

Abstract
The objective of this scoping review is to summarize the current use of pharmacokinetics for tailoring prophylaxis in hemophilia patients switching between clotting factor products. Patients with hemophilia may require switching of clotting factor concentrates due to a variety of factors, but there have been perceived risks associated with switching, such as inhibitor development or suboptimal protection due to inadequate dosing while titrating treatment. Studies that look at patients switching from one clotting factor concentrate to another are categorized in terms of their primary and/or secondary objectives, notably biosimilarity and comparative pharmacokinetic studies and inhibitor development studies. Research on how best to switch concentrates with respect to dosing regimen are lacking, and currently a trial‐and‐error approach is used for dosing the new factor concentrate. In the future, studies looking at the predictability of pharmacokinetics (PK) of a new factor concentrate based on individual PK knowledge of the original factor concentrate may offer clinical benefit by providing a safer switching approach and protocol.
Keywords: drug substitution, factor IX, factor VIII, hemophilia A, hemophilia B
Essentials.
We assessed use of pharmacokinetics (PK) to tailor hemophilia prophylaxis when switching factor products.
Identified studies primarily assessed biosimilarity and none used PK to inform switching.
Switching is common based on a review of the WAPPS database for both factor VIII and IX.
Evidence‐based switching methods (eg, population PK) may improve dosing during switching.
1. INTRODUCTION
The mainstay treatment of hemophilia involves administration of factor concentrates. In the past, factors VIII (FVIII) and IX (FIX) infusions were given during or soon after an acute bleed. This “on‐demand” treatment decreased the number of patients with joint deformities but also significantly lowered their morbidity and mortality, ultimately increasing their quality of life.1 This practice was soon to be found suboptimal and a study by Aledort et al demonstrated that severe hemophilia patients without inhibitors undergoing an on‐demand treatment regimen still experienced reduced orthopedic outcomes and increased deteriorated joints compared to those treated prophylactically.1, 2 Prophylactic FVIII or FIX infusion has now been accepted as the standard for treating hemophilia patients well before joint damage is apparent.2, 3, 4, 5, 6
Prophylaxis was conceived as repeatedly dosing the patient so as to obtain a measurable factor activity at all times. The challenge is that appropriate dosing regimens vary by patient and factor concentrate and should be individualized from a therapeutic and economic standpoint.6, 7, 8 A “trial‐and‐error” approach is usually adopted, which involves using a typical prophylactic dosing regimen of 20 to 50 IU/kg, a dose that should provide the average patient with hemophilia with enough clotting factor to achieve the goal of a trough activity ≥0.01 IU/mL at 48 hours. However, this trial‐and‐error approach fails to account for individual pharmacokinetic (PK) variability and, as per Iorio et al,9 may lead to suboptimal results.
The trial‐and‐error approach is used again when switching between factor concentrates. Common practice in this scenario is that either the dose is initially kept the same as before the switch and frequency is adjusted proportionally to the relative expected change in terminal half‐life, or the dose and frequency tested in the pivotal studies are used in a first instance. Current guidelines suggest initiating extended half‐life (EHL) products at the same dose as standard half‐life concentrates but reducing the infusion frequency from 3 to 2 times weekly, and subsequently adjusting the dose based on a population pharmacokinetic (PopPK) approach.10, 11 When a person with hemophilia switches between factor concentrates, the person is switching from a product with known PK, or at least with known outcomes (eg, dose required to reduce bleeding events), to one with unknown PK. Dosing a factor concentrate with unknown PK introduces the risk of underdosing or resource wastage, leading to increased risk of bleeds or unnecessary use of factor concentrate, respectively.
The decision to switch between factor concentrates depends on a variety of factors, and shared decision making while assessing the product's safety, efficacy, cost, and convenience is essential before introducing a new product. The availability of newer and safer FVIII concentrates has resulted in switching between different plasma‐derived or recombinant FVIII concentrates throughout the course of hemophilia treatment.12 Newer FVIII products report to have better PK in terms of longer half‐life and thus may provide the advantage of fewer infusions.12 Other reasons for switching FVIII products may include cost savings, via a tender‐based national plan coverage or otherwise, side effects, drug shortages, or hypersensitivity to the formulation.12
The optimal approach to dose selection when switching between factor concentrates remains unknown. To answer the question of what is known about the current use of PK for tailoring prophylaxis in people with hemophilia switching between factor concentrates, we conducted (1) a scoping literature review, searching for empirical evidence regarding optimal switching practice; and (2) a review of the Web‐Accessible Population Pharmacokinetics Service–Hemophilia (WAPPS‐Hemo) database available to explore the practice of switching as recorded in the real world. WAPPS‐Hemo is a globally accessible online tool allowing hemophilia treaters to estimate individual PK using a population PK approach based on a limited set of 2 to 3 plasma factor activity measurements and patient covariates (eg, age, weight, height). Patient covariates and PK profiles gathered by WAPPS‐Hemo are deidentified and stored in a database. This database is available for research purposes to the members of the WAPPS‐Hemo research network.13 The WAPPS‐Hemo database provides information on current practices regarding product switching, as patients who have had >1 infusion recorded and have used >1 factor concentrate can be tracked within the system.
2. METHODS
2.1. Scoping review
The scoping review process followed these steps: (1) identify possible eligible studies; (2) select relevant studies; (3) chart the data; and (4) collate, summarize, and report the results, as proposed by Arksey and O'Malley.14 Following the PCC mnemonic,15 studies included hemophilia A or B patients (Population) switching between different factor concentrates and including appropriate PK assessments (Concept) and without any limitation as to reasons for switching, socioeconomic setting, and underlying health care system characteristics (Context). Relevant studies were prospective in nature. A search strategy was developed using medical subject headings (MeSH). The literature search was independently performed in PUBMED (MEDLINE) in September 2018 by both JKY and ANE. Search terms included:
(“Hemophilia A”[MeSH] OR “Hemophilia B”[MeSH] OR “Factor IX”[MeSH] OR “Factor VIII”[MeSH]) AND switch*
(“Hemophilia A”[MeSH] OR “Hemophilia B”[MeSH]) AND “Cross‐Over Studies”[MeSH]
(“Hemophilia A”[MeSH] OR “Hemophilia B”[MeSH]) AND “Pharmacokinetics”
(“Hemophilia A”[MeSH] OR “Hemophilia B”[MeSH]) AND “Bioequivalence”
2.2. WAPPS data review
For this review, all patients within the WAPPS‐Hemo database were eligible for inclusion unless they had only 1 infusion or had only 1 type of factor concentrate recorded on multiple occasions (Figure 1). The WAPPS user agreement allows reuse of the data for modeling and other research purposes, as described in the WAPPS study protocols, approved by the ethics boards at McMaster University and the University of Waterloo and registered in clinicaltrial.gov (NCT02061072, NCT03533504).
Figure 1.
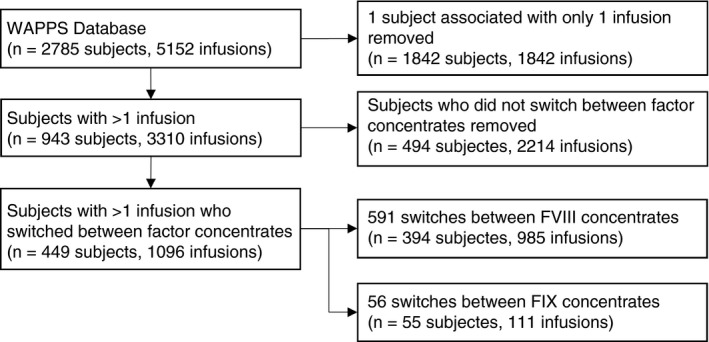
Study flow diagram of WAPPS data
3. RESULTS
3.1. Study selection
There were no research articles that specifically addressed the optimal approach to switching between factor concentrates. However, there were 39 peer‐reviewed scientific articles that fell within our inclusion criteria (Figure 2). Reviewer 1 identified 39 and reviewer 2 identified 38 that were identical to those selected by reviewer 1. Upon discussion of the missing article, the reviewers decided to include it as it met the inclusion criteria. The 39 articles were the only studies that could provide treaters with methods for evidence‐based switching using PK and were thus sorted based on their primary objective and appraised. Studies included bioequivalence or comparative PK studies, as well as inhibitor development studies during switching. All 39 studies are outlined in Table 1 (FVIII) and Table 2 (FIX).
Figure 2.
