

Abstract
The contact pathway factors XI (FXI) and XII (FXII) have been demonstrated to be largely dispensable for hemostasis, as their absence results in a mild to absent bleeding diathesis. A growing body of literature, however, suggests that the contact pathway contributes to the pathologic host response to certain infectious organisms that produces the often‐fatal syndrome known as sepsis. The contact pathway factors serve as a central node connecting inflammation to coagulation, and may offer a potentially safe therapeutic target to mitigate the morbidity and mortality associated with sepsis. Herein, we summarize published in vivo and in vitro data that have explored the roles of the contact pathway in sepsis, and discuss potential clinical applications of novel FXI‐ and FXII‐inhibiting drugs currently under investigation.
Keywords: contact activation, factor XI, factor XII, kallikrein‐kinin system, sepsis
Abbreviations
- FXI/FXIa
zymogen/activated coagulation factor XI
- FXII/FXIIa
zymogen/activated coagulation factor XII
Essentials.
The contact activation system serves as a common node linking inflammatory and coagulation cascades.
In vitro and in vivo studies suggest that contact pathway inhibition may be a therapeutic target in sepsis.
Contact inhibition in animal models blunts cytokine response and improves survival in bacterial sepsis.
Further studies in humans are required to evaluate efficacy in clinical practice.
1. INTRODUCTION
Despite advances in the delivery and quality of supportive care, judicious use of antimicrobial therapy, and global epidemiologic shifts, sepsis remains a leading cause of morbidity and mortality worldwide, leading to an estimated 5 million deaths annually.1 Sepsis is an infection‐induced systemic inflammatory response syndrome that typically progresses to organ hypoperfusion and death within hours to days when left untreated.2 The increasing focus on the role of the host reflected in the definition of sepsis, rather than the pathogen itself, has been a paradigm shift emphasized by society guidelines in recent years2 and highlights an unmet clinical need to explore novel methods targeting the unique pathobiology of this syndrome.
Physiologic pathways involved in the dysregulated host response in sepsis include the inflammatory cascade, complement system, and the coagulation system. While often considered isolated entities, these systems are in fact intimately linked, with perturbations in one affecting the others. These relationships can be collectively referred to as immunothrombosis and suggest a role for coagulation as a form of innate immune defense. Purported mechanisms of coagulation being protective against disseminated infection include trapping of pathogens and leukocytes within fibrin thrombi, as well as prevention of pathogens exiting the vascular space into the tissues.3 At its most extreme, progression of this immunothrombosis mechanism can lead to disseminated intravascular coagulation (DIC), characterized by systemic activation and consumption of coagulation factors, platelets, and fibrinogen, leading to both widespread thrombosis and bleeding, and contributing substantially to the morbidity of sepsis (Figure 1).
Figure 1.
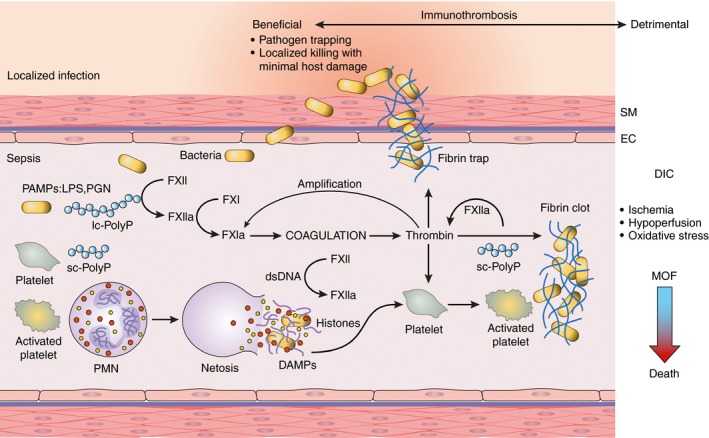
Bacteria‐induced contact activation leading to immunothrombosis and DIC. The coagulation factor XII (FXII) is activated by live bacteria and bacterial wall–derived pathogen‐associated molecular patterns (PAMPS), such as lipopolysaccharides (LPS) for gram‐negative or peptidoglycan (PGN) for gram‐positive bacteria or bacteria‐produced long‐chain polyphosphates (lc‐PolyP). Activation of FXII leads to thrombin generation, platelet activation, and fibrin formation, which in concert trap and contain pathogens. Activated platelets promote neutrophils to release their nuclear contents to form extracellular traps (netosis) as part of the routes of pathogen clearance; NETs are also potent platelet activators and cytotoxic to the host cells. Histones and double‐stranded DNA (dsDNA) act as damage‐associated pattern molecules (DAMPs) that together with platelet‐derived short‐chain polyphosphates (sc‐polyPs) contribute to contact activation via direct and indirect mechanisms. Failing the redundant pathways of pathogen clearance offered by the immune and coagulation systems, immunothrombosis can result in widespread microvascular thrombosis termed disseminated intravascular coagulation (DIC) followed by consumptive coagulopathy and bleeding. Thrombosis within the microvasculature of vital organs leads to ischemia, hypoperfusion, and oxidative stress, thus impairing organ function and leading to multiple organ failure (MOF). When more than two organs fail, death is the most likely outcome
2. HISTORICAL ATTEMPTS AT TARGETING IMMUNOTHROMBOSIS IN SEPSIS
Historical attempts to attenuate the activation of coagulation in patients with sepsis have been met with equivocal benefit and potential harm, most notably with the use of activated protein C (APC; drotrecogin alfa, Xigris). Drotrecogin alfa was theorized to confer a benefit in sepsis by repleting a natural anticoagulant (APC) that was known to be decreased in septic patients, as well as augmenting its cytoprotective effects via protease‐activated receptor (PAR)‐1 signaling.4 Early‐phase human trials of APC in sepsis were compelling due to significant reductions in inflammatory cytokine levels as well as statistically significantly improved mortality compared with placebo.5 A subsequent large, phase 3, placebo‐controlled trial of drotrecogin alfa in sepsis, however, failed to show this same mortality benefit after both acute and long‐term follow‐up, and the drug was subsequently removed from the market.6 Drotrecogin alfa also led to a significantly higher incidence of bleeding, including some instances of major hemorrhage, than placebo.7 Data on the use of heparin products as adjunctive therapy for sepsis, despite benefits reported in several meta‐analyses, must be interpreted with caution due to these analyses’ frequent inclusion of nonrandomized studies, variable definitions of sepsis, and the concomitant use of other investigational agents (APC, antithrombin, and others).3 In addition, bleeding remains a persistent, unwanted side effect of most traditional anticoagulants used in this setting. Given that no anticoagulant has ever shown a convincing benefit in sepsis, their routine use is not currently recommended in septic patients without another indication for anticoagulation.3
3. THE CONTACT ACTIVATION SYSTEM IN SEPSIS
One potential explanation for the lack of clear benefit and consistent harm with anticoagulation strategies trialed in sepsis to date include their nonspecific mechanisms of action, targeting multiple coagulation factors from the intrinsic, extrinsic, and common coagulation pathways. More specifically targeted anticoagulant strategies may therefore provide more favorable balance of benefits and risks in sepsis.
