

Abstract
Although the field of genetics has grown by leaps and bounds within the last decade due to the completion and availability of the human genome sequence, transcriptional regulation still cannot be explained solely by an individual’s DNA sequence. Complex coordination and communication between a plethora of well-conserved chromatin modifying factors are essential for all organisms. Regulation of gene expression depends on histone post translational modifications (HPTMs), DNA methylation, histone variants, remodeling enzymes, and effector proteins that influence the structure and function of chromatin, which affects a broad spectrum of cellular processes such as DNA repair, DNA replication, growth, and proliferation. If mutated or deleted, many of these factors can result in human disease at the level of transcriptional regulation. The common goal of recent studies is to understand disease states at the stage of altered gene expression. Utilizing information gained from new high-throughput techniques and analyses will aid biomedical research in the development of treatments that work at one of the most basic level of gene expression, chromatin. This chapter will discuss the effects of and mechanism by which histone modifications and DNA methylation affect transcriptional regulation.
1.1. DNA Methylation
1.1.1. CpG Islands
With respect to epigenetic research and a causal relationship to human disease, DNA methylation is the most characterized modification. The enzymatic addition of a methyl group to DNA is performed by DNA methyltransferase (DNMT) on the 5’-carbon of the pyrimidine ring in cytosine. Four human DNMTs have been characterized: DNMT1 (Bestor et al. 1988), DNMT2 (Yoder and Bestor 1998), DNMT3a and DNMT3b (Okano et al. 1999). De novo DNA methylation patterns are established early in development by DNMT3a and DNMT3b and maintained by DNMT1, which prefers to methylate hemi-methylated templates during DNA replication through its recruitment by proliferating cell nuclear antigen (PCNA). About 3% of cytosines are methylated in the human genome almost exclusively in the context of the dinucleotide, CpG. 5-methylcytosine (5-mC) is also found in very low abundance at the trinucleotide, CpNpG (Clark et al. 1995).
CpG dinucleotides are rarer than expected in the human genome (~1%) (Josse et al. 1961; Swartz et al. 1962) as a result of 5-mC deamination and subsequent mutation to thymine (Scarano et al. 1967). 70 to 80% of CpG dinucleotides are methylated and those dinucleotides that are unmethylated tend to cluster in islands (Ehrlich et al. 1982). Regions containing the normal expected density of CpG dinucleotides are called CpG islands (CGI), which are regions no smaller than 200 bp that contain a GC content of more than 55% and an expected GC content to observed GC content ratio greater than 0.65 (Takai and Jones 2002).
Approximately 60% of human gene promoters and first exons are associated with CGIs. CGIs at promoters are frequently hypomethylated corresponding to a permissive chromatin structure in order to poise genes for a transcriptional activation (Larsen et al. 1992; Antequera and Bird 1993) while some are hypermethylated during development, which stably silences the promoter (Figure 1.1a) (Straussman et al. 2009). Such programmed CGI methylation is important for genomic imprinting, which results in monoallelic expression through the silencing of a parental allele (Kacem and Feil 2009) and gene dosage compensation such as X-chromosome inactivation in females (Reik and Lewis 2005). Recently, Doi et al. has shown that limited gene expression in differing tissue types is caused by differential methylation of CpG island shores (2009), which are located within 2.0 kb of CGIs (Figure 1.1b) (Saxonov et al. 2006). Still, a fraction of CGIs are prone to methylation in some tissues due to aging, in promoters of tumor suppressor genes in cancer cells (Issa et al. 2000), and committed cell lines (Jones et al. 1990). The remaining 40% of CGIs are located intra- and intergenically. Intragenically located CGIs within the coding region of genes are methylated at trinucleotides CpXpG (Lister et al. 2009) and are commonly found in highly expressed, constitutively active genes (Figure 1.1c) (Zhang et al. 2006) while intergenic CGIs may be used for transcription of non-coding RNAs (Illingworth et al. 2008).
Figure 1.1.
