

SUMMARY
Immune cells constantly survey the host for pathogens or tumors and secrete cytokines to alert surrounding cells of these threats. In vivo, activated immune cells secrete cytokines for several hours, yet an acute immune reaction occurs over days. Given these divergent timescales, we addressed how cytokine-responsive cells translate brief cytokine exposure into phenotypic changes that persist over long timescales. We studied melanoma cell responses to transient exposure to the cytokine Interferon γ (IFNγ) by combining a systems-scale analysis of gene expression dynamics with computational modeling and experiments. We discovered that IFNγ is captured by phosphatidylserine (PS) on the surface of viable cells both in vitro and in vivo, then slowly released to drive long-term transcription of cytokine-response genes. This mechanism introduces an additional function for PS in dynamically regulating inflammation across diverse cancer and primary cell types, and has potential to usher new immunotherapies targeting PS and inflammatory pathways.
eTOC BLURB
Activated immune cells secrete cytokines for few hours, yet acute immune reactions unfold over a week or more. Oyler-Yaniv et al. resolve this timescale discrepancy by uncovering how cancer & immune cells rely on their surface phosphatidylserine to capture cytokines then slowly release them and elicit long-term cell-to-cell communication.
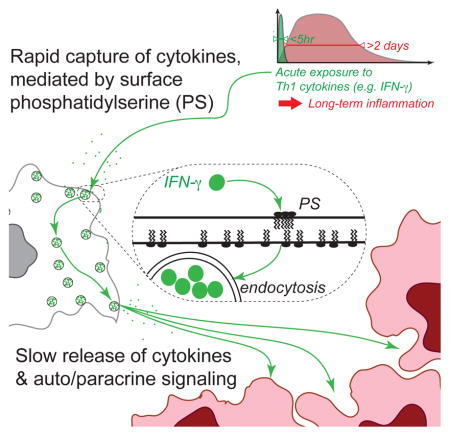
INTRODUCTION
Immune cells constantly survey the environment for pathogens or tumors, and then secrete cytokines to alert surrounding cells of the threat. Many different cytokines exist which serve to communicate a specific immune context. Paradoxically, cytokines are often secreted transiently (~hours) by activated immune cells, while the dynamics of an acute immune response typically unfold over a week (Helmstetter et al., 2015; Honda et al., 2014; Hosking et al., 2014; Manicassamy et al., 2010). These widely varied timescales pose two questions: how do cells translate short-lived cytokine exposure into long-term gene expression changes that persist for the duration of an immune reaction? More fundamentally, what mechanisms regulate the duration of cell responses to transient cytokine exposure?
Answering these questions requires addressing how cells transduce cytokine signals by phosphorylation of signaling molecules and activation of transcription factors. To do so, the strategy of combining mathematical modeling with time-course experiments (dynamics) has proven fruitful for dissecting gene regulatory mechanisms from the kinetics of transcription factor activation (Cai et al., 2008; Hoffmann et al., 2002; Lee et al., 2014; Nelson et al., 2004; Purvis et al., 2012). Understanding such dynamics has immediate practical implications. For example, tracking the signaling dynamics of the tumor suppressor p53 enabled identification of the optimal timing for administration of DNA damage to maximize tumor cell death (Chen et al., 2016; Lee et al., 2012). Given that the timing and order of chemotherapeutic intervention can be optimized, we speculated that there is also an optimal timing for immunotherapeutic intervention after tumor cell exposure to pro-inflammatory cytokines.
In this study, we focus on the pro-inflammatory cytokine, Interferon γ (IFNγ). This cytokine is produced by activated T cells and Natural Killer (Schroder et al., 2004) cells to prevent microbial infection, block primary tumor development, and enhance rejection of established tumors (Dighe et al., 1994; Shankaran et al., 2001). Upon T cell activation, IFNγ is produced for 3–10 hours (Helmstetter et al., 2015; Honda et al., 2014; Hosking et al., 2014). Although this cytokine is secreted transiently (~hours), it is crucial for host-pathogen or host-tumor defense over long timescales (~days/weeks). Therefore, we sought to determine the temporal duration of cancer cell responses to brief IFNγ exposure.
We studied the transcriptional dynamics of melanoma cells exposed to brief pulses of IFNγ and observed long-term up-regulation of antigen presentation genes in vitro. Enhanced transcription of these genes peaked 2 days after the initial signal was abolished, then decayed over one week. By combining mathematical modeling with diverse experimental approaches and an in vivo mouse model of thyroid cancer, we learned that IFNγ is captured by cell surface-exposed phosphatidylserine on viable cancer and primary cells. The cytokine is then slowly released to drive persistent transcription of IFNγ-response genes by autocrine and paracrine signaling. We named this mode of cell communication, cytokine “catch-and-release”. Our findings reveal an unexplored function for phosphatidylserine (PS) in regulating inflammatory responses in the tumor microenvironment and in other cell types, with potential applications in anti-cancer immunotherapies.
RESULTS
Transient exposure to IFNγ drives persistent up-regulation of antigen presentation
The timescale over which cells maintain phenotypic changes after exposure to a transient signal can span several orders of magnitude, as illustrated in Figure 1A. In the most basic model of gene regulation, the cell response tracks the external stimulus (see Methods S1): once the signal is abrogated, cells rapidly return to their original phenotypic state. In that context, slower mRNA decay would only modestly extend the response. Alternatively, phenotypic changes may persist indefinitely because of positive feedback (Justman et al., 2009) or chromatin modifications (Agarwal and Rao, 1998). Immune responses pose a unique challenge: to effectively respond to a threat, the immune system must achieve an intermediate timescale of response by extending phenotypic changes past the brief period of cytokine secretion, yet eventually return to homeostasis.
Figure 1. Transient IFNγ drives persistent T cell activation caused by up-regulation of antigen processing and presentation.

See also Figure S1. (A) The cell response to a transient stimulus spans several timescales. (B) Cartoon of experiment to test tumor antigenicity. CD8+ pmel T cells recognize an endogenous peptide antigen presented in the context of MHC-I by B16 mouse melanoma cells. (C) Diagram of T cell activation experiment. B16 cells were pulsed with IFNγ, washed, and cultured in fresh media. On subsequent days, B16 cells were co-cultured with pmel T cells. (D) Activation of T cells by IFNγ-pulsed B16 cells was quantified by cytokine secretion assays. Data are representative of 3 independent experiments. (E) B16 cells were pulsed with IFNγ (or mock-stimulated), washed, and cultured in fresh media. RNA was sequenced and the mRNA dynamics were clustered using the k-means algorithm. Lines represent the mean of duplicate RNA samples. (F) Genes from each cluster were analyzed using the gene ontology database. All pathways that were significantly enriched (p<0.001) were plotted and the fold enrichment is depicted by the shade of blue. (G) Diagram of experiment. B16 and EL4 cells were pulsed with IFNγ, washed, and cultured in fresh media. On subsequent days, cells were co-cultured with pmel T cells. An additional cohort of B16-pmel co-cultures received αMHC-I. (H) Activation of T cells by IFNγ-pulsed B16 and EL4 cells was quantified by cytokine secretion assays. Data are representative of 2 independent experiments.
We sought to quantify the timescale of a cell’s response to transient IFNγ. Exposure of cancer cells to IFNγ increases their recognition and killing by T cells (Dunn et al., 2002), so we designed an assay to quantify when IFNγ-pulsed cells are most sensitive to T cell recognition (Figure 1B). B16-F10 mouse melanoma cells were pulsed briefly (5h) with IFNγ, then washed and cultured in cytokine-free conditions (Figure 1C). B16-F10 cells (hereafter called B16 cells) are an immortalized mouse tumor line that originally derived from a spontaneous melanoma that arose in a C57Bl/6 mouse (Overwijk and Restifo, 2001). Subsequent to IFNγ exposure, pmel CD8+ T cells were added to the culture and their activation was quantified. These T cells specifically recognize the endogenously-expressed gp100 peptide antigen presented by major histocompatibility complex class I (MHC-I) on B16 melanoma cells (Overwijk et al., 2003).
IFNγ-pulsed B16 cells increased over time in their capacity to activate T cells, peaked 2 days post-exposure, then returned to pre-treatment levels by 5–7 days (Figure 1D). This implied that B16 cells persistently up-regulated cytokine-response genes for days after removal of IFNγ, yet retained the ability to return to homeostatic levels. To check whether the dynamics of IFNγ-response genes matched those of T cell activation, we sequenced mRNA from B16 cells for several days after IFNγ exposure. The resulting transcriptional dynamics were grouped based on their similarity to one another using k-means clustering (Figure 1E and S1A). Our analysis revealed three main transcriptional clusters, whose biological significance was defined by gene ontology analysis (Ashburner et al., 2000; Gene Ontology Consortium, 2015). Cluster 1 was significantly enriched with genes involved in the MHC-I and II antigen presentation pathways, which are critical for T cell activation (Figure 1F). Clusters 2 and 3 were of limited amplitude and not enriched for any specific pathway. We chose two candidate genes from cluster 1, H2-Db and H2-Kb, which encode both alleles of the mouse H2b MHC-I haplotype, to confirm that protein expression matched that of mRNA expression (Figure S1B).
These data suggested that persistent up-regulation in antigen presentation caused the observed pattern of T cell activation. To test this, we repeated our T cell activation assay with additional conditions (Figure 1G). In a second group of B16 cells, we blocked MHC-I – T cell receptor interaction with a neutralizing antibody. Additionally, EL4 cells, which possess the H2b haplotype but lack the peptide antigen gp100, were pulsed with IFNγ and cultured with T cells. T cell activation was completely abrogated when MHC-I was blocked, or in the absence of peptide antigen in the case of EL4 cells (Figure 1H). These data show that the long-term pattern of T cell activation after brief B16 exposure to IFNγ is peptide-specific, and dependent on the MHC-I antigen presentation pathway.
To summarize, B16 melanoma cells respond to transient IFNγ (5h) with sustained up-regulation of antigen presentation that peaks 2 days post-exposure. Thus, the cell response to IFNγ extends long past when the original cytokine source is gone.
