

Abstract
Environmental exposure to inorganic arsenic (iAs) has been shown to disturb glucose homeostasis, leading to diabetes. Previous laboratory studies have suggested several mechanisms that may underlie the diabetogenic effects of iAs exposure, including (i) inhibition of insulin signaling (leading to insulin resistance) in glucose metabolizing peripheral tissues, (ii) inhibition of insulin secretion by pancreatic β cells, and (iii) dysregulation of the methylation or expression of genes involved in maintenance of glucose or insulin metabolism and function. Published studies have also shown that acute or chronic iAs exposures may result in depletion of hepatic glycogen stores. However, effects of iAs on pathways and mechanisms that regulate glycogen metabolism in the liver have never been studied. The present study examined glycogen metabolism in primary murine hepatocytes exposed in vitro to arsenite (iAs3+) or its methylated metabolite, methylarsonite (MAs3+). The results show that 4-h exposures to iAs3+ and MAs3+ at concentrations as low as 0.5 and 0.2 pM, respectively, decreased glycogen content in insulin-stimulated hepatocytes by inhibiting insulin-dependent activation of glycogen synthase (GS) and by inducing activity of glycogen phosphorylase (GP). Further investigation revealed that both iAs3+ and MAs3+ inhibit insulin-dependent phosphorylation of protein kinase B/Akt, one of the mechanisms involved in the regulation of GS and GP by insulin. Thus, inhibition of insulin signaling (i.e., insulin resistance) is likely responsible for the dysregulation of glycogen metabolism in hepatocytes exposed to iAs3+ and MAs3+. This study provides novel information about the mechanisms by which iAs exposure impairs glucose homeostasis, pointing to hepatic metabolism of glycogen as one of the targets.
Keywords: Arsenic exposure, Glycogen metabolism, Hepatocytes, Insulin resistance, Glycogen synthase
Introduction
A growing number of laboratory and epidemiological studies have demonstrated that chronic exposure to inorganic arsenic (iAs) is associated with an increased prevalence or incidence of diabetes mellitus (Navas-Acien et al. 2006; Maull et al. 2012; Sung et al. 2015). However, the molecular mechanisms that underlie the development of diabetes in the context of iAs exposure are not well understood. Previous laboratory studies using animal or in vitro models have identified several pathways of glucose metabolism and several regulatory mechanisms that are targeted by iAs or its metabolites. These studies have shown that trivalent iAs, arsenite (iAs3+), and its methylated trivalent metabolites, methyl-arsonite (MAs3+) and dimethylarsinite (DMAs3+), inhibit insulin signaling in differentiated adipocytes, resulting in insulin resistance and impaired glucose uptake (Paul et al. 2007; Walton et al. 2004; Xue et al. 2011). The same arsenic species have also been shown to inhibit glucose-stimulated insulin secretion in isolated pancreatic islets or p cells (Douillet et al. 2013; Fu et al. 2010; Diaz-Villasenor et al. 2006, 2008). Consistent with these findings were results of population studies that found iAs exposure to be associated with insulin resistance (impaired insulin sensitivity) or β-cell dysfunction (Maull et al. 2012; Park et al. 2016). Additional mechanisms underlying the diabetogenic effects of iAs exposure may include: (i) inhibition of the differentiation of preadipocytes or myoblasts into adipocytes or myotubes (Wauson et al. 2002; Wang et al. 2005; Yen et al. 2010; Steffens et al. 2010), which could affect the capacity of adipose tissue and skeletal muscle to metabolize glucose, (ii) activation of gluconeogenesis in the fasting state (Liu et al. 2014; Huang et al. 2015), and (iii) altered methylation and/ or expression of genes associated with glucose metabolism and diabetes (Bailey et al. 2013; Diaz-Villasenor et al. 2007). Thus, multiple mechanisms may contribute to the development of diabetes associated with chronic exposure to iAs.
Surprisingly, little attention has been paid to the liver as a possible target in iAs-associated diabetes. Liver is a major glucose metabolizing organ involved in the regulation of glucose homeostasis (Klover and Mooney 2004), and also the organ that plays a central role in the metabolism of iAs (Drobna et al. 2009). The liver converts glucose into glycogen in fed state, while the breakdown of glycogen into glucose is critical for maintaining glucose homeostasis during fasting (Adeva-Andany et al. 2016). The metabolism of glucose in the liver is regulated by complex allosteric and hormonal mechanisms. Disruption of these mechanisms leads to impaired glucose homeostasis, which is often manifested by hyperglycemia and/or impaired glucose tolerance (Chung et al. 2014; Krssak et al. 2004; Lin and Accili 2011). Therefore, targeting hepatic glucose metabolism has become an important clinical practice in the treatment of type 2 diabetes (Rines et al. 2016).
The pathways of glucose metabolism in the liver include glycogenesis, glycogenolysis and gluconeogenesis (GNG). Glycogenesis is the pathway for glycogen synthesis during the fed state and glycogen synthase (GS) is the rate-limiting enzyme in this pathway (Dashty 2013). Both glycogenolysis and GNG are responsible for maintaining blood glucose levels in the fasting state. Glycogen phosphorylase (GP) is the rate-limiting enzyme in glycogenolysis, which breaks down glycogen to glucose (Agius 2015), while GNG synthesizes glucose from non-carbohydrate substrates (Sharabi et al. 2015). Phosphoenolpyruvate carboxykinase (PEPCK) is the rate-limiting enzyme in GNG. Insulin and glucagon regulate the rate-limiting enzymes in all these pathways through transcriptional mechanisms and/or by covalent modification, i.e., phosphorylation and dephosphorylation (Rognstad 1979; Rosella et al. 1993). Only few studies have examined effects of iAs on these pathways and regulatory mechanisms. These studies found that chronic exposures to iAs may increase fasting glycemia by stimulating GNG through upregulation of PEPCK expression (Liu et al. 2014; Tseng 2004). In addition, acute exposures to toxic doses of iAs have been shown to stimulate glycogen depletion (Reichl et al. 1988; Verma et al. 2004). However, little data are available on the effects of low, environmentally relevant exposures to iAs on the pathways of glycogen metabolism and on mechanisms underlying these effects. Similarly, no data are available on effects on these pathways and mechanisms of the toxic methylated metabolites of iAs that are produced in the course of iAs metabolism in the liver (Thomas et al. 2007).
The goal of the present study was to characterize effects of iAs and its toxic methylated metabolite, MAs3+, on glucose and glycogen metabolism in primary hepatocytes from mouse liver. Our results show that exposures to sub-cytotoxic concentrations of iAs3+ and MAs3+ decrease glycogen content in insulin-stimulated hepatocytes and that inhibition of GS and activation of GP, possibly due to insulin resistance, are the underlying mechanisms. These results outline a new mechanism by which arsenic exposure can disrupt glucose homeostasis, thus increasing risk of diabetes.
