) ISG expression after transfection of RN7SL1, exoRNA from ISG-R co-culture, or cellular RNA into 1833 cells with wild-type(WT)or RIG-I knockout (KO), or into 1833 RIG-I KO cells restored with either wild-type RIG-I (KO + WT) or RIG-IK858/861A (KO + MUT) (n = 3). Values are relative to mock control.
(I) ATP hydrolysis assay for recombinant RIG-I activation by increasing concentrations of RN7SL1 and mutants (right) or control RNAs (left) (n = 3). Error bars are SEM of biological replicates and *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3 and Table S1.
To confirm that RN7SL1 directly stimulates RIG-I and binds using expected structural features, we produced ribozyme-cleaved in vitro transcribed RN7SL1. Transfection of this synthetic RN7SL1 induced ISGs in breast cancer cells in a RIG-I-dependent manner, similar to ISG-R co-culture exoRNA (Figure 3H). This induction requires a 5’ppp as demonstrated by alkaline phosphatase treatment prior to RN7SL1 transfection (Figure S3C) and by the ability of WT but not RIG-IK858/861A to reconstitute RIG-I KO breast cancer cells (Figure 3H). RN7SL1 binding to RIG-I also requires secondary structure at the 5’ end. In contrast to WT RN7SL1, which exhibits extensive RNA secondary structure and binds recombinant RIG-I comparably to a Sendai virus-derived RNA (DVG396) (Mercado-Lopez et al., 2013), disrupting RN7SL1 secondary structure on the 5’ end but not the middle significantly inhibited RIG-I ATP hydrolysis (Figures 3I and S3D). Scrambling the sequence throughout the length of RN7SL1 diminished RIG-I binding to levels similar to a 300-bp glyceraldehyde 3-phosphate dehydrogenase (GAPDH) negative control RNA with a 5’ppp (GAPDH300). In total, these results indicate that RN7SL1 activates RIG-I using structural features expected of RIG-I ligands.
Exosomes from Activated Stromal Cells Contain Unshielded RN7SL1
Although RN7SL1 in ISG-R co-culture exosomes is transferred from stromal to breast cancer cells and activates RIG-I, it was unclear why this highly abundant cellular 5’ppp RNA does not persistently activate RIG-I in the cytoplasm. We reasoned that binding by RNA binding proteins (RBPs) might influence the ability of RN7SL1 to activate anti-viral signaling. To examine this, we treated cells or exosomes with micrococcal nuclease (MNase) with or without membrane permeabilization prior to sequencing (MNase-seq). This revealed variable MNase-dependent RNA degradation in cells and exosomes, which is indicative of differing degrees of RNA “shielding” by RBPs (Figure 4A). Examination of 5’ppp RNA shielding along with predicted RNA secondary structure as measured by normalized minimum free energy (MFE), demonstrated that RN7SL1 stands out as a 5’ppp exoRNA that has extensive secondary structure (low MFE) and is markedly shielded in cells but highly unshielded in co-culture exosomes (Figures 4A and S4A; Table S1). In contrast, RN7SL1 exoRNA from stromal mono-culture shows more shielding similar to cytoplasmic RN7SL1 (Figure 4B), and most other 5’ppp exoRNA have less predicted secondary structure and/or are significantly more shielded in exosomes compared to RN7SL1. RNA without a 5’ppp (i.e., 5’ cap mRNAand 5’-monophosphate rRNA) generally are equally unshielded in cells and exosomes (Figures 4A, 4B, and S4A). Unshielding of RN7SL1 exoRNA was also observed when other ISG-R breast cancer cells were co-cultured with stromal fibroblasts (Figure S4B) and when primary mouse lung fibroblasts were co-cultured with K14cre;p53F/F;Brca1F/F murine ISG-R breast cancer cells (Figures S4C–S4F). In contrast, exosomes from ISG-NR co-cultures demonstrated significantly less unshielding compared to exoRNA from ISG-R co-cultures (Figure S4B). This, along with diminished exosome transfer (Figure 1B), correlates with the lack of ISG induction after ISG-NR coculture. In total, these results suggest that cytoplasmic RN7SL1 is normally extensively shielded by RBPs, which may prevent its recognition by RIG-I. However, after interaction with ISG-R breast cancer cells, stromal cells selectively deploy unshielded RN7SL1 in exosomes.
Figure 4. SRP9 and SRP14 Regulate RN7SL1 Shielding in Cells and Exosomes and the Ability to Stimulate RIG-I.
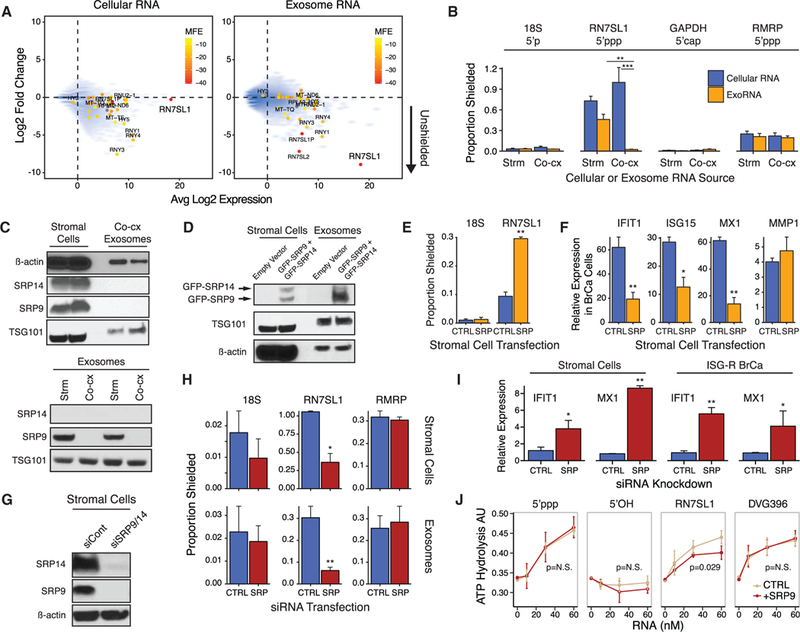
(A) Expression of cellular RNA (left) or exoRNA (right) from 1833 ISG-R co-culture versus degree of RNA binding protein (RBP) shielding. RBP shielding (y axis) is determined by fold change in RNA expression after MNase treatment with or without detergent (n = 2). Smaller y axis values indicate more unshielding. Transcripts identified by 5’ppp-seq are denoted by solid circles and color-coded based on normalized minimum free energy (MFE) whereby lower MFE predicts more extensive secondary structure.
(B) Extent of RBP-shielding of indicated RNAs in cells(Cellular RNA) or exosomes (ExoRNA) isolated from eitherMRC5 stromal mono-culture (Strm) or co-culture with 1833 breast cancer cells (Co-cx). Proportion shielded is determined by MNase treatment with and without detergent followed by qRT-PCR (MNase-qRT- PCR) (n = 3).
(C) Immunoblot of SRPs and indicated proteins in MRC5 stromal cells and exosomes from ISG-R co-culture (top), or in exosomes from stromal mono-culture (Strm) or ISG-R co-culture (Co-cx) (bottom). Each lysate is a biological replicate normalized to levels of RN7SL1 RNA.
(D) Immunoblot of indicated proteins in stromal cells and exosomes after 1833 ISG-R co-culture using MRC5 stromal cells transfected with GFP-SRP9 and GFP-SRP14.
(E and F) RBP-shielding for the indicated exoRNAs (E) or (F) relative expression of ISGs in sorted 1833 breast cancer cells after co-culture using MRC5 cells transfected with control vector (CTRL) orGFP-SRP9 and GFP-SRP14(SRP) (n = 3). Expression values are relative to 1833 cells in mono-culture. MMP1 is a non-ISG not expected to change.
(G and H) Expression ofSRP9 and SRP14protein in MRC5 cells after siRNA knockdown (G), and (H) resulting RBP-shielding for the indicated RNA in MRC5 cells (top) or exosomes (bottom) (n = 4).
(I) ISG induction in MRC5 cells after SRP9/14 knockdown or in 1833 cells after addition of conditioned media from MRC5 knockdown cells (n = 4).
(J) Stimulation of recombinant RIG-I ATP hydrolysis activity by RN7SL1 with or without addition of equimolar amounts of recombinant SRP9 (n = 3). Error bars are SEM of biological replicates and *p < 0.05, **p < 0.01. See also Figure S4.