JAK-STAT signaling drives persistent transcription
Downstream of the IFNγ receptor, the transcription factor STAT1 is phosphorylated (pSTAT1) and drives changes in gene expression. Persistent phosphorylation of STAT1 after IFNγ withdrawal could explain persistent up-regulation of antigen presentation genes. To test this, cells were exposed to IFNγ for 5h, and then washed to remove cytokine (Figure 2A). Cells were harvested periodically for intracellular phospho-flow cytometry. During IFNγ exposure, pSTAT1 peaks rapidly (Figure 2B). Immediately after washing, pSTAT1 drops to 10% of its maximum level, and then slowly decays back to baseline four days after initial cytokine exposure. Persistent phosphorylation of STAT1 requires JAK activity, because application of a small molecule inhibitor of the JAK 1/2 kinase (JAKi) (Hedvat et al., 2009) after IFNγ stimulation caused rapid decay of pSTAT1 (Figure S2A). We also tested whether JAK-STAT signaling is necessary for persistent transcription of antigen presentation genes by applying a JAK inhibitor to cells and quantifying h2kb transcript over time (Figure 2C). Cells that were JAK inhibited after IFNγ-stimulation peaked at the time when JAK inhibitor was administered (5h), and with a lower magnitude of response (Figure 2D).
Figure 2. Jak-STAT signaling drives persistent transcription.

See also Figure S2. (A) Diagram of experiment. B16 cells were pulsed with IFNγ, washed, and cultured in fresh media. Cells were harvested at indicated timepoints, fixed, permeabilized, and stained for pSTAT1. (B) Phosphorylation of STAT1 was measured by flow cytometry. Data are representative of at least 3 independent experiments. (C) Diagram of JAK inhibitor experiment. Cells were stimulated with IFNγ, washed, and cultured in fresh media. At twash, one cohort of cells received a JAK1/2 inhibitor. RNA was extracted at indicated timepoints. (D) h2kb transcripts were quantified by RT-qPCR. Data are representative of at least 2 independent experiments. (E) Diagram of αIFNγ experiment. Cells were stimulated as above, washed, and cultured in fresh media. At the time of wash, one cohort of cells received αIFNγ. (F) MHC-I (H2-Kb) was measured by flow cytometry. Data are representative of at least 3 independent experiments.
To establish that it is IFNγ itself that drives persistent up-regulation in antigen presentation, cells were stimulated with cytokine, washed, and an IFNγ neutralizing antibody was added (Figure 2E). Blocking IFNγ after the initial washout caused an earlier peak in the MHC-I protein H2-Kb relative to the control (Figure 2F). Given that IFNγ antibody blockade prevents sustained up-regulation of IFNγ response genes, we revisited our RNA seq data and observed that IFNγ does not induce its own production in B16 cells. We also confirmed that media harvested immediately after the last wash does not up-regulate MHC-I when cultured with naïve B16 cells (Figure S2C). Finally, neither α nor β interferons contributed to persistent up-regulation of the antigen presentation pathway (Figure S2B).
Together, our experiments demonstrate that transient exposure to IFNγ drives persistent JAK-STAT signaling, and persistent up-regulation of the antigen presentation pathway, independently of de novo IFNγ transcription.
IFNγ-exposed cells catch and release IFNγ in a receptor-independent manner
Our experiments suggested that IFNγ can be captured and released by cytokine-exposed cells. To test this, we set up a transwell assay (Figure 3A) in which one group of cells were pulsed with IFNγ, washed, and co-cultured in the bottom of a plate with unpulsed sensor cells. “Sensors” are B16 cells that have not been pre-treated with cytokines and are effectively naïve. They are denoted as sensors in this assay because they quantitatively report IFNγ exposure by up-regulating IFNγ-response genes, such as MHC-I. An additional group of un-stimulated sensors were spatially separated from cytokine-pulsed cells by culturing them in the top of a cytokine-permeable transwell insert. MHC-I on sensor cells was measured by flow cytometry after one day of co-culture. Sensor cells cultured in close proximity with IFNγ-pulsed cells up-regulated MHC-I 10-fold after 1 day of co-culture (Figure 3B). All MHC-I up-regulation was abrogated when cells were cultured in the presence of IFNγ blocking antibody. Thus, IFNγ-pulsed cells slowly release cytokine, which can then signal in a paracrine manner.
Figure 3. Cytokine-pulsed cells slowly release IFNγ.

See also Figure S3. (A) Diagram of transwell experiment. One cohort of B16 cells were pulsed with IFNγ, washed, and co-cultured with unstimulated B16 sensor cells in the bottom of a plate. Additional sensors were cultured in the top of a transwell. αIFNγ was added where indicated. (B) Sensor MHC-I (H2-Kb) expression was measured by flow cytometry. Experiment is representative of at least 3 independent experiments. (C) General experimental diagram for (D–F). One cohort of cells were pulsed with IFNγ, washed, and co-cultured with receptor-competent B16 sensor cells. αIFNγ was added where indicated. MHC-I was measured by flow cytometry. (D) B16, SK-Mel-2, H460, CH12, Hek293T, and RAW cells were pulsed with mouse IFNγ, washed, and co-cultured with unstimulated B16 sensor cells, Sensor MHC-I (H2-Kb) was measured by flow cytometry. (E) B16 IFNγR KO cells were pulsed with IFNγ, washed, and co-cultured with unstimulated B16 sensor cells. Sensor MHC-I (H2-Kb) was measured by flow cytometry. Data are representative of 2 independent experiments.
To determine whether cell-to-cell cytokine sharing is dependent on cell proximity or contact, we measured MHC-I expression on sensor cells after multiple days of co-culture. Sensor cells mixed in the bottom of the transwell continued to up-regulate MHC-I, and sensor cells in the top began to up-regulate MHC-I, eventually reaching levels comparable to mixed sensor cells (Figure S3A). This suggests that IFNγ is slowly released from IFNγ-pulsed cells and diffuses to sensor cells in the top of the transwell over time. Thus, cell-to-cell cytokine sharing likely depends on proximity, but not cell contact.
We then asked whether other tumor-derived and immortalized cell types exhibit cell-to-cell cytokine sharing. A panel of mouse and human cell lines were pulsed with mouse IFNγ, then washed and co-cultured with un-stimulated B16 sensor cells (Figure 3C). All cell types assayed, were capable of sharing IFNγ with B16 cells (Figure 3D). These experiments demonstrated that IFNγ is captured by diverse cell types, then slowly released.
We next addressed whether IFNγ release originated from de-binding from IFNγ receptors (IFNγR) after washing. We repeated our assay with B16 IFNγ receptor knockout (B16 IFNγR KO) cells (Figure 3C). These cells were pulsed with IFNγ, washed, and co-cultured with receptor competent B16 sensor cells. We again observed IFNγ-dependent up-regulation of MHC-I on sensor cells (Figure 3E). Hence IFNγ is captured and released by cells in an IFNγ receptor-independent manner.
Finally, we developed an assay to quantify how much cytokine cells could capture (Figure 5F). B16 IFNγR KO cells were incubated with 50pM recombinant IFNγ in a well-mixed setting for 5h. The culture supernatant was then harvested and the remaining cytokine was quantified using a bead-based ELISA. We found that B16 IFNγR KO could capture more than 60% of the soluble cytokine. We repeated this cytokine-capture assay with multiple concentrations of T cell-derived IFNγ and found that it was as efficiently captured by cells as recombinant IFNγ (Figure S3B).
Figure 5. Phosphatidylserine is necessary for IFNγ cell binding.

See also Figure S5. (A) Lipid-spotted strips were incubated with 50pM IFNγ, probed with αIFNγ, and developed. For lipid key, see figure S6A. (B) Live cells were stained with Annexin V or kept in buffer alone. Cells were gated first as live (Dapi-). Fluorescence was quantified by flow cytometry. Staining is representative of 3 independent experiments. (C) IFNγR KO B16 cells were adhered to a glass bottom dish, stained with Annexin V, and imaged with confocal microscopy. Staining is representative of 3 independent experiments. (D) IFNγR KO B16 cells were stained with Annexin V, and the plasma membrane was stained with cell mask orange. Cells were imaged with confocal microscopy before they could adhere to a dish (to make subcellular detection of PS clearer). Arrows point to PS localized to the plasma membrane. Staining is representative of 2 independent experiments. (E) IFNγR KO B16 cells were adhered to a glass bottom dish, stained with low-dose Annexin V, then incubated with IFNγ-A647, washed, and imaged with confocal microscopy. Data are representative of 2 independent experiments. (F) B16 IFNγR KO cells were incubated with 50pM cytokine in 50μL volume for 5 hours. After 5h, supernatant was collected and the cell-mediated depletion of cytokine was quantified by bead-based ELISA. (G) IFNγR KO B16 cells were pre-treated with Annexin V, or Annexin V binding buffer before performing the IFNγ cell capture assay. Data are representative of at least 3 independent experiments. (H) IFNγR KO B16 cells were depleted of cholesterol using MβCD, Saponin, Filipin, or lovastatin. Then the IFNγ cell capture assay was performed. Data are representative of 3 independent experiments. (I) IFNγR KO B16 cells were adhered to a glass bottom dish, incubated with BODIPY-cholesterol, then stained with Annexin V. Images are representative of 2 independent experients. (J) IFNγR KO B16 cells were adhered to a glass bottom dish, incubated with BODIPY-cholesterol, then incubated with IFNγ-A488. Cells were washed and then imaged. Images are representative of 3 independent experiments. (K) IFNγR KO B16 cells were incubated with IFNγ-A488, then washed and stained with cell mask orange and Hoechst. Images are representative of 3 independent experiments. In membrane close-up, arrows denote IFNγ associated with the plasma membrane, o symbols denote intracellular, membrane-adjacent IFNγ, and * symbols denote intracellular IFNγ.