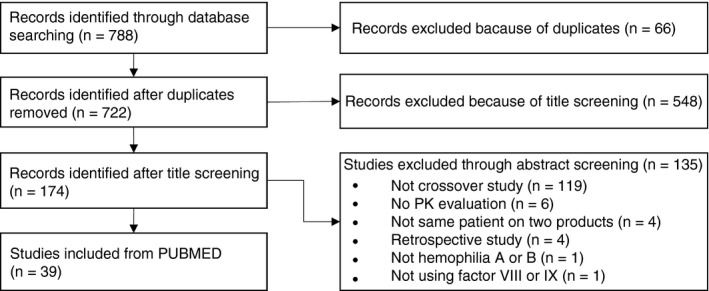
Study flow diagram of PUBMED search
Table 1.
Summary of studies of hemophilia patients switching between factor VIII concentrates
| Author | Products | Dose (IU/kg) | No. of subjects screened for PK |
Age range (mean) [median] |
Minimum washout period (d) | Primary objective |
|---|---|---|---|---|---|---|
| Biosimilarity or comparative PK studies | ||||||
| Di Paola17 |
(1) Advate (2) ReFacto |
50 ± 5 | 21 |
19‐72 (35.8) [30] |
3 | Compare PK of ReFacto and Advate to establish bioequivalence |
| Dmoszynska31 |
(1) Prior FVIII product (2) Optivate |
50 | 15 | 12‐65 | 3 | Investigate the PK of Optivate against other FVIII products |
| Fijnvandraat32 |
(1) rFVIII SQ (2) Octonativ M |
50 | 12 |
17‐64 (34) |
4 | Compare PK of rFVIII SQ and Octonativ M |
| Kessler18 |
(1) ReFacto (2 formulations) (2) Hemofil M |
50 | 19 |
18‐44 (26.3) |
5 | Compare PK of the 2 formulations of ReFacto with Hemofil M to establish bioequivalence |
| Klamroth23 |
(1) Advate (2) rFVIII single‐chain |
50 | 27 |
19‐60 (35.4) |
4 | Compare PK parameters of rFVIII single‐chain with full‐length rFVIII |
| Martinowitz22 |
(1) Advate (2) N8 |
50 | 25 |
13‐54 (24) |
4 | Compare PK profiles of N8 and Advate to establish bioequivalence |
| Morfini33 |
(1) pdFVIII (2) rFVIII |
25‐56 25‐45 |
17 |
15‐51 (27.7) [24.9] |
7 | Compare PK profiles of 2 different classes of FVIII concentrates |
| Morfini34 |
(1) Recombinate (2) Hemofil M |
50 | 47 |
6‐62 (26.4) |
7 | Compare PK profiles of Recombinate and Hemofil M |
| Morfini35 |
(1) Hemofil M (2) Monoclate HT (3) Monoclate P |
25 | 10 | ‐ | 7 | Compare in vivo behavior among the 3 products |
| Recht36 |
(1) Advate (2) Xyntha |
50 | 24 |
12‐60 [24] |
3 | Demonstrate PK equivalence of Advate |
| Shah19 |
(1) Advate (2) Kovaltry |
50 | 18 |
19‐64 (37.3) [36] |
3 | Compare PK profile of Advate and Kovaltry |
| Shirahata37 |
(1) BAY14‐2222 (2) Kogenate |
50 | 5 |
15‐43 (32) [35] |
5 | Compare PK profile of BAY14‐2222 and Kogenate |
| Biosimilarity or comparative PK and inhibitor development studies | ||||||
| Abshire38 |
(1) Kogenate (2) rFVIII‐FS |
50 | 35 | ‐ | 4 | Compare PK and safety of Kogenate and rFVIII‐FS |
| Coyle39 |
(1) rFVIII‐FS (2) BAY 94‐9027 |
25/50 25/60 |
14 |
21‐58 (36.1) |
3 | Assess PK and safety of BAY 94‐9027 |
| Kulkarni40 |
(1) Prior FVIII product (2) Turoctocog alfa |
‐ 25‐60 |
69 |
1‐11 (6.1) |
3 | Investigate safety, efficacy, and PK properties of turoctocog alfa |
| Mahlangu29 |
(1) Advate (2) rFVIIIFc |
50 | 30 |
12‐65 [29] |
‐ | Evaluate safety, efficacy, and PK of rFVIIIFc |
| Meunier41 |
(1) Prior FVIII product (2) N8‐GP |
‐ 60 |
24 |
0‐11 (6.0) |
‐ | Assess safety, efficacy, and PK of N8‐GP |
| Mullins42 |
(1) Advate (2) BAX855 |
60 ± 5 | 31 |
1‐11 (6) [6] |
‐ | Determine immunogenicity, PK, efficacy, safety, and quality of life using BAX855 |
| Powell43 |
(1) Kogenate (2) Kogenate with pegylated liposome carrier (13 or 22 mg/kg) |
35 | 26 | 12‐60 | 2 | Investigate the safety, tolerability, bioavailability, pharmacokinetics, and pharmacodynamics of Kogenate with pegylated liposome barrier compared with standard Kogenate |
| Schwartz44 |
(1) Koate‐HS (2) Recombinant FVIII |
50 20‐40 |
17 | ‐ | 7 | Compare PK of plasma‐derived and recombinant FVIII, assess efficacy of recombinant FVIII for home therapy, and assess efficacy for major surgical procedures and hemorrhage |
| Skotnicki45 |
(1) Vocento (2) Biostate‐RP |
50 | 17 |
18‐57 (36.5) [37] |
4 | Evaluate efficacy, safety, and PK of Voncento |
| Tiede46 |
(1) Prior FVIII product (2) N8‐GP |
‐ 25/50/75 |
26 |
20‐60 [36.5] |
4 | Evaluate safety and PK of N8‐GP in comparison with previous FVIII products |
| Young30 |
(1) Prior FVIII product (2) rFVIIIFc |
50 | 60 |
1‐11 [5] |
3 | Evaluate safety, efficacy, and PK of rFVIIIFc |
| Inhibitor development studies | ||||||
| Hsu47 |
(1) Kogenate (2) Koate‐HS |
50 ‐ |
12 |
23‐53 (37.8) |
7 | Evaluate safety and efficacy of Kogenate |
| Powell48 |
(1) Advate (2) rFVIIIFc |
25/65 25/65 |
19 |
23‐61 (34.6) |
3 | Evaluate safety and treatment‐emergent adverse events, development of antibodies, and laboratory monitoring |
‐, not specified; FVIII, factor VIII; pdFVIII, plasma‐derived factor VIII; PK, pharmacokinetics; rFVIII, recombinant factor VIII; SQ, subcutaneous.
Table 2.
Summary of studies of hemophilia patients switching between factor IX concentrates
| Author | Products | Dose (IU/kg) | No. of subjects screened for PK |
Age range (mean) [median] |
Minimum washout period (d) | Primary objective |
|---|---|---|---|---|---|---|
| Biosimilarity or comparative PK studies | ||||||
| Alamelu49 |
(1) Alphanine (2) Benefix |
50 | 9 |
15‐73 (41.2) [42] |
7 | Compare PK and pharmacodynamics properties of rFIX and pdFIX |
| Aznar50 |
(1) Immunine/Octanine (2) FIX Grifols |
65‐75 | 25 |
12‐38 (23.1) |
7 | Compare pharmacokinetic profile of FIX Grifols to available Immunine or Octanine |
| Ewenstein51 |
(1) Benefix (2) Mononine |
50 | 43 |
7‐75 [18.5] |
7 | Assess PK properties of the 2 products and address how variables affect in vivo recovery and half‐life |
| Goudemand52 |
(1) FIX‐SD‐15 (2) FIX‐SD |
60 | 11 | ‐ | 10 | Compare PK and coagulation activation markers of FIX‐SD‐15 and FIX‐SD |
| Liebman53 |
(1) Alphanine (2) Mononine |
40 | 12 | ‐ | 7 | Evaluate kinetics of FIX activity and protein |
| Lissitchkov54 |
(1) Benefix (2) Alphanine |
65‐75 | 22 |
15‐45 (27) |
7 | Compare PK between Benefix and Alphanine |
| Martinowitz55 |
(1) Benefix (2) IB1001 |
75 ± 5 | 32 | 15‐64 | 5 | Compare PK of IB1001 with those of Benefix and assess consistency of PK parameters |
| Thomas56 |
(1) Conventional FIX (2) High‐purity FIX |
75 | 19 | ‐ | 7 | Compare PK of high‐purity FIX to conventional FIX |
| Windyga57 |
(1) Benefix (2) BAX326 |
75 ± 5 | 86 | 12‐65 | 5 | Characterize PK profile of BAX326 and determine PK equivalence with Benefix |
| Biosimilarity or comparative PK and inhibitor development studies | ||||||
| Collins58 |
(1) Benefix (2) IB1001 |
75 ± 5 | 32 |
14.8‐64.5 (32.7) [29.9] |
5 | Establish PK noninferiority of IB1001 to Benefix, safety, and efficacy |
| Kenet59 |
(1) Prior FIX product (2) rFIX‐FP |
50 | 27 |
1‐11 (5.9) |
‐ | Evaluate PK, efficacy, and safety of rFIX‐FP |
| Inhibitor development studies | ||||||
| Negrier60 |
(1) Prior FIX product (2) N9‐GP |
‐ 25/50/100 |
20 |
21‐55 [30] |
7 | Determine safety by evaluating adverse events, antibody formation against FIX and N9‐GP, physical examination, and clinical laboratory assessments |
| Powell61 |
(1) Benefix (2) rFIXFc |
50 | 22 | ‐ | 5 | Determine annualized bleeding rate and development of inhibitors |
| Solano Trujillo62 |
(1) Immunine (2) BAX326 |
20‐40 75 ± 5 |
44 | 1‐55 | ‐ | Document exposure to Immunine and monitor for inhibitor development |
‐, not specified; FIX, factor IX; FVIII, factor VIII; pdFX, plasma‐derived factor X; PK, pharmacokinetics; rFIX, recombinant factor X.