SRP9 and SRP14 Control RN7SL1 Shielding and RIG-I Binding
Because the ability of stromal RN7SL1 to stimulate RIG-I maybe masked by RBP shielding, we reasoned that the expression of this RBP may be associated with RN7SL1 stimulatory activity. Specifically, this RBP would be present in the cytoplasm and exosomes from mono-cultured stromal cells but be absent in stromal exosomes produced after ISG-R co-culture. RN7SL1 typically complexes with signal recognition particle (SRP) proteins to control co-translational protein translocation (Akopian et al., 2013). Two SRP proteins, SRP9 and SRP14, normally bind the 5’ end of RN7SL1, potentially obscuring the 5’ppp. Accordingly, relatively high levels of SRP9 and SRP17 were detected in cellular extracts, and SRP9 was present in exosomes from stromal cell mono-cultures (Figure 4C). In contrast, neither SRP9 nor SRP14 were detectable in exosomes from ISG-R co-culture (Figure 4C). Therefore, we transiently overexpressed GFP-tagged SRP9 and SRP14in stromal cells prior to co-culture to determine if this could drive these SRP proteins into exosomes (Figures S4G–S4J). Indeed, expression of tagged SRP9, and to a lesser extent tagged SRP14, was increased in co-culture exosomes (Figure 4D), and this led to a significant increase in shielding of RN7SL1 exoRNA but not in 18S rRNA (Figure 4E). Consequently, stroma-mediated ISG induction in co-cultured breast cancer cells was reduced, while expression of non-ISGs such as MMP1 was not affected (Figure 4F). Conversely, transient knockdown of SRP9/14 in mono-cultured stromal cells enhanced unshielding of cytoplasmic and exoso-mal RN7SL1 (Figures 4G and 4H), resulting in ISG induction both in stromal cells and in mono-cultured ISG-R breast cancer cells upon addition of CM produced after SRP9/14 knockdown (Figure 4I). We were also able to purify recombinant SRP9 (Figure S4K). Addition of SRP9 to in vitro transcribed RN7SL1 partially inhibited ATP hydrolysis by recombinant RIG-I but did not influence Sendai virus-derived RNA (DVG396) or unrelated 5’ppp or 5’OH control RNAs (Figure 4J). These results suggest that RBP shielding of cellular RN7SL1 by SRP9/14 restricts inappropriate recognition by RIG-I in the cytoplasm. After interaction with ISG-R breast cancer cells, RN7SL1 devoid of SRP9/14 is deployed into stromal exosomes, resulting in unshielded RN7SL1 that can activate RIG-I.
NOTCH1-MYC Couples Stromal Activation with Deployment of Unshielded RN7SL1 ExoRNA
We sought to investigate the regulation of RN7SL1 unshielding and why unshielded RN7SL1 is markedly higher in exosomes produced after ISG-R co-culture compared to ISG-NR co-culture or stromal cell mono-culture. Genome-wide transcriptomic profiling of stromal cells after ISG-R co-culture compared to mono-culture revealed an extensive transcriptional upregulation characteristic of stromal cell activation (Figure 5A). This was accompanied by ISG induction in both cell types. Stromal activation also resulted in an approximate doubling of RN7SL1 levels, which was already highly abundant in cells, as well as other POL3 transcripts (Figure 5B). In contrast, ISG-NR co-culture led to only modest transcriptional responses in stromal and breast cancer cells and no increase in RN7SL1. Transcriptional changes in stromal cells activated by ISG-R co-culture included genes associated with the NOTCH juxtacrine signaling pathway (Figures S5A and S5B). This was particularly notable given that ISG induction in ISG-R co-culture required cell-cell contact (Figure S5C). Indeed, cleavage of NOTCH1 to liberate its transcription activation domain (NICD1) occurred in stromal cells after ISG-R co-culture, while cleavage of other NOTCH family members was absent or marginal compared to breast cancer cells (Figure 5C). The appearance of NICD1 in activated stromal cells was accompanied by upregulation of MYC target genes (Figures S5A and S5B), which is consistent with MYC being a NOTCH transcriptional target (Weng et al., 2006), and by the nuclear accumulation of MYC (Figure 5D, top), which also required cell-cell contact (Figure S5D) and was inhibited by blocking NOTCH activation with a gamma secretase inhibitor (GSI) (Figure 5D, bottom). Thus, stromal cells are activated by ISG-R breast cancer cells, resulting in coordinated NOTCH1-MYC signaling, increased levels of cytoplasmic RN7SL1, and induction of ISGs in both cell types.
Figure 5. NOTCH1-MYC Signaling Couples Stromal Activation with Unshielding of POL3-Driven Stromal RN7SL1.
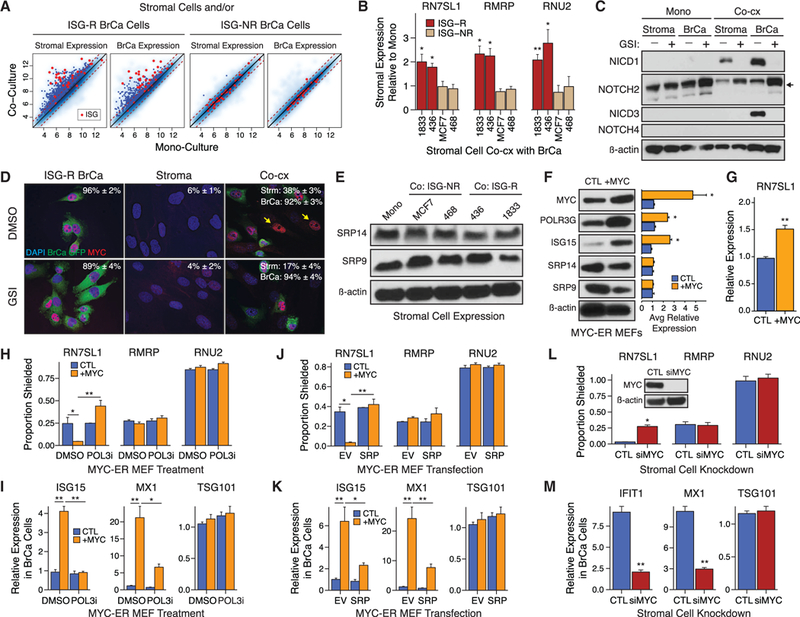
(A) Gene expression in MRC5 stromal cells (panels 1 and 3), ISG-R 1833 breast cancer cells (panel 2), or ISG-NR breast cancer cells (panel 4) after mono-culture versus co-culture. Genes significantly upregulated after co-culture are indicated by blue dots. Cancer-associated ISGs are marked red.
(B) RN7SL1 levels in MRC5 stromal cells after co-culture with ISG-R or ISG-NR breast cancer cells.
(C) Immunoblot of NOTCH family members ortheiractivation domain (NICD1, NICD3) in MRC5 stromal or ISG-R 1833 breast cancer cells. Activation of NOTCHI/ NOTCH3 is indicated by appearance of NICD1/NICD3 in absence of GSI, while activation of NOTCH2 is indicated by accumulation of cleavage product (arrow) with GSI.
(D) Immunofluorescence and quantitation of MYC+ nuclei in MRC5 stromal or 1833 breast cancer cells with or without GSI.
(E) SRP9 and SRP14 protein levels in MRC5 stromal cells after mono-culture or after ISG-R or ISG-NR co-culture.
(F and G) Immunoblot for the indicated proteins with quantification of biological replicates(right) (F), or(G) cellular RN7SL1 expression in MYC-ER MEFs treated with vehicle control (CTL) or 4OHT (+MYC).
(H-K) RBP-shielding of exoRNA isolated from MYC-ER MEFs (H), or (I) ISG expression in ISG-R breast cancer cells after addition of MYC-ER MEF exosomes. MYC-ER MEFs were treated with vehicle control (CTL) or 4OHT (+MYC) with or without POL3 inhibitor (POL3i) (n = 3). TSG101 is a non-ISG not expected to change. Experiments in (H) and (I) were repeated in (J) and (K) but with overexpression of SRP9/14 (SRP) or empty vector (EV) (n = 3).
(L) RBP-shielding of exoRNA isolated from ISG-R co-cultures using MRC5 cells treated with control (CTL) or MYC siRNA(n = 3). Inset: stromal MYC protein after knockdown.
(M) ISG expression in 1833 breast cancer cells after addition of conditioned media from ISG-R co-cultures using siRNA targeting MYC (siMYC)-treated MRC5 cells (n = 3).
Error bars are SEM of biological replicates and *p < 0.05, **p < 0.01. See also Figure S5.
Previous studies have established that MYC can enhance POL3 transcriptional activity (Gomez-Roman et al., 2003). Because RN7SL1 is a POL3 transcript, this suggests that NOTCH1-MYC signaling may couple stromal activation to unshielding of RN7SL1 and ISG induction by increasing RN7SL1 to alter its stoi-chiometry with SRP9/14. Indeed, although RN7SL1 expression dynamically responds to stromal activation, SRP9 and SRP14 levels remain invariant in stromal cells regardless of culture conditions (Figure 5E). Therefore, to test if MYC activation is sufficient to unshield RN7SL1 in a POL3- and SRP9/14-dependent manner, we employed mouse embryonic fibroblasts (MEFs) with a MYC-ER construct allowing for inducible MYC activation with 4-OHT (Figure S5E). Induction of MYC in mono-cultured MEFs was sufficient to increase the POL3 subunit POLR3G and RN7SL1, while SRP9/ 14 protein remained constant (Figures 5F and 5G). Consequently, MYC activation generated unshielded RN7SL1 in exosomes (Figure 5H) and induced ISGs in stromal MEFs (Figure 5F) and in breast cancer cells after addition of the stromal exosomes (Figure 5I). Importantly, both unshielding of RN7SL1 exoRNA and exosome-mediated ISG induction were inhibited by either treatment of stromal MEFs with a POL3 inhibitor (Figures 5H and 5I) or by increasing cytoplasmic expression of SRP9/14 using a lentiviral transgene (Figures 5J, 5K, and S5F). Conversely, transient knockdown of MYC in MRC5 stromal cells prior to ISG-R co-culture interfered with unshielding of RN7SL1 exoRNA and prevented ISG induction in breast cancer cells after co-culture (Figures 5L and 5M). With all manipulations, shielding status of other 5’ppp RNA (such as RNU2 and RMRP) and RNA without a 5’ppp was not affected, nor was expression of non-ISGs (Figures 5H–5M). Thus, these data suggest that stromal NOTCH1-MYC signaling can enhance POL3-driven RN7SL1 levels while cytoplasmic SRP9/14 levels remain constant. Modulating the stoichiometry between RN7SL1 and its RBPs regulates RN7SL1 unshielding, coupling stromal activation with deployment of unshielded RN7SL1 in exosomes.