The contact activation system (CAS) is the most upstream component of the intrinsic pathway of coagulation, consisting of coagulation factors XI (FXI), XII (FXII), prekallikrein (PK) and high‐molecular‐weight kininogen (HK). Upon activation, the contact system promotes not only coagulation but also platelet and leukocyte recruitment, complement activation, and bradykinin generation.8 Thus, the contact system represents a specific node linking the inflammatory and coagulopathic responses that contribute to the pathogenesis of sepsis, and represents a biologically plausible potential target upon which to focus research on novel therapeutics. A growing body of literature has examined the specific physiologic roles of contact factors, epidemiologic data on contact factor deficiencies in humans, and numerous animal models of both genetic and drug‐induced factor deficiencies. The general conclusion of this work is that that the contact system has a limited role in hemostasis but can generate and amplify pathologic thrombosis and inflammation, and potentially could be safely inhibited to confer benefits in various disease states.
The CAS is activated upon blood exposure to an increasing number of recognized substances, including negatively charged artificial surfaces, various infectious pathogens, long‐chain polyphosphates (polyPs), and cell‐free DNA/RNA. In sepsis in particular, CAS activation seems to be triggered by contact with negatively charged surfaces, including platelet‐derived polyP and bacterial cell components such as peptidoglycans, teichoic acid, and lipopolysaccharide.9 Upon exposure to an activating surface, FXII changes conformation to become the serine protease FXIIa. This enzyme activates PK to form kallikrein (PKa), which, via reciprocal activation of FXIIa, rapidly increases the serum concentration of both enzymes.
The reciprocal activation of FXIIa and PKa catalyzes downstream activation of both coagulation and inflammation. FXIIa activates FXI to FXIa and triggers the intrinsic pathway of the coagulation cascade through the subsequent activation of FIX, FX, and prothrombin. FXIIa‐induced activation of PKa also results in cleavage of HK to release the vasoactive substance bradykinin, which leads to endothelial permeability, leukocyte recruitment, and cytokine generation.9
Of the contact factors, FXI is most prominently poised at the intersection of these physiologic pathways, via its role in the activation of both downstream and upstream inflammatory and coagulation cascades. In addition to activation by FXIIa, FXI can also be activated by thrombin in the setting of trauma‐induced bleeding via the tissue factor pathway of coagulation, and FXIa can reciprocally activate FXII, leading to bradykinin generation.10, 11 Certain bacterial cell components, including peptidoglycan and polyP, are also known to activate FXII and promote FXI activation.12, 13, 14 We have shown that bacterial‐type long‐chain polyP promotes platelet activation, microaggregate formation, and consumption in blood flow in a contact activation pathway–dependent manner in vitro.15, 16 In a murine model, long‐chain polyP promoted platelet deposition and fibrin generation in the lungs in an FXII‐dependent manner. In a nonhuman primate model of bacterial sepsis, pretreatment of animals with an antibody blocking FXI activation by FXIIa reduced Staphylococcus aureus–induced platelet and fibrinogen consumption.15 This work suggests that bacterial components, including long‐chain polyP, promote platelet activation in an FXII‐dependent manner in flowing blood, which may contribute to sepsis‐associated thrombotic processes, consumptive coagulopathy, and thrombocytopenia.
Contact activation affects common surface‐activated coagulation assays, including the activated partial thromboplastin time (aPTT) and activated clotting time, but has not been found to correlate meaningfully with true hemostatic potential. Indeed, the activation of FXI by FXIIa is considered dispensable for normal hemostasis but does play a prominent role in thrombin generation in pathologic settings.17 This has been shown clinically, as FXI deficiency is associated with a mild bleeding diathesis typically only in the face of sufficient traumatic or surgical challenge despite markedly elevated aPTT values, unlike the frequently spontaneous hemorrhage seen in FVIII and FIX deficiencies.18 Even more compelling is the apparent lack of abnormal bleeding of any type seen in congenital FXII deficiency,19 though these individuals have rarely been encountered. The Ashkenazi Jewish population has a relatively high rate of congenital FXI deficiency, and a large, retrospective cohort study of FXI‐deficient patients in Israel demonstrated significantly reduced rates of both venous and arterial thrombotic events over a decade of follow‐up.20 Epidemiologic data on the effect of FXII deficiency on thrombosis are scarce and remain inconclusive.21
Certain animal populations with congenital FXI deficiency, including Holstein cattle, may have an increased susceptibility to pulmonary infection, but the contact proteins do not seem to play a significant role in innate immunity in humans.22 The various data describing the ways in which the CAS plays a multimodal role in organisms' responses to infection are outlined through the remainder of this text and in Table 1.