3.2. Biosimilarity/bioequivalence or comparative PK studies
Strictly speaking, the term bioequivalence should not be used for drugs produced by biotechnology; the term biosimilarity is more appropriate.16 However, bioequivalence was the terminology used in many of the studies as many were published prior to the 2014 European Medicines Agency's guidance.16 Irrespective of the term used, studies assessing biosimilarity/bioequivalence did not usually enhance a switching protocol as a primary objective; however, their standardized dosing protocol allowed for comparison of individual PK profiles between the 2 brands under study. Thus, this section focuses on biosimilarity and comparative PK studies as both types compared population PK.
There were a limited number of studies that were biosimilarity or comparative PK studies (n = 34) (Tables 1 and 2). Biosimilarity refers to a lack of statistically significant differences in drug exposure between 2 drug products. In multiple crossover studies, biosimilarity was assessed by using a PK analysis to derive the maximum plasma factor activity (Cmax) following infusion and the area under the plasma concentration vs. time curve (AUC).17, 18, 19 To establish biosimilarity, the ratio of the logarithmic geometric mean values of Cmax and AUC must fall within the interval of 80% to 125% based on a 90% confidence interval.17, 18
All of the studies looking at comparing PK between 2 brands used PK end points, as suggested by the International Society of Thrombosis and Haemostasis and American and European regulatory bodies.12, 13, 14 The test dose before and after the switch was almost always identical, usually with a weight‐based dosing of 50 IU/kg of the factor concentrates. Using the same dose for different concentrates is a requisite for biosimilarity studies. All trials studied included a washout period of between 2 and 7 days before starting the trial and between different factor concentrates (Tables 1 and 2).
Biosimilarity/bioequivalence testing employs various types of statistics that are dependent upon the trial design. Most trial designs for biosimilarity testing of clotting factors employed a 2 × 2 × 2 crossover design. All biosimilarity and comparative PK studies observed average biosimilarity or average mean PK parameter differences and did not examine individual differences. Average biosimilarity assesses the PK between‐subject variability (BSV) but does not directly assess the within‐subject variability (WSV) over time. This may be reasonable given the a priori knowledge that clotting factor concentrates demonstrate a high PK BSV and low WSV within 1 brand,6 and therefore the assessment of individual biosimilarity may not be necessary. Individual biosimilarity assesses for both the mean and variability of PK metrics and also the ratio of the 2 drug products on an individual basis and is recognized when both the average biosimilarity is established and the subject‐by‐formulation effect is insignificant.20 Average biosimilarity is important to assess mean PK differences in a population, but individual biosimilarity is highly impactful if the goal is to give prescribers confidence that biosimilarity will occur when a patient on one of the drug products is switched to the other.
In order for a drug to be therapeutically equivalent to another product, it requires the same active pharmaceutical ingredient (API), dosage form, strength, route of administration, and established bioequivalence.21 Because clotting factors are not identical, as they are biologics, the PK BSV and WSV of the 2 brands may not hold; this is not the case with small molecules, where the API systemic disposition is exactly the same between 2 drug products. As a result, the individual concentration‐time profile of 1 factor concentrate can be different as compared to another factor concentrate of the same dose and frequency. If individual biosimilarity for 2 factor concentrates is established, they can be used interchangeably, and the PK of one factor concentrate is therefore predictive of the other. However, no study confirming individual patient biosimilarity has been completed because it is difficult to achieve. In a study by Di Paola et al,17 patients who switched from Advate to ReFacto had very different individual PK parameters even though the average PK parameters were similar. Similar findings were observed with Martinowitz et al22 and Klamroth et al (Figure 3).23 The conclusion that 2 factor concentrates are bioequivalent does not mean that individuals will achieve the same concentration‐time profile if the same dose is given. Likewise, similar average half‐life between 2 factor concentrates does not mean that the half‐life between 2 factor concentrates in any given individual will be similar; some individuals in Figure 3 had drastic differences in their PK across factor concentrates.
Figure 3.
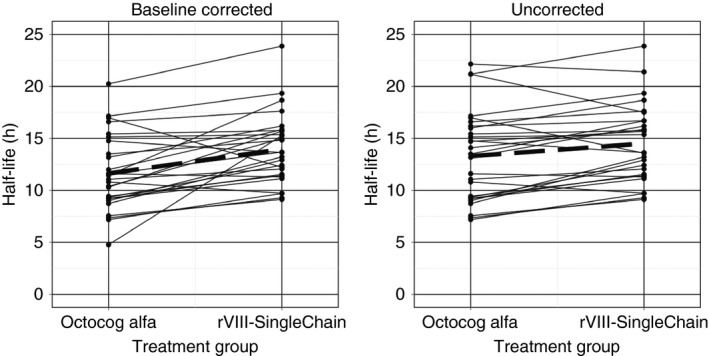
Example of individual PK parameters after switching
No study involving switching between factor concentrates where PK was assessed used this information to predict a proper dosing regimen.
3.3. Inhibitor development studies
The second type of study included patients serially taking at least 2 clotting factor concentrates and had the objective of examining inhibitor development. Inhibitors are antibodies that neutralize clotting factors. These inhibitors are generally measured using the Nijmegen modification of the Bethesda assay.24, 25 Once inhibitors develop in a patient with hemophilia, it becomes much more difficult to treat them, resulting in an increase in morbidity and mortality in the affected population.24, 26
Eighteen articles were identified in which their primary outcome was focusing on inhibitor development after switching factor concentrate products (Tables 1 and 2). It was previously thought that switching between factor concentrates was associated with an increased risk of inhibitor development,27 but recent studies have not shown consistent results.27, 28 Although PK data may have been used in their statistical analysis, dosing regimens of each factor concentrate were not tailored based on PK. It was unclear whether the dose provided to the patient after switching was the optimal dosing regimen. Without knowledge of the dosing regimen in patients with hemophilia, it was also unclear whether the overdosing or underdosing of factor concentrate had an effect on inhibitor risk.
No inhibitor study that incorporated PK into its assessment was usable to inform methods for PK‐tailored dosing.
3.4. WAPPS‐Hemo data
As of September 15, 2018, there were >250 centers enrolled worldwide with >3000 patients and >6300 infusions recorded. Infusion data was gathered for the purposes of determining the incidence of switching between factor concentrates.
A total of 2785 patients were taken from the WAPPS data platform. The methodology is presented in Figure 1. Of the 2785 subjects, 449 (16%) had infusions on ≥2 concentrates, with a total of 647 switches. A summary of patient demographics is presented in Table 3.
Table 3.
Demographics from WAPPS patients who have switched between factor concentrates
| Parameter | Whole cohort | FVIII | FIX |
|---|---|---|---|
| Subjects (n) | 449 | 394 | 55 |
| Switches (n) | 647 | 591 | 56 |
| Age (y) | 1‐78 | 1‐78 | 2‐68 |
| Body weight (kg) | 10‐150 | 10‐150 | 13‐117 |
| As of September 2018. | |||
FVIII, factor VIII; FIX, factor IX; WAPPS, Web‐Accessible Population Pharmacokinetics Service.