Unshielded Stromal RN7SL1 Deployed in Exosomes Has Immune and Tumor Cell Functions
Cardinal features of stromal fibroblasts in the tumor microenvironment include the ability to promote inflammation, cancer progression, and metastasis. To examine if exosomes from activated stromal cells and unshielded RN7SL1 can directly influence inflammation by functioning as DAMPs, we intravenously injected either exosomes from MYC-activated MEFs or liposome-encapsulated unshielded RN7SL1. This was followed by flow cytometry of splenic myeloid/dendritic cells (DCs) combined with unbiased cluster analysis to avoid arbitrary classification of myeloid/DC populations, which consists of a spectrum of different subsets (Becher et al., 2014). For this, we overlaid expression of lineage-related and activation markers on a tSNE low-dimensional map and determined the proportion of cells in distinct clusters (Figure 6A). Injection of exosomes from stromal cells without MYC activation had modest effects on the composition and activation of myeloid/DC subsets compared to injection of PBS (Figures 6B and 6C). In contrast, exosomes from MYC-activated MEFs increased the proportion of cells in myeloid/DC subsets expressing high levels of various maturation/activation markers such as CD40, CD86, PDL1, and MHCII. Similar results were observed with liposome-encapsulated unshielded RN7SL1 and the viral RNA mimic poly I:C but not with GAPDH300 control RNA (Figures 6B and S6A). Thus, these data suggest that MYC-activated stromal cells can deploy unshielded RN7SL1 in exosomes to function as a DAMP.
Figure 6. Unshielded RN7SL1 in Exosomes Functions as a DAMP and Promotes Breast Cancer Progression and Metastasis.
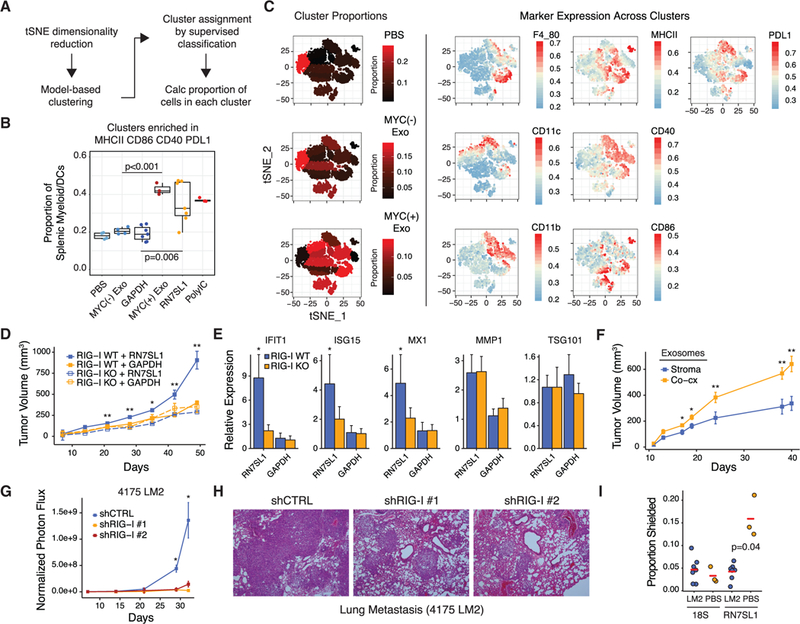
Fora Figure360 presentation of Figure 6, see http://dx.doi.org/10.1016/j.cell.2017.06.031#mmc4.
(A) Schema for unbiased flow cytometry analysis of splenic myeloid/DC populations using tSNE dimensionality reduction, cluster identification, and supervised classification.
(B) Summary of the proportion of cells in splenic myeloid/DC clusters enriched in MHCII, CD86, CD40, and PDL1 after injection of the indicated exosomes or RNA.
(C) Representative data for exosome injected groups. See Figure S6 for other groups. Shown are proportions of cells in each cluster as represented by the color gradient on the tSNE plot (left), which maps cells to a two-component dimensionality reduced space. Expression of the indicated markers is overlaid on the tSNE plot to visualize color-coded mean fluorescence intensities across clusters (right).
(D and E) Tumor growth curves (D) and (E) expression of ISGsfor 1833 ISG-R breast cancer cells with or without RIG-I knockout xenografted into athymic mice and intratumorally injected three times per week with the indicated liposome-encapsulated RNA (n = 5 per group). ISGs were measured by qRT-PCRwith human- specific primers. TSG101 and MMP1 are non-ISGs not expected to change.
(F) Tumors growth curves of 1833 tumors injected with exosomes from MRC5 stromal cells or 1833 ISG-R co-culture (n = 5 per group).
(G and H) Normalized photon flux (G) or (H) H&E staining of mouse lungs after tail vein injection of luciferase-labeled 4175 LM2 ISG-R breast cancer cells expressing one of two shRNAs to RIG-I or a control shRNA (n = 5 per group).
(I) RBP-shielding of mouse RN7SL1 or 18S rRNA from serum exosomes 2 weeks after tail vein injection with 4175 LM2 breast cancer cells or with PBS. Mouse-specific primers were used.
Error bars are SEM of biological replicates and *p < 0.05, **p < 0.01. See also Figure S6.
Figure360: an author presentation of Figure 6.
To examine whether unshielded RN7SL1 in exosomes can contribute to tumor growth and metastasis, liposome-encapsulated unshielded RN7SL1 or GAPDH300 control RNA was injected intratumorally into subcutaneous 1833 ISG-R breast cancer xenografts. Only RN7SL1 enhanced tumor growth in a RIG-I-dependent manner as no effect was observed in RIG-I KO tumors or with GAPDH300 (Figure 6D). Examination of the tumor confirmed an increase in ISG expression, but not in unrelated genes like TSG101, specifically in tumors injected with RN7SL1 and expressing WT RIG-I (Figure 6E). Similar results were obtained with injection of exosomes from ISG-R co-culture compared to exosomes isolated from stromal cells alone (Figure 6F). To examine metastatic progression, we utilized 4175 LM2 human breast cancer cells, which are an ISG-R lung metastatic derivative of MDA-MB-231 (Minn etal., 2005). Inhibiting RIG-I expression in 4175 cells using two independent small hairpin RNAs (shRNAs) (Figure S6B) resulted in a significant defect in lung metastatic colonization, indicating the importance of RIG-I signaling in breast cancer cells (Figures 6G and 6H). Compared to non-tumor bearing mice, interrogation of exoRNA from serum of mice with wild-type 4175 lung metastases revealed more unshielding of mouse RN7SL1, but not 18S rRNA, as measured using mouse-specific primers (Figure 6I). This suggests that lung metastases can increase circulating unshielded RN7SL1 in exosomes originating from mouse stromal cells. In total, these findings demonstrate that unshielded RN7SL1 in exosomes and RIG-I activation in breast cancer cells can influence breast cancer growth and metastasis.
Stroma from Human Tumors Show Evidence for NOTCH-MYC Regulation of Anti-viral Signaling and Production of Unshielded RN7SL1 in Exosomes
To provide evidence for NOTCH1-MYC signaling in stromal cells and associated anti-viral signaling in primary human breast cancers, we utilized a previously published transcriptomic dataset of laser-captured micro-dissected tumors (Finak et al., 2008). This revealed that the average expression of NOTCH target genes (metagene) in stromal cells strongly correlated with a stromal metagene for MYC targets (Figure 7A). This stromal MYC metagene associated with an ISG metagene in stromal and breast cancer cells and with myeloid and lymphoid metagenes in the stromal compartment. Consistent with these gene expression changes from primary tumors, ISG-R co-cultures with a panel of carcinoma-associated fibroblasts (CAFs) isolated after surgical resection of primary human breast cancers (Tchou et al., 2012) confirmed induction of ISGs (Figure 7B), activation of stromal NOTCH1 and MYC (Figure 7C), and production of unshielded RN7SL1 in exosomes (Figure 7D). In contrast, fibroblasts isolated from the contralateral breast of one of three breast cancer patients (N4) did not exhibit these properties, corroborating the association between NOTCH1-MYC and generation of unshielded RN7SL1 in exosomes.
Figure 7. Stromal NOTCH1-MYC Signaling, Inflammatory Gene Expression, and Unshielded RN7SL1 ExoRNA from Primary Human Breast Cancers.