Materials and methods
Antibodies and other reagents
Primary antibodies were from Cell Signaling Technology (Danvers, MA, USA) unless otherwise noted. Secondary antibodies and SuperSignal West Pico chemiluminescent substrate were from Thermo Scientific (Waltham, MA, USA). Human recombinant insulin, sodium-D-lactate, sodium pyruvate, Avertin (2–2-2-tribromoethanol), phosphatase inhibitor cocktails 1 and 2, and Percoll were from Sigma-Aldrich (St. Louis, MO, USA). Protease inhibitor tablets were from Roche (Indianapolis, IN, USA). Type I collagenase was from Worthington Biochemical Corporation (Lakewood, NJ, USA). Cell culture media and reagents were purchased from Invitrogen (Carlsbad, CA, USA). UDP-[14C] glucose was from American Radiolabeled Chemicals, Inc. (St. Luis, MO, USA).
Hepatocyte isolation, culture and treatment with arsenicals
Hepatocytes were isolated from livers of anesthetized 8–15-week-old C57BL/6J mice by collagenase perfusion (Zhang et al. 2012, 2014) and seeded in 6-well or 12-well culture plates at density of 5.0 × 105/well or 2.0 × 105/well, respectively. The cell monolayers were cultured overnight in William’s medium E (WME) (Invitrogen Carlsbad, CA, USA) with 1% penicillin/streptomycin and 2 mM glutamine, and with or without 10% fetal bovine serum (Hyclone Laboratories, South Logan, UT, USA). The hepatocytes were then exposed for 4 h to iAs3+ (sodium arsenite, > 99% pure from Sigma) or MAs3+ (methylarsine oxide, 98% pure) provided by Dr. William Cullen, University of British Columbia, Vancouver, Canada. Additional treatments are detailed below.
Glycogen and free glucose assay
For this assay, the hepatocytes were cultured in serum-free WME overnight and then exposed to iAs3+ or MAs3+ for 4 h, with or without addition of 100 nM insulin for the last 2 h. Glycogen content was measured using a Glycogen Assay Kit from BioVision Inc. (Milpitas, CA, USA), following the manufacturer’s instructions. In this assay, glycogen is enzymatically hydrolyzed to glucose, which is then oxidized to form an intermediate that reduces a colorless probe to a colored product with absorbance at 450 nm. When the glycogen hydrolyzing enzymes are omitted from the reaction mixture, this assay measures free intracellular glucose.
GS activity assay
Glycogen synthase activity was determined by measuring incorporation of the 14C-glucosyl moiety of UDP-[14C]glucose into glycogen (Thomas et al. 1968; Hue et al. 1975; Nuttall and Gannon 1989). Hepatocytes (5.0 × 105) were homogenized in 0.5 ml of a buffer containing 100 mM NaF, 20 mM EDTA, 0.5% glycogen, 1% protease inhibitor, 1% phosphatase inhibitor cocktail, and 50 mM glycylglycine (pH 7.4). The homogenate was centrifuged at 9000×g for 10 min. Twenty microliters of the supernate was mixed with 100 μl reaction buffer containing 0.25 mM UDP-[14C]glucose (~0.01 μCi/μmol), 1% glycogen, 10 mM Na2SO4, and 60 mM glycylglycine (pH 7.4). The mixture was incubated at 25 °C for 10 min (the reaction velocity was constant for up to 20 min). An aliquot of the reaction mixture (75 μl) was spotted on a filter paper. The paper was let to dry and then extracted twice with cold 66% ethanol—20 min for the first and 10 min for the second extraction. The filter paper was then washed with acetone for 5 min and dried. The radioactivity associated with the filter paper was measured by TRICARB 1900 TR liquid scintillation analyzer (PerkinElmer, Waltham, MA, USA).
GP activity assay
Glycogen phosphorylase activity was measured by determining the level of free phosphate that is released during the reversed reaction, i.e., synthesis of glycogen from glucose-1-phosphate (Hue et al. 1975; Saheki et al. 1985; Stalmans and Hers 1975). Hepatocytes were lysed in a buffer containing 100 mM NaF, 20 mM EDTA, 0.5% glycogen, 1% protease inhibitor, 1% phosphatase inhibitor cocktail, and 50 mM glycylglycine (pH 7.4); supernate obtained after 9000×g centrifugation was used for the assay. The reaction mixture contained 100 μl of the supernate, 100 μl of a reaction incubation buffer (2% glycogen, 100 mM glucose-1-phosphate) and 1 mM caffeine (to measure GPa activity) or 2 mM AMP (to measure total GP activity). After a 20-min incubation at room temperature (the reaction velocity was constant for up to 30 min), the reaction was terminated by addition 50 μl of 20% SDS. The concentration of inorganic phosphate was measured in 20 μl aliquots of the reaction mixtures by a colorimetric assay using a molybdate reagent (Saheki et al. 1985) and Synergy HT plate reader (BioTek, Winooski, VT, USA).
Serine/threonine phosphatase activity assay
Serine/threonine phosphatase activity was assayed using the RediPlate™ 96 EnzChek® Serine/Threonine Phosphatase Assay Kit (Molecular Probes, Eugene, OR, USA) which measures inorganic phosphate produced by dephosphorylation of a synthetic substrate (provided in the kit). This assay is designed to measure activities of various serine/threonine phosphatases. The specificity of the assay depends on the reaction mixture composition. We used the reaction mixture that is recommended by the manufacturer for protein phosphatase 1 activity assay, i.e., the mixture containing 2 mM dithiothreitol (DTT) and 200 μM MnCl2. The 9000×g supernate was prepared from hepatocytes as described above; 50 μl of the supernate was added into the reaction mixture and incubated in the dark for 20 min at room temperature (the reaction velocity was constant for up to 40 min). Fluorescence was measured at 358/452 nm using Synergy HT plate reader.
Cell viability assay
The 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Styblo et al. 2000) was used to monitor viability of hepatocytes exposed for 4 h to iAs3+ or MAs3+ in the presence of insulin during the last 2 h of the exposure.
Immunoblot analysis
Hepatocytes were lysed in buffer containing 20 mM Tris-HCl (pH 7.5), 0.1 mM Na3VO4, 25 mM NaF, 25 mM glycerophosphate, 2 mM EGTA, 1 mM DTT, 0.5 mM phe- nylmethylsulfonyl fluoride, 0.3% Triton X-100, 1% protease inhibitor mix, and 1% phosphatase inhibitor cocktail. The lysates were separated by SDS-PAGE and transferred on PVDF membrane by electroblotting. Immunoblotting was performed to quantify target proteins. Horseradish peroxidase-conjugated secondary antibodies (ThermoFisher) were detected with SuperSignal West Pico chemiluminescent substrate by exposure to X-ray film. The film images were digitalized using an Epson scanner (Perfection 2400) and quantified using program Image J image processing program (NIH).
Statistical analysis
A minimum of three biological replicates (i.e., hepatocytes from at least three mice) were used for each assay or immunoblot analysis, with two or three technical replicates each. Results from the technical replicates were averaged to obtain a representative value for each biological replicate. The values in figures are expressed as mean and SD for the biological replicates. Two types of statistical analyses were carried out. Differences between control (unexposed) cells and control cells treated with insulin were assessed by unpaired one-tail Student’s t test (as the effects of insulin can be predicted using published data). Differences between insulin-treated control cells and insulin-treated cells exposed to different concentrations of iAs3+ or MAs3+ were evaluated by one-way ANOVA followed by Student-Newman-Keuls multiple comparisons test. For both types of analyses, differences with p < 0.05 were considered statistically significant and were marked in the figures describing experimental results; p values for marginally significant differences were also marked where appropriate.