To summarize, we uncovered a cytokine catch-and-release phenomenon whereby IFNγ-exposed cells capture it in an IFNγR-independent manner, and then slowly release it, enabling autocrine and paracrine communication. This allows IFNγ to act over long timescales by persisting in the environment after the original cytokine source is removed.
Cytokine catch-and-release is well approximated by a slow, single-step mechanism
In this section, we introduce a quantitative model to probe the essential dynamic features of cytokine catch-and-release.
We first quantified the strength of IFNγ cell capture, by measuring the concentration (IFNγ50) of IFNγ at which cells fulfill 50% of their catch and release capacity. We prepared fluorescently-labeled IFNγ (IFNγfluo) and confirmed its full functionality (Figure S3D,E, and supplemental experimental procedures sections 2.9 and 2.10). B16 IFNγR KO cells were then exposed to varied concentrations of IFNγfluo for several hours, washed and the amount of IFNγfluo captured per cell was measured by flow cytometry. We confirmed that IFNγ capture and release had reached a steady state (Data not shown). The data were well fitted with a Hill function, with a coefficient of 1, suggesting that IFNγ catch-and-release can be coarse grained as a single-step process, with an equilibrium concentration IFNγ50 of 340±30 pM (Figure 4B).
Figure 4. A mathematical model, including a slow catch and release process, recapitulates experimental results.

See also Figure S4. (A) Schematic of model. IFNγ binds to the IFNγR and is captured by the cells in a receptor-independent manner. After removal of exogenous IFNγ, release from cells drives persistent signaling through the IFNγR, until it is consumed. (B) B16 IFNγR KO cells were exposed to different doses of IFNγfluo, washed, and fluorescence was quantified by flow cytometry. The data were fit with a Hill function and the IFNγ50 was computed from the fit. Data are representative of 3 independent experiments. (C) The model was run keeping the IFNγ50 constant, and varying the catch and release rates. pSTAT1 was calculated from the EC50 of signaling (Figure S4A) and the concentration of free IFNγ generated from cell release (Figure S4D). Curves colored in orange and red resemble the pSTAT1 curves observed experimentally (Figure 2B). Inset shows the time of pSTAT1 peak versus the halflife of cell release. (D) B16 IFNγR KO cells were pulsed with IFNγfluo in a large volume of well mixed RPMI. The media was then replaced with an excess of unlabeled IFNγ and the decay of cell fluorescence was quantified by flow cytometry. The data were fit with an exponential decay function and the decay rate was computed from the fit. Data are representative of 3 independent experiments. (E) The model was updated with the IFNγ-cell release rate. The cell catch rate was inferred from: (IFNγ50=krelease/kcatch). The model was fit to the pSTAT1 curve obtained after removal of exogenous IFNγ (Figure 2B). (F) The pSTAT1 curve obtained from the model in figure 5E was used to calculate the mRNA trajectory and compared to several candidate genes from Figure 1E, cluster 1.
We then used a mathematical model to better understand the cells’ long-term responses to IFNγ observed experimentally (for modeling details see Methods S1). We modeled two modes of IFNγ binding (via its receptor and by cell capture) using mass action kinetics (Figure 4A). Once exogenous IFNγ is removed from the system, IFNγ can be released from cells, rebinds to IFNγR and induces signaling until it is consumed. We then used our model to predict the release rate of IFNγ from the cell. As a modeling target, we generated the pSTAT1 profile after removal of exogenous IFNγ from the system. The pSTAT1 profile was generated using the EC50 of IFNγ signaling (Figure S4A) and the concentration of free IFNγ generated by the model (Figure S4D). We kept IFNγ50 constant to its measured value (Figure 4B) and varied the catch and release rates over several orders of magnitude (Figure 4C). Our model predicted that the timescale for IFNγ cell release (τrelease) would need to be surprisingly slow, between 4 and 40 hours to account for our experimental data (i.e. pSTAT1 peak time around 1 day - Figure 2B and 4C-inset).
We tested this prediction by performing a well-mixed competition experiment. B16 IFNγR KO cells were pulsed with IFNγfluo, and then the media was replaced with an excess of unlabeled IFNγ. The decay in cell fluorescence was quantified by flow cytometry and fitted as an exponential decay (Figure 4D). We estimated τrelease to be 5.1±0.8h, in accordance with our model prediction. We used this experimentally-determined value to demonstrate that our coarse-grained model accounts for our pSTAT1 measurements (Figure 4E). Next, we used the pSTAT1 profile generated by the model to infer the mRNA dynamics: these compared well to the dynamics of gene expression for the antigen presentation cluster (Figure 4F, 1E).
Our experiments verified that the cell release of IFNγ is extremely slow compared to typical molecular de-binding rates. Illustrating this is an alternative example of cytokine sequestration where Interleukin 10 (IL10) interacts with the extracellular matrix (ECM) protein heparan sulfate (Salek-Ardakani et al., 2000). The halflife of IL10-heparan sulfate de-binding is ~30s, which contrasts starkly with our measurement of 5h for IFNγ cell release (Figure 4D). Hence, cell catch-and-release of IFNγ operates on a timescale 600-fold slower than typical ECM sequestration of cytokines, enabling this new mode of cell-to-cell communication (Figure S4E). Using the experimentally-derived timescales for IFNγ cell catch and release, our model quantitatively accounts for the dynamics of pSTAT1, mRNA, and cell-to-cell cytokine sharing (Figure 4E,F and S4G,H). Ultimately, our model shows that coarse-graining IFNγ catch-and-release to a slow, one-step process is sufficient to explain all of our experimental observations.
Cell surface-exposed phosphatidylserine mediates IFNγ catch-and-release
We then sought to identify by what mechanism cells achieve this extraordinarily slow catch-and-release. We used our IFNγ cell-capture assay (Figure 5F) to demonstrate that cytokine catch and release was not mediated by surface proteins, heparan sulfate, chondroitin sulfate, gangliosides or clathrin-dependent endocytosis (Figure S3C). We then used IFNγfluo to reveal by confocal microscopy a punctate pattern for IFNγ during cytokine catch and release (Figure S3F), suggesting a role for membrane microdomains. To test whether IFNγ directly bound to lipids, we performed an immunoblot using strips spotted with a panel of different lipids. IFNγ bound strongly and directly to several different lipids including cardiolipin (CL), phosphatidylserine (PS) and phosphatidyl inositol (4) phosphate (PI(4)P) (Figure 5A). This is consistent with a previous observation that IFNγ could bind PS in liposome membranes (Yoshimura and Sone, 1987). We then stained live cells with lipid-specific reagents and used flow cytometry to test the presence of these lipids on the outer leaflet of the plasma membrane. While CL and PI(4)P were undetectable, PS was ubiquitously on the surface of live cells and was detected by both of the PS-binding proteins, Annexin V and MFG-E8 (Figure 5B and S5A–B, D–E, and supplemental experimental procedures sections 2.4 and 2.5), making it the best candidate to mediate IFNγ cell capture.
In healthy cells, PS is primarily localized on the inner leaflet of the membrane (van Meer et al., 2008). However, there are certain contexts where PS accumulates on the outer leaflet of non-apoptotic cells, such as primary macrophages and monocytes, and activated T and B cells (Figure S6B) (Appelt et al., 2005; Callahan et al., 2000; Dillon et al., 2000; Fischer et al., 2006). as well as many primary tumors, tumor vascular endothelium, and tumor cell lines (Figure S5D) (Blanco et al., 2014; Chu et al., 2013; Dong et al., 2009; Judy et al., 2012; Lima et al., 2009; Riedl et al., 2011).
We imaged PS using Annexin V staining and observed a punctate pattern similar to that of IFNγ (Figure 5C, S3F), and distinctly different than that of a dead cell (Figure S5C). To resolve the subcellular localization of PS (membrane versus cytosolic), we imaged cells in suspension after staining with Annexin V and with plasma-membrane labeling cell mask orange. This strategy revealed punctate PS embedded in the plasma membrane (Figure 5D). We then imaged IFNγ and Annexin V together. To circumvent potentially-competing interactions of IFNγ by Annexin V with PS, we stained cells with a very low dose of Annexin V and found the patterns of IFNγ and Annexin V to be super-imposable (Figure 5E). To determine whether PS is necessary for IFNγ cell capture, cells were pre-incubated with Annexin V (60nM) and tested for their ability to capture IFNγ (Figure 5F). Annexin V blocked IFNγ cell capture relative to cells treated with the buffer control by more than 80% (Figure 5G). Similar blocking was obtained with the PS-binding protein MFG-E8 (Figure S5F). Taken together, these data reveal that PS directly binds IFNγ, co-localizes with IFNγ on cells, and is necessary for cell capture of IFNγ.
The punctate plasma membrane distribution of PS resembled lipid rafts, leading us to test the potential role of cholesterol (as a key component of these rafts) in IFNγ cell capture. Cholesterol was depleted using several different approaches, including methyl β cyclodextrin (MβCD) (Mahammad and Parmryd, 2014), filipin (Maxfield and Wüstner, 2012), saponin (Simons and Ikonen, 1997), and statin treatment (Istvan, 2001), and then we repeated the IFNγ cell capture assay (Figure 5F). MβCD sequesters membrane cholesterol to transiently deplete the plasma membrane, filipin and saponin both bind to cholesterol, and statins inhibit activity of HMG-CoA reductase, an enzyme in the cholesterol biosynthetic pathway. In all cases where cholesterol was depleted, IFNγ capture was reduced (Figure 5H). We introduced fluorescent cholesterol (BODIPY-cholesterol) to cells (Ilnytska et al., 2013), and co-stained with Annexin V, revealing co-localization between the two (Figure 5I). BODIPY-cholesterol also co-localized with IFNγfluo (Figure 5J). Importantly, IFNγ did not bind directly to cholesterol as revealed by the IFNγ lipid blot, suggesting that cholesterol plays an indirect role in cytokine capture (Figure 5A).