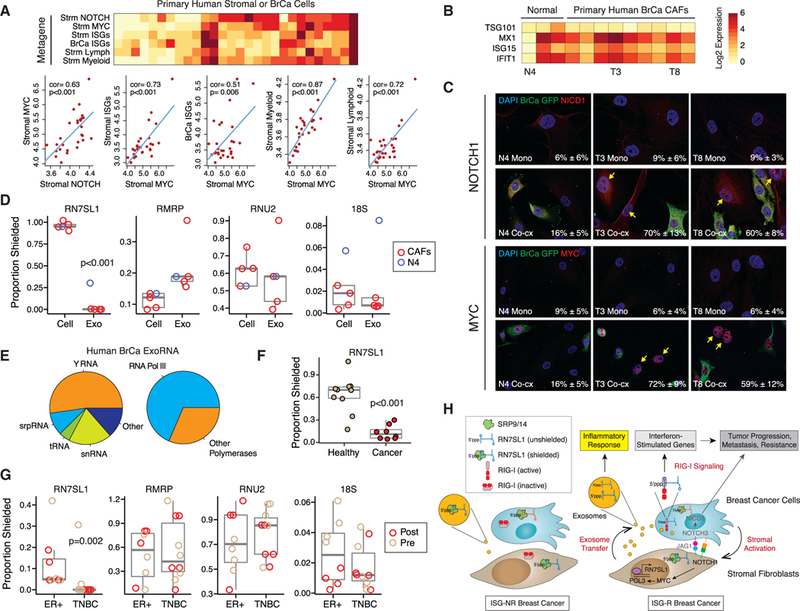
(A) Heatmap and correlation plots of metagenes for NOTCH, MYC, ISGs, or myeloid or lymphoid genes In stromal (Strm) or breast cancer (BrCa) cells from laser- captured micro-dissected human tumors. Red is high expression and yellow is low. Univariate correlations and p values are shown. Significance was confirmed by gene set enrichment analysis compared to random genes.
(Band C) ISG expression after co-culture of 1833ISG-R breast cancer cells with either CAFs from surgically resected primary human breast cancers or fibroblasts from the contralateral breast (Normal). (B) Select fibroblasts from each group are labeled below the heatmap and (C) used to examine activated NOTCH1 (NICD1) and nuclear MYC expression after mono-culture (Mono) or co-culture with GFP-labeled 1833 breast cancer cells (Co-cx). Percentage of NICD1(+) or MYC(+) stromal cells averaged from multiple high-powered fields is indicated in the lower right corner (n = 2).
(D) RBP-shielding of cellular RNA(Cell) from primary CAFs or exoRNA (Exo) isolated from 1833 ISG-R co-cultures. Values from N4 fibroblast (blue) isolated from normal breast is shown for comparison.
(E) Average distribution of exoRNA in each RNA class (left) or by POL3 regulation (right) from serum exosomes of breast cancer patients (n = 2). Only the top 200 highest expressed non-rRNA transcripts are considered.
(F and G) RBP-shielding of exoRNA from cancer patients or normal volunteers without cancer (Healthy) (F), or (G) from a cohort of breast cancer patients stratified by ER(+) or triple negative breast cancer (TNBC) subtype. For the breast cancer cohort, samples collected before (Pre) or after (Post) surgery are labeled. See Tables S2 and S3 for patient details.
(H) Model for coupling stromal activation to regulation and deployment of unshielded RN7SL1 exoRNA. See text for details. Genes labeled in gray are primarily described in our previous study (Boelens et al., 2014).
To determine if unshielded RN7SL1 can be detected in patient exosomes, we obtained serum from two cohorts of cancer patients (Tables S2 and S3). ExoRNA-seq from two patients confirmed that RN7SL1 and POL3 transcripts are present at high levels among the non-rRNA transcripts (Figure 7E). For shielding status, we focused on samples primarily collected after surgical resection of gross tumor to facilitate assessment of RN7SL1 exoRNA from stromal cells and to better allow comparison to normal controls without cancer. Compared to healthy controls, RN7SL1 exoRNA was significantly less shielded in cancer patients, suggesting that RN7SL1 from remaining can-cerized stroma is more unshielded than from normal cells (Figure 7F). These results were confirmed using exosomes from breast cancer patients after primary tumor resection, with a similar pattern noted using pre-surgical exosomes as well (Figure 7G). Interestingly, more unshielded RN7SL1 exoRNA was detected in patients with triple negative breast cancer (TNBC) compared to ER-positive tumors. This is consistent with the fact that ISG-R breast cancers are enriched in the TNBC subtype, while ISG-NR tumors tend to be ER-positive (Boelens et al., 2014). Together, these findings suggest that in primary human cancers, activated stromal cells can deploy unshielded stromal RN7SL1 in exosomes to propagate anti-viral signaling in the tumor microenvironment (Figure 7H).
DISCUSSION
Endogenous RNA must avoid recognition by PRRs under non-pathological conditions. Therefore, our discovery that RN7SL1 is a cancer-associated DAMP presented a conceptual problem. Specifically, given its abundance in the cytoplasm, it was unclear how RN7SL1 could both function as a DAMP in exosomes but at the same time avoid recognition by RIG-I while in the cytoplasm. Indeed, it has long been recognized that RNA modification and subcellular localization may be insufficient to prevent inappropriate activation by endogenous and abundant POL3 5’ppp transcripts, arguing that unknown mechanisms must exist (Hornung et al., 2006). Our findings on how differential RBP shielding of endogenous RN7SL1 can control DAMP activity and PRRactiva-tion provide an explanation for how this discrimination can be achieved. These data also indicate that the stimulatory effects of high affinity RNA ligands for RIG-I measured in vitro, may be superseded in vivo by RBP shielding. Thus, control of RBP shielding may be a critical regulatory layer that prevents inappropriate PRR activation, especially of abundant RNAs, while concurrently allowing for a readily available and rapidly deployable DAMP.
Our work suggests that one signal for the deployment of unshielded RN7SL1 exoRNA as a cancer-associated DAMP is stromal activation by cancer cells. We show that ISG-R breast cancers, which are enriched in the TNBC subtype, activate stromal cell NOTCH1 and its target MYC. This juxtacrine signaling is consistent with the requirement for cell-cell contact between stromal and breast cancer cells to induce ISGs and therapy resistance. Indeed, abnormal cell-cell contact between epithelial cells and fibroblasts, which are often separated by a basement membrane, typically occurs under pathological situations such as wounding or invasive carcinoma. Thus, heterotypic interaction itself may represent a “damage” signal that initiates DAMP release by the stromal compartment. MYC activation is accompanied by pronounced transcriptional upregulation characteristic of cellular activation. Interestingly, POL3 activity is augmented by MYC (Gomez-Roman et al., 2003) and by nearby RNA polymerase II (POL2) occupancy (Oler et al., 2010). This suggests that high MYC and POL2 transcriptional output not only from NOTCH1 activation but potentially other oncogenic/ mitogenic signals (Moroishi et al., 2016) occurring after interaction with ISG-R breast cancer cells may enhance POL3-driven RN7SL1 levels in stromal cells. Because SRP9/14 levels do not significantly change with stromal cell activation, the dynamic upregulation of RN7SL1 alters its stoichiometry with SRP proteins. This results in accumulation of unshielded RN7SL1 in the cytoplasm and secreted exosomes, explaining why ISG induction occurs in both stromal cells and breast cancer cells and how it can function as a DAMP. Although experimentally modulating either RN7SL1 (through MYC or POL3) or SRP9/14 (through knockdown or ectopic expression) supports the notion that simple mass action between RN7SL1 and its RBPs controls unshielding, we cannot rule out additional more complex regulatory mechanisms. Thus, juxtracrine signaling involving oncogenic pathways such as NOTCH-MYC may represent a ‘‘damage’’ signal that couples inappropriate stromal and cancer cell interaction with the unshielding and release of RN7SL1 as an exoRNA DAMP.
Activated stromal cells can regulate a multitude of important features of cancer, including inflammation, progression, metastasis, and therapy resistance. The deployment of unshielded stromal RN7SL1 systemically and into the tumor microenvironment may regulate some of these stromal-mediated effects. We show that exosomes from MYC-activated stromal cells and unshielded RN7SL1 can increase the proportion of myeloid/DC populations expressing maturation and activation markers in the spleen, which is a typical inflammatory property of damage- and pathogen-associated molecular patterns. In human tumors, stromal MYC metagene expression also correlates with myeloid and lymphoid genes in the stromal compartment. However, given the multitude of factors within a tumor microenvironment that might alter or facilitate immune cell function, whether the effects of unshielded RN7SL1 exoRNA favor immunosuppression (e.g., MDSC development) or immune stimulation (e.g., T cell activation) requires further investigation. In cancer cells, our previous work demonstrates that RIG-I activation by exoRNA can amplify NOTCH3 signaling, resulting in expansion of tumor-initiating cells and therapy resistance. Building on these findings, we show that transfer of unshielded RN7SL1 exoRNA to breast cancer cells and/or RIG-I signaling promotes both tumor growth and metastasis. Interestingly, cell-cell interaction between breast cancer and astrocytes can facilitate brain metastasis through transfer of the nucleoside second-messenger cGAMP via gap junctions (Chen etal., 2016). Other instances of DAMPsand RNA activating stromal or host cell PRRs to enhance metastasis have also been described (Liu et al., 2016). Thus, the horizontal dissemination of DAMP signals can influence multiple properties of aggressive cancers. Among a subset of breast cancers, these DAMP signals can originate from stromal activation by juxtacrine NOTCH1-MYC that results in the unshielding of RN7SL1 in exosomes. Evidence for NOTCH1 -MYC signaling in stromal cells and CAFsfrom breast cancer patients, and the presence of unshielded RN7SL1 in exosomes from patient blood, highlight the potential clinical relevance for coupling stromal activation to exoRNA DAMP signaling.