Results
Exposure to iAs3+ and MAs3+ decreased glycogen and increased free glucose content in hepatocytes treated with insulin
We examined glycogen content in control hepatocytes cultured in basal WME medium and in hepatocytes exposed to iAs3+ or MAs3+ for 4 h, with or without addition of 100 nM insulin for the last 2 h of the exposure. In control hepatocytes that were not exposed to arsenicals, treatment with insulin resulted in an increase in glycogen content, while free glucose content decreased (Fig. 1a-d). Treatment with either iAs3+ (0.5, 1, and 2 μM) or MAs3+ (0.2, 0.5 and 1 μM) decreased glycogen content in the insulin-stimulated hepatocytes, thus offsetting the effect of insulin (Fig. 1a, b). Notably, exposure to 1 μM MAs3+ decreased glycogen content by 57% as compared to a 43% decrease in hepatocytes exposed to 2 μM iAs3+. While decreasing glycogen content, both iAs3+ (1 and 2 μM) and MAs3+ (1 μM) increased free glucose levels in insulin-stimulated hepatocytes. At these concentrations, neither iAs3+ nor MAs3+ affected cell viability measured by MTT assay (Fig. 1e, f). Cell viability was impaired only in hepatocytes exposed to 10 or 50 μM iAs3+, the concentrations that were much higher than those used for assessment of the effects of iAs3+ or MAs3+ on glycogen content and on the glycogen metabolizing enzymes in this and the following experiments.
Fig. 1.
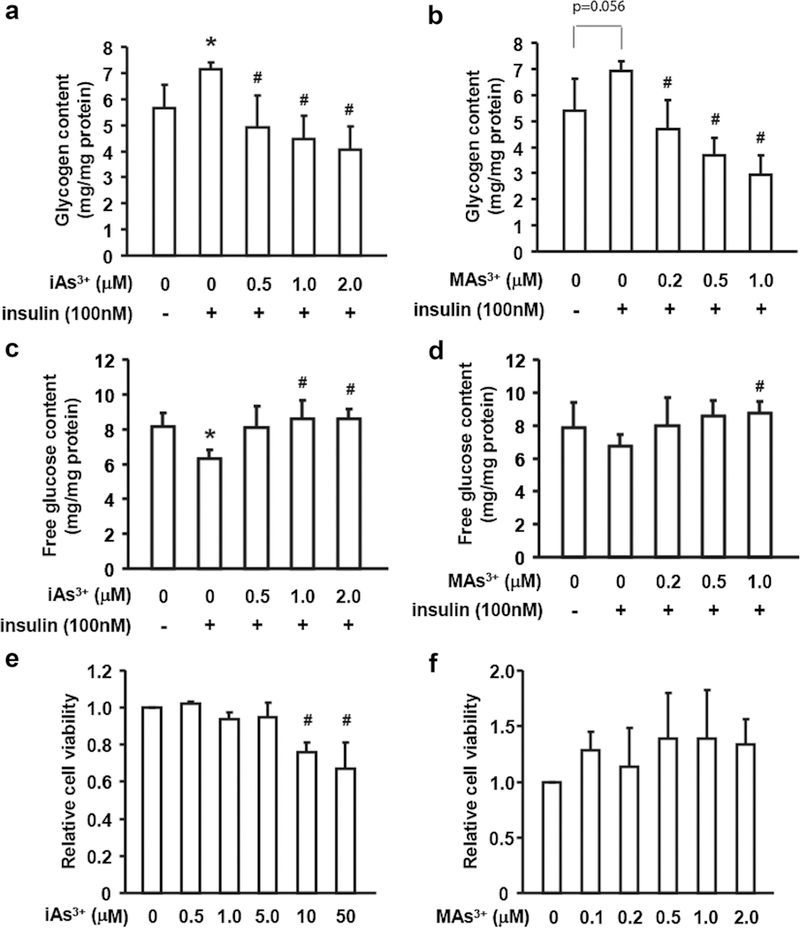
Exposures to iAs and MAs3+ decreased glycogen content and increased free glucose concentration in primary murine hepatocytes. Hepatocytes were exposed to iAs3+ (a, c) or MAs3+ (b, d) for 4 h with or without insulin treatment (100 nM) for the last 2 h of the exposure. Glycogen (a, b) and free glucose (c, d) were measured in cell lysates. MTT assay was used to assess cell viability in hepatocytes exposed to iAs3+ (e) or MAs3+ (f) for 4 h with insulin treatment (100 nM) for the last 2 h of the exposure. Values are expressed as mean and SD for three independent experiments. *Statistically significant differences (p < 0.05) between control hepatocytes and control hepatocytes treated with insulin, and #between insulin-treated control hepatocytes and insulin-treated hepatocytes exposed to arsenicals; p values for marginally significant differences are also shown
Exposure to iAs3+ and MAs3+ did not induce PEPCK expression in hepatocytes treated with insulin
It has been previously reported that chronic exposure to iAs3+ stimulates glucose production in GNG by inducing PEPCK expression (Liu et al. 2014; Tseng 2004). Thus, activation of GNG could be responsible for, or could contribute to the increased free glucose levels in hepatocytes treated with arsenicals. To assess possible role of GNG, we measured PEPCK protein levels in hepatocytes exposed to 2 μM iAs3+ or 1 μM MAs3+ for 4 h, with or without addition of 100 nM insulin for the last 2 h of the exposure. In spite of only 2-h treatment, insulin suppressed PEPCK protein level by 20%; a similar decrease was found in insulin-treated hepatocytes exposed to iAs3+ (Supplemental Fig. 1). Exposure to MAs3+ decreased PEPCK protein level by 65%. These results suggest that the increase in free glucose in insulin-stimulated hepatocytes exposed to iAs3+ or MAs3+ was not due to stimulation of GNG and was likely associated with changes in glycogen metabolism.
Exposure to iAs3+ and MAs3+ inhibited GS activity and stimulated GP activity in hepatocytes treated with insulin
To determine whether the decrease in glycogen content in hepatocytes exposed to iAs3+ or MAs3+ was due to inhibition of glycogen synthesis or activation of glycogenolysis, we measured activities of GS and GP, the enzymes that catalyze the rate-limiting reactions in these pathways. The results show that treatment with insulin increased GS activity; however, exposures to iAs3+ and MAs3+ reversed the effect of insulin, decreasing GS activity in insulin-stimulated hepatocytes (Fig. 2a, b). GS activity in hepatocytes exposed to 1 μM iAs3+ and 1 μM MAs3+ and treated with insulin decreased by 55 and 45%, respectively. To further explore these findings, we compared GS activities in lysates of insulin-treated control hepatocytes in a regular reaction mixture and in a mixture, to which iAs3+ or MAs3+ was added at final concentrations up to 10 μM. No differences in GS activities were found (data not shown) suggesting that inhibition of GS in hepatocytes exposed to iAs3+ or MAs3+ in culture and treated with insulin was not due to a direct interaction of the arsenicals with the enzyme, but rather due to effects on insulin-dependent mechanisms that regulate GS activity.