Table 1.
Effects of FXI inhibition on coagulation and inflammation
| Processes | Findings |
|---|---|
| Coagulopathy |
In in vitro studies, FXI was found to be directly activated by thrombin independent of FXII.47, 48
In a human in vivo model of low‐grade endotoximea, thrombin generation and FXI activation were detected in the absence of FXII activation.37 In a perforation‐induced peritoneal sepsis model, mice anticoagulated with a selective antibody (14E11) that inhibited FXI activation by FXIIa led to suppression of systemic thrombin‐antithrombin complex formation.26 In a cecal puncture– and ligation‐induced sepsis model, FXI‐deficient mice exhibited lower FXII activation in the presence of microbial molecules (eg, polyphosphate), revealing the role of activated FXIa in feedback activation of FXII in the presence of polyanions.25 In human blood studies ex vivo and in primates in vivo, presence of microbial molecules (eg polyphosphates, potentiated thrombin generation and fibrin formation in FXII‐FXI axis–dependent manner.15 |
| Platelet consumption |
In a perforation‐induced peritoneal sepsis model, mice anticoagulated with a selective antibody (14E11) that inhibited FXI activation by FXIIa led to reduced platelet consumption in the circulation and deposition in the blood vessels.26
In human blood studies ex vivo and in primates in vivo, FXI potentiated thrombin generation at a local site of thrombus formation and led to platelet microaggregate formation and single platelet consumption in flowing blood distal to the local site of thrombus formation.15, 16 |
| Cytokine production and inflammation |
In a cecal puncture–induced sepsis model, FXI‐deficient mice had a survival advantage with significant smaller increases in plasma levels of TNF‐α and IL‐10 and delayed IL‐1β and IL‐6 responses.23, 25
In a cecal puncture–induced sepsis model, FXI‐deficient mice expressed reduced serum levels of an acute phase inflammatory protein, amyloid P.25, 26 In a perforation‐induced peritoneal sepsis model, mice anticoagulated with a selective antibody (14E11) that inhibited FXI activation by FXIIa led to suppression of systemic IL‐6 and TNF‐α levels.25 In nonhuman primate studies, inhibition of FXII‐ and FXII‐mediated activation of FXI have both been shown to blunt the cytokine response and improve measurable physiologic outcomes after inoculation with live or heat‐inactivated bacterium.15, 34 |
| Neutrophil function |
Coagulation FXI was found to immunolocalize with human neutrophil membrane bound by kininogen.49
In a cecal puncture–induced sepsis model, FXI‐deficient mice had decreased lymphocyte accumulation in infected tissues.23 In in vitro assays, purified polymorphonuclear leukocytes exposed to activated FXIa exhibited less chemokine‐induced chemotaxis and calcium release.49 In Streptococcus pneumoniae and Klebsiella pneumoniae models, FXI‐deficient mice had lower levels of phagocytosis.29 |
FXI, factor XI; FXII, factor XII; IL, interleukin; TNF‐α, tumor necrosis factor‐α.
These data make selective inhibition of the CAS an attractive therapeutic possibility in sepsis, with the potential to mitigate severe inflammation without compromising hemostasis or the host immune response. A growing body of work utilizing murine and primate models has demonstrated that inhibition of FXI can reduce pathologic inflammation and improve survival in the setting of systemic bacterial infections in vivo. In the following sections, we discuss the contribution of the contact system in immunothrombosis as shown by studies of FXI and FXII inhibition in several mammal models of infection, and consider future clinical applications of this knowledge in humans.
4. MURINE STUDIES
The catalytic role of FXI in immunothrombosis suggests that its absence may lead to a less severe host inflammatory response and improved morbidity and mortality in the setting of systemic infection. Evidence from several animal models supports this hypothesis.