The ability to horizontally transfer DAMPs is a shared feature between host-virus infection and tumor-stromal cell interaction. This ability of stromal cells in the tumor microenvironment to borrow cues from virally infected cells is an example of “virus mimicry.” Indeed, virions have been described to contain not only RN7SL1 in the absence of SRP proteins but multiple other endogenous non-viral RNAs (Telesnitsky and Wolin, 2016). It has been postulated that these non-viral RNAs in virions may stimulate innate immune signaling. Our results would support this notion and suggest that RN7SL1 in virions may act as a potent activator of RIG-I like it does in exosomes. Alternatively, in addition to containing viral RNA, exosomes secreted by infected cells may also package unshielded RN7SL1 capable of RIG-I activation. Therefore, whether in virons or in exosomes, cells under viral attack may help to ensure a broad anti-viral response by packaging endogenous DAMPs alongside viral RNA PAMPs. In support of this concept, cells infected by viruses can package cGAMP into secreted virions to trigger a STING- dependent interferon response in recipient cells (Bridgeman et al., 2015; Gentili et al., 2015). Thus, RBP unshielding and horizontal transfer of DAMPs to propagate anti-viral signaling may be a key feature of both host-viral and tumor-stromal interaction.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β-actin | Cell Signaling | Cat#4970; RRID: AB_2223172 |
| SRP9 | Proteintech | Cat#11195–1-AP; RRID: AB_2239820 |
| SRP14 | Proteintech | Cat#11528–1-AP; RRID: AB_2194708 |
| TSG101 | Proteintech | Cat#14497–1-AP; RRID: AB_2208090 |
| GFP | Abcam | Cat#ab6673; RRID: AB_305643 |
| GFP | Abcam | Cat#ab13970; RRID: AB_300798 |
| RPC32 | Santa Cruz | Cat#sc-21754; RRID: AB_675824 |
| RIG-I | Cell Signaling | Cat#3743; RRID: AB_2269233 |
| ISG15 | Santa Cruz | Cat#sc-50366; RRID: AB_2126309 |
| FLAG | Sigma | Cat#F1804; RRID: AB_262044 |
| HA | Santa Cruz | Cat#sc-7392; RRID: AB_627809 |
| c-MYC | Abcam | Cat#ab32072; RRID: AB_731658 |
| NICD1 | Cell Signaling | Cat#4147; RRID: AB_2153348 |
| NICD1 | Abcam | Cat#ab8925; RRID: AB_306863 |
| NOTCH2 | Cell Signaling | Cat#5732; RRID: AB_10693319 |
| NICD3 | Chris Siebel | N/A |
| NOTCH4 | Cell Signaling | Cat#2423; RRID: AB_2151366 |
| CD11c-FITC | BioLegend | Cat#117306; RRID: AB_313775 |
| CD44-PerCp-Cy5.5 | BioLegend | Cat#103031; RRID: AB_2076206 |
| MHCII-BV421 | BioLegend | Cat#107631; RRID: AB_10900075 |
| CD40-BV711 | BD Biosciences | Cat#740700; |
| CD11b-APC | BD Biosciences | Cat#553312; RRID: AB_398535 |
| CD45.2-AF700 | BioLegend | Cat#109822; RRID: AB_493731 |
| CD86-APC-Cy7 | BioLegend | Cat#105030; RRID: AB_2244452 |
| PDL1-PE | BD Biosciences | Cat#558091; RRID: AB_397018 |
| CD8a-PE-ef610 | eBioscience | Cat#61–0081-80; RRID: AB_2574523 |
| F4/80-PE-Cy7 | BioLegend | Cat#123114; RRID: AB_893478 |
| Bacterial and Virus Strains | ||
| Sendai virus Cantell | Mercado-López et al., 2013,Carolina Lopez | N/A |
| One Shot Stbl3 Chemically Competent E. coli | ThermoFisher | Cat#C737303 |
| DH5α Competent Cells E. coli | ThermoFisher | Cat#18265017 |
| BL21 Competent E. coli | NEB | Cat#C2530H |
| Biological Samples | ||
| Patient serum | Obtained through the UPENN RadOnc Biosample Repository or the Breast Program Translational Resource Center. Informed consent and HIPAA authorization was procured for all study subjects. | N/A |
| Serum from healthy volunteers | Innovative Research | Cat#IPLA-SERS |
| Cancer-associated fibroblasts | Tchou et al., 2012, Julia Tchou | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant RIG-I | Devarkar et al., 2016, Joseph Marcotrigiano | N/A |
| Recombinant SRP9 | This paper | N/A |
| DAPT | Sigma | Cat#D5942 |
| TEV Protease | Sigma | Cat#T4455 |
| Glutathione Sepharose 4 Fast Flow | GE Healthcare | Cat#17–5132-01 |
| L-Glutathione | Sigma | Cat#G4251 |
| 4-Thiouridine | Sigma | Cat#T4509 |
| Micrococcal Nuclease | NEB | Cat#M0247S |
| ProLong Diamond Antifade Mountant with DAPI | ThermoFisher | Cat#P36962 |
| D-Luciferin | PerkinElmer | Cat#122796 |
| RNA Polymerase III Inhibitor (ML-60218) | Millipore | Cat#557403 |
| 4-Hydroxytamoxifen | Sigma | Cat#H7904 |
| 5-(and-6)-carboxyf luorescein diacetate, succinimidyl ester (CFSE) | ThermoFisher | Cat#C34554 |
| Critical Commercial Assays | ||
| Click-iT RNA Alexa Fluor® 488 Imaging Kit | ThermoFisher | Cat#C10329 |
| Anti-FLAG M2 Magnetic Beads | Sigma | Cat#M8823 |
| EnzChek Phosphate Assay Kit | ThermoFisher | Cat#E6646 |
| NEBNext Ultra Directional RNA Library Prep Kit | NEB | Cat#E7420L |
| MEGAshortscript T7 Transcription Kit | ThermoFisher | Cat#AM1354 |
| Dynabeads MyOne Streptavidin C1 | ThermoFisher | Cat#65001 |
| SYTOX Red Dead Cell Stain | ThermoFisher | Cat#S34859 |
| Vybrant Multicolor Cell-Labeling Kit | ThermoFisher | Cat#V22889 |
| Deposited Data | ||
| ExoRNA-seq, MNase-seq, 5’ppp-seq, Patient ExoRNA-seq | This paper | GEO: GSE93078 |
| ISG-R and ISG-NR gene expression data | Boelens et al., 2014 | GEO: GSE60998 |
| Stromal and breast cancer gene expression data from laser-capture micro-dissection | Finak et al., 2008 | GEO: GSE9014 |
| Experimental Models: Cell Lines | ||
| MDA-MB-231 (1833) | Kang et al., 2003, Yibin Kang | N/A |
| MDA-MB-231 (4175 LM2) | Minn et al., 2005 | N/A |
| MDA-MB-436 | ATCC | ATCC HTB-130 |
| HCC1937 | ATCC | ATCC CRL-2336 |
| MCF7 | ATCC | ATCC HTB-22 |
| MDA-MB-468 | ATCC | ATCC HTB-132 |
| MRC5 | ATCC | ATCC CCL-171 |
| BJ | ATCC | CRL-2522 |
| KB1P | Liu et al., 2007, Jos Jonkers | N/A |
| MYC-ER MEF | Gomez-Roman et al., 2003, Costas Koumenis | N/A |
| Adult Lung Fibroblasts | Harvested from adult lung of C57BL/6 mouse | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River | Stock#027 |
| Mouse: NU/J (athymic nude, nu/nu) | The Jackson Laboratory | Stock#002019 |
| Oligonucleotides | ||
| ERCC RNA Spike-In Mix | ThermoFisher | Cat#4456740 |
| See Table S5 for all primers used in qRT-PCR | This paper | Table S5 |
| See Table S6 for all adaptors used for 5’ triphosphate RNA sequencing. | This paper | Table S6 |
| See Table S7 for all gene targeting sequences by RNAi and CRISPR/Cas9 | This paper | Table S7 |
| See Table S8 for all in vitro transcribed RNA sequences. | This paper | Table S8 |
| Recombinant DNA | ||
| Plasmid: pGFH-9 | Katharina Strub | Addgene #39538 |
| Plasmid: pGFH-14c | Katharina Strub | Addgene #39541 |
| Plasmid: pUltra-hot | Malcom Moore | Addgene #48139 |
| Plasmid: pUltra-hot-SRP9-14 | This paper | N/A |
| Plasmid: pOZ-N-FH | Roger Greenberg | N/A |
| Plasmid: pOZ-N-RIG-I-WT | This paper | N/A |
| Plasmid: pOZ-N-RIG-I-MUT | This paper | N/A |
| Plasmid: pCMV-dR8.2 dvpr | Bob Weinberg | Addgene #8455 |
| Plasmid: pCMV-VSV-G | Bob Weinberg | Addgene # 8454 |
| Plasmid: PX458 | Feng Zhang | Addgene #48138 |
| Plasmid: pCT-CD81-RFP | SBI | Cat#CYTO125-PA-1 |
| Plasmid: pET His6 GST TEV LIC cloning vector (1G) | Scott Gradia | Addgene #29655 |
| Plasmid: pET His6 GST TEV-SRP9 | This paper | N/A |
| Software and Algorithms | ||
| R language and environment for statistical computing and graphics | https://www.r-project.org | |
| Bioconductor and packages | https://www.bioconductor.org | |
| CRAN | https://cran.r-project.org/web/packages/ | |
| cutadapt | https://pypi.python.org/pypi/cutadapt | |
| bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | |
| STAR | https://github.com/alexdobin/STAR/releases | |
| Subread | http://subread.sourceforge.net | |
| DESeq2 | https://www.rdocumentation.org/packages/DESeq2 | |
| flowCore | http://bioconductor.org/packages/release/bioc/html/flowCore.html | |
| Rtsne | https://cran.r-project.org/web/packages/Rtsne/index.html | |
| mclust | https://cran.r-project.org/web/packages/mclust/vignettes/mclust.html | |
| RNAstructure | http://rna.urmc.rochester.edu/RNAstructure.html | |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact Andy Minn (andyminn@upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mouse studies were completed in accordance with University Laboratory Animal Resources and Institutional Animal Care and Use Committee of the University of Pennsylvania regulations. Five to seven-week-old female C57BL/6 (Stock# 027) were obtained from Charles River Laboratory. Five to seven-week-old female athymic nude (Stock#002019) were obtained from The Jackson Laboratory. Mice were maintained in pathogen-free conditions.