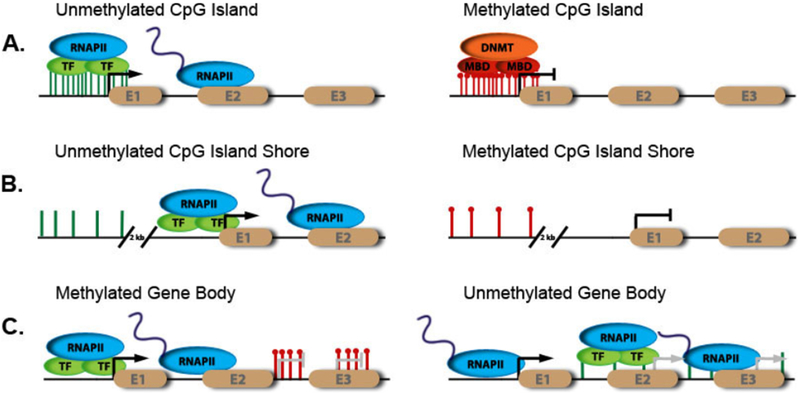
Various sites and effects of DNA methylation throughout the genome
DNA methylation is found at inter- and intragenic regions throughout the genome. DNA methylation dependent transcriptional activity is contingent on CpG dinucleotide genic location and density. Normal methylation events and subsequent effects are shown on the left. (a) CpG islands at promoters are normally unmethylated resulting in gene expression. However, aberrant hypermethylation at the same promoter results in corepressor complex recruitment and subsequent gene repression. (b) Intragenic regions characterized by scattered CpG dinucleotides located 2kb upstream of the promoter called CpG island shores are regulated in the same manner as (a). (c) DNA methylation within the gene body prevents initiation of transcription from spurious sites in the gene. If unmethylated, these sites become transcriptional start sites resulting in an incorrect product. (Portela and Estellar 2010)
1.1.2. Transcriptional Regulation
More often than not, DNA methylation is usually associated with gene silencing due to 1) the occlusion of DNA binding proteins that act as or recruit transcriptional activators or 2) the recruitment of methyl-binding proteins (MBPs), which recruit transcriptional corepressor complexes (Figure 1.1a). Transcriptional activators and repressors recruit histone modifying and chromatin remodeling complexes that can remodel chromatin, which ultimately changes the transcriptional activity of a gene. Modifications made by such complexes and subsequent effects on transcription will be discussed later.
Even previous to DNA methylation, DNMTs can be recruited to DNA via DNA binding transcription factors, which results in specific promoter DNA methylation and regulatory gene repression. For example, studies (Di Croce et al. 2002) showed that DNMTs interact with the oncogenic transcription factor formed by the fusion of promyelocytic leukemia protein and retinoic acid receptor (PML-RAR), found in acute promyelocytic leukemia. DNMT recruitment to the RARβ2 gene promoter by PML-RAR results in promoter hypermethylation and subsequent gene silencing (Di Croce et al. 2002). A similar mechanism has been described for Myc, a DNA binding transcription factor. Myc interacts with DNMT3a and is recruited to the p21 gene promoter resulting in subsequent DNA methylation and p21 gene repression (Brenner et al. 2005). In addition, p53 also interacts with DNMT3a and represses p53’s transactivator function at the p21 gene promoter but in a DNA methylation independent manner (Wang et al. 2005). Both mechanisms elucidate cancer promoting pathways that intersect with DNA methylation and cause repression of expression of p21, a cyclin dependent kinase inhibitor. Moreover, one study (Hervouet et al. 2009) showed that DNMT3a/b interacts with 79 different DNA binding transcription factors. Some interactions were exclusive to each DNMT while some were shared between both (Hervouet et al. 2009). The diversity of interactions further illustrates the importance of DNA methylation on gene expression regulation through DNMT recruitment via DNA binding transcription factors.
Once DNA is methylated, DNA methyl-binding proteins (MBP) can bind to DNA and recruit transcriptional corepressors such as histone deacetylase (HDAC) complexes, polycomb proteins, and chromatin remodeling complexes. One family consists of MBPs, which possess a conserved methyl-CpG-binding-domain (MBD) and includes MBD1, MBD2, MBD3, MBD4, and MeCP2. MeCP2 is the founding member of the MBD family and contains a MBD in addition to an adjacent transcriptional repressor domain (TRD) (Klose and Bird 2006). The TRD interacts with Sin3 corepressor complex containing HDAC1 and 2 (Nan et al. 1998). MBD1 also contains three zinc-binding domains (CxxC), which has been shown to be responsible for its ability to bind unmethylated CpG sites (Jorgensen et al. 2004). MBD1 and 2 both contain a TRD that recruits different transcriptional corepressor complexes containing HDACs. MBD3 contains a MBD but does not bind methylated DNA due to two amino acid substitutions (Hendrich and Tweedie 2004) but is associated with the nucleosome remodeling and histone deacetylase (NuRD) corepressor complex, which contains HDACs necessary for transcriptional silencing. MBD4 is a thymidine glycosylase DNA repair enzyme that excises mismatched thymines that have resulted from 5-methylcytosine deamination in the context of CpG dinucleotides (Hendrich et al. 1999).