Fig. 2.
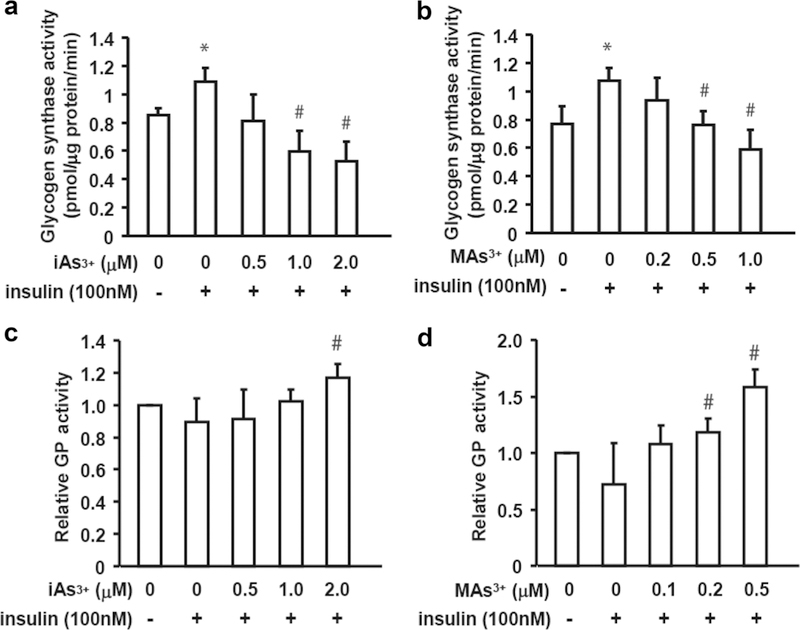
Exposures to iAs and MAs3+ inhibited the insulin-dependent activation of glycogen synthase and activated glycogen phosphorylase. Hepatocytes were exposed to iAs3+ (a, c) or MAs3+ (b, d) for 4 h with or without insulin treatment (100 nM) for the last 2 h of the exposure. Glycogen synthase (a, b) activity and the relative glycogen phosphorylase activity (i.e., GPa activity/total GP activity) (c, d) were measured in cell lysates. Values are expressed as mean and SD for three independent experiments. *Statistically significant differences (p < 0.05) between control hepatocytes and control hepatocytes treated with insulin, and #between insulin-treated control hepatocytes and insulin-treated hepatocytes exposed to arsenicals
The GP assay used in this study measured activities of GPa (the activated, phosphorylated form of GP) and the total GP activity, i.e., sum of the activities of GPa and GPb (the less active-dephosphorylated form). Here, treatment with insulin, a negative regulator of GP (Aiston et al. 2003), decreased the relative GP activity (i.e., the ratio of GPa/total GP), but this effect was not statistically significant (Fig. 2c, d). The relative GPa activity in insulin-treated hepatocytes exposed to 2 μM iAs3+ was significantly higher (1.3-fold) than in control insulin-treated hepatocytes; the effects of lower concentrations of iAs3+ were not statistically significant. In comparison, exposure to MAs3+ increased GP activity in insulin-treated cells at concentrations as low as 0.2 μM (1.7-fold) and 0.5 μM (2.2.-fold).
Further experiments carried out in this study focused on GS, which was significantly affected by exposure to both iAs3+ and MAs3+.
Exposure to iAs3+ and MAs3+ inhibited GS dephosphorylaton in hepatocytes treated with insulin
The activity of GS in hepatocytes is regulated by phosphorylation. Specifically, phosphorylation on Ser-641 by glycogen synthase kinase 3 (GSK3) results in deactivation of GS and decreases glycogen synthesis (Rylatt et al. 1980; Imazu et al. 1984a, b). To determine whether altered GS phosphorylation was responsible for the decrease in GS activity in hepatocytes exposed to iAs3+ or MAs3+, we examined GS/Ser-641 phosphorylation status using immunoblot. As expected, insulin treatment decreased Ser-641 phosphorylation (Fig. 3). However, exposure to iAs3+ and MAs3+ reversed the effect of insulin, increasing Ser-641 phosphorylation by 2- and 2.2-fold in hepatocytes exposed to 2 μM iAs3+ and 1 μM MAs3+, respectively.
Fig. 3.
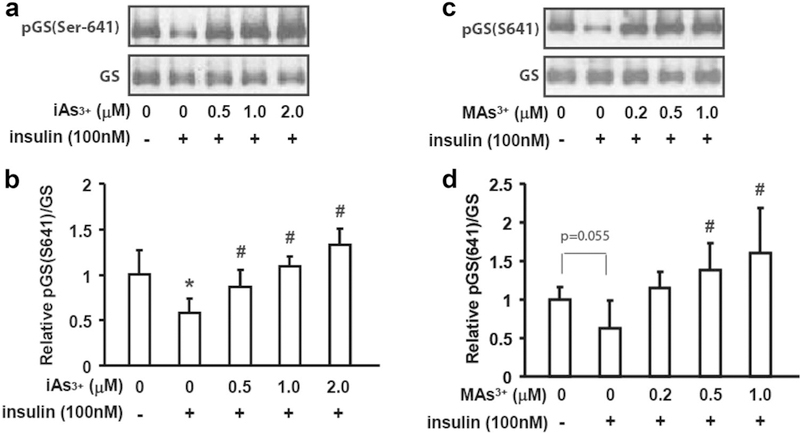
Exposures to iAs3+ and MAs3+ inhibited the insulin-dependent dephosphorylation of glycogen synthase. Hepatocytes were exposed to iAs3+ (a, b) or MAs3+ (c, d) for 4 h with or without insulin treatment (100 nM) for the last 15 min of the exposure. The total glycogen synthase (GS) levels and the phosphorylation of glycogen synthase on Ser-641 [pGS(Ser-641)] were assessed by immuno- blot (a, c). The immunoblot data were quantified using the ratio of pGS(Ser-641)/GS (b, d). Representative data from four independent experiments are shown; values in b and d are expressed as mean and SD. *Statistically significant differences (p < 0.05) between control hepatocytes and control hepatocytes treated with insulin, and #between insulin-treated control hepatocytes and insulin-treated hepatocytes exposed to arsenicals; p values for marginally significant differences are also shown
Exposure to iAs3+ and MAs3+ inhibited insulin-dependent phosphorylation of protein kinase B, but had no effects on GSK3 phosphorylation
Glycogen synthase kinase 3 that phosphorylates GS on Ser-641 is regulated by insulin via phosphoinositide 3-kinase (PI3K)-protein kinase B (PKB/Akt) signaling pathway (Welsh et al. 1996). The phosphorylation of GSK3α on Ser-21 by phosphorylated PKB/Akt that results in downregulation of GSK3 kinase activity (Srivastava and Pandey 1998) is the mechanism by which insulin prevents phosphorylation deactivation of GS and inhibition of glycogen synthesis (Cross et al. 1995). We examined PKB/Akt and GSK3 phosphorylation status in hepatocytes exposed to iAs3+ and MAs3+ using immunoblot. We found that the insulin-dependent phosphorylation of Akt at Ser-473 and Thr-308, the two sites responsible for full activation of Akt (Alessi et al. 1996), is severely inhibited by exposure to either iAs3+ or MAs3+ (Fig. 4a-d). As expected, insulin treatment increased the phosphorylation of GSK3α/Ser-21; however, exposures to iAs3+ and MAs3+ had no effects on GSK3α/Ser-21 phosphorylation in hepatocytes treated with insulin (Fig. 4e-h).