In terms of FVIII products, there were a total of 394 patients and 591 switches, accounting for 91% of total switches on WAPPS‐Hemo. FVIII products, classified based on their molecular structure, are presented in Table 4. Of the 591 switches, the majority of the switches (n = 293) occurred from second‐ and third‐generation recombinant full‐length products (50%). There were 208 switches (35%) to EHL products, 73 switches (12%) to B‐domain–deleted products, 229 switches (39%) to another recombinant full‐length product, and 81 switches (14%) to plasma‐derived products.
Table 4.
Number of hemophilia patients from WAPPS‐Hemo switching between FVIII concentrates
| FVIII products | Switch to | Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Plasma‐derived | Plasma‐derived with vWF | First‐gen rec. full‐length | Second‐gen rec. full‐length | Third‐gen rec. full‐length | Second‐gen rec. BDD | Third‐gen rec. BDD | Fourth‐gen rec. BDD | Third‐gen EHL rec. BDD‐PEGylated | Fourth‐gen EHL rec. Fc‐Fusion | Third‐gen EHL rec. single‐chain | |||
| Switch from | Plasma‐derived | 2 | 8 | 6 | 9 | 2 | 3 | 1 | 0 | 0 | 6 | 0 | 37 |
| Plasma‐derived with vWF | 4 | 26 | 6 | 20 | 5 | 2 | 0 | 0 | 2 | 10 | 1 | 76 | |
| First‐gen rec. full‐length | 4 | 11 | 0 | 15 | 18 | 7 | 6 | 1 | 0 | 2 | 0 | 64 | |
| Second‐gen rec. full‐length | 5 | 16 | 16 | 24 | 57 | 6 | 2 | 4 | 2 | 56 | 3 | 191 | |
| Third‐gen rec. full‐length | 0 | 0 | 8 | 5 | 6 | 3 | 13 | 4 | 12 | 50 | 1 | 102 | |
| Second‐gen rec. BDD | 1 | 2 | 5 | 7 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 17 | |
| Third‐gen rec. BDD | 0 | 0 | 0 | 0 | 2 | 1 | 5 | 2 | 2 | 19 | 1 | 32 | |
| Fourth‐gen rec. BDD | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 5 | |
| Third‐gen EHL rec. BDD‐PEGyl | 0 | 0 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | |
| Fourth‐gen EHL rec. Fc‐Fusion | 0 | 1 | 0 | 4 | 9 | 0 | 6 | 6 | 30 | 0 | 0 | 56 | |
| Third‐gen EHL rec. single‐chain | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 5 | 0 | 7 | |
| Total | 16 | 65 | 41 | 86 | 102 | 22 | 34 | 17 | 49 | 153 | 6 | 591 | |
BDD, B‐domain deleted; EHL, extended half‐life; FVIII, factor VIII; rec, recombinant;vWF, von Willebrand factor; WAPPS, Web‐Accessible Population Pharmacokinetics Service.
In terms of FIX products, there were a total of 55 patients and 56 switches, accounting for 9% of total switches on WAPPS‐Hemo. FIX products, classified based on their molecular structure, are presented in Table 5. Of the 56 switches, the majority of switches in WAPPS‐Hemo occurred when switching from any FIX product to a recombinant Fc‐fusion protein FIX product (n = 34), accounting for 61% of all FIX switches.
Table 5.
Number of hemophilia patients from WAPPS‐Hemo switching between FIX concentrates
| FIX products | Switch to | Total | |||||
|---|---|---|---|---|---|---|---|
| Plasma‐derived | Recombinant | Recombinant glycoPEGylated | Recombinant Fc‐fusion protein | Recombinant albumin fusion protein | |||
| Switch from | Plasma‐derived | 4 | 1 | 0 | 11 | 1 | 17 |
| Recombinant | 0 | 1 | 1 | 22 | 7 | 31 | |
| Recombinant glycoPEGylated | 0 | 0 | 0 | 0 | 0 | 0 | |
| Recombinant Fc‐fusion protein | 1 | 0 | 1 | 0 | 5 | 7 | |
| Recombinant albumin fusion protein | 0 | 0 | 0 | 1 | 0 | 1 | |
| Total | 5 | 2 | 2 | 34 | 13 | 56 | |
FIX, factor IX; WAPPS, Web‐Accessible Population Pharmacokinetics Service.
4. PHARMACOKINETIC TARGETS WHEN SWITCHING
While literature states the average of PK parameters (eg, half‐life) when switching between factor concentrates, the range of such PK parameters can be highly variable. A study by Mahlangu et al29 compared the terminal half‐life of the recombinant FVIII Fc fusion protein, Eloctate, with a standard‐acting FVIII concentrate (Advate) in a phase 3 study to determine the safety, efficacy, and PK. On average, the half‐life of Eloctate was 1.5 times that of Advate at a dose of 50 IU/kg.29, 30 This provides valuable information about the population, although it is clear from the breadth of factor concentrate brands being switched to and from, as identified in the WAPPS‐Hemo database, that this type of study cannot be completed for all scenarios. A study by Young et al30 demonstrated that the individual half‐life ratios of FVIII and Eloctate ranged from 0.79 to 2.98. Such high half‐life variability within an individual across FVIII products makes the application of the mean population difference irrelevant for use in individual dosing recommendations.
Of particular note was the lack of evidence that standard‐acting factor concentrates have shorter half‐lives than long‐acting factor concentrates at the individual level. In the study by Klamroth et al,23 the majority of patients had increased half‐life when switching from octocog alfa to a recombinant FVIII single‐chain concentrate; however, this was not the case for 4 of 27 subjects. The potential risk of assuming an increase in half‐life when switching from a standard‐acting to a long‐acting concentrate may lead to increased risk of bleeds due to underdosing. Without assessing individual PK parameters, the current approach of using population‐level information to switch between factor concentrates may not yield expected results.
It would be desirable to estimate dosing regimens across a switch using an individualized approach. In an ideal scenario, where population PK tailored prophylaxis was widely adopted, patients planning on switching to a different factor concentrate would have information regarding their own PK estimates on their current factor concentrate. In theory, combining the knowledge of the individual's PK of a factor concentrate prior to the switch (origin concentrate) with the knowledge of the population PK characteristics of the concentrate after the switch (destination concentrate) may potentially lead to the ability to predict individual PK estimates of the destination concentrate. The accuracy and precision of such an approach have not yet been studied, and empirical demonstration of the feasibility of the process is first required. However, the perspective of enabling better estimation of individual PK on the destination concentrate is undoubtedly appealing. This is an example of a research project that could be performed with the rich WAPPS‐Hemo database that contains many hemophilia subjects who have switched between different factor concentrates.
5. STUDY LIMITATIONS
The volume of literature we expected to find in this specific field was limited. As such, we have not registered the protocol or used the Peer Review of Electronic Search Strategies checklist when conducting our search strategy. We cast a wide net with regards to our search terms, but we are aware that this will limit the internal and external validity of our results.
6. CONCLUSION
Hemophilia treatment requires accurate and individualized dosing regimens to provide safe, effective, and cost‐effective medication use. Although studies looking at bioequivalence/biosimilarity or assessing PK between 2 factor concentrates have led to PK comparisons, these studies lack the information required to predict an optimal dosing regimen for hemophilia patients starting on a new product. Studies that have examined the development of inhibitors did not mention the use of PK parameters to optimize a dosing regimen. As such, there exists no literature on the role or use of PK in optimizing factor concentrate dosing during product switching.
Given these limitations, further research is required to utilize PK parameters from the origin product to predict the PK of the destination product in patients with hemophilia. Due to similarity in PK parameters, especially across FVIII products,6 dose regimen predictability may be feasible using population PK methods and Bayesian forecasting. For example, standard‐acting FVIII concentrates may be compared with other standard‐acting FVIII concentrates and, in the same way, with newer long‐acting FVIII concentrates.
With the introduction of newer and longer‐acting concentrates, the use of PopPK methods will be an integral part in determining and predicting accurate dosing regimens for patients. The use of PopPK can change the current trial‐and‐error approach into a safer dosing regimen that makes use of prior PK knowledge to ensure patient safety and mindful resource consumption.
RELATIONSHIP DISCLOSURE
AI's institution has received project‐based funding via research or service agreements with Bayer, CSL Behring, Grifols, NovoNordisk, Octapharma, Pfizer, Roche, Shire (formerly Baxter and Baxalta), and Spark Therapeutics. The other co‐authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
JKY and ANE performed the research. All authors revised and approved the final and submitted version of the manuscript.
ACKNOWLEDGMENTS
We thank Caitlin Carter, Librarian, School of Pharmacy, University of Waterloo, Waterloo, Ontario, Canada, for assistance with this scoping review.
APPENDIX 1.