In addition to cholesterol-dependence, we were intrigued by the extremely slow IFNγ cell release rate (Figure 4). This release timescale is much slower than that for molecular de-binding. We asked whether the IFNγ release rate reflects simple de-binding from PS, or whether IFNγ is endocytosed and released after binding PS on the cell surface. We found that some of the captured IFNγfluo was adjacent to and even associated with the plasma-membrane, while some was clearly intracellular (Figure 5K). These results support a mechanism for cytokine catch-and-release, where IFNγ is captured on the cell surface in a PS-dependent manner, endocytosed, then slowly released.
Cytokine catch-and-release enables communication between spatio-temporally separate cells, in multiple cellular settings, for Th1-driving cytokines
In this section, we test the functional significance of the slow catch-and-release of cytokines in diverse cellular contexts. T cells produce IFNγ for only a short period of time (Helmstetter et al., 2015; Honda et al., 2014; Hosking et al., 2014). However, if IFNγ is captured by cells in the vicinity where it was produced, its slow release time could permit signaling to different cells that migrate into the environment later. Thus cells that are separated by both space and time could communicate via cytokine catch-and-release.
We tested whether the IFNγ captured by PS on tumor cells was sufficient to activate macrophages. B16 IFNγR KO cells were pulsed with IFNγ, washed and co-cultured with either WT or IFNγR KO BALB/c bone marrow-derived macrophages (BMDM). Macrophage activation was quantified by expression of inducible Nitric Oxide Synthase (iNOS) (Figure 6A). IFNγ-pulsed melanoma cells up-regulated iNOS expression in WT but not IFNγR KO BMDM (Figure 6B). Therefore, catch and release of IFNγ is sufficient to activate macrophages.
Figure 6. Cytokine catch-and-release could enable communication between spatio-temporally separate cells and is also observed for IL12.

See also figure S6. (A) Diagram of experiment. B16 IFNγR KO cells were pulsed with IFNγ, washed, and then co-cultured with either wildtype or IFNγR KO BMDM. (B) Macrophage activation was assessed by iNOS expression by flow cytometry. Data are representative of at least 3 independent experiments. (C) Schematic of experiment. B16 cells were stimulated with IFNγ for 5 hours and then washed. After washing, one cohort of cells received TNFα and another received TNFα and αIFNγ. (D) Cell viability was assessed by DAPI incorporation. Data are representative of 2 independent experiments. (E) Catch-and-release communication could enable activated T cells to release IFNγ, even after IFNγ production has been shut down. (F) Naïve or activated T cells were split into three cohorts where cohort 1 was left untreated, cohort 2 was pulsed with IFNγ for 4 hours, and cohort 3 was treated with a combination of drug inhibitors to abrogate IFNγ production. All cohorts were then co-cultured with B16 sensor cells and sensor MHC-I (H2-Kb) up-regulation was quantified by flow cytometry after 1 day. Data are representative of 3 independent experiments. (G) Lipid-spotted strips were incubated with 5nM IL12, probed with antibodies directed against IL12, and developed. Blot is representative of 3 independent experiments. (H) IFNγR KO B16 cells were pre-treated with Annexin V, or Annexin V binding buffer before performing the IL12 cell capture assay. Data are representative of at least 3 independent experiments. (I) B16 IFNγR KO cells were pulsed with 10nM IL12, then washed and co-cultured with BALB/c wildtype BMDM. Production of IFNγ was quantified by bead-based ELISA. Data are representative of 3 independent experiments. N.D. stands for not detected. (J) Thyroid tumors or healthy thyroids were isolated and dissociated into single cell suspensions and cultured for 1h with a combination of drug inhibitors to abrogate IFNγ production. Tumors or healthy thyroids were then co-cultured with labeled B16 (IFNγR+/+ or −/−) for 48 hours. MHC-I was quantified by flow cytometry across live DAPI negative cells. n of tumor-bearing and healthy thyroids = 3 each. The cytokine sharing assay was repeated three independent times.
Next, since dual administration of IFNγ and tumor necrosis factor α (TNFα) are known to cooperatively induce cell death (Braumüller et al., 2013; Liu et al., 2011), we asked if catch-and-release communication could enhance cell death in response to sequential treatment with these cytokines. B16 IFNγR KO cells were transiently exposed to IFNγ or mock-exposed. After washing, IFNγ-pulsed or un-pulsed cells were co-cultured with B16 IFNγR competent cells. One cohort of cells was left untreated, one was incubated with TNFα, and the final cohort was incubated with TNFα and an anti-IFNγ blocking antibody (Figure 6C). After 24 hours, cell death was quantified. In conditions where persistent IFNγ signaling was permitted, TNFα boosted cell death (Figure 6D). This shows that cytokines can act additively on cells despite sequential secretion.
Constitutive presentation of PS on the plasma membrane outer leaflet appears distinctive of tumor cells and implies that tumor cells would be more efficient at capturing IFNγ than healthy primary tissues. To test this, we harvested different primary tissues from IFNγR KO mice and tested their ability to capture IFNγ compared to B16 IFNγR KO melanoma cells using our IFNγ cell capture assay (Figure 5F). This experiment revealed that many primary tissues were relatively inefficient at IFNγ cell capture compared to B16 melanoma cells (Figure S6C).
Although constitutively externalized PS is characteristic of tumor cells, there are contexts where PS flips transiently to the outer leaflet occurs in healthy cells. For example, PS has been observed on the outer leaflet of live monocytes and macrophages, and activated T and B cells (Figure S6B) (Appelt U et al., 2005; Callahan et al., 2000; Dillon et al., 2000; Fischer et al., 2006). Consistent with the outer leaflet localization of PS in activated lymphocytes, activated but not naïve T cells were capable of capturing IFNγ in our cytokine cell capture assay (Figure 5F and S6D and supplemental experimental procedures section 2.8). We then asked whether activated T cells capture IFNγ and are capable of sharing IFNγ after de novo IFNγ production has been shut down (Figure 6E). Naïve or activated C57Bl/6 IFNγR KO T cells were split into three cohorts. Cohort 1 was left untreated. Cohort 2 was incubated with IFNγ for 4 hours and then washed to replicate our previous IFNγ sharing assays. Cohort 3 was incubated with a combination of drug inhibitors designed to shut down IFNγ production. We used the combination of Cyclosporin A (CsA), an inhibitor of the Nuclear Factor of Activated T cells (NFAT), Dasatinib SRC kinase inhibitor (SRCi), and MEK kinase inhibitor (MEKi). All three cohorts were then co-cultured with wild type B16 sensor cells and H2-Kb expression was quantified on sensor cells after 1 day. We verified that our drug inhibitor combination completely shut down IFNγ production in activated T cells (Figure S6F). Naïve T cells did not up-regulate MHC-I on B16 sensor cells regardless of the condition (Figure 6F). However, for activated T cells, all three conditions up-regulated MHC-I on B16 cells in an IFNγ-dependent manner. This experiment reveals that activated T cells are capable of releasing IFNγ even after de novo IFNγ production has been shut down.
Interleukin 12 and 23 also bind to phosphatidylserine and IL12 participates in catch-and-release communication
We tested whether other cytokines could also bind PS by repeating our lipid blot screen. Lipid strips were probed with Interleukin 2 (IL2), IL4, IL10, IL12p70, IL17a, IL23 and TNFα. Of these cytokines, only IL12 and IL23 exhibited lipid-binding activity (Figure 6G, S6A, and data not shown).
Using our cytokine cell capture assay (Figure 5F), we confirmed that IL12 is captured by cells in a PS-dependent manner (Figure 6H). To assess whether IL12 can be caught and released in sufficient amount to mediate signaling to other cells, we performed a cell-to-cell IL12 sharing assay. B16 IFNγR KO cells were pulsed with IL12, then washed and co-cultured with BALB/c BMDM for 24 hours. Since IL12 can promote IFNγ secretion from macrophages (Munder et al., 1998), we assayed accumulation of IFNγ in the culture supernatant. We observed IL12-dependent accumulation of IFNγ in cultures where macrophages were co-cultured with IL12-pulsed B16 cells (Figure 6I). This demonstrates that IL12 can also participate in PS-mediated catch-and-release communication, similarly to IFNγ.
IFNγ accumulates in tumors in vivo and can participate in cytokine catch-and-release communication
We tested the functional significance of IFNγ catch-and-release by tumor cells in vivo. We hypothesized that IFNγ would be released from thyroid tumors, but not healthy thyroids. We made use of the Thrb(PV/PV)Pten(+/−) thyroid tumor model based on a mutant dominant-negative thyroid receptor beta (TRbPV) together with haploinsufficiency for the Pten gene (Guigon et al., 2009). This thyroid tumor model develops spontaneous metastatic thyroid follicular carcinoma. We harvested primary thyroid tissues from tumor-bearing mice or healthy littermates (experimental procedures for details) to test ex vivo whether they would release functionally significant concentrations of IFNγ (the cytokine having been captured in vivo before harvest). This assay was set up in the presence of Cyclosporin A and SRC kinase inhibitor to abrogate IFNγ production by immune infiltrates ex vivo (Figure S6E–F).
First, we quantified IFNγ transcripts from thyroid tumors by RT-qPCR and confirmed that Ifng mRNA is not produced (Figure S6E). Then, we co-cultured tumors or healthy thyroids with dye-labeled IFNγR-competent or IFNγR KO B16 cells and MHC-I was measured by flow cytometry after 2 days. MHC-I served as the sensitive read-out for IFNγ release (see Figure 3D). When co-cultured with thyroid tumor cells, B16 cells up-regulated MHC-I (Figure 6J). This up-regulation was not observed when B16 cells were co-cultured with healthy thyroid cells. Moreover, the effect we observed was mostly dependent on IFNγ as B16 IFNγR KO cells up-regulated MHC-I to a much lesser degree. We emphasize that the IFNγ shared by thyroid tumor cells ex vivo originated from a natural in vivo source. This means that physiologic concentrations of cytokines are sufficient to facilitate cytokine catch-and-release in vivo. These data demonstrate that cytokine catch-and-release can operate in vivo.