The second family of MBPs includes Kaiso, zinc finger and BTB (for BR-C, ttk, and bab) domain containing (ZBTB) 4 and ZBTB38 (Zollman et al. 1994). These are atypical MBPs, because they depend on a zinc-finger domain to recognize methylated DNA and a POZ (for Pox virus and Zinc finger) (Bardwell et al. 1994)/BTB domain to repress transcription through its interaction with nuclear receptor co-repressor-1 (N-CoR) (Prokhortchouk and Defossez 2008). Another study (Iioka et al. 2009) showed that Kaiso can regulate transcription factor activity by modulating the interaction between β-catenin and HDAC1 activity. The third family of MBPs includes ubiquitin-like plant homeodomain and RING finger (UHRF)-domain containing protein 1 and 2. Both contain SET and RING associated (SRA) domains, which preferentially bind to DNMT1’s substrate, hemi-methylated DNA (Bostick et al. 2007). Furthermore, UHRF1 has been shown to colocalize with DMNT1, which suggests that this family of MBPs may help target DMNT1 to DNA (Bostick et al. 2007).
DNA methylation is usually associated with transcriptional silencing, and one of the most well known cases where differential DNA methylation induces and suppresses expression is genomic imprinting at the H19/IGF2 locus. Genomic imprinting is a form of gene regulation in which an allele is expressed from one of the two parental homologous genes. H19 and IGF2 are reciprocally imprinted so that H19 is expressed from the maternally inherited allele and IGF2 from the paternally inherited allele (Bell and Felsenfeld 2000). Transcriptional regulation of these genes is dependent on a differentially methylated DNA domain (DMD) or imprinting control region (ICR) located upstream of H19 and downstream of IGF2. The DMD/ICR is methylated on the paternal allele but not the maternal allele (Bell and Felsenfeld 2000; Hark et al. 2000; Szabo et al. 2000; Kanduri et al. 2000). CCCTC-binding factor (CTCF) binds to the unmethylated ICR of the maternal allele, which blocks an enhancer region located downstream of H19 from activating transcription of IGF2 (Hark et al. 2000). CTCF binding also protects against de novo methylation and subsequent repression at the H19 locus on the maternal allele (Rand et al. 2004). This is one of the most basic examples of how differentially methylated regions can determine levels of gene expression. Mutations or deletions in the H19 promoter, ICR, or enhancer can lead to growth defects such as Beckwith-Wiedemann Syndrome or Silver-Russell dwarfism (Delaval et al. 2006).
With the advent of microarrays and high-throughput technologies, an explosion of gene expression profile comparisons in normal and diseased cells has occurred. Many studies have pursued genes of interest by comparing the DNA methylation status of a gene’s 5’ promoter region (Weber et al. 2005; Hatada et al. 2006), and presently, more comprehensive results are available as more direct solutions to discovering gene expression controlled by DNA methylation are established. Using Arabidopsis thaliana as a model system, Zhang et al. analyzed and compared whole genome methylome tiling arrays gathered from immunoprecipitating 5-mC or chromatin crosslinked MBPs in normal and mutant cells (Zhang et al. 2006). Another study (Javierre et al. 2010) compared the DNA methylome of monozygotic twins who were differently affected by the disease, systemic lupus erythematosus (SLE) (Javierre et al. 2010). In comparison to the healthy twin, the twin affected by SLE had a decrease in promoter DNA methylation for many genes involved in immune system function including IFNGR2, MMP14, LCN2, CSF3R PECAM1, CD9, AIM2, and PDX1. These genes had also previously been shown to participate in the development of SLE (Javierre et al. 2010).
1.1.3. 5-Hydroxymethylcytosine
In the previous sections, 5-methylcytosine (5-mC) was discussed extensively. 5-mC can be converted to 5-hydroxymethylcytosine (5-hmC) by an oxidation reaction carried out the ten-eleven-translocation (TET) family of proteins (Tahiliani et al. 2009). 5-hmC was first discovered in bacteriophage DNA in 1952 (Wyatt and Cohen 1952) and has since been found to be enriched in mouse brain (Kriaucionis and Heintz 2009), embryonic stem cells (Tahiliani et al. 2009), and human tissues (Li and Lui 2011).
Levels of 5-hmC are dynamically regulated by TET1-3 in stem cells and seem to be higher in pluripotent cells. Knockdown of TET1 and TET2 causes a decrease in 5-hmC levels and an increase in 5-mC at stem cell related gene promoters (Ficz et al. 2011). These genes are subsequently silenced. TET3 is highly expressed in zygotes and oocytes (Wossidlo et al. 2011) and a recent study (Iqbal et al. 2011) has shown that after fertilization, 5-mC is converted to 5-hmC in the male but not the female pronucleus. This data (Iqbal et al. 2011) suggests an alternative to the global demethylation theory during cellular dedifferentiation where genome-wide 5-mC may be converted to 5-hmC by TET3 and differentiation is promoted by a decrease in TET3 and an increase in TET1 and 2 (Koh et al. 2011; Walter 2011). The mechanisms behind 5-hmC’s role in cellular differentiation (Ito et al. 2010), carcinogenesis, (Li and Liu 2011) and association with actively transcribed genes is a mystery (Ficz et al. 2011). One clue provided is that 5-hmC prevents the binding of MBDs (Valinluck et al. 2004) and DNMTs (Valinluck and Sowers 2007).