When challenged with high doses of intraperitoneal Listeria monocytogenes, FXI knockout mice were found to develop a less severe coagulopathy and lower serum cytokine levels than their wild‐type (WT) counterparts.23 FXI knockout mice likewise displayed significantly higher serum fibrinogen levels, platelet counts, and reduced thrombin‐antithrombin complex (TAT) levels after inoculation with high‐dose L. monocytogenes as compared to controls. Plasma markers of inflammation, including interleukin (IL)‐6 and hepatic mRNA encoding IL‐6 and IL‐10, were detected at significantly lower levels in the FXI‐deficient animals. The attenuated inflammation in the FXI‐deficient mice resulted in measurable improvements in end‐organ outcomes as well: whereas WT mice were found on histologic analysis to have large areas of hepatic necrosis, FXI‐deficient mice had relatively scant necrosis and less inflammatory cell infiltration in the liver parenchyma. Finally, FXI‐deficient mice demonstrated significantly improved survival: while only 30% of WT mice survived to day 10 after inoculation, 67% of FXI‐deficient mice were alive on day 20.24
The protective effect of FXI deficiency appears to extend to polymicrobial infections as well. After inducing peritoneal sepsis via cecal ligation and puncture (CLP), FXI‐deficient mice experienced a substantially smaller decrease in platelet count than WT mice, suggesting a milder consumptive coagulopathy.23 Prothrombin time, a marker of plasma clotting factor levels, increased markedly in WT mice after CLP but remained unchanged in FXI‐deficient animals, and correspondingly TAT levels were reduced in FXI‐deficient mice compared to WT mice. Mice that lacked FXI clearly developed a less pronounced pathologic coagulopathy and relatively attenuated inflammatory response compared to WT controls, and this translated directly to a significant survival advantage. At 1 week after CLP, 46% of FXI‐deficient mice were alive compared to just 13% of WT mice. This experimental sepsis model was later replicated to further define the changes in inflammatory cytokine levels and coagulation profiles in FXI knockout mice. Four hours after CLP, FXI‐deficient mice demonstrated significantly lower plasma levels of tumor necrosis factor‐α (TNF‐α), IL‐1β, IL‐6, and IL‐10 compared to WT mice. Survival at 7 days was also significantly higher in the FXI knockout mice than WT in this follow‐on study (39% vs 6%).25
Antibody‐mediated FXI inhibition has also been applied to the CLP polymicrobial sepsis model with similarly favorable results. Tucker et al26 investigated the use of a murine monoclonal antibody, 14E11, that selectively inhibits FXI activation by FXIIa in WT mice undergoing CLP, comparing this to administration of APC and vehicle (control). At 1 week, 80% of mice treated with 14E11 survived, compared to 45% survival for vehicle‐treated animals and 15% for APC‐treated mice. A survival benefit was also seen in mice treated with 14E11 at 6 and 12 hours after surgery. Overall, the 14E11‐treated group had a 30% improvement in overall survival compared to the vehicle‐treated arm. Perhaps surprisingly, treatment of WT mice with 6 mg/kg APC was detrimental and both decreased survival and increased bleeding in this model, reflective of the clinical observation that APC administration may not produce a significant benefit in septic patients.
The effect of 14E11 on inflammation and coagulopathy was directly measured. Mice treated immediately with 14E11 had a 31% higher average platelet count 36 hours after surgery compared to vehicle‐treated controls. On postmortem organ evaluation, only 40% of 14E11‐treated mice had evidence of hepatic vascular microthrombi compared to 75% of the control group, further supporting the close link between coagulation and inflammation in polymicrobial sepsis. Though levels of inflammatory cytokines TNF‐α and IL‐6 increased after CLP in all treatment groups, the 14E11‐treated group had significantly lower levels of both inflammatory markers than vehicle‐treated controls.26 The attenuated inflammatory response seen in 14E11‐treated mice, with the associated reduction in platelet consumption, TAT levels, and hepatic microvascular thrombosis, suggests that inhibition of thrombin generation and the resultant tissue ischemia may be one functional link between sepsis‐induced coagulopathy and mortality.
While the end points of these key studies focused on markers of inflammation and coagulation, others have examined the effect of contact pathway inhibition on other physiologic parameters. Work by Kossmann et al27 found that antisense oligonucleotide‐mediated reduction in FXI levels abrogated angiotensin II and nephrectomy‐mediated arterial hypertension in mice. While this mechanism could be desirable to mitigate morbidity and mortality associated with cardiovascular diseases, it is unclear to what degree this might affect blood pressure in the setting of sepsis, and indeed in other animal models, contact factor inhibition attenuates sepsis‐associated hypotension. Clearly, the contact pathway's role in cardiovascular physiology requires further study.
An additional key finding by Luo et al highlights that selective inhibition of the contact system but not the extrinsic or common pathways of anticoagulation may be beneficial during sepsis. In a mouse model of Yersinia enterocolitica sepsis, mice engineered to express low levels of tissue factor (TF) or fibrin and those treated with the vitamin K antagonist warfarin demonstrated increased hepatic bacterial burden and reduced survival compared to WT or FXI knockout mice.23 It may be that fibrin is an essential component of innate immunity in certain bacterial infections and that inhibition of the TF‐mediated extrinsic pathway or common pathways reduces fibrin production to a lethal degree. Contact pathway inhibition may provide a milder reduction in fibrin production, balancing the beneficial and detrimental aspects of innate immune response more favorably. These points may explain why previous trials of heparin products during sepsis have yielded mixed results.
An additional critical point learned from mouse sepsis models is the potential for pathogen‐specific immunothrombotic phenotypes. Virulence of 2 different gram‐negative bacteria, E. coli and Y. enterocolitica, is affected by fibrin to variable degrees, so a “one‐size‐fits‐all” approach to anticoagulation may in fact be harmful with certain infections. Indeed, differences in virulence factors between gram‐negative and gram‐positive bacteria are even more pronounced, making further exploration of the contribution of thrombosis to the pathogenicity of various organisms essential.
Though these data linking the absence or inhibition of FXI to attenuated inflammation and reduced coagulopathy are compelling, it must also be remembered that improved outcomes in mice do not necessarily imply clinical relevance for humans. FXI is physically associated with the vascular endothelium in mice, whereas it primarily circulates in the plasma of primates, and FXI‐deficient mice do not display any obvious impairment in hemostasis, unlike humans.28 FXI knockout mice actually display an exaggerated local inflammatory response in the lungs when exposed to inhaled dust mites compared to WT mice,29, 30 though a similar effect has not been documented in humans. More broadly, inflammatory and coagulation mechanisms in mice do not seem to correlate strongly with responses in primates,31 which has led to further evaluation of the role of FXI and FXII inhibition in nonhuman primate models.