DISCUSSION
We discovered a mechanism of cell-to-cell communication mediated by phosphatidylserine (PS) on cancer cells: catch-and-release of cytokines. Cytokine catch-and-release is not unique to mouse melanoma cells, but is a general feature of diverse primary and cancer cell types (Figure 3D, 6J, and S6C–D). Furthermore, the cytokines IL23 and IL12 also bind PS and IL12 can participate in catch-and-release communication (Figure 6G–I and S6A). These results suggest that PS may be a key factor in tumor Th1 polarization, especially given that sequential waves of IFNγ and IL12 are required for Th1 lineage differentiation (Schulz et al., 2009).
What biochemical characteristics enable binding of IFNγ, IL12, and IL23 to PS? All three cytokines bind to negatively-charged phospholipids (Figure 5A, 6G, and S6A), suggesting that positively charged regions of these cytokines mediate binding. Examination of IFNγ, IL12, and IL23 structures reveals, polycationic (positively-charged) domains (Saesen et al., 2013; Wolf et al., 1991). Similarly, the bacterial endolysin PlyC binds strongly to PS via a positively charged region of the protein, to penetrate infected cells and lyse intracellular Streptococci (Shen et al., 2016). IFNγ is also internalized and recycled after binding PS on the cell surface (5K). Internalization is independent of the clathrin-dynamin pathway, but may rely on caveolae-mediated endocytosis as cholesterol depletion disrupts IFNγ cell capture (Figure S3C and 5H–J) (Nabi and Le, 2003; Rothberg et al., 1992). The biochemistry of PS IFNγ interactions may answer the question as to why IFNγ is recycled rather than processed after endocytosis. The fate of internalized ligands often depends on the stability of the complex at endosomal (acidic) pH (French et al., 1995; Reddy et al., 1996; Sarkar et al., 2002).
Externalization of PS on tumor cells confers a fitness advantage by establishing an immunosuppressive tumor microenvironment (Birge et al., 2016; Scott et al., 2001). This effect is mediated by ligation of PS by receptors present on dendritic cells and macrophages and likely evolved to prevent immune reaction against autoantigens liberated by dying cells. Perhaps unsurprisingly, inhibiting PS using monoclonal antibodies has been shown to specifically target cancer cells and potentiate anti-tumor immunotherapy in both pre-clinical animal models and early-phase clinical trials (Bondanza et al., 2004; Chalasani et al., 2015; Digumarti et al., 2014; Gray et al., 2016). Based on our results, we propose that the fitness advantage conferred by PS externalization on tumor cells may be mitigated by a fitness cost due to PS-mediated localization and persistence of Th1 cytokines. Despite excitement surrounding the promise of PS-targeting antibodies, the latest late-phase clinical trials have indicated underwhelming results (Gerber et al., 2016). Our result that PS modifies tumor responses to inflammatory cytokines should inform the design of strategies targeting PS to maximize their therapeutic potential. Maximizing tumor cell responses to IFNγ goes hand-in-hand with immunotherapeutic strategies as dysfunctional responses to this cytokine are associated with resistance to anti-CTLA4 therapy (Gao et al., 2016).
To conclude, our results uncovered a novel function for PS in regulating tumor cell inflammatory responses to cytokines. We anticipate that our results will motivate new treatment strategies targeting PS and inflammatory pathways to improve upon the current options in cancer immunotherapy.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Grégoire Altan-Bonnet (gregoire.altan-bonnet@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57Bl/6 and C57Bl/6 IFNγR KO mice were purchased from The Jackson Laboratories. BALB/c wildtype and BALB/c IFNγR KO bone marrow was provided by Dr. Romina Goldszmid (NCI, NIH). Splenocytes from pmel-1 mice were provided by Drs. Jedd Wolchok and Taha Merghoub (MSKCC). Thyroid tumors and normal thyroids from Thrb(PV/PV) Pten(+/−) mice and littermate wild type controls were provided by Dr. Sheue-Yann Chen (NCI,NIH). Both male and female mice aged 8–15 weeks were used in our experiments. All mice were maintained in SPF conditions at an Association for Assessment and Accreditation of Laboratory Animal Care accredited animal facility (both NIH and MSKCC).
Cell lines and culture conditions
B16-F10 (mouse, ATCC CRL-6475), SK-Mel-2 (human, male, ATCC HTB-68), H460 (human, male, ATCC HTB-177), CH12 (mouse, gift of Dr. Jayanta Chaudhuri, MSKCC), HEK293T (human, ATCC CRL-3216), and RAW (mouse, male, ATCC TIB-71) cells (with the exception of HEK293T) were maintained in RPMI 1640 media supplemented with heat-inactivated 10% fetal calf serum, 2mM L-glutamine, 10mM HEPES, 0.1mM non-essential amino acids, 1mM sodium pyruvate, 100μg/ml of penicillin, 100μg/ml of streptomycin and 50μM β-mercaptoethanol. For imaging experiments, cells were cultured in phenol-free RPMI with identical supplementation. HEK293T cells were maintained in DME with identical supplementation. All cell lines used were maintained at 37°C with 5% CO2. Adherent cells were detached using 0.05% EDTA in PBS with agitation.
METHOD DETAILS
RNA sequencing and bioinformatics pipeline
B16 cells were pulsed with 10nM IFNγ or mock-pulsed for 5h, washed and 3.5×104 cells were seeded in a 96-well plate. RNA was harvested using the RNeasy mini kit. cDNA library preparation, quality control, and sequencing were performed by the Integrated Genomics core facility (MSKCC). RNA sequencing was done using the Illumina Hi-seq 2000 platform. Genome alignment was done by the Bioinformatics core facility (MSKCC).
For both cytokine and mock-stimulated conditions, genes that occupied the bottom 20th percentile based on variance over time were eliminated. Genes remaining in the IFNγ-stimulated condition were eliminated if they did not change by greater than log2(4)-fold at any time point compared to t0. Genes remaining in the mock-stimulated condition were eliminated if they did not change by greater than log2(5)-fold at any time point compared to t0. Genes remaining in the mock-stimulated condition that were present in the IFNγ-stimulated condition were removed from the IFNγ dataset.
For Gene Ontology (GO), genes in each cluster were analyzed with the PANTHER Overrepresentation Test (release 4/30/2015) using the GO database (release 8/6/2015) for mouse biological pathways.
Cluster analysis was performed using the k-means algorithm. The initial cluster centroid was chosen randomly, and the distance metric which was minimized was the Euclidean distance between the sample points and the cluster centroid. The algorithm was iterated until the distance metric was minimized according to the MATLAB kmeans function. The appropriate number of clusters was determined using the elbow method. In other words, the algorithm was run for increasing number of clusters and the number of clusters was plotted versus the distance. The point at which this curve forms an elbow is the point at which the distance between data and model no longer appreciably falls with each added cluster. After the elbow, newly-added centroids cluster transcriptional trajectories based on small differences or experimental noise and fail to convey new features of the dynamics.
T cell stimulation assays
Pmel splenocytes were harvested on day -4 prior to co-culture, activated with 5ng/ml PMA and 500ng/ml Ionomycin, and maintained in 10nM Interleukin 2 (IL2). On days -7 through -1, B16 cells were pulsed with 10nM IFNγ for 5h, washed and cultured in fresh media. On day 0, 1×105 B16 cells cells were co-cultured with an equivalent number of CD8+ pmel T cells for 7h in a 96 well v-bottom plate. The fraction of IFNγ+ activated cells were quantified by cytokine secretion assay. MHC-T cell receptor interaction was blocked using 5μg/ml αH2-Db. EL4 cells were stimulated with IFNγ as B16 cells.
Cytokine stimulation and cell signaling
Cells were stimulated with 10nM IFNγ for 5h, and then washed 3× with 10ml warm RPMI. 3.5×104 IFNγ-pulsed cells were seeded in a 96 well plate and MHC-I was measured with αH-2Kb or αH-2Db by flow cytometry. IFNγ signaling was blocked with 10μg/ml αIFNγ.
For intracellular phospho-flow cytometry, cells were stimulated with 10nM IFNγ or mock-stimulated and washed. At each timepoint, cells were fixed and permeabilized with 1.6% paraformaldehyde (PFA) for 10min on ice, followed by 90% Methanol for at least 20min on ice, then stained with αpSTAT1, then αRabbit IgG.
To determine the EC50 of signaling, 5×104 B16 cells were stimulated with indicated concentrations of IFNγ for 20 minutes, then fixed and permeabilized. Cells were stained for pSTAT1 and fluorescence was quantified with flow cytometry. To determine the EC50 of signaling, the data were fitted with a Hill function with coefficient = 1.
For JAK inhibition assay, 5×104 B16 cells were stimulated with 10nM IFNγ for 20 minutes before application of 10μM of the JAK1/2 inhibitor AZD1480 (JAKi), and then fixed and permeabilized. Cells were stained for pSTAT1 and fluorescence was quantified with flow cytometry. To quantify the rates of pSTAT1 decay after JAKi, the data were fitted with a double exponential decay function.
Lipid labeling
For cardiolipin staining, live or fixed and permeabilized cells were incubated with αCardiolipin, then αHuman IgG. For PI(4)P staining, cells were incubated with αPI(4)P, then αMouse IgM. As a negative control, cells were incubated with the appropriate secondary antibody only. Cells were stained with the indicated antibodies for 30 min at room temperature in FACS buffer (4% fetal calf serum and 0.1% sodium azide in PBS), or, in FACS buffer lacking sodium azide. Live cells were identified by DAPI exclusion. Just prior to flow cytometric analysis, 0.1μg/ml DAPI was added.
Annexin V binding was carried out according to the manufacturer instructions. Cells were washed once in PBS, and then incubated for 15 min at room temperature with a 1:25 dilution of fluorescent Annexin V in Annexin binding buffer (100mM HEPES, 140mM NaCl, 25mM CaCl2, pH 7.4). To block PS, a high dose of unlabeled Annexin V was used: 60nM. For co-imaging of fluorescent IFNγ and PS, fluorescent Annexin V was titrated down to a non-competitive concentration (1:250 dilution).