1.2. Histone Modifications
1.2.1. Types of Modifications
As mentioned in the previous section, methylated DNA can recruit different transcriptional activator and repressor complexes. In most cases, these complexes contain histone modifying and chromatin remodeling enzymes that regulate chromatin structure, which ultimately changes the transcriptional activity of a gene. Such complexes are not just recruited by DNA methylation but also by various post-translational modifications (PTMs) of the proteins that make up chromatin. In this section, the effects of histone modifications and chromatin remodeling on gene expression will be discussed.
Chromatin is the organization of the eukaryotic genome into a condensed form due the function of many proteins and RNAs. The fundamental unit of the highly ordered chromatic fiber is the nucleosome, which consists of 146 base pairs of DNA wrapped around an octamer of core histones that contains two of each histone H2A, H2B, H3, and H4. A linker histone, H1, binds to DNA as it enters and exits its 1.65 turns around the nucleosome (Luger et al. 1997). Naturally, the condensed structure forms a barrier to cell processes that require accessibility to DNA such as DNA replication, damage repair, and transcription (Workman et al. 1998). Covalent HPTMs such as acetylation, methylation, phosphorylation, ubiquitination, SUMOylation, ADP-ribosylation, deimination, and the non-covalent proline isomerization (Kouzarides et al. 2007) can affect the condensation of chromatin as to organize the genome into transcriptionally active and inactive regions termed euchromatin and heterochromatin (Heitz 1929) respectively and recruit effector proteins (Jenuwein 2001).
The core histones are highly conserved basic proteins composed of a globular domain and highly flexible N-terminal tails that protrude from the DNA wrapped nucleosome (Luger et al. 1997). All histone N-terminal tails and globular domains are subject to modification and more is known about the smaller covalent modifications methylation, acetylation, and phosphorylation. Lysine residues can be mono-, di-, and trimethylated while arginine residues can only be mono- or symmetrically or asymmetrically dimethylated (Bannister and Kouzarides 2011). The interactions between chromatin associated proteins that bind HPTMs can act synergistically or antagonistically with one another resulting in various gradients of transcriptional activation and repression across the genome. The term “histone code” was coined in order to convey that chromatin modifying proteins ultimately determine phenotype rather than simple possession of a certain genetic code (Strahl and Allis 2000; Jenuwein and Allis 2001). HPTMs specific roles in gene expression and cellular activities are shown in Table 1.1 (adapted from Kouzarides and Berger 2007; Wang et al. 2008).
Table 1.1.
Transcriptional and cellular role of histone modifications
| Modification | Histone Residues Modified | Role in Cell Activity and Transcription | Histone Modification Readers |
|---|---|---|---|
| Acetylated Lysine (Kac) | H3 (K4,9,14,18,23,27,36,56) H4 (K5,8,12,16,19) H2A (K5,9) H2B (K5,6,7,12,16,17,20,120) |
Activation DNA Damage Repair |
Bromodomain Tandem PHD |
| Phosphorylated Serine/Threonine (S/Tph) | H3 (S10,28 and T3,11,45) H4 (S1,47) H2A (S1) |
Apoptosis Activation Mitosis (Baker et al. 2010) |
14-3-3 Domain |
| Methylated Lysine (Kme) | H3 (K4,23,36,79) H3 (K9, 27) and H4 (K20) |
Activation Repression |
MBT PHD Tudor Chromodomain WD40 |
| Methylated Arginine (Rme) | H3 (R2,17,26) and H4 (R3) H3 (R8) |
Activation Repression |
Tudor (Yang et al. 2010; Chen et al. 2011) ADD (Zhao et al. 2009) |
| Ubiquitylated Lysine (Kub) | H2A (K119) H2B (K120) |
Repression Activation (Zhu et al. 2005) |
Cps35 (Lee et al. 2007; Zheng et al. 2010) |
| Sumoylated Lysine (Ksu) | H4 (?) | Repression (Shiio and Eisenman 2003) |
Euchromatin is characterized by high levels of acetylation and high levels of H3K4me1/2/3, H3K36me3 and H3K79me1/2/3. On the other hand, heterochromatin is characterized by low levels of acetylation and high levels of H3K9me2/3, H3K27me2/3 and H4K20me3 (Table 1.1) (Li et al. 2007). More recently, a group (Wang et al. 2008) performed chromatin immunoprecipitation sequencing (ChIP-seq) on 39 different core histone acetylations and methylations at 3,286 promoter regions. As shown in previous studies (Turner 1992), acetylated histones consistently correlate with increased gene transcription. However, certain modifications localized to specific gene regions rather than just at transcriptional start sites (TSS). H2AK9ac, H2BK5ac, H3K9ac, H3K18ac, H3K27ac, H3K36ac and H4K91ac were mainly located in the region surrounding the TSS, whereas H2BK12ac, H2BK20ac, H2BK120ac, H3K4ac, H4K5ac, H4K8ac, H4K12ac and H4K16ac were prominent in the promoter and transcribed regions of active genes (Wang et al. 2008).