5. NONHUMAN PRIMATE MODELS
Original work by Pixley et al32 and Jansen et al33 in the 1990s showed the beneficial effects of FXII inhibition in a baboon model of E. coli sepsis.32 Wild‐type baboons showed a significant decrease in HK and increase in alpha‐2 macroglobulin‐kallikrein complexes in the blood compared to baboons given a dose of a FXII‐inhibiting antibody (C6B7). Most impressively, terminal hypotension was attenuated in the group that received C6B7; while both control baboons and those treated with the FXII inhibitor C6B7 developed initial hypotension, the control group subsequently developed irreversible lethal hypotension, while shock was reversible in those with inhibited FXII. These findings suggested that the upstream role of FXII in regulating bradykinin may participate in the pathologic shock that develops in severe sepsis. A similar study also showed that pretreatment with the FXII inhibitor C6B7 before E. coli inoculation led to reduced complement activation, neutrophil degranulation, and levels of tissue plasminogen activator and IL‐6 compared to untreated baboons.33
More recent nonhuman primate work by Silasi et al34 investigated the effects of the monoclonal anti‐FXI antibody 3G3, a humanized variant of the mouse anti‐FXI antibody 14E11 discussed above, in baboons with S. aureus bacteremia. Baboons challenged with an infusion of heat‐inactivated S. aureus were randomized to pretreatment with 3G3 30 minutes before the bacterial infusion or to an untreated control arm. All animals treated with the anti‐FXI antibody reached the 7‐day survival end point compared to none in the control arm.
The mechanisms of this survival advantage were investigated by assessment of the effects of 3G3 treatment on parameters of inflammation, coagulation, and organ dysfunction. Administration of 3G3 significantly reduced the consumptive coagulopathy induced by S. aureus infusion, as reflected in markedly lower aPTT and protease‐serpin complex levels compared to control animals. Baboons pretreated with 3G3 also exhibited higher fibrinogen and HK levels, suggestive of reduced fibrin formation and HK cleavage, a marker of bradykinin production. The platelet count in 3G3‐treated animals did not fall as low as in control animals and recovered more quickly. Postmortem analysis showed no microvascular thrombi in the lungs, kidneys, and livers of 3G3‐treated baboons. These findings indicated that inhibition of FXI attenuates the consumptive coagulopathy of sepsis in a primate model without inducing bleeding.34
Infusion of S. aureus caused hemodynamic compromise in baboons not treated with 3G3, manifested by tachycardia and hypotension, respiratory distress, hypoxemia, and fever. Pretreatment with 3G3 resulted in fewer respiratory complications and less tachypnea, improved peripheral perfusion, and reduced fever compared to untreated controls. The treatment arm also experienced less pronounced elevations in plasma transaminases, pancreatic amylase, and serum creatinine, indicating attenuation of end‐organ dysfunction. Baboons treated with 3G3 also had delayed and less severe increase in lactate dehydrogenase and circulating nucleosome levels, reflecting reduced cell death in comparison with controls. The 3G3‐treated baboons had normal organ appearance on histopathologic evaluation, compared to extensive organ damage in untreated animals.34
Inhibition of FXI with 3G3 resulted in a marked blunting of the cytokine storm associated with bacteremia and sepsis. Plasma levels of IL‐6, IL‐8, monocyte chemoattractant protein‐1, granulocyte‐macrophage colony‐stimulating factor, and IL‐1β were lower in treated animals than in controls, reflecting reduced cytokine production. Treated animals also displayed decreased complement cascade activation reflected by lower levels of circulating C3b and C5b‐9 terminal complement complex.34 How FXI activation contributes to cytokine production and complement activation is not completely understood but likely involves downstream effects of decreased microvascular thrombosis, hypoperfusion, and oxidative stress. It is known that FXIIa and cleaved HK can promote cytokine production by monocytes.35, 36 Coagulation proteases such as thrombin and FXa can signal via protease‐activated receptors to induce cytokine production and increase vascular permeability. By decreasing FXIIa and kallikrein, 3G3 likely reduced generation of bradykinin, a potent vasodilator and inducer of vascular permeability.
Together, these nonhuman primate experiments suggest that targeting the contact pathway may effectively blunt the pathologic host immune response seen in sepsis. As novel agents targeting FXI and FXII accumulate clinical data, the ability to perform pilot trials in septic humans is becoming increasingly plausible.
6. HUMAN DATA
Minnema and colleagues performed an informative experiment in which they infused 8 healthy volunteers with a low dose of endotoxin in an attempt to measure contact pathway activation. They found a significant increase in FXIa, peaking at 1 and 2 hours after endotoxin infusion, followed by a gradual increase in FXIa‐FXIa inhibitor complexes. Thrombin generation peaked at 3 to 4 hours after infusion, and in contrast, markers of activation of prekallikrein and FXII remained undetectable.37 While data specific to sepsis are limited, several studies have shown increased contact pathway activation using blood samples from septic patients. Using assays measuring FXIIa‐C1‐inhibitor, kallikrein‐C1‐inhibitor, and FXIa‐inhibitor complexes, several studies have shown FXII activation and kallikrein formation in both adult and pediatric patients with sepsis.38, 39, 40, 41 Likewise, recent studies have suggested that the reduced form of coagulation factor XI is associated with worse outcomes and coagulopathy in septic patients.42
While these data suggest that the CAS contributes to the pathology of sepsis in humans, further study is needed before any major conclusions are drawn to determine if targeting this pathway could offer a meaningful benefit to septic patients. As multiple novel contact pathway inhibitors enter clinical testing, additional clinical investigations may begin to address this question.43, 44, 45
7. LIMITATIONS AND REMAINING QUESTIONS
The evidence supporting a positive effect of contact pathway inhibition on outcomes in sepsis is noteworthy mechanistically and in preclinical animal models. However, the therapeutic possibilities of FXI and FXII inhibition in murine and primate models do not necessarily translate to clinical efficacy in humans. The discovery that FXI knockout mice display an exaggerated pulmonary inflammatory response to inhaled dust mites, and that these mice have worse overall survival upon exposure to Streptococcus pneumoniae and Klebsiella pneumoniae compared to wild‐type animals,29 raised concerns that FXI inhibition may have unintended consequences in certain infectious contexts. In evaluating these concerns, Salomon et al46 found that people with partial and complete FXI deficiency did not experience a significantly higher incidence of pneumonia, severe pneumonia, or short‐term mortality compared to those with normal FXI activity. This indicates that the physiologic role of FXI in humans may differ from that in certain other mammal species.