To stain cells with MFG-E8, Cells were incubated with 1μg/ml recombinant mouse MFG-E8 for 30 minutes at room temperature in live cell FACS buffer, then washed. Cells were then incubated with 1μg/ml biotinylated αmouse MFG-E8 for 30 min at room temperature then washed. Finally, cells were incubated with 1μg/ml fluorescent conjugated streptavidin for 30 min at room temperature. DAPI was added just prior to flow cytometric analysis.
Transwell assay
To perform the transwell assay, B16 cells were stimulated for 5h with 10nM IFNγ then washed 3× with 10ml warm culture media. Prior to cytokine stimulation, B16 cells were labeled with the dye cell trace violet (CTV). Cells were washed once with PBS, then stained for 10 min at 37°C with a 1:1000 dilution of CTV in PBS. CTV-labeled cells were then washed 2× in warm culture media. 1×105 IFNγ-pulsed CTV-labeled cells were co-cultured with 2×104 un-stimulated, un-labeled B16 cells. An additional 2×104 un-stimulated, un-labeled B16 cells were cultured on top of a 0.4μm pore size transwell. We denote un-labeled, un-stimulated B16 cells “sensors” because we quantify cell-to-cell cytokine sharing by their up-regulation of IFNγ-response genes. MHC-I (H2-Kb) on sensor cells was quantified after 24h by flow cytometry.
Conditioned media assay
To test whether IFNγ remained in the media after washing, B16 cells were pulsed with 10nM IFNγ for 5h, and then centrifuged. 10ml fresh RPMI was added to the cells in between centrifugation three consecutive times to wash. Immediately after the last wash, RPMI was harvested (denoted ”conditioned media”). A fresh cohort of un-stimulated B16 cells were cultured with either media alone, media supplemented with 10nM IFNγ, or conditioned media for 24h at 3.5×104 cells/well. After 24h, MHC-I (H2-Kb) was measured by flow cytometry.
Cytokine sharing assays
For cell-to-cell cytokine sharing experiments with different cell types, cells were pulsed with 10nM mouse IFNγ for 5h and washed. 3.5×104 IFNγ-pulsed B16, SK-Mel-2, H460, CH12, HEK293T, or RAW cells were co-cultured with 2×104 CTV-labeled B16 sensor cells. Sensor cell MHC-I (H2-Kb) was measured after 24h.
To test for the role of IFNγ receptors, B16 IFNγR KO cells were pulsed with 10nM IFNγ for 5h, and then washed. 3.5×104 IFNγ-pulsed cells were co-cultured with 2×104 CTV-labeled B16 IFNγR competent sensor cells. Sensor cell MHC-I (H2-Kb) was measured after 24h.
Confocal microscopy
Live-cell confocal microscopy was performed on a Zeiss LSM510META laser scanning confocal microscope equipped with lasers emitting 458, 488, 514, 565 and 633nm. A 63X oil immersion objective with 1.4 numerical aperture was used with pinhole set at 1.2 Airy units. IFNγ-A647 and IFNγ-A488 (IFNγfluo) were generated using a microscale protein labeling kit. Carrier-free IFNγ was dissolved in PBS at a concentration of 1mg/ml. Sodium bicarbonate was added to a final concentration of 100mM. Next Alexa Fluor 647 or 488 ester dye was prepared in water, added to a molar ratio of 19 and the mixture was incubated for 15 min at room temperature, shielded from light. The unconjugated dye particles were separated from the mixture by centrifuging through the kit-provided gel resin. The degree of labeling and final protein concentration were determined using a Nanodrop spectrophotometer. Where indicated, cells were adhered to human fibronectin-coated glass-bottom dishes. Cells were incubated with 10nM IFNγfluo for 5h then washed. Annexin V staining was done as described above, or with 10-fold less than recommended when co-staining with IFNγfluo. To stain with Cell Mask Orange (CMO), cells were first exposed to 10nM fluorescent IFNγ for 5h, then washed and incubated with a 1:3×105 of CMO and Hoechst 33342 for 5 min at 37°C. To introduce fluorescent cholesterol, cells were rinsed with warmed serum-free culture media and incubated for 5 min at 37°C with 20 μg/ml BODIPY-cholesterol diluted in serum-free culture media. Cells were then incubated at 37°C for 5 hours with 10nM fluorescent IFNγ. Before imaging, cells were washed 3× with 10 ml warm culture media. For details about the number of cells analyzed in our imaging experiments see supplemental experimental procedures section 2.12.
Quantification of cytokine catch-and-release dynamics
To verify IFNγfluo, B16 IFNγR KO cells were incubated with 10nM IFNγ-A647 for 3.5h while tumbling in RPMI, washed, and cell fluorescence was quantified by flow cytometry. To assess IFNγ-A647 specificity, cells were first pre-treated with 10nM unlabeled, recombinant IFNγ for 3.5h. After 3.5h, 10nM IFNγ-A647 was spiked in and cells were incubated for an additional 3.5h. Prior to flow cytometric analysis, cells were washed in FACS buffer. To check whether residual dye leftover from the protein microscale labeling prep could bind non-specifically to cells, we prepared a protein-free sample of Alexa647 carboxylic acid succinimidyl ester dye treated exactly as the IFNγ-A647 was treated and exposed an equal quantity to cells for 3.5h.
To quantify the IFNγ half max, B16 IFNγR KO cells were incubated with the indicated concentration of fluorescent IFNγ for 7h, and then washed. Cell fluorescence was quantified by flow cytometry and then fitted with a Hill function with coefficient=1. To ensure that IFNγ cell capture had reached steady-state, the half max measurement was compared after incubations of 6 and 8h.
To quantify the IFNγ release rate, B16 IFNγR KO cells were pulsed overnight with 10ml of 10nM fluorescent IFNγ in well-mixed conditions. Cells were then washed and the media was replaced with 10ml of 10nM unlabeled IFNγ with rotation. Cell fluorescence was quantified by flow cytometry and fitted with a single exponential decay function to compute the decay rate.
Cytokine capture assays
To perform the IFNγ and IL12 cell capture assays, 3×105 B16 IFNγR KO cells were incubated for 5h with 50pM IL12 or IFNγ in 50μl total volume with mixing. Supernatant was collected and the amount of IL12 or IFNγ remaining was quantified by bead based ELISA. Cell capture was calculated as the difference between the number of molecules in the cell-free sample and the number of molecules in samples with cells. (# moleculescell free- # moleculeswith cells). The amount of cell capture in experimental samples was then converted into a percentage of the control cell capture.
To inhibit glycolipid biosynthesis, B16 IFNγR KO cells were cultured for 3 days in the presence of 10μM Fumonisin B, 10μM PDMP, or an equivalent concentration of DMSO as a vehicle control (Rusnati et al., 2002). An additional group of cells were left untreated. After 3 days, the IFNγ cell-capture assay was performed in the presence of each drug or DMSO.
To inhibit clathrin/dynamin-dependent endocytosis, B16 IFNγR KO cells were pre-treated with 30μM Dynasore for 30 min. After 30 min, the IFNγ cell-capture assay was performed in the presence of Dynasore.
To generate T cell-derived IFNγ, C57Bl/6 IFNγR KO splenocytes were incubated with PMA and Ionomycin overnight. Culture supernatant was harvested and the amount of IFNγ generated was quantified by bead based ELISA. Equivalent concentrations of recombinant and T cell-derived IFNγ were incubated with B16 IFNγR KO cells to assess IFNγ cell capture after 5h of incubation.
To block PS with the peptide MFG-E8, B16 IFNγR KO cells were pre-incubated for 15 min at room temperature with 50nM recombinant mouse MFG-E8 in warm culture media. The IFNγ cell-capture assay was performed in the presence of MFG-E8.
For the following treatments, the IFNγ cell capture assay was incubated for 2 instead of 5h to ensure that pericellular matrix proteoglycans and proteins did not re-populate the cell after enzymatic removal.
To digest proteoglycans, B16 IFNγR KO cells were pre-treated for 1h in PBS with either 10U/mL heparinase I (hepI), 5U/ml heparinase III (hepIII), or 1.95μg/ml chondroitin ABC lyase (chABC) (Brooks et al., 2000; Lortat-Jacob and Grimaud, 1991). Cells were washed, and then the IFNγ cell-capture assay was performed.
To digest cell surface proteins, B16 IFNγR KO cells were treated as is described in (Suzuki et al., 1995). Briefly, cells were incubated with 0.01% pronase in PBS for 15 minutes. Cells were pelleted and pronase solution was replaced. Cells were incubated for an additional 15 minutes. To stop digestion, an equal volume of fetal calf serum was added to cells. Cells were then washed and the IFNγ cell-capture assay was performed.
To isolate primary cells, the heart, liver, kidneys, and spleen were isolated from C57Bl/6 IFNγR KO mice and crushed into a single cell suspension. Then 5×106 cells were incubated with 20pM IFNγ for 5h as described above. For the T cell IFNγ cell capture assay, splenocytes were harvested from C57Bl/6 IFNγR KO mice and crushed into a single cell suspension. Splenocytes were then either maintained naive in 1nM IL7 for 2 days, or activated with PMA (5ng/ml) and Ionomycin (500ng/ml) for 2 days. Then 5×106 cells were incubated with 10pM IFNγ for 5h as described above.
To block PS, cells were washed in PBS, then pre-treated with 60nM Annexin V in annexin binding buffer. After 15 min, cells were resuspended in 50μl of 60nM Annexin V in binding buffer + 3% fetal calf serum (to block non-specific binding of IFNγ to cells) and 50pM of the relevant cytokine before carrying out the cytokine capture assay.
Cholesterol depletion
To deplete cholesterol with MβCD (methyl β cyclodextrin), B16 IFNγR KO cells were washed once in PBS, and then incubated for 45 min at 37°C in 5mM M βCD in 25mM HEPES. Cells were then washed twice in 5ml warm culture medium and the IFNγ cell capture assay was performed as described below. Control cells were incubated with 25mM HEPES for 45 min prior to the IFNγ cell capture assay. To deplete cholesterol with saponin, cells were incubated for 10 min at 37°C with 0.1% saponin diluted in warm culture media. To deplete cholesterol with filipin, cells were incubated for 15 min at 37°C with 50 μg/ml filipin III. To deplete cells of cholesterol using a statin treatment, cells were incubated with 20μM Lovastatin for 4 days prior to performing the IFNγ cell capture assay.