Co‐Investigator Contact Information
The following clinicians have agreed to participate in this phase of the WAPPS‐Hemo project by signing a co‐investigator agreement (as of August 10, 2018):
Alfonso Iorio, McMaster University, Hamilton, Ontario, Canada
Sanjay Ahuja, University Hospital Health Systems Cleveland, Ohio, USA
Ma Teresa Álvarez Román, Hospital Universitario La Paz, Madrid, Spain
Ma E. Arrieta, Hospital Público Descentralizado Dr. Guillermo Rawson, San Juan, Argentina
Mikko Arola, Tampere University Hospital, Tampere, Finland
Giovanni Barillari, Center for Hemorrhagic and Thrombotic Diseases, University Hospital of Udine, Italy
Vinod Balasa, Valley Children's Healthcare, Fresno, California, USA
Mark Belletrutti, University of Alberta, Edmonton, Alberta, Canada
Ruben Berrueco Moreno, Hospital de Sant Joan de Déu, Barcelona, Spain
Philippe Beurrier, Chu, Angers, France
Cristoph Bidlingmaier, Pediatric Hemophilia Center, Munich, Germany
Victor Blanchette, Sick Children's Hospital, Toronto, Ontario, Canada
Jan Blatny, University Hospital Brno, Brno, Czech Republic
Santiago Bonanad, University & Polytechnic Hospital La Fe, Valencia, Spain
Kelsey Brose, Saskatchewan Bleeding Disorders Program, Saskatoon, Saskatchewan, Canada
Deborah Brown, Gulf States Hemophilia, Houston, Texas, USA
Paulette C. Byant, St. Jude Affiliate Clinic, NH Hemby Children's Hospital, Charlotte, NC, USA
Mariana Canaro, Congenital Coagulopathies Unit of Balearic Islands, Spain
Manuela Carvalho, Centro Hospitalar S. João, Oporto, Portugal
Cristina Catarino, Hospital de Santa Maria, Lisbon, Portugal
Meera Chitlur, Children's Hospital of Michigan, Detroit, Michigan, USA
Erin Cockrell, St. Joseph's Hospital, Tampa, Florida, USA
Pratima Chowdary, Royal Free Hospital, London, UK
Marjon Cnossen, Erasmus MC‐Sophia Children's Hospital, Rotterdam, Netherlands
Peter Collins, Arthur Bloom Centre, Cardiff, UK
Michial Coppens, Academic Medical Center Amsterdam, Amsterdam, The Netherlands
Stacy Croteau, Dana‐Farber/Boston Children's Hospital, Boston, USA
Dorina Cultrera, Policlinico‐Vittorio Emanuele, Catania, Italy
Raimundo de Cristofaro, Università Cattolica S. Cuore, Rome, Italy
Emmauelle de Raucourt, Clinic des Anticoagulants (CAC), Clichy, France
Dominique Desprez, Hospital de HautePierre, CHU de Strasbourg, France
Amy Dunn, Nationwide Children's Hospital, Columbus, Ohio, USA
Magda El‐Ekiabi, Shabrawishi Hospital Blood Bank, Egypt
Barbara Faganel Kotnik, Haemophilia Comprehensive Care Center, Slovenia
Kathleen Fischer, University Medical Center (UMC) Utrecht, The Netherlands
Brigit Frotscher, CHU Nancy, France
Susana Garbiero, Centro Asistencial Regional de Hemoterapia (CARDHE), Bahia Blanca, Argentina
Raquel Garrido Ruiz, Hospital Universitario de Puerto Real, Cádiz, Spain
Joan Gill, Blood Research Institute, Milwaukee, Wisconsin, USA
Carmen Gomez del Castillo, Complexo Hospitalario Universitario, Coruña, Spain
Saskia Gottstein, Vivantes Clinic in Friedrichshain, Berlin, Germany
Giuseppe Lassandro and Paola Giordano, U.O. Pediatria Generale e Specialistica B. Trambusti, Bari, Italy
Daniel Hart, Royal London Hospital, London, UK
Inga Hegemann, Zurich University Hospital, Zurich, Switzerland
Cedric Hermans, St‐Luc University Hospital, Brussels, Belgium
Baolai Hua, Peking Union Medical College Hospital, Beijing, China
Nina Hwang, Center for Inherited Blood Disorders (CIBD), Orange, California, USA
Shannon Jackson, St. Paul's Hospital, Vancouver, British Columbia, Canada
Paula James, SE Ontario Regional Inherited Bleeding Disorders Program, Kingston, Canada
Olga Katsarou, Laiko General Hospital, Athens, Greece
Kaan Kavakli, Ege University Hospital, Izmir, Turkey
Christine Kempton, AFLAC Cancer Center and Blood Disorders Services, Atlanta, Georgia, USA
Karim Kentouche, Universitätsklinikum Jena, Jena, Germany
Osman Khan, Oklahoma Center for Bleeding and Clotting Disorders, Oklahoma City, Oklahoma, USA
Rainer Kobelt, University Children's Hospital, Berne, Switzerland
Rebecca Kruse‐Jarres, Harborview Medical Center, Seattle, Washington, USA
Edward Laane, North Estonia Medical Center, Tallinn, Estonia
Eric Larson, Maine Hemophilia and Thrombosis Centre, Scarborough, Maine, USA
Riitta Lassila, Coagulation Disorders in Helsinki University Hospital, Finland
Adrienne Lee and Man‐Chiu Poon, Foothills Medical Centre, Calgary, Alberta, Canada
Jennifer Lissick, Children's of Minnesota, Minneapolis, Minnesota, USA
Satu Langstrom, Hospital of Helsinki and Uusimaa, Helsinki, Finland
Johnny Mahlangu, University of the Witwatersrand, Johannesburg, South Africa
Michael Makris, Royal Hallamshire Hospital, Sheffield, UK
Emmanuela Marchesini, Department of Medicine, Perugia, Italy
Jose Mateo, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain
Pacual Marco Vera, Hospital General Universitario de Alicante, Alicante, Spain
Marta Martorell, Hospital Universitari Vall d'Henron, Barcelona, Spain
Tadashi Matsushita, Nagoya University Hospital, Nagoya, Japan
Simon McCrae, Royal Adelaide Hospital, Adelaide, Australia
Eva Mignot‐Castellano, Hospital Regional Universitario de Málaga, Spain
Caitlin Montcrieff, Rhode Island Hemostasis and Thrombosis Center, Rhode Island, USA
Philip Maes, Koningin Mathilde Moeder‐ en kindcentrum, Edegem, Belgium
Veerle Mondelars and Marlies Bekart, Gent University Hospital, Gent, Belgium
Elena Mora, Hematologia y Oncologia del Oriente, Bogota, Colombia
Juan Cristóbal Morales, Complejo Asistencial Dr. Sótero del Río, Santiago, Chile
Guillaume Mourey and Marie Ann Bertrand, CHRU de Besançon, France
Mariasanta Napolitano and Sergio Siragusa, Policlinico di Palermo, Palermo, Italy
Claude Negrier, Edouard Herriot University Hospital, Lyon, France
Daniela Neme, Fundación de la Hemofilia, Buenos Aires, Argentina
Ritta Niinimaki, Oulu University Hospital, Oulu, Finland
Johannes Oldenburg and Thilo Albert, Universitätsklinikum Bonn, Germany
Deborah Ornstein, Dartmouth‐Hitchcock Medical Center, Lebanon, New Hampshire, USA
Margarete Ozelo, INCT do Sangue Hemocentro UNICAMP, Sao Paulo, Brazil
John Carl Panetta and Ellis J. Neufeld, St. Jude's Hospital, Memphis, Tennessee, USA
Stephanie P'Ng, Fiona Stanley Hospital Haemophilia Treatment Centre, Perth, Australia
Kathelijne Peerlinck, Hemophilia center Leuven, Leuven, Belgium
Berardino Pollio, Centre for Hemorrhagic and Thrombotic Diseases, Torino, Italy
Claire Pouplard and Yves Gruel, Universite Francois‐Rabelais, Tours, France
Alessandra Prezotti, Centro de Hemoterapia e Hematologia do Espirito Santo, Vitoria, Brasil
Vicky Price, IWK, Halifax, Nova Scotia, Canada
Fitri Primacakti, Dr.Cipto Mangunkusumo General Hospital, Jakarta, Indonesia
Mathieu Puyade, Site du CHU, Poitiers, France
Paolo Radossi, Haematology and Haemophilia Centre, Castelfranco Veneto, Italy
Leslie Raffini, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, USA
Margaret Ragni, Hemophilia Center of Western Pennsylvania, Philadelphia, Pennsylvania, USA
Savita Rangarajan, Southern Hemophilia Network, Basingstoke, UK
Mark T. Reding, Center for Bleeding and Clotting, Minneapolis, Minnesota, USA