Human
Serum from patients with cancer were obtained through the UPENN RadOnc Biosample Repository and were also procured from The Breast Program: Translational Cancer Resource (TRACR). TRACR is a breast cancer Biobank and a shared resource for Translational research at the Perelman School of Medicine at the University of Pennsylvania and the Abramson Cancer Center. Informed consent and HIPAA authorization for the TRACR Biobank was procured for all study subjects.
Cell culture
Cell culture was completed as previously described (Boelens et al., 2014). Cell lines are listed in the Key Resources Table and Table S4. All cell lines were confirmed to be mycoplasma-free with repeated testing. All human breast cancer and stromal cell lines were cultured at37°C in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin, and 2 mM l-glutamine. The KB1P mouse breast cancer cell lines from K14cre;p53F/F;Brca1F/F mice (Liu et al., 2007) were cultured in RPMI.
METHOD DETAILS
Cell sorting and treatment
All co-culture experiments were performed in DMEM with exosome-depleted FBS. Breast cancer cells were labeled with 7.5 μM 5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester (CFSE) and mixed 1:1 with stromal cells. Cell populations with a purity of at least 98% were used for RNA or protein isolation. MYC-ER was activated with 4-Hydroxytamoxifen at 250 nM (vehicle was ethanol). For GSI treatment, 10 μM DAPT (Sigma) or DMSO vehicle control was used.
Cell death assays
Sytox cell death assays were completed as previously described (Boelens et al., 2014). In brief, mono- or co-cultures were irradiated after 48 hr with 10 Gy using a Cs-137 Gammacell 40 EXACTOR. Cell death of CFSE-labeled breast cancer cells was measured at 96 hr post-radiation by flow cytometry using Sytox-Red (Invitrogen). Relative cell death was calculated by comparing mono and co-culture cell death.
Cell culture exosome isolation
Cell cultures used to isolate exosomes were grown in exosome-depleted media prepared by ultracentrifugation of FBS for 3 hr at 100,000 × g. Exosomes were isolated from conditioned media collected at 48–72 hr by serial high speed ultracentrifugation as previously described (Théry et al., 2006). Exosomes for protein isolation were purified using 10% final concentration of polyeth- ylene-glycol and centrifugation at 8,000 × g. Purity was examined by electron microscopy negative staining and protein analysis by immunoblotting. Exosomes were quantified by NanoSight NS500 as previously described (Boelens et al., 2014). For exosome injection experiments, protein was quantified by Lowry method, and equivalent volume of 10 μg of exosomes were injected. Forexo- some depletion, conditioned media was ultracentrifuged for 8–16 hr.
Serum exosome collection
Blood was collected using yellow top Vacutainer (BD) and centrifuged at 3000 rpm for 10 min. The samples were then frozen at −80°C until use. Serum from healthy donors was obtained commercially (Innovative Research). For exosomes from human or mouse serum, 500 μl of serum was spun at 2000 × g for 15 min, filtered through a 0.22 μm filter, and then purified by serial high speed ultracentrifugation.
EU labeling
Stromal cells were labeled with 100 μM 5-Ethynyl Uridine (EU) for 24 hr, and breast cancer cells were labeled with DiD (1:200) for 10 min at 37°C. Both cells types were then washed and co-cultured for 8 or 24 hr on glass coverslips. EU was then visualized byAlexa Fluor 488 azide (Alexa Fluor 488 5-carboxamido-(6-azidohexanyl), bis(triethylammonium salt)).
4sU RNA transfer
Stromal cells were labeled with 200 μM 4sU (4-Thiouracil) for 24 hr, washed, and either left in mono-culture or co-cultured with breast cancer cells. Conditioned media was isolated after 24 hr and added to mono-cultured breast cancer cells. Breast cancer cells were harvested 24 hr later and RNA extracted. 4sU-labeled RNA was specifically biotinylated with EZ-Link HPDP-Biotin (ThermoFisher) for 2.5 hr at room temperature. Free biotin was removed by phenol-chloroform RNA extraction and stromal 4sU-labeled RNA was enriched with Dynabeads MyOne Streptavidin C1 magnetic beads (ThermoFisher) per manufacturer’s instructions. Stromal 4sU-enriched RNA was then eluted with 1,4-Dithiothreitol (Sigma) and further purified by phenol-chloroform RNA extraction for downstream analysis.
4sU-FLAG-RIP
Stromal cells were labeled with 200 μM 4sU (4-Thiouracil), washed, and co-cultured with breast cancer cells with RIG-I CRISPR KO, RIG-I KO with re-expression of FLAG-tagged RIG-I or RIG-IK858/861A for 48 hr. Co-cultures were harvested and 100 mg of wet cell pellet was lysed by sonication (five, one-second bursts, medium output) in RSB-200 buffer (20 mM Tris pH 7.5, 200 mM NaCl, 2.5 mM MgCl2, 0.5% NP-40, 0.1% Triton X-100, 0.2 U/uL RNase Inhibitor, and one tablet of protease inhibitors). Post-lysis, FLAG-RIG-I was immunoprecipitated with prebound and washed FLAG-M2 beads (Sigma) using 30 uL of beads per 100 mg of wet cell pellet for 2–3 hr at 4°C. Beads were then washed three times with RSB-200. RNA was extracted with TRIzol reagent utilizing linear acrylamide as a carrier. 4sU-labeled RNA was then enriched as described above.
Gene targeting and expression
Gene knockdown by siRNA was completed using SMARTPool siRNAs (ThermoFisher) and transfected using 20 nM siRNA and RNAiMax (Invitrogen) transfection reagent. For stable knockdowns, shRNAs were cloned into the pGIPZ vector and transduced by virus using pCMV-VSV-G and pHR8.2ΔR envelope and packaging vectors in HEK293T cells. Transduced cells were selected using 1–2 μg/ml of puromycin. Wild-type and K858A/K861A binding mutant of RIG-I was cloned into the pOZ-N-FH vector (a kind gift from Roger Greenberg). Transduced cells were then selected with IL-2 receptor magnetic beads and expression was confirmed by western blot for FLAG, HA, and RIG-I. RIG-I restoration was functionally confirmed by RIG-I pathway activation in response to Sendai virus infection. SRP9 and SRP14 were transiently transfected with pGFH-9 (Addgene plasmid # 39538) and pGFH-14c (Addg- ene plasmid # 39541), both gifts from Katharina Strub. SRP9/14 lentivirus was produced by subcloning GFP-SRP9 and GFP-SRP14 from pGFH-9 and pGFH-14c into pUltra-hot (Addgene plasmid # 24130), a gift from Malcom Moore. Transduced cells were selected by sorting on mCherry expressing populations and confirmed by western blot. Gene knockout by CRISPR was accomplished using pSpCas9(BB)-2A-GFP (PX458), a gift from Feng Zhang (Addgene plasmid # 48138). To knock out RIG-I, two distinct guide RNAs cloned into the pSpCas9(BB)-2A-GFP backbone were transiently transfected into breast cancer cells. After 48 hr, single cells were sorted into 96 well single cell clones based on highest GFP expression. Clones were confirmed to have no RIG-I expression by immunoblot and pooled. RIG-I KO in the pooled clones were functionally confirmed by RIG-I pathway activation in response to Sendai virus infection.