Lipid immunoblotting
To perform lipid immunoblots, blots were first blocked by incubating strips for 1h at room temperature with blocking buffer (0.1% Tween-20 and 3% fatty acid free bovine serum albumin dissolved in PBS). 50pM IFNγ or 5nM of the relevant cytokine (IL2, IL4, IL10, IL12, IL17, IL23, or TNFα) was diluted in blocking buffer and strips were incubated with gentle agitation overnight at 4°C. Strips were washed in blocking buffer, then incubated for 1h at room temperature with the relevant antibody diluted to 1μg/ml in blocking buffer. Blots were washed again and then incubated with anti-Rat HRP for 30 min at room temperature. Blots were developed using ECL western blotting substrate.
Macrophage assays
To perform Macrophage experiments, bone marrow was harvested from BALB/c wildtype, and BALB/c IFNγR KO mice and differentiated for 7d in 10ng/ml M-CSF in Teflon bags. B16 IFNγR KO cells were pulsed with 10nM IFNγ (or mock-pulsed) for 5h, washed, and 3.5×104 cells were co-cultured with 2×104 macrophages. Cells were fixed, permeabilized, and stained for CD11b, and iNOS.
B16 IFNγR KO cells were pulsed with 10nM IL12 (or mock-pulsed) for 5h, washed, and 3.5×104 cells were co-cultured with 2×104 macrophages. Where indicated, αIL12 was added. Culture supernatant was harvested and IFNγ was quantified by bead-based ELISA.
TNFα-IFNγ cytokine sharing assays
To perform TNFα experiments, 3.5×104 B16 IFNγR KO cells were pulsed with 10nM IFNγ for 7h then washed. IFNγ-pulsed cells were then co-cultured with 2×104 un-stimulated B16 IFNγR competent cells labeled with the dye cell trace far red (CTFR). Cells were labeled with CTFR as with CTV. To relevant wells, either 10nM TNFα or 10nM TNFα and 20μg/ml αIFNγ was added. Live, CTFR-labeled cells were identified by DAPI exclusion after 24h of co-culture.
T cell IFNγ release experiments
C57Bl/6 IFNγR KO splenocytes were harvested and cultured in RPMI complemented with 1nM mouse IL7 and rested or activated for 2 days with a combination of αCD3 and αCD28. Cells were then incubated for 4hr with 1nM of mouse recombinant IFNγ or with a combination of drug inhibitors (1μM Cyclosporin A, 1μM Dasatinib SRC inhibitor, 1μM MEK inhibitor (PD325901). Cells were then collected and centrifuged on a FICOLL gradient. T cells (2×104) were co-cultured with 2×104 B16 sensor cells for 30h and sensor MHC-I (H2-Kb) was quantified by flow cytometry.
Thyroid cytokine sharing assay
To perform Thyroid tumor experiments, thyroids were first dissected from Thrb(PV/PV)Pten(+/−) mice or from wild type littermates. Mice were monitored and euthanized when they displayed breathing difficulties due to airway constriction and/or due to metastasis. Tissues were mechanically dissociated into single-cell suspensions, passed through a 70μm sieve, then cultured with 1μM Cyclosporin A and 1μM Dasatinib SRC inhibitor for 1h at 37°C. Then the indicated number of tumor or normal thyroid cells were co-cultured with 2×104 CFSE-labeled B16 (IFNγR+/+ or −/−) cells for 48h in culture medium with 1μM Cyclosporin A and 1μM Dasatinib SRC inhibitor. Cells were then harvested, washed in FACS buffer lacking sodium azide, and stained for MHC-I (H2-Kb). DAPI was used to distinguish live and dead cells. Cell fluorescence was analyzed by flow cytometry.
To perform qPCR on thyroid samples, 2 million cells were collected, washed in PBS, and re-suspended in RLT lysis buffer. Total RNA was isolated using the total RNA purification kit and cDNA was produced. PCR was carried out using the iTaq Universal SYBR green super mix. PCR was run and monitored on a Light Cycler 96 with 3 technical replicates for each sample. We estimated the amount of Ifng mRNA by the ΔΔCT method where the fold change in gene expression was equal to 2−(ΔΔCT) and the housekeeping gene was Gapdh. Naive T cells served as the negative control. As a positive control, we used primary B10.A splenocytes whose T cells were activated over 48h in vitro using 3μg/ml anti-CD3 and anti-CD28 cross-linking antibodies in culture media supplemented with 1nM IL7. As a negative control, we used naive primary B10.A splenocytes cultured for 48h with 1nM IL7. We also prepared primary T cells (activated over 48h) to which 1μM of Cyclosporin A and 1μM Dasatinib SRC kinase inhibitor were added 1h before collection.
To perform cytokine secretion assays, primary B10.A splenocytes were activated over 48h in vitro using 3μg/ml anti-CD3 and anti-CD28 cross-linking clones. Cells were treated with 1μM of Cyclosporin A and 1μM Dasatinib SRC kinase inhibitor, or with 1μM Dasatinib SRC kinase inhibitor and 1μM MEK inhibitor (PD325901) 1h before collection. An IFNγ cytokine secretion assay was then performed per the manufacturer’s instructions. Briefly, cells were collected from in vitro culture, washed in complete medium, incubated on ice with the cytokine secretion assay capture reagent in 50μl for 5min, re-suspended in 15ml culture medium (with or without drug inhibition) and incubated with rotation for 45min. Cells were then collected, washed, and stained for IFNγ secretion using the cytokine secretion assay detection reagent, anti-CD8, anti-CD4, and DAPI. Dead cells were excluded by DAPI inclusion and fluorescence was analyzed by flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are shown in figures are represented as mean ± standard error of the mean (error bars).
DATA AND SOFTWARE AVAILABILITY
The accession number for the sequencing data reported in this paper is GEO: GSE85535 Other raw data files are available via Mendeley Data: http://dx.doi.org/10.17632/5yygbbg542.1
ADDITIONAL RESOURCES
A detailed description of the mathematical model used in this paper can be found in Methods S1.
Supplementary Material
HIGHLIGHTS.
Transient IFNγ exposure elicits long-lived inflammatory responses in cancer cells.
Long-lived inflammatory responses are caused by persistent cytokine signaling.
Long-lived IFNγ signaling is mediated by catch-and-release of cytokines.
Cytokines are captured by cell surface exposed phosphatidylserine, and then recycled.
Acknowledgments
We thank the Integrated Genomics and Bioinformatics core facilities at MSKCC for RNA-sequencing and genome alignments respectively; Romina Goldszmid, Jedd Wolchok and Taha Merghoub, Jackie Bromberg, Mark Connors, Marianita Santiana for sharing critical reagents; Tamas Balla, Howard Young, and Anton Zilman for helpful discussions; Robin Winkler-Pickett for assuring a smooth lab move to the NIH; Ron Germain for critically reading the manuscript; and all members of the G.A-B. lab. J.O-Y is grateful to Carlos Carmona-Fontaine for microscopy help and insightful discussion. This work was supported by the U.S.-Israel Binational Science Foundation (#2012327 to G.A-B. and O.K.), by the US National Institutes of Health (R01-AI083408 to G.A-B), by the Intramural Research programs of the NHLBI, NIH (N.A-B) and of the Center for Cancer Research, NCI, NIH (G.A-B).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, J.O-Y. and G.A-B.; Investigation, J.O-Y., M.S., N.K.M., and Y-H.C.; Software, J.O-Y., A.O-Y., G.A-B., and O.K.; Writing-Original Draft, J.O-Y., G.A-B., and N.A-B.; Resources, N.A-B., and S-Y.C.; Funding Acquisition, G.A-B., O.K., and N.A-B.; Supervision G.A-B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal S, Rao a. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity. 1998;9:765–775. doi: 10.1016/s1074-7613(00)80642-1. [DOI] [PubMed] [Google Scholar]
- Appelt U, Sheriff A, Gaipl US, Kalden JR, Voll REHM. Viable, apoptotic and necrotic monocytes expose phosphatidylserine: cooperative binding of the ligand Annexin V to dying but not viable cells and implications for PS-dependent clearance. Cell Death Differ. 2005;12:194–196. doi: 10.1038/sj.cdd.4401527. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: Tool for The Unification of Biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, Barcinski M, Brekken RA, Huang X, Hutchins JT, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23:1–17. doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco VM, Chu Z, Vallabhapurapu SD, Sulaiman MK, Kendler A, Rixe O, Warnick RE, Franco RS, Qi X. Phosphatidylserine-selective targeting and anticancer effects of SapC-DOPS nanovesicles on brain tumors. Oncotarget. 2014;5:7105–7118. doi: 10.18632/oncotarget.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondanza A, Zimmermann VS, Rovere-Querini P, Turnay J, Dumitriu IE, Stach CM, Voll RE, Gaipl US, Bertling W, Pöschl E, et al. Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J Exp Med. 2004;200:1157–1165. doi: 10.1084/jem.20040327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–365. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- Brooks B, Briggs DM, Eastmond NC, Fernig DG, Coleman JW. Presentation of IFN-gamma to nitric oxide-producing cells: a novel function for mast cells. J Immunol. 2000;164:573–9. doi: 10.4049/jimmunol.164.2.573. [DOI] [PubMed] [Google Scholar]
- Cai L, Dalal CK, Elowitz MB. Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature. 2008;455:485–490. doi: 10.1038/nature07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Williamson P, Schlegel Ra. Surface expression of phosphatidylserine on macrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ. 2000a;12:645–653. doi: 10.1038/sj.cdd.4400690. [DOI] [PubMed] [Google Scholar]
- Callahan MK, Williamson P, Schlegel Ra. Surface expression of phosphatidylserine on macrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ. 2000b;7:645–653. doi: 10.1038/sj.cdd.4400690. [DOI] [PubMed] [Google Scholar]