The innate heterogeneity of sepsis is a major barrier to identifying a standardized approach to CAS inhibition. The diversity of infectious organisms, baseline host characteristics, and degree of inflammatory responses not only introduce discrepant effects of contact pathway blockade, but may also result in potential harm with CAS inhibition in certain patient subsets. Conversely, there may be specific patient populations, such as those expected to mount robust inflammatory responses, in whom CAS inhibition could significantly improve outcomes in sepsis. Further studies will help identify subsets of patients most likely to benefit from this intervention.
As previously discussed, one area warranting further attention is pathogen‐specific host responses, and the effects of contact inhibition on these responses. Initial studies of S. aureus bacteremia by our group used heat‐inactivated bacteria; heat inactivation neutralizes the ability of S. aureus to generate and secrete potent exotoxins, but their presence in natural infections with live bacteria might change the immunothrombotic phenotype and the effects of various methods of contact pathway inhibition. This hypothesis is part of ongoing work by our group. A further limitation of animal sepsis models to date is that methods of pathogen inoculation for study likely do not reflect natural acquisition and dissemination of these pathogens. Acknowledging this limitation, future studies should explore whether inoculum size, route, and rate of administration alter the effects of contact inhibition in sepsis.
The optimal timing of CAS inhibition is also unclear. Inhibition of the contact pathway is likely to produce benefit if sepsis is identified in its early stages, potentially preventing DIC and avoiding the later stages characterized by consumption of clotting factors and platelets. The effects of this intervention in patients with disseminated infection but without consumptive coagulopathy is not known. Furthermore, contact inhibition may prove to be harmful in certain immunocompromised populations by further blunting an already diminished host response.
Finally, while contact pathway inhibition in general holds promise, it remains to be clarified which specific component(s) of this pathway should be targeted to optimize clinical outcomes. Phase 1 clinical trial data have now been published on 4 different monoclonal antibodies targeting FXI, all with unique mechanisms.43, 44, 45 Our group has worked extensively with 14E11/3G3, a monoclonal antibody inhibiting FXIIa‐mediated activation of FXI. The benefits of this specific contact pathway–inhibiting mechanism have been shown in several of the animal models discussed above. It is not known if more direct FXIa active enzymatic site inhibition would produce similar results. FXII inhibition in baboon sepsis models yielded results similar to FXI inhibition, but whether such an approach would be effective in humans is also unclear. Targeting FXII could alter the ratio of coagulation/kinin‐kallikrein inhibition seen with FXI inhibition, potentially to even greater positive clinical effect. The kinin‐kallikrein axis specifically may also hold promise, as evidenced by its critical role in the pathophysiology of hereditary angioedema (HAE) as well as animal models showing it has similar effects as FXI and FXII inhibition on immunothrombosis in sepsis. A novel kallikrein‐inhibiting antibody, lanadelumab, was recently approved by the US Food and Drug Administration for treatment of HAE, indicating the safety and feasibility of this strategy.
8. CONCLUSIONS
The role of the contact activation system in catalyzing downstream inflammation and coagulation, together with the apparent minor roles of FXI and FXII in normal hemostasis, make antagonism of this pathway a promising therapeutic target in sepsis. This enthusiasm should be tempered, however, by the historical lessons learned from attempts to use heparin to improve outcomes in septic patients, as well as several lingering questions regarding the precise physiologic role of the contact factors. Ongoing preclinical investigation by our group and others will help to clarify many of the remaining questions discussed above, and continued early‐phase clinical study of FXI‐ and FXII‐inhibiting monoclonal antibodies will provide additional valuable insight into safety of this strategy. With these steps, clinical trials of contact inhibition in human subjects with infections will become increasingly feasible and could meet a critically unmet clinical need in the improved treatment of sepsis.
RELATIONSHIP DISCLOSURE
Dr. Shatzel is a consultant for Aronora, Inc, a pharmaceutical company with interests in targeting factors XI and XII for therapeutic purposes. The remaining authors report nothing to disclose.
AUTHOR CONTRIBUTIONS
VR performed literature review, collection of data, composition of the primary manuscript, and editing. JZR performed literature review, collection of data, composition of the primary manuscript, and editing. SRO participated in collection of data and composition of the primary manuscript. OMC participated in composition of the primary manuscript and editing. JJS participated in collection of data, composition of the primary manuscript, and editing. FL contributed to the primary text and participated in revision of the manuscript. JZR, OMC, and JJS were directly involved in the primary research that is summarized in this review article.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health, National Institute of General Medical Sciences (GM116184 [O.J.T.M.]).
Raghunathan V, Zilberman‐Rudenko J, Olson SR, Lupu F, McCarty OJT, Shatzel JJ. The contact pathway and sepsis. Res Pract Thromb Haemost. 2019;3:331–339. 10.1002/rth2.12217
Vikram Raghunathan and Jevgenia Zilberman‐Rudenko contributed equally.