Recombinant protein production and purification
Recombinant SRP9 was produced by subcloning the SRP9 cDNA from pGFH-9 plasmid into the pET His6 GST TEV LIC cloning vector (1G), a gift from Scott Gradia (Addgene plasmid # 29655). Recombinant protein was produced in BL21 competent E. coli and captured with Glutathione Sepharose beads (GE Healthcare). GST-tagged TEV Protease (Sigma) was used to cleave GST-SRP9.
In vivo mouse studies
All mouse studies were completed in accordance with ULAR and IACUC regulations. For exosome injection studies, 1 × 106 1833 breast cancer cells were injected with Matrigel (Corning) into the flanks of 6–8 week old athymic nude mice and 10 μg of mono- or co-culture exosomes were directly injected into the tumors 3 times a week. For RNA injection studies, 50 ng of 7SL or GAPDH300 RNA encapsulated into RNAiMax liposomes were directly injected into the tumors 3 times a week. Subcutaneous tumor growth was measured by caliper. For lung colonization studies, 2 × 105 luciferase-labeled 4175 breast cancer cells were injected in the tail vein. Injections were confirmed by immediate imaging using a Xenogen IVIS 100 system. Serum was isolated from mice by cardiac puncture. For immune studies, 50 μg of exosomes or 1 μg of RN7SL1 or GAPDH300 RNA encapsulated in RNAiMax liposomes were injected retro-orbitally. Spleens were harvested at 18 hr for FACS analysis.
Flow cytometry
Single-cell suspensions were prepared from mouse spleens, and red blood cells were lysed using ACK Lysis Buffer (Life Technologies). Live/dead cell discrimination was performed using Live/Dead Fixable Aqua Dead Cell Stain Kit (Life Technologies). Cell surface staining was done for 30 min at 4°C. All data acquisition was done using an LSR II (BD).
Exosome RNA sequencing
Exosome RNA was extracted with TRIzol and library preparation was completed using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) modified so that the RNA was not fragmented prior to library preparation. ERCC controls (Invitrogen) were added into all exosome RNA samples. Libraries were sequenced on Illumina HiSeq 2500 with 100 base paired end reads.
MNase qRT-PCR and RNA sequencing
Either whole cells or whole exosomes were incubated at 37°C for 30 min in MNase Buffer (25 mM Tris-HCl, 2.5 mM CaCl2, 50 mM NaCl, 1X PBS), with or without MNase and with or without 0.1% Triton X-100. Pre-MNase treatment, 10 ng of DVG396 RNA was spiked-in to control for differences in MNase activity with or without detergent. Post-MNase treatment, TRIzol LS reagent was used to purify RNA using linear acrylamide as a carrier, and ERCC Controls (Invitrogen) were spiked-in to account for differences in efficiency of RNA extraction. For RNA sequencing studies, libraries were prepared from purified RNA using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) without further RNA fragmentation. Libraries were sequenced on Illumina HiSeq 2500 with 100 base paired end reads.
5’-Triphosphate RNA sequencing
To enrich for 5’-triphosphate RNA, 0.1–2 μg of exosomal RNA was prepared by first degrading 5’monophosphate RNA with Terminator 5’-Phosphate-Dependent Exonuclease (Epicenter), then converting 5’-triphosphate to 5’p with RNA 5’ Polyphosphatase (Epicenter), to allow for specific ligation of RNA adaptor P5_RNA to RNAs that originally have 5’-triphosphate. Then, cDNAs were synthesized by using a primer with 5’ random 9-mer (P7_N9), and amplified with NEBNext PCR reagents (NEB) by using the same protocol as other RNA-seq libraries. Libraries were sequenced on Illumina HiSeq 2500 with 100 base paired end reads.
In vitro transcription
In vitro transcription was performed using of PCR amplified cDNA templates that contained Hepatitis Delta Virus Ribozyme to ensure homogeneous 3’ ends of the transcripts of interest (see Table S8 for sequences). In vitro transcription was completed with the MEGAshortscript T7 Transcription Kit (ThermoFisher) per manufacturer’s instructions. RNA was DNase treated and phenol/chloro-form purified. After thermocycling to ensure ribozyme cleavage, correct size transcripts were gel purified. RNA secondary structure of RN7SL1 mutants were predicted using RNAstructure.
RIG-I ATPase assays
RIG-I ATPase assays were performed as previously described (Devarkar et al., 2016). In brief, increasing amounts of RNA (10–60 nM) were added to a constant quantity of RIG-I (5 nM) in the presence of 1 mM ATP. ATP hydrolysis was measured with the EnzChek Phosphate Assay Kit (ThermoFisher) after 60–90 min at 37°C. ATP hydrolysis was then measured by absorbance of 360 nm compared to background. A 19-mer 5’-triphosphate dsRNA (Invivogen) and DVG396 were used as positive controls and a 19-mer 5’OH dsRNA (Invivogen) and an in vitro transcribed 300bp ssRNA stretch of GAPDH (GAPDH300) were used as negative controls.
Protein analysis
Protein was extracted using 2XSDS lysis buffer, separated by 4%−12% SDS-PAGE, transferred to a PVDF membrane, blocked with 5% nonfat milk in PBS-Tween (0.01%), and probed with the antibodies described. Protein was visualized using ECL (SuperSignal West Pico, Thermo).
Immunofluorescence
Cells were plated on coverslips for 24–48 hr and subsequently fixed with 4% paraformaldehyde. Cells were then permeabilized with PBS containing 0.5% saponin. After PBS wash, cells were blocked with 1% BSA in PBST with glycine, and followed by primary and secondary antibody incubation in a humidified chamber. Coverslips were mounted with Prolong Diamond Antifade Mountant with DAPI (ThermoFisher) and imaged on a Leica DM6000 Widefield microscope.
qRT-PCR
Total RNA was isolated and purified from cells using TRIzol reagent (Invitrogen). cDNA was synthesized using the High Capacity RNA-to-cDNA kit (ABI) according to manufacturer’s instructions. qRT-PCR was performed using Power SYBR Green PCR MasterMix (ABI) on the TaqMan 7900 (ABI).
QUANTIFICATION AND STATISTICAL ANALYSIS
EU labeling quantification and 4sU RNA transfer
Percentage of cells double positive for EU and DiD after co-culture with EU labeled stromal cells that matched breast cancer cell morphology were scored as EU+ breast cancer cells. For 4sU RNA experiments, transfer of stromal-derived RNA was determined by quantification of total 4sU-labeled RNA in recipient breast cancer cells compared to total RNA or by qRT-PCR.
Flow cytometry clustering analysis
For unbiased cluster identification and assignment, fluorescence values from FCS files were transformed using the Gating-ML 2.0 compliant logicle transformation from the flowCore Bioconductor package. After downsampling the data, t-Distributed Stochastic Neighbor Embedding (tSNE) was used to perform dimensionality reduction as implemented in the Rtsne package. Clusters were identified by model-based clustering using the mclust package and a value for the number of clusters was selected based on a plateauing of the Bayesian Information Criterion. Cluster assignments were then used as class labels to train a random forest classifier using the randomForestSRC R package to assign all cells from all samples to one of the clusters. The proportion of cells in each cluster was determined for all samples. For independent biological replicates, clusters with similar features for myeloid/ DC activation markers were determined using Pearson correlation, allowing results from independent experiments to be combined.
Microarray data analysis
Gene expression data for ISG-R and ISG-NR breast cancer cells (Table S4) co-culture with MRC5 fibroblasts have been described (Boelens et al., 2014) and available at the GEO: GSE60998. Pre-processing, filtering, and differential gene expression analysis were performed as previously described (Boelens et al., 2014). Gene set analysis was performed using the GSA R package and Reactome gene sets downloaded from the Molecular Signatures Database v5.1 (http://software.broadinstitute.org/gsea/msigdb). The gene set for upregulated cancer associated ISGs has been previously described (Weichselbaum et al., 2008). For analysis of stromal and breast cancer cell gene expression (GEO: GSE9014), metagenes were calculated from normalized data by averaging the expression of the Reactome target genes for MYC or for NOTCH, or by averaging the expression of the cancer-associated ISGs. For calculation of myeloid and lymphoid metagenes, a previously described collection of immune specific genes was used (Abbas et al., 2005).
RNA-seq and 5’-triphosphate RNA sequencing data analysis
For exosome RNA-seq and MNase RNA-seq analysis, reads were trimmed first using cutadapt v1.9 with parameters -q 10 -m 30 -O 4. Trimmed reads were then aligned to ERCC controls, rRNAs sequences as well as RN7SL1 by using bowtie2. The remaining reads were aligned to the GRCh38 reference genome using STAR v2.4.0k with parameters-outFilterMultimapNmax 100-outFilterMismatchNmax 999-outFilterMismatchNoverLmax 0.06. Primary aligned reads were counted against GENCODE annotation v21 and RepeatMasker annotation (UCSC Genome Browser) using Subread v1.4.6 with parameters -s 2 -minReadOverlap 10. The DESeq2 R package was used for differential gene expression analysis. ERCC controls were used for inter-sample normalization. For 5’-triphosphate RNA sequencing only the first reads of the paired end reads were used in data analysis. Reads were trimmed and aligned the same as RNA-seq analysis. Primary reads that matched the 5’ end of annotated features were counted and only reads present in all replicates were examined (excludes low abundance and/or inconsistently represented transcripts).
qRT-PCR gene expression and MNase qRT-PCR analysis
Relative gene expression levels after qRT-PCR were defined using the ΔΔCt method and normalizing to 18S rRNA, β-Actin and GAPDH. Percent shielding of RNA was quantified by ΔΔCt method normalizing to DVG396 spike-in and MNase without detergent.