Fig. 4.
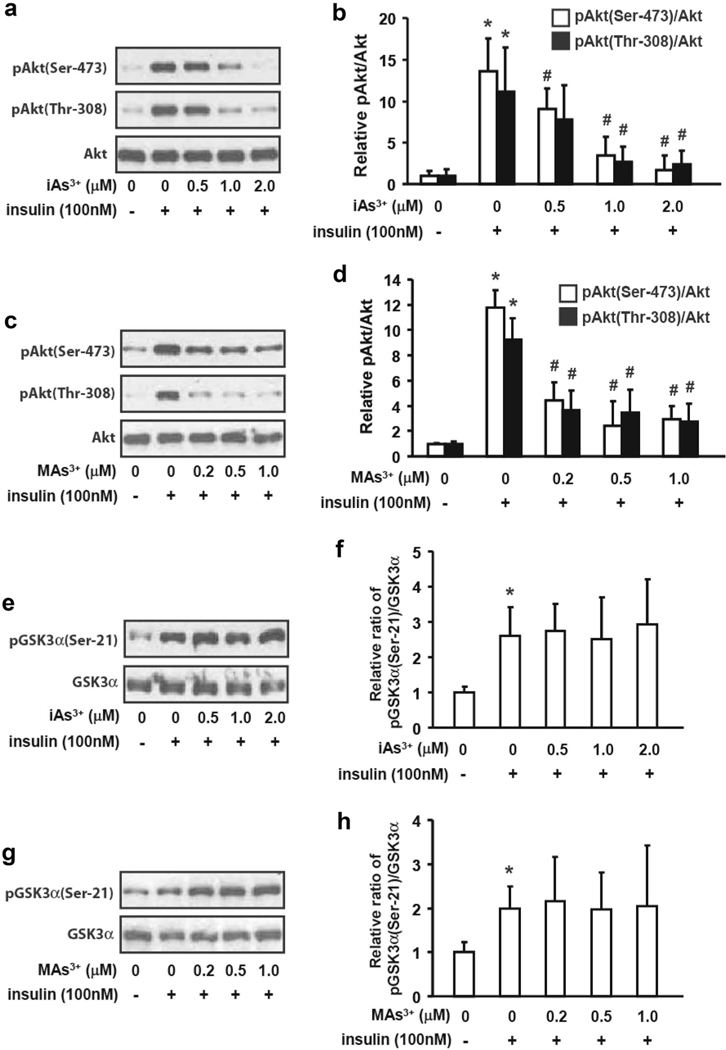
Exposures to iAs + and MAs3+ inhibited the insulindependent phosphorylation of PKB/Akt bud had no effects on GSK3 phosphorylation. Hepatocytes were exposed to iAs3+ (a, b, e, f) or MAs3+ (c, d, g, h) for 4 h with or without insulin treatment (100 nM) for the last 15 min of the exposure. The total Akt and phospho-Akt [pAkt(Ser-473) and pAkt(Thr-308)] levels (a, c), and total GSK3α and phosphor- GSK3α [pGSKa(Ser-21)] (e, g) were assessed by immunoblot. The immunoblot data were quantified using the ratios of pAkt/Akt (b, d) and pGSK3α(Ser-21)/GSK3α (f, h). Representative data from three independent experiments are shown; values in b, d, f and h are expressed as mean and SD. *Statistically significant differences (p < 0.05) between control hepatocytes and control hepatocytes treated with insulin, and #between insulin-treated control hepatocytes and insulin-treated hepatocytes exposed to arsenicals
Exposure to iAs3+ and MAs3+ did not change serine/threonine phosphatase activity in hepatocytes
Another mechanism by which insulin activates GS involves activation of protein phosphatase 1 (PP1). PP1 is a serine/ threonine phosphatase that dephosphorylates (activates) GS (DePaoli-Roach et al. 2003; Roach et al. 1998). We examined serine/threonine phosphatase activity in lysates from hepatocytes treated with iAs3+ and MAs3+ using a commercial kit from Molecular Probes and a reaction mixture containing 2 mM DTT and 200 μM MnCl2 which, according to the manufacturer, should select specifically for PP1 activity. Using this assay mixture, we found no difference between the serine/threonine phosphatase activities in control hepatocytes and in hepatocytes exposed to either iAs3+ or MAs3+ (Supplemental Fig. 2). Notably, treatment with insulin did not increase the phosphatase activity in control hepatocytes suggesting that, despite the manufacturer’s claim, the assay may not be specific for PP1 and that other, insulin-independent serine/threonine phosphatases may contribute to the activity measured by this assay. However, because DTT chelates trivalent arsenic (Spuches et al. 2005), and thus may restore phosphatase activity in hepatocyte lysates after exposure to iAs3+ or MAs3+, we repeated the assay using the reaction mixture in which DTT was replaced with a non-thiol reductant tris(2-carboxyethyl) phosphine (TCEP). Notably, even with TCEP-containing reaction mixture no effects of iAs3+ or MAs3+ on the phosphatase activity were found (Supplemental Fig. 2).
Discussion
Hepatic glycogenolysis and gluconeogenesis are the main sources of glucose for energy production between meals and during fasting. Dysregulation of glycogen metabolism may affect the energy-dependent metabolic and physiological processes in the body, and may also contribute to impaired glucose homeostasis as one of the attributes of diabetes (Krssak et al. 2004). Most of the previously published studies of the effects of iAs exposure on glycogen metabolism used acute treatments with high doses of iAs. For example, a significant decrease in hepatic glycogen content was reported in livers of mice and rats that were given a single or multiple doses of sodium arsenite or As2O3 (3–15 mg/kg b.w.) orally or by injection (Reichl et al. 1988; Verma et al. 2004; Berry and Smythe 1959; Reichl et al. 1991; Kawaguchi 1981; Albores et al. 1996; Singh et al. 2017; Huang et al. 2015). Similarly, repeated injections of As2O3, 2.5 mg/kg b.w. per day for five consecutive days, decreased glycogen content in livers of guinea pigs (Reichl et al. 1988). Only one study has examined effects of chronic iAs exposure at environmentally relevant levels (Huang et al. 2015). This study found that exposure to iAs3+ in drinking water, 0.05 or 0.5 mg As/L for 6 weeks, decreased hepatic glycogen content in mice.