Robin Reid, Mary M Gooley Hemophilia Center, Rochester, New York, USA
Jose Restrepo and Jose Ramirez, Centre Medico Imbanaco, Cali, Colombia
Michael Recht, Oregon Health & Science University, Portland, Oregon, USA
Manuel Rodriguez Lopez, Hospital Alvaro Cunquiero, Vigo, Spain
Arlette Ruiz‐Sàez, Centro Nacional de Hemofilia, Caracas, Venezuela
Mahasen Saleh, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
Amy Shapiro, Indiana Hemophilia and Thrombosis Center, Indianapolis, Indiana, USA
Anjali Sharathkumar, University of Iowa Children's Hospital, Iowa City, Iowa, USA
Anna Selmeczi, Hungarian Defence Forces, Budapest, Hungary
Mindy Simpson, Rush University, Chicago, Illinois, USA
Tami Singleton, Tulane University School of Medicine, New Orleans, USA
Maria Sol Cruz, Hemophilia Salta, Salta, Argentina
Veronica Soto, Hospital Roberto del Río, Santiago, Chile
MacGregor Steele, Southern Alberta Pediatric Bleeding Disorder Program, Calgary, Canada
Werner Streif, Medical University of Innsbruck (MUI), Innsbruck, Austria
Hao Wei Sun and Bruce Ritchie, Kaye Edmonton Clinic, Edmonton, Alberta, Canada
Jing Sun and Xiaqin Feng, Nanfang Hospital, Guangzhou, China
Takashi Suzuki and Asuza Nagao, Ogikubo Hospital, Tokyo, Japan
Cliff Takemoto, Johns Hopkins University, Baltimore, Maryland, USA
Heather Tapp, Woman's and Childrens Hospital, North Adelaide, Australia
Jerry Teitel, Toronto & Central Ontario Hemophilia Program, Toronto, Ontario, Canada
Alan Tinmouth, Ottawa Regional Adult Bleeding Disorders Program, Ottawa, Ontario, Canada
Courtney Thornburg, Rady Children's Hospital, San Diego, California, USA
Alberto Tosseto, Divisione di Ematologia Ospedale S. Bortolo, Vicenza, Italy
Oliver Turnstall, Bristol Haematology and Oncology Centre, Bristol, UK
Catherine Vezina, Montreal Children's Hospital, Montreal, Quebec, Canada
Beth Warren, Children's Hospital Colorado, Aurora, Colorado, USA
Allison Wheeler, Vanderbilt University Medical Center, Nashville, TN, USA
Juan D. Wilches Gutierrez, IPS ESPECIALIZADA, Bogota, Colombia
John K.M. Wu, BC Children's Hospital, Vancouver, Canada
Tung Wynn, University of Florida, Gainesville, Florida, USA
Renchi Yang, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Guy Young, Children's Hospital, Los Angeles, California, USA
Ezio Zanon, Centro Emofilia di Pavola, Padua, Italy
Irena Zupan, University Medical Centre Ljubljana, Ljubljana, Slovenia
Yu JK, Iorio A, Edginton AN; on behalf of the WAPPS co‐investigators . Using pharmacokinetics for tailoring prophylaxis in people with hemophilia switching between clotting factor products: A scoping review. Res Pract Thromb Haemost. 2019;3:528–541. 10.1002/rth2.12204
[Article corrected on October 8, 2019, after first online publication on May 20, 2019: Investigator Claire Pouplard's name was misspelled as “Claire Poulard.” This has been corrected.]
Contributor Information
Andrea N. Edginton, Email: aedginto@uwaterloo.ca.
the WAPPS co‐investigators:
Sanjay Ahuja, Ma Teresa Álvarez Román, Ma E. Arrieta, Mikko Arola, Giovanni Barillari, Vinod Balasa, Mark Belletrutti, Ruben Berrueco Moreno, Philippe Beurrier, Cristoph Bidlingmaier, Victor Blanchette, Jan Blatny, Santiago Bonanad, Kelsey Brose, Deborah Brown, Paulette C. Byant, Mariana Canaro, Manuela Carvalho, Cristina Catarino, Meera Chitlur, Erin Cockrell, Pratima Chowdary, Marjon Cnossen, Peter Collins, Michial Coppens, Stacy Croteau, Dorina Cultrera, Raimundo de Cristofaro, Emmauelle de Raucourt, Dominique Desprez, Amy Dunn, Magda El‐Ekiabi, Barbara Faganel Kotnik, Kathleen Fischer, Brigit Frotscher, Susana Garbiero, Raquel Garrido Ruiz, Joan Gill, Carmen Gomez del Castillo, Saskia Gottstein, Giuseppe Lassandro, Paola Giordano, Daniel Hart, Inga Hegemann, Cedric Hermans, Baolai Hua, Nina Hwang, Shannon Jackson, Paula James, Olga Katsarou, Kaan Kavakli, Christine Kempton, Karim Kentouche, Osman Khan, Rainer Kobelt, Rebecca Kruse‐Jarres, Edward Laane, Eric Larson, Riitta Lassila, Adrienne Lee, Man‐Chiu Poon, Jennifer Lissick, Satu Langstrom, Johnny Mahlangu, Michael Makris, Emmanuela Marchesini, Jose Mateo, Pacual Marco Vera, Marta Martorell, Tadashi Matsushita, Simon McCrae, Eva Mignot‐Castellano, Caitlin Montcrieff, Philip Maes, Veerle Mondelars, Marlies Bekart, Elena Mora, Juan Cristóbal Morales, Guillaume Mourey, Marie Ann Bertrand, Mariasanta Napolitano, Sergio Siragusa, Claude Negrier, Daniela Neme, Ritta Niinimaki, Johannes Oldenburg, Thilo Albert, Deborah Ornstein, Margarete Ozelo, John Carl Panetta, Ellis J. Neufeld, Stephanie P'Ng, Kathelijne Peerlinck, Berardino Pollio, Claire Pouplard, Yves Gruel, Alessandra Prezotti, Vicky Price, Fitri Primacakti, Mathieu Puyade, Paolo Radossi, Leslie Raffini, Margaret Ragni, Savita Rangarajan, Mark T. Reding, Robin Reid, Jose Restrepo, Jose Ramirez, Michael Recht, Manuel Rodriguez Lopez, Arlette Ruiz‐Sàez, Mahasen Saleh, Amy Shapiro, Anjali Sharathkumar, Anna Selmeczi, Mindy Simpson, Tami Singleton, Maria Sol Cruz, Veronica Soto, MacGregor Steele, Werner Streif, Hao Wei Sun, Bruce Ritchie, Jing Sun, Xiaqin Feng, Takashi Suzuki, Asuza Nagao, Cliff Takemoto, Heather Tapp, Jerry Teitel, Alan Tinmouth, Courtney Thornburg, Alberto Tosseto, Oliver Turnstall, Catherine Vezina, Beth Warren, Allison Wheeler, Juan D. Wilches Gutierrez, John K.M. Wu, Tung Wynn, Renchi Yang, Guy Young, Ezio Zanon, and Irena Zupan
REFERENCES
- 1. Iorio A, Marchesini E, Marcucci M, Stobart K, Chan AK. Clotting factor concentrates given to prevent bleeding, bleeding‐related complications in people with hemophilia A or B. Cochrane Database Syst Rev. 2011:Cd003429. [DOI] [PubMed] [Google Scholar]
- 2. Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor‐VIII‐deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med. 1994;236:391–9. [DOI] [PubMed] [Google Scholar]
- 3. Oldenburg J. Prophylaxis in bleeding disorders. Thromb Res. 2011;127(suppl 1):S14–7. [DOI] [PubMed] [Google Scholar]
- 4. Berntorp E, Astermark J, Baghaei F, Bergqvist D, Holmstrom M, Ljungberg B, et al. Treatment of haemophilia A and B and von Willebrand's disease: summary and conclusions of a systematic review as part of a Swedish health‐technology assessment. Haemophilia. 2012;18:158–65. [DOI] [PubMed] [Google Scholar]
- 5. Fischer K, Collins PW, Ozelo MC, Srivastava A, Young G, Blanchette VS. When and how to start prophylaxis in boys with severe hemophilia without inhibitors: communication from the SSC of the ISTH. J Thromb Haemost. 2016;14:1105–9. [DOI] [PubMed] [Google Scholar]
- 6. McEneny‐King A, Iorio A, Foster G, Edginton AN. The use of pharmacokinetics in dose individualization of factor VIII in the treatment of hemophilia A. Expert Opin Drug Metab Toxicol. 2016;12:1313–21. [DOI] [PubMed] [Google Scholar]
- 7. Ar MC, Vaide I, Berntorp E, Bjorkman S. Methods for individualising factor VIII dosing in prophylaxis. Eur J Haematol Suppl. 2014;76:16–20. [DOI] [PubMed] [Google Scholar]
- 8. Bjorkman S, Berntorp E. Pharmacokinetics of coagulation factors: clinical relevance for patients with haemophilia. Clin Pharmacokinet. 2001;40:815–32. [DOI] [PubMed] [Google Scholar]