DATA AND SOFTWARE AVAILABILITY
Software
The R language and environment for statistical computing and graphics (https://www.r-project.org) was used for the majority of the statistical and bioinformatics analysis. The R packages used for analyses described in the methods were obtained from Bioconductor (https://www.bioconductor.org) and/or from CRAN (https://cran.r-project.org/web/packages/). Additional software and packages for processing, alignment, and analysis of sequencing data are listed in the Key Resources Table.
RNA sequencing and microarray gene expression data
The ExoRNA-seq, MNase-seq, 5’ppp-seq, and patient ExoRNA-seq data have been deposited at the GEO (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GEO: GSE93078. Gene expression data for ISG-R and ISG-NR breast cancer cells co-culture with MRC5 fibroblasts have been described (Boelens et al., 2014) and deposited under GEO: GSE60998. Data from stromal and breast cancer cell laser-capture micro-dissection samples have been described (Finak et al., 2008) and are also available at GEO: GSE9014.
Supplementary Material
Highlights.
Cytoplasmic RNA is shielded by RNA binding proteins to prevent RIG-I recognition
NOTCH-MYC signaling in cancer fibroblasts deploys unshielded RN7SL1 RNA in exosomes
Unshielded RN7SL1 in exosomes acts as a DAMP and activates breast cancer RIG-I
RNA unshielding couples stromal activation to inflammation and cancer progression
ACKNOWLEDGMENTS
The authors thank JoannaTober and Bihui Xu for experimental assistance, and Noah Goodman and Natalie Shihfor helpwith clinical samples (ACC Breast Program Translational Cancer Resource). B.Y.N.was supported by agrant from the NIH(F31CA189707),J.M. byagrantfromtheNIH(R01GM111959), J.T. byaNCI Cancer Center Support Grant (2-P30-CA-016520–35) and the Breast Cancer Alliance Research Foundation, and A.M.D. by funds from FFANY/QVC. A.J.M. was supported by the Melanoma Research Alliance, Parker Institute for Cancer Immunotherapy, and Basser Research Center for BRCA. A.J.M. is a Department of Defense Era of Hope Scholar (W81XWH-09–1-0339) and funded by the Department of Defense (W81XWH-14–1-0450) and the NIH/NCI (R01CA172651).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and eight tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.06.031.
A video abstract is available at http://dx.doi.org/10.1016/j.cell.2017.06.031#mmc3.
REFERENCES
- Abbas AR, Baldwin D, Ma Y, Ouyang W, Gurney A, Martin F, Fong S, van Lookeren Campagne M, Godowski P, Williams PM, et al. (2005). Immune response in silico (IRIS): immune-specific genes identified from a compendium of microarray expression data. Genes Immun. 6, 319–331. [DOI] [PubMed] [Google Scholar]
- Akopian D, Shen K, Zhang X, and Shan SO (2013). Signal recognition particle: an essential protein-targeting machine. Annu. Rev. Biochem. 82, 693–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KWW, Low D, Ruedl C, Riccardi-Castagnoli P, Poidinger M, et al. (2014). High-dimensional analysis of the murine myeloid cell system. Nat. Immunol. 15, 1181–1189. [DOI] [PubMed] [Google Scholar]
- Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, and Lyden D (2016). Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell 30, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JJ, Cowing-Zitron C, Nakatsuji T, Muehleisen B, Muto J, Bor-kowski AW, Martinez L, Greidinger EL, Yu BD, and Gallo RL (2012). Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat. Med. 18, 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, et al. (2014). Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 159, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgeman A, Maelfait J, Davenne T, Partridge T, Peng Y, Mayer A, Dong T, Kaever V, Borrow P, and Rehwinkel J (2015). Viruses transfer the antiviral second messenger cGAMP between cells. Science 349, 1228–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob LS, Patwa R, Shah H, Xu K, et al. (2016). Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F, Khan AG, Patel SS, and Marcotrigiano J (2016). Structural basis for m7G recognition and 2’-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. USA 113, 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, Hartley JL, Crow YJ, and Stetson DB (2014). The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol. 15, 839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. (2008). Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 14, 518–527. [DOI] [PubMed] [Google Scholar]
- Gentili M, Kowal J, Tkach M, Satoh T, Lahaye X, Conrad C, Boyron M, Lombard B, Durand S, Kroemer G, et al. (2015). Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 349, 1232–1236. [DOI] [PubMed] [Google Scholar]
- Gomez-Roman N, Grandori C, Eisenman RN, and White RJ (2003). Direct activation of RNA polymerase III transcription by c-Myc. Nature 421, 290–294. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann K-K, Schlee M, et al. (2006). 5’-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997. [DOI] [PubMed] [Google Scholar]
- Hung T, Pratt GA, Sundararaman B, Townsend MJ, Chaivorapol C, Bhangale T, Graham RR, Ortmann W, Criswell LA, Yeo GW, and Behrens TW (2015). The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science 350, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C,Guise TA, and Massague J (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549. [DOI] [PubMed] [Google Scholar]
- Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, Kerkhoven RM, van Vliet MH, Wessels LF, Peterse JL, et al. (2007). Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl. Acad. Sci. USA 104, 12111–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, Huang B, Xu X, Zheng J, and Cao X (2016). Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell 30, 243–256. [DOI] [PubMed] [Google Scholar]
- Mercado-López X, Cotter CR, Kim WK, Sun Y, Muñoz L, Tapia K, and López CB (2013). Highly immunostimulatory RNA derived from a Sendai virus defective viral genome. Vaccine 31, 5713–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, and Massagué J (2005). Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hayashi T, Pan WW, Fujita Y, Holt MV, Qin J, Carson DA,and Guan KL (2016). The Hippo pathway kinases LATS½ suppress cancer immunity. Cell 167, 1525–1539.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, Chandler KJ, Cassiday PA, Nelson CA, Hagedorn CH, Graves BJ, and Cairns BR (2010). Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat. Struct. Mol. Biol. 17, 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranoa DRE, Parekh AD, Pitroda SP, Huang X, Darga T, Wong AC, Huang L, Andrade J, Staley JP, Satoh T, et al. (2016). Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7, 26496–26515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H,Liang G, Jones PA, Pugh TJ, et al. (2015). DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Owen DM, Jiang F, Marcotrigiano J, and Gale M Jr. (2008). Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M, and Hartmann G (2016). Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 16, 566–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth-Wagner C, Ludwig J, Bruder AK, Herzner A-M, Zillinger T, Goldeck M, Schmidt T, Schmid-Burgk JL, Kerber R, Wolter S, et al. (2015). A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1–2’O-methylated self RNA. Immunity 43, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I,Goubar A, Baracco EE, Remédios C, et al. (2014). Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 20, 1301–1309. [DOI] [PubMed] [Google Scholar]
- Tchou J, Kossenkov AV, Chang L, Satija C, Herlyn M, Showe LC, and Pure E (2012). Human breast cancer associated fibroblasts exhibit subtype specific gene expression profiles. BMC Med. Genomics 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telesnitsky A, and Wolin SL (2016). The host RNAs in retroviral particles. Viruses 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C, Amigorena S, Raposo G, and Clayton A (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. Chapter 3, Unit 3.22. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, Juranek S, Sheng G, Micura R, Tuschl T, et al. (2010). Structural and functional insights into 5’-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 17, 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Kho-darev N, Su AW, Shaikh AY, Roach P, Kreike B, et al. (2008). An interferon-related gene signature for DNAdamage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 105, 18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, et al. (2006). c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 20, 2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ (2011). Transcription by RNA polymerase III: more complex than we thought. Nat. Rev. Genet. 12, 459–463. [DOI] [PubMed] [Google Scholar]
- Wu L, Pan J, Thoroddsen V, Wysong DR, Blackman RK, Bulawa CE, Gould AE, Ocain TD, Dick LR, Errada P, et al. (2003). Novel small-mole-cule inhibitors of RNA polymerase III. Eukaryot. Cell 2, 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software
The R language and environment for statistical computing and graphics (https://www.r-project.org) was used for the majority of the statistical and bioinformatics analysis. The R packages used for analyses described in the methods were obtained from Bioconductor (https://www.bioconductor.org) and/or from CRAN (https://cran.r-project.org/web/packages/). Additional software and packages for processing, alignment, and analysis of sequencing data are listed in the Key Resources Table.
RNA sequencing and microarray gene expression data
The ExoRNA-seq, MNase-seq, 5’ppp-seq, and patient ExoRNA-seq data have been deposited at the GEO (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GEO: GSE93078. Gene expression data for ISG-R and ISG-NR breast cancer cells co-culture with MRC5 fibroblasts have been described (Boelens et al., 2014) and deposited under GEO: GSE60998. Data from stromal and breast cancer cell laser-capture micro-dissection samples have been described (Finak et al., 2008) and are also available at GEO: GSE9014.