REFERENCES
- 1. Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of global incidence and mortality of hospital‐treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med. 2016;193:259–72. [DOI] [PubMed] [Google Scholar]
- 2. Singer M, Deutschman CS, Seymour CW, Shankar‐Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA. 2016;315:801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li X, Ma X. The role of heparin in sepsis: much more than just an anticoagulant. Br J Haematol. 2017;179:389–98. [DOI] [PubMed] [Google Scholar]
- 4. Levi M, van der Poll T. Recombinant human activated protein C: current insights into its mechanism of action. Crit Care. 2007;11(suppl 5):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernard GR, Vincent J‐L, Laterre P‐F, LaRosa SP, Dhainaut J‐F, Lopez‐Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. [DOI] [PubMed] [Google Scholar]
- 6. Ranieri VM, Thompson BT, Barie PS, Dhainaut J‐F, Douglas IS, Finfer S, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366:2055–64. [DOI] [PubMed] [Google Scholar]
- 7. Gentry CA, Gross KB, Sud B, Drevets DA. Adverse outcomes associated with the use of drotrecogin alfa (activated) in patients with severe sepsis and baseline bleeding precautions. Crit Care Med. 2009;37:19–25. [DOI] [PubMed] [Google Scholar]
- 8. Long AT, Kenne E, Jung R, Fuchs TA, Renne T. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost. 2016;14:427–37. [DOI] [PubMed] [Google Scholar]
- 9. Pathak M, Kaira BG, Slater A, Emsley J. Cell receptor and cofactor interactions of the contact activation system and factor XI. Front Med. 2018;5:66–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ivanov I, Shakhawat R, Sun MF, Dickeson SK, Puy C, McCarty OJ, et al. Nucleic acids as cofactors for factor XI and prekallikrein activation: different roles for high‐molecular‐weight kininogen. Thromb Haemost. 2017;117:671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tillman BF, Gruber A, McCarty OJT, Gailani D. Plasma contact factors as therapeutic targets. Blood Rev. 2018;32:433–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. 2011;118:6963–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Popescu NI, Silasi R, Keshari RS, Girton A, Burgett T, Zeerleder SS, et al. Peptidoglycan induces disseminated intravascular coagulation in baboons through activation of both coagulation pathways. Blood. 2018;132:849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Puy C, Tucker EI, Wong ZC, Gailani D, Smith SA, Choi SH, et al. Factor XII promotes blood coagulation independent of factor XI in the presence of long‐chain polyphosphates. J Thromb Haemost. 2013;11:1341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zilberman‐Rudenko J, Reitsma SE, Puy C, Rigg RA, Smith SA, Tucker EI, et al. Factor XII activation promotes platelet consumption in the presence of bacterial‐type long‐chain polyphosphate in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2018;38:1748–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zilberman‐Rudenko J, Itakura A, Wiesenekker CP, Vetter R, Maas C, Gailani D, et al. Coagulation factor XI promotes distal platelet activation and single platelet consumption in the bloodstream under shear flow. Arterioscler Thromb Vasc Biol. 2016;36:510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schmaier AH. Physiologic activities of the contact activation system. Thromb Res. 2014;133(suppl 1):S41–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menegatti M, Peyvandi F. Treatment of rare factor deficiencies other than hemophilia. Blood. 2019;133:415–24. [DOI] [PubMed] [Google Scholar]
- 19. Lammle B, Wuillemin WA, Huber I, Krauskopf M, Zurcher C, Pflugshaupt R, et al. Thromboembolism and bleeding tendency in congenital factor XII deficiency–a study on 74 subjects from 14 Swiss families. Thromb Haemost. 1991;65:117–21. [PubMed] [Google Scholar]
- 20. Preis M, Hirsch J, Kotler A, Zoabi A, Stein N, Rennert G, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129:1210–5. [DOI] [PubMed] [Google Scholar]
- 21. Endler G, Marsik C, Jilma B, Schickbauer T, Quehenberger P, Mannhalter C. Evidence of a U‐shaped association between factor XII activity and overall survival. J Thromb Haemost. 2007;5:1143–8. [DOI] [PubMed] [Google Scholar]
- 22. Liptrap RM, Gentry PA, Ross ML, Cummings E. Preliminary findings of altered follicular activity in Holstein cows with coagulation factor XI deficiency. Vet Res Commun. 1995;19:463–71. [DOI] [PubMed] [Google Scholar]
- 23. Tucker EI, Gailani D, Hurst S, Cheng Q, Hanson SR, Gruber A. Survival advantage of coagulation factor XI–deficient mice during peritoneal sepsis. J Infect Dis. 2008;198:271–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo D, Szaba FM, Kummer LW, Johnson LL, Tucker EI, Gruber A, et al. Factor XI–deficient mice display reduced inflammation, coagulopathy, and bacterial growth during listeriosis. Infect Immun. 2012;80:91–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bane CE Jr, Ivanov I, Matafonov A, Boyd KL, Cheng Q, Sherwood ER, et al. Factor XI deficiency alters the cytokine response and activation of contact proteases during polymicrobial sepsis in mice. PLoS One. 2016;11:e0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJ, Gailani D, et al. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood. 2012;119:4762–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kossmann S, Lagrange J, Jackel S, Jurk K, Ehlken M, Schonfelder T, et al. Platelet‐localized FXI promotes a vascular coagulation‐inflammatory circuit in arterial hypertension. Sci Transl Med. 2017;9 pii:eaah4923. [DOI] [PubMed] [Google Scholar]