- Chalasani P, Marron M, Roe D, Clarke K, Iannone M, Livingston RB, Shan JS, Stopeck AT. A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer Med. 2015;4:1051–1059. doi: 10.1002/cam4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Forrester W, Lahav G. Schedule-dependent interaction between anticancer treatments. Science. 2016;351:1204–1208. doi: 10.1126/science.aac5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Abu-Baker S, Palascak MB, Ahmad SA, Franco RS, Qi X. Targeting and Cytotoxicity of SapC-DOPS Nanovesicles in Pancreatic Cancer. PLoS One. 2013:8. doi: 10.1371/journal.pone.0075507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dighe aS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1994;1:447–456. doi: 10.1016/1074-7613(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Digumarti R, Bapsy PP, Suresh AV, Bhattacharyya GS, Dasappa L, Shan JS, Gerber DE. Bavituximab plus paclitaxel and carboplatin for the treatment of advanced non-small-cell lung cancer. Lung Cancer. 2014;86:231–236. doi: 10.1016/j.lungcan.2014.08.010. [DOI] [PubMed] [Google Scholar]
- Dillon SR, Mancini M, Rosen A, Schlissel MS. Annexin V binds to viable B cells and colocalizes with a marker of lipid rafts upon B cell receptor activation. J Immunol. 2000;164:1322–1332. doi: 10.4049/jimmunol.164.3.1322. [DOI] [PubMed] [Google Scholar]
- Dong HP, Holth A, Kleinberg L, Ruud MG, Elstrand MB, Tropé CG, Davidson B, Risberg B. Evaluation of cell surface expression of phosphatidylserine in ovarian carcinoma effusions using the annexin-V/7-AAD assay: Clinical relevance and comparison with other apoptosis parameters. Am J Clin Pathol. 2009;132:756–762. doi: 10.1309/AJCPAVFA8J3KHPRS. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- Fischer K, Voelkl S, Berger J, Andreesen R, Pomorski T, Mackensen A. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108:4094–4101. doi: 10.1182/blood-2006-03-011742. [DOI] [PubMed] [Google Scholar]
- French AR, Tadaki DK, Niyogi SK, Lauffenburger DA. Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J Biol Chem. 1995;270:4334–4340. doi: 10.1074/jbc.270.9.4334. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-?? Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404. e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–56. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber DE, Spigel DR, Giorgadze D, Shtivelband M, Ponomarova OV, Shan JS, Menander KB, Belani CP. Docetaxel Combined with Bavituximab in Previously Treated, Advanced Nonsquamous Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2016;17:169–176. doi: 10.1016/j.cllc.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Gray MJ, Gong J, Hatch MMS, Nguyen V, Hughes CCW, Hutchins JT, Freimark BD. Phosphatidylserine-targeting antibodies augment the anti-tumorigenic activity of anti-PD-1 therapy by enhancing immune activation and downregulating pro-oncogenic factors induced by T-cell checkpoint inhibition in murine triple-negative breast cancers. Breast Cancer Res. 2016;18:50. doi: 10.1186/s13058-016-0708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, et al. The JAK2 Inhibitor AZD1480 Potently Blocks Stat3 Signaling and Oncogenesis in Solid Tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstetter C, Flossdorf M, Peine M, Kupz A, Zhu J, Hegazy AN, Duque-Correa MA, Zhang Q, Vainshtein Y, Radbruch A, et al. Individual T Helper Cells Have a Quantitative Cytokine Memory. Immunity. 2015;42:108–122. doi: 10.1016/j.immuni.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- Honda T, Egen JG, Lämmermann T, Kastenmüller W, Torabi-Parizi P, Germain RN. Tuning of Antigen Sensitivity by T Cell Receptor-Dependent Negative Feedback Controls T Cell Effector Function in Inflamed Tissues. Immunity. 2014;40:235–247. doi: 10.1016/j.immuni.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosking MP, Flynn CT, Whitton JL. Antigen-specific naive CD8+ T cells produce a single pulse of IFN-γ in vivo within hours of infection, but without antiviral effect. J Immunol. 2014;193:1873–1885. doi: 10.4049/jimmunol.1400348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilnytska O, Santiana M, Hsu NY, Du WL, Chen YH, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe. 2013;14:281–293. doi: 10.1016/j.chom.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istvan ES. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science (80-) 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- Judy BF, Aliperti LA, Predina JD, Levine D, Kapoor V, Thorpe PE, Albelda SM, Singhal S. Vascular endothelial-targeted therapy combined with cytotoxic chemotherapy induces inflammatory intratumoral infiltrates and inhibits tumor relapses after surgery. Neoplasia. 2012;14:352–359. doi: 10.1593/neo.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justman Qa, Serber Z, Ferrell JE, El-Samad H, Shokat KM. Tuning the activation threshold of a kinase network by nested feedback loops. Science. 2009;324:509–512. doi: 10.1126/science.1169498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee REC, Walker SR, Savery K, Frank DA, Gaudet S. Fold change of nuclear NF-??B determines TNF-induced transcription in single cells. Mol Cell. 2014;53:867–879. doi: 10.1016/j.molcel.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima LG, Chammas R, Monteiro RQ, Moreira MEC, Barcinski MA. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 2009;283:168–175. doi: 10.1016/j.canlet.2009.03.041. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang L, Kikuiri T, Akiyama K, Chen C, Xu X, Yang R, Chen W, Wang S, Shi S. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-γ and TNF-α. Nat Med. 2011;17:1594–1601. doi: 10.1038/nm.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortat-Jacob H, Grimaud JA. Interferon-?? binds to heparan sulfate by a cluster of amino acids located in the C-terminal part of the molecule. FEBS Lett. 1991;280:152–154. doi: 10.1016/0014-5793(91)80225-r. [DOI] [PubMed] [Google Scholar]
- Mahammad S, Parmryd I. Methods in Membrane Lipids: Second Edition. 2014. Cholesterol depletion using methyl-??-cyclodextrin; pp. 91–102. [DOI] [PubMed] [Google Scholar]
- Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, García-Sastre A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A. 2010;107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, Wüstner D. Analysis of Cholesterol Trafficking with Fluorescent Probes. Methods Cell Biol. 2012;108:367–393. doi: 10.1016/B978-0-12-386487-1.00017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J Exp Med. 1998;187:2103–2108. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161:673–677. doi: 10.1083/jcb.200302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, Ihekwaba AEC, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- Overwijk WW, Restifo NP. B16 as a Mouse Model for Human Melanoma. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 Dynamics Control Cell Fate. Science (80-) 2012;336:1440–1444. doi: 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy CC, Niyogi SK, Wells a, Wiley HS, Lauffenburger Da. Engineering epidermal growth factor for enhanced mitogenic potency. Nat Biotechnol. 1996;14:1696–1699. doi: 10.1038/nbt1296-1696. [DOI] [PubMed] [Google Scholar]
- Riedl S, Rinner B, Asslaber M, Schaider H, Walzer S, Novak A, Lohner K, Zweytick D. In search of a novel target - Phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim Biophys Acta - Biomembr. 2011;1808:2638–2645. doi: 10.1016/j.bbamem.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RGW. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- Rusnati M, Urbinati C, Tanghetti E, Dell’Era P, Lortat-Jacob H, Presta M. Cell membrane GM1 ganglioside is a functional coreceptor for fibroblast growth factor 2. Proc Natl Acad Sci U S A. 2002;99:4367–4372. doi: 10.1073/pnas.072651899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saesen E, Sarrazin SS, Laguri C, Sadir R, Maurin D, Thomas A, Imberty A, Lortat-Jacob H. Insights into the mechanism by which interferon-gamma basic amino acid clusters mediate protein binding to heparan sulfate. J Am Chem Soc. 2013;135:9384–9390. doi: 10.1021/ja4000867. [DOI] [PubMed] [Google Scholar]
- Salek-Ardakani S, Arrand JR, Shaw D, Mackett M. Heparin and heparan sulfate bind interleukin-10 and modulate its activity. Blood. 2000;96:1879–1888. [PubMed] [Google Scholar]
- Sarkar CA, Lowenhaupt K, Horan T, Boone TC, Tidor B, Lauffenburger DA. Rational cytokine design for increased lifetime and enhanced potency using pH-activated “histidine switching”. Nat Biotechnol. 2002;20:908–913. doi: 10.1038/nbt725. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon- y: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Schulz EG, Mariani L, Radbruch A, Höfer T. Sequential Polarization and Imprinting of Type 1 T Helper Lymphocytes by Interferon-γ and Interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Scott RS, McMahon EJ, Pop SM, Reap Ea, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- Shankaran V, Ikeda H, Bruce aT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- Shen Y, Barros M, Vennemann T, Gallagher DT, Yin Y, Linden SB, Heselpoth RD, Spencer DJ, Donovan DM, Moult J, et al. A bacteriophage endolysin that eliminates intracellular streptococci. Elife. 2016:5. doi: 10.7554/eLife.13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Punt JA, Granger LG, Singer A. Asymmetric signaling requirements for thymocyte commitment to the CD4+ versus CD8+ T cell lineages: A new perspective on thymic commitment and selection. Immunity. 1995;2:413–425. doi: 10.1016/1074-7613(95)90149-3. [DOI] [PubMed] [Google Scholar]
- Wolf SF, Temple Pa, Kobayashi M, Young D, Dicig M, Lowe L, Dzialo R, Fitz L, Ferenz C, Hewick RM. Cloning of cDNA for natural killer cell stimulatory factor, a heterodimeric cytokine with multiple biologic effects on T and natural killer cells. J Immunol. 1991;146:3074–3081. [PubMed] [Google Scholar]
- Yoshimura T, Sone S. Different and synergistic actions of human tumor necrosis factor and interferon-gamma in damage of liposome membranes. J Biol Chem. 1987;262:4597–4601. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data reported in this paper is GEO: GSE85535 Other raw data files are available via Mendeley Data: http://dx.doi.org/10.17632/5yygbbg542.1