The present study builds on these findings. Our goal was to identify mechanisms that underlie the effects of iAs exposure on hepatic glycogen by examining the enzymes and pathways regulating glycogen metabolism in primary hepatocytes exposed to iAs3+ and to its toxic methylated metabolite, MAs3+. We found that iAs3+ exposure decreased glycogen content in insulin-stimulated hepatocytes, which is consistent with results of the previous in vivo studies in laboratory animals. In addition, we showed that exposure to MAs3+ also reduced glycogen content, and that the decline in glycogen levels was associated with increased intracellular concentration of free glucose. Neither iAs3+ nor MAs3+ induced expression of PEPCK, the rate-limiting enzyme in GNG pathway. Thus, the decrease in glycogen content and the increase in glucose content in hepatocytes exposed to by iAs3+ and MAs3+ were either due to impairment in insulin-dependent utilization of glucose for glycogen synthesis or due to stimulation of glycogen breakdown to produce free glucose. Further investigation revealed that iAs3+ and MAs3+ altered both pathways by inhibiting insulin-stimulated GS activity and by simultaneously increasing GP activity. Because GS activity was inhibited by low concentrations of both iAs3+ and MAs3+ while only MAs3+ was a potent activator of GP, our further investigation focused on GS as the common target for both arsenicals.
The hepatic GS is regulated by several protein kinases: AMP-activated kinase (AMPK) phosphorylates GS on Ser-7, casein kinase 2 (CK2) on Ser-10, and GSK3 on Ser-641, Ser-645, Ser-649 and Ser-653 (Ros et al. 2009; Imazu et al. 1984b; Rylatt et al. 1980). Mutagenesis studies have shown that phosphorylation on Ser-7, Ser-10, Ser-641, Ser-645, Ser-649 and Ser-653 plays a key role in GS regulation (Ros et al. 2009). GSK3 is negatively regulated by insulin (Welsh et al. 1996) suggesting that inhibition of insulin signaling resulting in GS phosphorylation by GSK3 and in downregulation of GS activity could be responsible for the decrease in glycogen content in hepatocytes exposed iAs3+ and MAs3+. To test this hypothesis, we examined the phosphorylation of GS on Ser-641, one of the targets of GSK3 kinase activity. As expected, insulin suppressed the phosphorylation of GS/Ser-641 in control hepatocytes, but exposures to iAs3+ and MAs3+ reversed the effect of insulin, resulting in a significant increase in Ser-641 phosphorylation. This result is consistent with inhibition of insulin signaling by iAs3+ and MAs3+, which has been previously reported in other studies, including studies carried out in our laboratory (Paul et al. 2007; Walton et al. 2004).
To further explore this mechanism, we examined effects of iAs3+ and MAs3+ on the insulin-activated signal transduction pathway, specifically on phosphorylation of PKB/Akt and of its downstream target, Ser-21 in GSK3α isoform. We found that treatment with insulin stimulated PKB/Akt phosphorylation in control hepatocytes and, in turn, GSK3α/Ser-21 phosphorylation also increased. The insulin-dependent phosphorylation of PKB/Akt was inhibited by both iAs3+ and MAs3+. This finding is consistent with results of our previous study in adipocytes that found both iAs3+ and MAs3+ to be potent inhibitors of phosphoinositide-dependent kinase-1 (PDK-1), which is responsible for PKB/Akt phosphorylation in the insulin signal transduction pathway (Paul et al. 2007). However, neither iAs3+ nor MAs3+ altered the insulin/PKB/Akt-dependent phosphorylation of GSK3α/ Ser-21. Notably, although the exposure to iAs3+ and MAs3+ inhibited insulin-dependent PKB/Akt phosphorylation, the levels of the phosphorylated PKB/Akt in the insulin-stimulated hepatocytes exposed to arsenicals were still higher than in control hepatocytes that were not treated with insulin (Fig. 4). Thus, it is possible that the residual PKB/Akt activity in arsenic-exposed and insulin-stimulated cells was sufficient to maintain GSK3α/Ser-21 phosphorylation at the level found in insulin-stimulated control cells. It is unclear, however, how the exposure to arsenical increased GS phosphorylation if GSK was phosphorylated, i.e., inactive.
Another mechanism by which insulin regulates GS activity involves activation of PP1 that dephosphorylates/ activates GS (Sharabi et al. 2015; Aiston et al. 2003). PP1 belongs to a larger family of protein phosphatases that also include PP2A, PP2B and PP2C (Cohen 1989). PP1 is a serine/threonine phosphatase with a relatively broad spectrum of substrates. The catalytic subunit of PP1 lacks substrate specificity. Affinity of PP1 towards specific substrates is conferred by a set of regulatory proteins that bind to the catalytic subunit of the enzyme (Peti et al. 2012). Thus, although GS is considered one of the primary targets of PP1, measuring the GS specific PP1 activity is difficult because various forms of PP1 with different substrate specificities are present in biological samples. The choice of methods/kits is very limited. In this study, we used a commercial 96 EnzChek® Serine/Threonine Phosphatase Assay Kit from Molecular Probes (R-33700). This kit uses 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) as a substrate for several Ser/Thr phosphatase, including PP1, PP2A, PP2B and PP2C. DiFMUP is dephosphorylated to produce fluorescent 6,8-difluoro-4-methylumbelliferyl (DiFMU). The manufacturer recommends specific components for the assay mixture to measure activities of particular Ser/Thr phosphatases. We used the composition that, according to the manufacturer, selects for PP1 activity, using first DDT (recommended) and then TCEP as a reductant. However, the fact that we did not observe any increase in the phosphatase activity in control hepatocytes treated with insulin, the PP1 activator (Supplemental Fig. 2), strongly suggests that the assay conditions were not PP1 specific. Thus, the role of PP1 in the inhibition of GS activity in hepatocytes exposed to iAs3+ and MAs3+ remains unclear.
In summary, the present study provides additional evidence that exposure to iAs may result in a reduction or depletion of glycogen stores in the liver and suggests that MAs3+, the intermediary metabolite formed in the pathway for iAs methylation, can significantly contribute to this outcome. This study is the first to show that exposure to iAs3+ and MAs3+ decreases glycogen content in hepatocytes by inhibiting the insulin-dependent phosphorylation (activation) of GS. MAs3+ and, to a lesser extent iAs3+, also stimulated GP activity. Thus, both GS inhibition and GP activation contribute to glycogen depletion, in particular in hepatocytes exposed to MAs3+. While we were not able to determine the roles of GSK3 or PP1 in the inhibition of GS activity by iAs3+ and MAs3+, our results point to inhibition of insulin signaling (i.e., insulin resistance) as the key underlying mechanism. Future studies should validate these findings in laboratory animal models and should also examine potential roles of other kinases that phosphorylate hepatic GS, specifically AMPK and CK2.
Supplementary Material
Acknowledgements
This work was supported by Grants from the National Institute of Health (R01ES022697 and DK 056350). The authors thank Dr. Rosalind Coleman (Department of Nutrition, University of North Carolina at Chapel Hill) for her helpful suggestions regarding the methods used in this study and result interpretation.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00204-017-2076-9) contains supplementary material, which is available to authorized users.