Another group (Karlic et al. 2010) analyzed ChIP-seq data produced by the Zhao lab in order to create a model that could predict levels of gene expression based on HPTM levels present at promoters. They found that actively transcribed genes are characterized by high levels of H3K4me3, H3K27ac, H2BK5ac and H4K20me1 in the promoter and H3K79me1 and H4K20me1 along the gene body. Moreover, they found high levels of H4K20me1 and H3K27ac at promoters that contained high CpG content and H3K4me3 and H3K79me1 at promoters with low CpG content (Karlic et al. 2010). Although there is no model that explains the HPTM difference at the two types of promoters, one can guess that the difference is caused by different regulatory mechanisms and possibly, changes in DNA methylation. In agreement with this theory, a recent paper (Ernst and Kellis 2010) used previous ChIP-seq data for HPTMs, CTCF, RNA Polymerase II (RNAPII), and the histone variant, H2A.Z, to describe 51 distinct chromatin states. Each state is described by the enrichment of different HPTMs and chromatin associated proteins across the genome. Moreover, biological states of cells (cell cycle, developmental, T-cell activation, etc.) were predicted using the 51 epigenetic states (Ernst and Kellis 2010). Another interesting study (Mikkelsen et al. 2007) showed that embryonic stem cells contained a bivalent pattern of HPTMs at promoters of genes that regulate development. Surprisingly, they found H3K4me3, an activation mark, and H3K27me3, a repressive mark, co-localizing at these promoters in stem cells (Mikkelsen et al. 2007). These bivalent domains can resolve into four different chromatin states: 1) marked with both H3K4me3 and H3K27me3; 2) marked with neither H3K4me3 nor H3K27me3; 3) marked with H3K4me3 alone; 4) marked with H3K27me3 alone. Maintenance or loss of both marks results in a poised transcriptional state while preservation of H3K27me3 alone or H3K4me3 alone results in inactive and active transcription respectively (Cui et al. 2009). This data (Mikkelsen et al. 2007) suggests that HPTM bivalency at promoters allows for plasticity during cellular differentiation and development (Bernstein et al. 2006).
1.2.2. Transcriptional Regulation
As presented in the previous section, various HPTMs correlate with gene expression and repression (Figure 1.2). Until recently, elucidating the mechanisms by which HPTMs interact with one another to control transcriptional activity has been complicated due to the layered complexity of combinatorial HPTMs and HPTM crosstalk. However, analysis of recently acquired ChIP-seq data and associated gene expression profiles has speedily facilitated decipherment of the histone code and its effect on transcriptional activity (Figure 1.2 and 1.3) (Barski et al. 2007; Wang et al. 2008; Heintzman et al. 2007; Mikkelsen et al. 2007). Three broad effects on transcription can be attributed to HPTMs: 1) HPTMs can prevent certain chromatin binding proteins from binding. For example, H3S10ph prevents heterochromatin protein 1 (HP1) from binding H3K9me3 (Kouzarides and Berger 2007); 2) HPTMs can recruit certain chromatin binding proteins, which can enhance or inhibit gene activation. For example, H3K9me3, a marker for mammalian heterochromatin, is bound by the chromodomain of HP1 resulting in chromatin condensation and occlusion of DNA and nucleosomal binding sites utilized by coactivators, transcription factors, and RNAPII (Kouzarides and Berger 2007); 3) HPTMs can act in cis by affecting transcription through alteration of chromatin structure. For example, H4K16ac alone prevents the formation of a higher ordered compacted chromatin structure resulting in chromatin decondensation and increased transcriptional activity (Shogren-Knaak et al. 2006).
Figure 1.2.