- 9. Iorio A, Edginton AN, Blanchette V, Blatny J, Boban A, Cnossen M, et al. Performing and interpreting individual pharmacokinetic profiles in patients with hemophilia A or B: rationale and general considerations. Res Pract Thromb Haemost. 2018;2(3):535–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collins P, Chalmers E, Chowdary P, Keeling D, Mathias M, O'Donnell J, et al. The use of enhanced half‐life coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. Haemophilia. 2016;22:487–98. [DOI] [PubMed] [Google Scholar]
- 11. Ragni MV, Croteau SE, Morfini M, Cnossen MH, Iorio A. Pharmacokinetics and the transition to extended half‐life factor concentrates: communication from the SSC of the ISTH. J Thromb Haemost. 2018;16:1437–41. [DOI] [PubMed] [Google Scholar]
- 12. Coppola A, Marrone E, Conca P, Cimino E, Mormile R, Baldacci E, et al. Safety of switching factor VIII products in the era of evolving concentrates: myths and facts. Semin Thromb Hemost. 2016;42:563–76. [DOI] [PubMed] [Google Scholar]
- 13. Iorio A, Keepanasseril A, Foster G, Navarro‐Ruan T, McEneny‐King A, Edginton AN, et al. Development of a Web‐Accessible Population Pharmacokinetic Service—Hemophilia (WAPPS‐Hemo): study protocol. JMIR Res Protoc. 2016;5:e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arksey H, O'Malley L. Scoping studies: towards a methodological framework. Int J Soc Res Methodol. 2005;8:19–32. [Google Scholar]
- 15. The Joanna Briggs Institute.Joanna Briggs Institute Reviewers’ Manual: 2015 edition/Supplement. 2015 [cited 10 May 2019]. Available from: http://joannabriggs.org/assets/docs/sumari/Reviewers-Manual_Methodology-for-JBI-Scoping-Reviews_2015_v2.pdf.
- 16. Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: non‐clinical and clinical issues. European Medicines Agency; 2014. [Google Scholar]
- 17. Di Paola J, Smith MP, Klamroth R, Mannucci PM, Kollmer C, Feingold J, et al. ReFacto®1 and Advate®2: a single‐dose, randomized, two‐period crossover pharmacokinetics study in subjects with haemophilia A. Haemophilia. 2007;13:124–30. [DOI] [PubMed] [Google Scholar]
- 18. Kessler CM, Gill JC, White GC, Shapiro A, Arkin S, Roth DA, et al. B‐domain deleted recombinant factor VIII preparations are bioequivalent to a monoclonal antibody purified plasma‐derived factor VIII concentrate: a randomized, three‐way crossover study. Haemophilia. 2005;11:84–91. [DOI] [PubMed] [Google Scholar]
- 19. Shah A, Solms A, Garmann D, Katterle Y, Avramova V, Simeonov S, et al. Improved pharmacokinetics with BAY 81‐8973 versus antihemophilic factor (recombinant) plasma/albumin‐free method: a randomized pharmacokinetic study in patients with severe hemophilia A. Clin Pharmacokinet. 2017;56:1045–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Federal Drug Administration Center for Drug Evaluation and Research . Guidance for industry: Statistical approaches to establishing bioequivalence. 2001 [cited 10 May 2019]. Available from: https://www.fda.gov/media/70958/download
- 21. Midha KK, McKay G. Bioequivalence; its history, practice, and future. AAPS J. 2009;11:664–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martinowitz U, Bjerre J, Brand B, Klamroth R, Misgav M, Morfini M, et al. Bioequivalence between two serum‐free recombinant factor VIII preparations (N8 and ADVATE(R)) – an open‐label, sequential dosing pharmacokinetic study in patients with severe haemophilia A. Haemophilia. 2011;17:854–9. [DOI] [PubMed] [Google Scholar]
- 23. Klamroth R, Simpson M, von Depka‐Prondzinski M, Gill JC, Morfini M, Powell JS, et al. Comparative pharmacokinetics of rVIII‐SingleChain and octocog alfa (Advate®) in patients with severe haemophilia A. Haemophilia. 2016;22:730–8. [DOI] [PubMed] [Google Scholar]
- 24. Iorio A, Puccetti P, Makris M. Clotting factor concentrate switching and inhibitor development in hemophilia A. Blood. 2012;120:720–7. [DOI] [PubMed] [Google Scholar]
- 25. Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser‐Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII: C inhibitors: improved specificity and reliability. Thromb Haemost. 1995;73:247–51. [PubMed] [Google Scholar]
- 26. Darby SC, Keeling DM, Spooner RJ, Wan Kan S, Giangrande PL, Collins PW, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977‐99. J Thromb Haemost. 2004;2:1047–54. [DOI] [PubMed] [Google Scholar]
- 27. Hay CRM, Palmer BP, Chalmers EA, Hart DP, Liesner R, Rangarajan S, et al. The incidence of factor VIII inhibitors in severe haemophilia A following a major switch from full‐length to B‐domain‐deleted factor VIII: a prospective cohort comparison. Haemophilia. 2015;21:219–26. [DOI] [PubMed] [Google Scholar]
- 28. Parra Lopez R, Nemes L, Jimenez‐Yuste V, Rusen L, Cid AR, Charnigo RJ, et al. Prospective surveillance study of haemophilia A patients switching from moroctocog alfa or other factor VIII products to moroctocog alfa albumin‐free cell culture (AF‐CC) in usual care settings. J Thromb Haemost. 2015;114:676–84. [DOI] [PubMed] [Google Scholar]
- 29. Mahlangu J, Powell JS, Ragni MV, Chowdary P, Josephson NC, Pabinger I, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123:317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Young G, Mahlangu J, Kulkarni R, Nolan B, Liesner R, Pasi J, et al. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia A. J Thromb Haemost. 2015;13:967–77. [DOI] [PubMed] [Google Scholar]
- 31. Dmoszynska A, Hellmann A, Baglin T, O'Shaugnessy D, Trelinski J, Kuliczkowski K, et al. Pharmacokinetics of Optivate((R)), a high‐purity concentrate of factor VIII with von Willebrand factor, in patients with severe haemophilia A. Haemophilia. 2011;17:185–90. [DOI] [PubMed] [Google Scholar]
- 32. Fijnvandraat K, Berntorp E, ten Cate JW, Johnsson H, Peters M, Savidge G, et al. Recombinant, B‐domain deleted factor VIII (r‐VIII SQ): pharmacokinetics and initial safety aspects in hemophilia A patients. Thromb Haemost. 1997;77:298–302. [PubMed] [Google Scholar]
- 33. Morfini M, Marchesini E, Paladino E, Santoro C, Zanon E, Iorio A. Pharmacokinetics of plasma‐derived vs. recombinant FVIII concentrates: a comparative study. Haemophilia. 2015;21:204–9. [DOI] [PubMed] [Google Scholar]
- 34. Morfini M, Longo G, Messori A, Lee M, White G, Mannucci P. Pharmacokinetic properties of recombinant factor VIII compared with a monoclonally purified concentrate (Hemofil M). The Recombinate Study Group. Thromb Haemost. 1992;68:433–5. [PubMed] [Google Scholar]
- 35. Morfini M, Mannucci PM, Longo G, Cinotti S, Messori A. Comparative evaluation of the pharmacokinetics of three monoclonal factor VIII concentrates. Thromb Res. 1991;61:285–90. [DOI] [PubMed] [Google Scholar]
- 36. Recht M, Nemes L, Matysiak M, Manco‐Johnson M, Lusher J, Smith M, et al. Clinical evaluation of moroctocog alfa (AF‐CC), a new generation of B‐domain deleted recombinant factor VIII (BDDrFVIII) for treatment of haemophilia A: demonstration of safety, efficacy, and pharmacokinetic equivalence to full‐length recombinant factor VIII. Haemophilia. 2009;15:869–80. [DOI] [PubMed] [Google Scholar]