References
- Adeva-Andany MM, Gonzalez-Lucan M, Donapetry-Garcia C, Fernandez-Fernandez C, Ameneiros-Rodriguez E (2016) Glycogen metabolism in humans. BBA Clin 5:85–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agius L (2015) Role of glycogen phosphorylase in liver glycogen metabolism. Mol Aspects Med 46:34–45 [DOI] [PubMed] [Google Scholar]
- Aiston S, Coghlan MP, Agius L (2003) Inactivation of phosphorylase is a major component of the mechanism by which insulin stimulates hepatic glycogen synthesis. Eur J Biochem 270:2773–2781 [DOI] [PubMed] [Google Scholar]
- Albores A, Cebrian ME, Garcia-Vargas GG, Connelly JC, Price SC, Hinton RH, Bach PH, Bridges JW (1996) Enhanced arsenite-induced hepatic morphological and biochemical changes in phenobarbital-pretreated rats. Toxicol Pathol 24:172–180 [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15:6541–6551 [PMC free article] [PubMed] [Google Scholar]
- Bailey KA, Wu MC, Ward WO, Smeester L, Rager JE, Garcia-Vargas G, Del Razo LM, Drobna Z, Styblo M, Fry RC (2013) Arsenic and the epigenome: interindividual differences in arsenic metabolism related to distinct patterns of DNA methylation. J Biochem Mol Toxicol 27:106–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry LJ, Smythe DS (1959) Carbohydrate metabolism in normal and altitude-exposed mice following arsenite poisoning. Am J Physiol 197:37–40 [DOI] [PubMed] [Google Scholar]
- Chung ST, Hsia DS, Chacko SK, Rodriguez LM, Haymond MW (2014) Increased gluconeogenesis in youth with newly diagnosed type 2 diabetes. Diabetologia 58:596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P (1989) The structure and regulation of protein phosphatases. Annu Rev Biochem 58:453–508 [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789 [DOI] [PubMed] [Google Scholar]
- Dashty M (2013) A quick look at biochemistry: carbohydrate metabolism. Clin Biochem 46:1339–1352 [DOI] [PubMed] [Google Scholar]
- DePaoli-Roach AA, Vilardo PG, Kim JH, Mavila N, Vemuri B, Roach PJ (2003) Determination of mammalian glycogen synthase phosphatase activity. Methods Enzymol 366:17–34 [DOI] [PubMed] [Google Scholar]
- Diaz-Villaseñor A, Burns AL, Hiriart M, Cebrián ME, Ostrosky- Wegman P (2007) Arsenic-induced alteration in the expression of genes related to type 2 diabetes mellitus. Toxicol Appl Pharmacol 225:123–133 [DOI] [PubMed] [Google Scholar]
- Díaz-Villaseñor A, Sánchez-Soto MC, Cebrián ME, Ostrosky-Wegman P, Hiriart M (2006) Sodium arsenite impairs insulin secretion and transcription in pancreatic beta-cells. Toxicol Appl Pharmacol. 214(1):30–34 [DOI] [PubMed] [Google Scholar]
- Díaz-Villasenor A, Burns AL, Salazar AM, Sordo M, Hiriart M, Cebrián ME, Ostrosky-Wegman P (2008) Arsenite reduces insulin secretion in rat pancreatic beta-cells by decreasing the calcium-dependent calpain-10 proteolysis of SNAP-25. Toxicol Appl Pharmacol. 231(3):291–299 [DOI] [PubMed] [Google Scholar]
- Douillet C, Currier J, Saunders J, Bodnar WM, Matousek T, Styblo M (2013) Methylated trivalent arsenicals are potent inhibitors of glucose stimulated insulin secretion by murine pancreatic islets. Toxicol Appl Pharmacol 267:11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobna Z, Walton FS, Paul DS, Xing W, Thomas DJ, Styblo M (2009) Metabolism of arsenic in human liver: the role of membrane transporters. Arch Toxicol 84:3–16 [DOI] [PubMed] [Google Scholar]
- Fu J, Woods CG, Yehuda-Shnaidman E, Zhang Q, Wong V, Collins S, Sun G, Andersen ME, Pi J (2010) Low-level arsenic impairs glucose-stimulated insulin secretion in pancreatic beta cells: involvement of cellular adaptive response to oxidative stress. Environ Health Perspect 118:864–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CF, Yang CY, Chan DC, Wang CC, Huang KH, Wu CC, Tsai KS, Yang RS, Liu SH (2015) Arsenic exposure and glucose intolerance/insulin resistance in estrogen-deficient female mice. Environ Health Perspect 123:1138–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue L, Bontemps F, Hers H (1975) The effects of glucose and of potassium ions on the interconversion of the two forms of glycogen phosphorylase and of glycogen synthetase in isolated rat liver preparations. Biochem J 152:105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imazu M, Strickland WG, Chrisman TD, Exton JH (1984a) Phosphorylation and inactivation of liver glycogen synthase by liver protein kinases. J Biol Chem 259:1813–1821 [PubMed] [Google Scholar]
- Imazu M, Strickland WG, Exton JH (1984b) Multiple phosphorylation of rat-liver glycogen synthase by protein kinases. Biochim Biophys Acta 789:285–293 [DOI] [PubMed] [Google Scholar]
- Kawaguchi I (1981) Studies on As2O3-induced hyperglycemia (author’s transl). Nihon Yakurigaku Zasshi 78:213–222 [PubMed] [Google Scholar]
- Klover PJ, Mooney RA (2004) Hepatocytes: critical for glucose homeostasis. Int J Biochem Cell Biol 36:753–758 [DOI] [PubMed] [Google Scholar]
- Krssak M, Brehm A, Bernroider E, Anderwald C, Nowotny P, Dalla Man C, Cobelli C, Cline GW, Shulman GI, Waldhausl W, Roden M (2004) Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 53:3048–3056 [DOI] [PubMed] [Google Scholar]
- Lin HV, Accili D (2011) Hormonal regulation of hepatic glucose production in health and disease. Cell Metab 14:9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Guo X, Wu B, Yu H, Zhang X, Li M (2014) Arsenic induces diabetic effects through beta-cell dysfunction and increased gluconeogenesis in mice. Sci Rep 4:6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, Silbergeld EK, Styblo M, Tseng CH, Thayer KA, Loomis D (2012) Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect 120:1658–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas-Acien A, Silbergeld EK, Streeter RA, Clark JM, Burke TA, Guallar E (2006) Arsenic exposure and type 2 diabetes: a systematic review of the experimental and epidemiological evidence. Environ Health Perspect 114:641–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuttall FQ, Gannon MC (1989) An improved assay for hepatic glycogen synthase in liver extracts with emphasis on synthase R. Anal Biochem 178:311–319 [DOI] [PubMed] [Google Scholar]