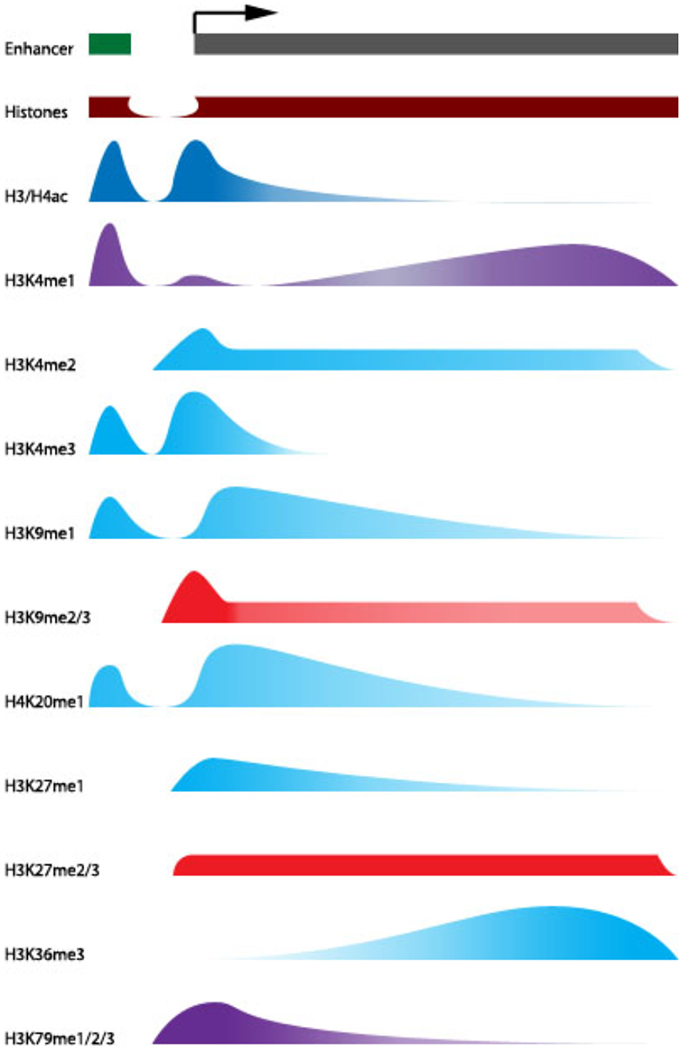
Localization of histone modifications across genes as it relates to transcriptional regulation
Patterns of histone modification enrichment are shown across an arbitrary enhancer and gene. The enhancer is shown as the smaller region succeeded by a gap denoting a nucleosome-free region and transcriptional start site as shown by the arrow. Data used to compile the profiles are from GWAS on histone modifications. The correlative effects of the modifications on gene expression are indicated by the labels: (+) expression, (−) repression, and (+/−) studies show enrichment in both expression and repressed genes. (Wang et al. 2008; Barski et al. 2007; Li et al. 2007)
Figure 1.3.
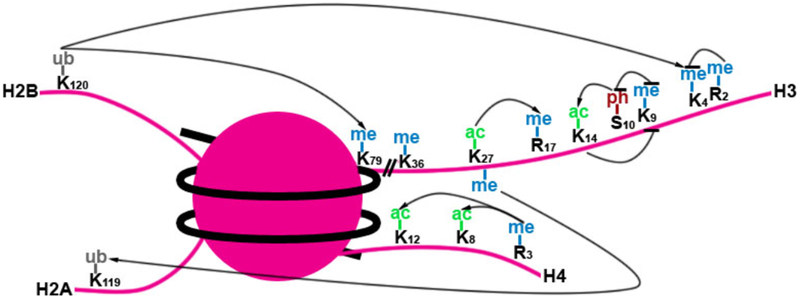
Histone modification crosstalk
Various post-translational histone modifications affect the binding of certain domains and catalysis of other HPTMs. Arrows indicate a positive effect and bars indicate inhibitory effects on other HPTMs. (Bannister and Kouzarides 2011)
The following sections will focus on the effects of histone (de)acetylation and methylation on gene expression. It should be noted that much of the mechanistic research done on transcriptional regulation and HPTMs is pioneered through the use of yeast model systems because genetic manipulation and high-yield results have been easier to obtain as compared to humans. Importantly, many yeast proteins have correlative homologs that serve in the same manner as they do in mammals. However, there are some differences between the two eukaryotic organisms. For example, yeast do not possess the repressive mark H3K27me and in some cases, homologous complexes may contain different chromatin targeting proteins.
1.2.3. Histone Acetylation and Deacetylation
Histone acetylation at conserved lysine residues is the most intensely studied HPTM and was the first modification linked to transcriptional activity (Hebbes, Thorne, and Crane-Robinson 1988). It was not until 1996, that a direct molecular link was made between acetylation and transcription. The first nuclear histone acetyltransferase (HAT) discovered, p55, was orthologous to a previously isolated transcriptional coactivator in Saccharomyces cerevisiae, Gcn5 (Brownell et al. 1996). HATs catalyze the addition of acetyl-coA to the ε-amino group on lysine side chains resulting in charge neutralization and affinity reduction between negatively charged DNA and basic histones. Acetylation ultimately creates an “open” chromatin structure (Shogren-Knaack et al. 2006) poised for active transcription through exposure of DNA-binding sites (Vettese-Dadey et al. 1996). There are two types of HATs: type-A (nuclear) and type-B (cytoplasmic). This discussion will only focus on type-A as they catalyze reactions related to active transcription (Bannister and Kouzarides 2011).