- 28. Mohammed BM, Cheng Q, Matafonov A, Monroe DM, Meijers JCM, Gailani D. Factor XI promotes hemostasis in factor IX–deficient mice. J Thromb Haemost. 2018;16:2044–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stroo I, Yang J, de Boer JD, Roelofs JJ, van‘t Veer C, Castellino FJ, et al. Factor XI deficiency enhances the pulmonary allergic response to house dust mite in mice independent of factor XII. Am J Physiol Lung Cell Mol Physiol. 2017;312:L163–l171. [DOI] [PubMed] [Google Scholar]
- 30. Gailani D, Mohammed BM, Cheng Q. Factor XI and pulmonary infections. Haemophilia. 2018;24:519–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, et al. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti‐factor XII antibody to block contact activation in baboons. J Clin Investig. 1993;91:61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jansen PM, Pixley RA, Brouwer M, de Jong IW, Chang AC, Hack CE, et al. Inhibition of factor XII in septic baboons attenuates the activation of complement and fibrinolytic systems and reduces the release of interleukin‐6 and neutrophil elastase. Blood. 1996;87:2337–44. [PubMed] [Google Scholar]
- 34. Silasi R, Keshari RS, Lupu C, Van Rensburg WJ, Chaaban H, Regmi G, et al. Inhibition of contact‐mediated activation of factor XI protects baboons against S aureus–induced organ damage and death. Blood Adv. 2019;3:658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toossi Z, Sedor JR, Mettler MA, Everson B, Young T, Ratnoff OD. Induction of expression of monocyte interleukin 1 by Hageman factor (factor XII). Proc Natl Acad Sci U S A. 1992;89:11969–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khan MM, Bradford HN, Isordia‐Salas I, Liu Y, Wu Y, Espinola RG, et al. High‐molecular‐weight kininogen fragments stimulate the secretion of cytokines and chemokines through uPAR, Mac‐1, and gC1qR in monocytes. Arterioscler Thromb Vasc Biol. 2006;26:2260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Minnema MC, Pajkrt D, Wuillemin WA, Roem D, Bleeker WK, Levi M, et al. Activation of clotting factor XI without detectable contact activation in experimental human endotoxemia. Blood. 1998;92:3294–301. [PubMed] [Google Scholar]
- 38. Nuijens JH, Huijbregts CC, Cohen M, Navis GO, de Vries A, Eerenberg AJ, et al. Detection of activation of the contact system of coagulation in vitro and in vivo: quantitation of activated Hageman factor‐C‐1‐inhibitor and kallikrein‐C‐1‐inhibitor complexes by specific radioimmunoassays. Thromb Haemost. 1987;58:778–85. [PubMed] [Google Scholar]
- 39. Wuillemin WA, Minnema M, Meijers JC, Roem D, Eerenberg AJ, Nuijens JH, et al. Inactivation of factor XIa in human plasma assessed by measuring factor XIa–protease inhibitor complexes: major role for C1‐inhibitor. Blood. 1995;85:1517–26. [PubMed] [Google Scholar]
- 40. Wuillemin WA, Fijnvandraat K, Derkx BH, Peters M, Vreede W, ten Cate H, et al. Activation of the intrinsic pathway of coagulation in children with meningococcal septic shock. Thromb Haemost. 1995;74:1436–41. [PubMed] [Google Scholar]
- 41. Nuijens JH, Huijbregts CC, Eerenberg‐Belmer AJ, Abbink JJ, Strack van Schijndel RJ, Felt‐Bersma RJ, et al. Quantification of plasma factor XIIa‐Cl(–)‐inhibitor and kallikrein‐Cl(–)‐inhibitor complexes in sepsis. Blood. 1988;72:1841–8. [PubMed] [Google Scholar]
- 42. Mor‐Cohen R, Zucker M, Grissom C, Brown SM, Seligsohn U, Campbell RA, et al. The reduced form of coagulation factor XI is associated with illness severity and coagulopathy in critically‐ill septic patients. J Thromb Thrombolysis. 2019;47:186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lorentz CU, Verbout NG, Wallisch M, Hagen MW, Shatzel JJ, Olson SR, et al. Contact activation inhibitor and factor XI antibody, AB023, produces safe, dose‐dependent anticoagulation in a phase 1 first‐in‐human trial. Arterioscler Thromb Vasc Biol. 2019;39:799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koch AW, Schiering N, Melkko S, Ewert S, Salter J, Zhang Y, et al. MAA868‐a novel FXI antibody with a unique binding mode‐shows durable effects on markers of anticoagulation in humans. Blood. 2019;133:1507–16. [DOI] [PubMed] [Google Scholar]
- 45. Thomas D, Thelen K, Kraff S, Schwers S, Schiffer S, Unger S, et al. BAY 1213790, a fully human IgG1 antibody targeting coagulation factor XIa: first evaluation of safety, pharmacodynamics, and pharmacokinetics. Res Pract Thrombo Haemost. 2019;3:242–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salomon O, Preis M, Abu Shtaya A, Kotler A, Stein N, Saliba W. Factor XI deficiency is not associated with an increased risk of pneumonia and pneumonia‐related mortality. Haemophilia. 2018;24:634–40. [DOI] [PubMed] [Google Scholar]
- 47. Gailani D, Broze GJ Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–12. [DOI] [PubMed] [Google Scholar]
- 48. Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII: factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266:7353–8. [PubMed] [Google Scholar]
- 49. Henderson LM, Figueroa CD, Muller‐Esterl W, Bhoola KD. Assembly of contact‐phase factors on the surface of the human neutrophil membrane. Blood. 1994;84:474–82. [PubMed] [Google Scholar]