- Park SK, Peng Q, Bielak LF, Silver KD, Peyser PA, Mitchell BD (2016) Arsenic exposure is associated with diminished insulin sensitivity in non-diabetic Amish adults. Diabetes Metab Res Rev. 32(6):565–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul DS, Harmon AW, Devesa V, ThoMAs DJ, Styblo M (2007) Molecular mechanisms of the diabetogenic effects of arsenic: inhibition of insulin signaling by arsenite and methylarsonous acid. Environ Health Perspect 115:734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti W, Nairn AC, Page R (2012) Structural basis for protein phosphatase 1 regulation and specificity. FEBS J 280:596–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichl FX, Szinicz L, Kreppel H, Forth W (1988) Effect of arsenic on carbohydrate metabolism after single or repeated injection in guinea pigs. Arch Toxicol 62:473–475 [DOI] [PubMed] [Google Scholar]
- Reichl FX, Kreppel H, Szinicz L, Fichtl B, Forth W (1991) Effect of glucose treatment on carbohydrate content in various organs in mice after acute As2O3 poisoning. Vet Hum Toxicol 33:230–235 [PubMed] [Google Scholar]
- Rines AK, Sharabi K, Tavares CD, Puigserver P (2016) Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat Rev Drug Discov 15:786–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach PJ, Cheng C, Huang D, Lin A, Mu J, Skurat AV, Wilson W, Zhai L (1998) Novel aspects of the regulation of glycogen storage. J Basic Clin Physiol Pharmacol 9:139–151 [DOI] [PubMed] [Google Scholar]
- Rognstad R (1979) Rate-limiting steps of metabolic pathways. J Biol Chem 254:1875–1878 [PubMed] [Google Scholar]
- Ros S, Garcia-Rocha M, Dominguez J, Ferrer JC, Guinovart JJ (2009) Control of liver glycogen synthase activity and intracellular distribution by phosphorylation. J Biol Chem 284:6370–6378 [DOI] [PubMed] [Google Scholar]
- Rosella G, Zajac JD, Kaczmarczyk SJ, Andrikopoulos S, Proietto J (1993) Impaired suppression of gluconeogenesis induced by overexpression of a noninsulin-responsive phosphoenolpyruvate carboxykinase gene. Mol Endocrinol 7:1456–1462 [DOI] [PubMed] [Google Scholar]
- Rylatt DB, Aitken A, Bilham T, Condon GD, Embi N, Cohen P (1980) Glycogen synthase from rabbit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur J Biochem 107:529–537 [PubMed] [Google Scholar]
- Saheki S, Takeda A, Shimazu T (1985) Assay of inorganic phosphate in the mild pH range, suitable for measurement of glycogen phosphorylase activity. Anal Biochem 148:277–281 [DOI] [PubMed] [Google Scholar]
- Sharabi K, Tavares CD, Rines AK, Puigserver P (2015) Molecular pathophysiology of hepatic glucose production. Mol Aspects Med 46:21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Dutta SR, Passi D, Bharti J (2017) Benefits of alcohol on arsenic toxicity in rats. J Clin Diagn Res 11:BF01–BF06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spuches AM, Kruszyna HG, Rich AM, Wilcox DE (2005) Thermodynamics of the As(III)-thiol interaction: arsenite and mono-methylarsenite complexes with glutathione, dihydrolipoic acid, and other thiol ligands. Inorg Chem 44:2964–2972 [DOI] [PubMed] [Google Scholar]
- Srivastava AK, Pandey SK (1998) Potential mechanism(s) involved in the regulation of glycogen synthesis by insulin. Mol Cell Biochem 182:135–141 [PubMed] [Google Scholar]
- Stalmans W, Hers HG (1975) The stimulation of liver phosphorylase b by AMP, fluoride and sulfate. A technical note on the specific determination of the a and b forms of liver glycogen phosphorylase. Eur J Biochem 54:341–350 [DOI] [PubMed] [Google Scholar]
- Steffens AA, Hong GM, Bain LJ (2010) Sodium arsenite delays the differentiation of C2C12 mouse myoblast cells and alters methylation patterns on the transcription factor myogenin. Toxicol Appl Pharmacol 250:154–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ (2000) Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in human cells. Arch Toxicol 74:289–299 [DOI] [PubMed] [Google Scholar]
- Sung TC, Huang JW, Guo HR (2015) Association between arsenic exposure and diabetes: a meta-analysis. Biomed Res Int 2015:368087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Schlender KK, Larner J (1968) A rapid filter paper assay for UDPglucose-glycogen glucosyltransferase, including an improved biosynthesis of UDP-14C-glucose. Anal Biochem 25:486–499 [DOI] [PubMed] [Google Scholar]
- Thomas DJ, Li J, Waters SB, Xing W, Adair BM, Drobna Z, Devesa V, Styblo M (2007) Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp Biol Med (Maywood). 232:3–13 [PMC free article] [PubMed] [Google Scholar]
- Tseng CH (2004) The potential biological mechanisms of arsenic-induced diabetes mellitus. Toxicol Appl Pharmacol. 197(2):67–83 [DOI] [PubMed] [Google Scholar]
- Verma RJ, Vasu A, Saiyed AA (2004) Arsenic toxicity in mice and its possible amelioration. J Environ Sci (China) 16:447–453 [PubMed] [Google Scholar]
- Walton FS, Harmon AW, Paul DS, Drobna Z, Patel YM, Styblo M (2004) Inhibition of insulin-dependent glucose uptake by trivalent arsenicals: possible mechanism of arsenic-induced diabetes. Toxicol Appl Pharmacol 198:424–433 [DOI] [PubMed] [Google Scholar]
- Wang ZX, Jiang CS, Liu L, Wang XH, Jin HJ, Wu Q, Chen Q (2005) The role of Akt on arsenic trioxide suppression of 3T3-L1 preadipocyte differentiation. Cell Res 15:379–386 [DOI] [PubMed] [Google Scholar]
- Wauson EM, Langan AS, Vorce RL (2002) Sodium arsenite inhibits and reverses expression of adipogenic and fat cell-specific genes during in vitro adipogenesis. Toxicol Sci 65:211–219 [DOI] [PubMed] [Google Scholar]
- Welsh GI, Wilson C, Proud CG (1996) GSK3: a SHAGGY frog story. Trends Cell Biol 6:274–279 [DOI] [PubMed] [Google Scholar]
- Xue P, Hou Y, Zhang Q, Woods CG, Yarborough K, Liu H, Sun G, Andersen ME, Pi J (2011) Prolonged inorganic arsenite exposure suppresses insulin-stimulated AKT S473 phosphorylation and glucose uptake in 3T3-L1 adipocytes: involvement of the adaptive antioxidant response. Biochem Biophys Res Commun 407:360–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH (2010) Arsenic inhibits myogenic differentiation and muscle regeneration. Environ Health Perspect 118:949–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wendel AA, Keogh MR, Harris TE, Chen J, Coleman RA (2012) Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc Natl Acad Sci U S A 109:1667–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Cooper DE, Grevengoed TJ, Li LO, Klett EL, Eaton JM, Harris TE, Coleman RA (2014) Glycerol-3-phosphate acyltransferase-4-deficient mice are protected from diet-induced insulin resistance by the enhanced association of mTOR and rictor. Am J Physiol Endocrinol Metab 307:E305-E315 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.