Type-A HATs are further divided into five families including the GCN5-related N-acetyltransferases (GNATs); the MOZ, Ybf2/Sas3, Sas2 and Tip60 (MYST)-related HATs; p300/CREB-binding protein (CBP) HATs; the general transcription factor HATs including the TFIID subunit TBP-associated factor-1 (TAF1); and the nuclear hormone-related HATs SRC1 and ACTR (SRC3) (Nagy and Tora 2007). They are often part of larger protein complexes and are recruited by DNA binding activators. For instance, in yeast, Gcn5 is part of the Spt-Ada-Gcn5-Acetyltransferase (SAGA) and Adaptor (ADA) complexes (Grant et al. 1997). In SAGA, Gcn5 is associated with three protein families known to be involved in gene expression: Spt, Ada, and a subset of TAFs (Grant et al. 1998). SAGA is recruited to active promoters via the SAGA subunit, Tra1’s interaction with acidic activator domains of transcriptional activators and subsequent recruitment of the TATA-binding protein (TBP) by the subunit, Spt3 (Grant et al. 1998; Larschan and Winston 2001; Brown et al. 2001; Reeves and Hahn 2005). Similar complex subunits have been found to be associated with Gcn5 human homologs, p300/CBP associated factor (P/CAF) and hGcn5 (Ogryzko et al. 1998; Martinez et al. 1998; Nagy and Tora 2007). Human Gcn5 is found in the SAGA complex homolog Spt3-Taf9-Gcn5-Acetyltransferase (STAGA) complex and is recruited to promoters by the Tra1 human homolog, Transactivation/transformation domain associated protein (TRRAP) via its interaction with the transactivation domain of c-Myc (McMahon et al. 2000; Lui et al. 2003).
Furthermore, Gcn5, P/CAF, and p300 contain a bromodomain that bind acetyl-lysine. Taf1 contains two bromodomains (Jacobson et al. 2000). The exact function of bromodomains has yet to be elucidated. However, it is speculated that once HAT complexes are targeted to the promoter and perform acetylation, subsequent coactivators can stably bind to acetylated histone rich promoter regions via bromodomains, which would facilitate an acetylation cascade. Consistent with this hypothesis, SAGA requires the functional bromodomains of Gcn5 and the remodeling complex proteins Swi2/Snf2 for stable promoter occupancy, efficient HAT activity, and increase in gene expression resulting from an “open” chromatin conformation, and subsequent gene activation (Hassan et al. 2002). It should also be noted that HATs also acetylate non-histone proteins including the tumor suppressor p53 and various transcription factors (Glozak et al. 2005), which ultimately regulates gene expression.
Like many bromodomains, DPF3b, a novel acetyl-lysine reader and BAF remodeling complex associated subunit also binds ambiguously to acetylated H3 and H4 (Lange et al. 2008) via its tandem plant homeodomain (PHD) fingers (Zeng et al. 2010). One PHD finger only has affinity for acetylated H3K14, which increases full-length DPF3b’s affinity for acetylated H3 and H4 (Zeng et al. 2010). Loss of DPF3b affects both skeletal and heart muscle development through transcriptional deregulation of other transcriptional factors (Lange et al. 2008).
On the other hand, histone deacetylases (HDACs) reverse the reaction carried out by HATs by removing acetyl marks on lysine to restore the positive charge. They fall into four classes: Class I (HDAC1, 2, 3, and 8), II (HDAC4, 5, 6, 7, 9, 10), III or Sir2-related enzymes, and class IV, which contains one member, HDAC11. Class III HDACs require the cofactor NAD+ for its activity (Yang and Set 2007). HDAC1 and 2 are found in the mammalian complexes Sin3A/B, NuRD, and corepressor for RE1 silencing transcription factor/neural-restrictive silencing factor (CoREST) while HDAC3 is found in nuclear receptor corepressor/silencing mediator for retinoid and thyroid hormone receptors (N-CoR/SMRT) (Yang and Seto 2008). Some of these corepressor complexes contain methyl-lysine binders that help target complexes to specific site on chromatin. For instance, a subunit of the Sin3a complex, ING2, contains a PHD finger domain that binds H3K4me3 (Champagne and Kutateladze 2009) in response to DNA damage. Once Sin3a is recruited, HDAC1 activity is stimulated, which stabilizes nucleosomes resulting in the repression of cell proliferation genes as a response to genotoxic events (Shi et al. 2006).
1.2.4. Histone Methylation
Histone methylation is performed on the residues lysine and arginine by histone methyltransferase (HMT) enzymes. Lysines can be mono-, di-, and trimethylated while arginines can be mono- and symmetrically or asymmetrically dimethylated. There are over twenty sites of methylation that have been identified on the core histones. Given all the possible combinations of histone methylation, it is one of the most complex HPTMs to study in a static model. The modifications most relevant to transcriptional regulation have been listed in Table 1.1 and a few of the most studied histone methylations will be discussed in this section. Figure 1.2 summarizes the transcriptional effects and genomic enrichment of the HPTMs discussed below.