- 37. Shirahata A, Fukutake K, Takamatsu J, Shima M, Yoshioka A. Pharmacokinetics, prophylactic effects, and safety of a new recombinant FVIII formulated with sucrose (BAY 14‐2222) in Japanese patients with hemophilia A. Int J Hematol. 2000;72:101–7. [PubMed] [Google Scholar]
- 38. Abshire TC, Brackmann HH, Scharrer I, Hoots K, Gazengel C, Powell JS, et al. Sucrose formulated recombinant human antihemophilic factor VIII is safe and efficacious for treatment of hemophilia A in home therapy – International Kogenate‐FS Study Group. Thromb Haemost. 2000;83:811–6. [PubMed] [Google Scholar]
- 39. Coyle TE, Reding MT, Lin JC, Michaels LA, Shah A, Powell J. Phase I study of BAY 94‐9027, a PEGylated B‐domain‐deleted recombinant factor VIII with an extended half‐life, in subjects with hemophilia A. J Thromb Haemost. 2014;12:488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kulkarni R, Karim FA, Glamocanin S, Janic D, Vdovin V, Ozelo M, et al. Results from a large multinational clinical trial (guardian3) using prophylactic treatment with turoctocog alfa in paediatric patients with severe haemophilia A: safety, efficacy and pharmacokinetics. Haemophilia. 2013;19:698–705. [DOI] [PubMed] [Google Scholar]
- 41. Meunier S, Alamelu J, Ehrenforth S, Hanabusa H, Abdul Karim F, Kavakli K, et al. Safety and efficacy of a glycoPEGylated rFVIII (turoctocog alpha pegol, N8‐GP) in paediatric patients with severe haemophilia A. Thromb Haemost. 2017;117:1705–13. [DOI] [PubMed] [Google Scholar]
- 42. Mullins ES, Stasyshyn O, Alvarez‐Roman MT, Osman D, Liesner R, Engl W, et al. Extended half‐life pegylated, full‐length recombinant factor VIII for prophylaxis in children with severe haemophilia A. Haemophilia. 2017;23:238–46. [DOI] [PubMed] [Google Scholar]
- 43. Powell JS, Nugent DJ, Harrison JA, Soni A, Luk A, Stass H, et al. Safety and pharmacokinetics of a recombinant factor VIII with pegylated liposomes in severe hemophilia A. J Thromb Haemost. 2008;6:277–83. [DOI] [PubMed] [Google Scholar]
- 44. Schwartz RS, Abildgaard CF, Aledort LM, Arkin S, Bloom AL, Brackmann HH, et al. Human recombinant DNA‐derived antihemophilic factor (factor VIII) in the treatment of hemophilia A. recombinant Factor VIII Study Group. N Engl J Med. 1990;323:1800–5. [DOI] [PubMed] [Google Scholar]
- 45. Skotnicki A, Lissitchkov TJ, Mamonov V, Buevich E, Kuliczkowski K, Goranov S, et al. Efficacy, safety and pharmacokinetic profiles of a plasma‐derived VWF/FVIII concentrate (VONCENTO(R)) in subjects with haemophilia A (SWIFT‐HA study). Thromb Res. 2016;137:119–25. [DOI] [PubMed] [Google Scholar]
- 46. Tiede A, Brand B, Fischer R, Kavakli K, Lentz SR, Matsushita T, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first‐in‐human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11:670–8. [DOI] [PubMed] [Google Scholar]
- 47. Hsu HC, Chen YF, Ho CH. Human recombinant DNA‐derived antihemophilic factor (factor VIII) in the treatment of hemophilia A. Zhonghua Yi Xue Za Zh. 1999;62:450–4. [PubMed] [Google Scholar]
- 48. Powell JS, Josephson NC, Quon D, Ragni MV, Cheng G, Li E, et al. Safety and prolonged activity of recombinant factor VIII Fc fusion protein in hemophilia A patients. Blood. 2012;119:3031–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alamelu J, Bevan D, Sorensen B, Rangarajan S. Pharmacokinetic and pharmacodynamic properties of plasma‐derived vs. recombinant factor IX in patients with hemophilia B: a prospective crossover study. J Thromb Haemost. 2014;12:2044–8. [DOI] [PubMed] [Google Scholar]
- 50. Aznar JA, Cabrera N, Matysiak M, Zawilska K, Gercheva L, Antonov A, et al. Pharmacokinetic study of a high‐purity factor IX concentrate (Factor IX Grifols) with a 6‐month follow up in previously treated patients with severe haemophilia B. Haemophilia. 2009;15:1243–8. [DOI] [PubMed] [Google Scholar]
- 51. Ewenstein BM, Joist JH, Shapiro AD, Hofstra TC, Leissinger CA, Seremetis SV, et al. Pharmacokinetic analysis of plasma‐derived and recombinant F IX concentrates in previously treated patients with moderate or severe hemophilia B. Transfusion. 2002;42:190–7. [DOI] [PubMed] [Google Scholar]
- 52. Goudemand J, Peynet J, Chambost H, Negrier C, Briquel ME, Claeyssens S, et al. A cross‐over pharmacokinetic study of a double viral inactivated factor IX concentrate (15 nm filtration and SD) compared to a SD factor IX concentrate. Thromb Haemost. 1998;80:919–24. [PubMed] [Google Scholar]
- 53. Liebman HA, Rosenwald‐Zuckerman T, Retzios A, Yasmin S, Kasper CK. Kinetics of factor IX activity differ from that of factor IX antigen in patients with haemophilia B receiving high‐purity factor IX replacement. Haemophilia. 1999;5:174–80. [DOI] [PubMed] [Google Scholar]
- 54. Lissitchkov T, Matysiak M, Zavilska K, Laguna P, Gercheva L, Antonov A, et al. Head‐to‐head comparison of the pharmacokinetic profiles of a high‐purity factor IX concentrate (AlphaNine(R)) and a recombinant factor IX (BeneFIX(R)) in patients with severe haemophilia B. Haemophilia. 2013;19:674–8. [DOI] [PubMed] [Google Scholar]
- 55. Martinowitz U, Shapiro A, Quon DV, Escobar M, Kempton C, Collins PW, et al. Pharmacokinetic properties of IB1001, an investigational recombinant factor IX, in patients with haemophilia B: repeat pharmacokinetic evaluation and sialylation analysis. Haemophilia. 2012;18:881–7. [DOI] [PubMed] [Google Scholar]
- 56. Thomas DP, Hampton KK, Dasani H, Lee CA, Giangrande PL, Harman C, et al. A cross‐over pharmacokinetic and thrombogenicity study of a prothrombin complex concentrate and a purified factor IX concentrate. Br J Haematol. 1994;87:782–8. [DOI] [PubMed] [Google Scholar]
- 57. Windyga J, Lissitchkov T, Stasyshyn O, Mamonov V, Rusen L, Lamas JL, et al. Pharmacokinetics, efficacy and safety of BAX326, a novel recombinant factor IX: a prospective, controlled, multicentre phase I/III study in previously treated patients with severe (FIX level <1%) or moderately severe (FIX level ≤2%) haemophilia B. Haemophilia. 2014;20:15–24. [DOI] [PubMed] [Google Scholar]
- 58. Collins PW, Quon DVK, Makris M, Chowdary P, Kempton CL, Apte SJ, et al. Pharmacokinetics, safety and efficacy of a recombinant factor IX product, trenonacog alfa in previously treated haemophilia B patients. Haemophilia. 2018;24:104–12. [DOI] [PubMed] [Google Scholar]
- 59. Kenet G, Chambost H, Male C, Lambert T, Halimeh S, Chernova T, et al. Long‐acting recombinant fusion protein linking coagulation factor IX with albumin (rIX‐FP) in children. Results of a phase 3 trial. Thromb Haemost. 2016;116:659–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Negrier C, Knobe K, Tiede A, Giangrande P, Moss J. Enhanced pharmacokinetic properties of a glycoPEGylated recombinant factor IX: a first human dose trial in patients with hemophilia B. Blood. 2011;118:2695–701. [DOI] [PubMed] [Google Scholar]
- 61. Powell JS, Pasi KJ, Ragni MV, Ozelo MC, Valentino LA, Mahlangu JN, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med. 2013;369:2313–23. [DOI] [PubMed] [Google Scholar]
- 62. Solano Trujillo MH, Stasyshyn O, Rusen L, Serban M, Lamas JL, Perina FG, et al. Safe switching from a pdFIX (Immunine(R)) to a rFIX (Bax326). Haemophilia. 2014;20:674–81. [DOI] [PubMed] [Google Scholar]