1.2.4.1. H3K4
H3K4 methylation is usually enriched at the enhancers and promoters of actively transcribed genes (Wang et al. 2008; Santos-Rosa et al. 2002). H3K4me1 is highly enriched at enhancers (Wang et al. 2005). H3K4me2 is commonly found in the body of active genes while H3K4me3 is largely observed at the 5’ ORF of genes (Pokholok et al. 2005). Methylation of H3K4 results from the recruitment of various H3K4 HMT enzymes by transcriptional machinery, specifically RNAPII. Once RNAPII is poised for active transcription through phosphorylation of serine-5 of the carboxy-terminal domain (CTD) by TFIIH (Phatnani and Greenleaf 2006), the Set1 containing H3K4 HMT complex, COMPASS, is recruited by the PAF complex (Ng et al. 2003; Wood et al. 2003). RNAPII is released into an early elongating complex where H2BK120 (K123 in yeast) is ubiquitylated, which is required for further Set1 activity. Sometime during elongation, RNAPII is phosphorylated at serine-2 resulting in the release of Set1 (reviewed in Martin and Zhang 2005).
Furthermore PAF also interacts with chromodomain containing protein Chd1 (Simic et al. 2003). Proteins possessing methyl-binding domains, called chromodomains, are recruited to the H3K4me3 enriched promoter. SAGA also interacts with Chd1, which has two chromodomains, one which helps recruit SAGA to sites of H3K4me2/3 (Pray-Grant et al. 2005). As discussed earlier, SAGA recruitment results in an acetylation cascade that further promotes transcriptional activation. In humans, the HMT containing mixed-lineage-leukemia (MLL) complex is recruited by the H3K4me2 binding domain, WDR5. WDR5 interacts preferentially with H3K4me2 through its WD40-repeat domain (Wysocka et al. 2005). MLL can then convert H3K4me2 to H3K4me3.
1.2.4.2. H3K36
Unlike the 5’ localization of H3K4 methylation, H3K36 methylation is highly enriched in the coding region and 3’ ORF of genes. As mentioned in the previous section, once the CTD of RNAPII is phosphorylated at Serine-2 by Ctk1 and Bur1 kinases (Keogh et al. 2003; Qui et al. 2009), Set1 is released and chromatin is primed for transcriptional elongation through recruitment of Set2 (Xiao et al. 2003; Krogan et al. 2003). Set2 HMT catalyzes H3K36 methylation and specifically binds to phosphorylated Serine-2 of RNAPII’s CTD (Hampsey and Reinberg 2003). This form of RNAPII is found in the transcribed regions of genes and the 3’ end of genes, which correlates with H3K36me2/3 localization (Xiao et al. 2003; Krogan et al. 2003; Hampsey and Reinberg 2003; Li et al. 2003). The passage of RNAPII during transcriptional elongation results in histone displacement and positioning behind RNAPII. These histones are hyperacetylated and subsequently methylated by Set2 (Hampsey and Reinberg 2003; Carrozza et al. 2005; Joshi and Struhl 2005; Keogh et al. 2005).
H3K36me2 is recognized by the chromodomain of Eaf3 and PHD finger of Rco1, which are subunits of the Rpd3S HDAC complex (Joshi and Struhl 2005; Govind et al. 2010). During transcriptional elongation, Rpd3S is recruited via the serine-2/serine-5-diphosphorylated CTD repeats followed by H3K36me2 binding by Eaf3 and Rco1 (Keogh et al. 2005, Govind et al. 2010). Once Eaf3 and Rci1 are recruited by H3K36me2, Rpd3 is transferred from the phosphorylated CTD to H3 where its HDAC activity creates a hypoacetylated environment within gene bodies and at the 3’ end. (Li et al. 2007; Govind et al. 2010). Deletion of Rco1 or Eaf3 results in hyperacetylation of ORFs and the production of aberrant transcripts that are presumably initiated from cryptic promoters that are usually silenced by the Set2-Rpd3 pathway after RNAPII progression (Carrozza et al. 2005; Joshi and Struhl 2005; Keogh et al. 2005).
1.3.4.3. H3K79
Unlike the previously discussed HPTMs, H3K79 methylation occurs in the globular domain of H3 and within the core of the nucleosome. It is found within the coding regions of genes and is usually associated with active chromatin. H3K79 methylation is catalyzed by the HMT, Dot1. Dot1 is the first lysine HMT that has been identified that’s lacks an identifiable SET domain (Feng et al. 2002). Dot1 is required to prevent the spread of HDACs into active chromosomal regions (van Leeuwen et al. 2002). There is no protein that links H3K79 methylation to transcriptional regulation. However, mammalian hDot1L has been implicated in mediating the leukemogenic fusion protein MLL (for mixed lineage leukemia)-AF10. It was found that hDot1L is recruited to MLL–AF10 target genes, such as HOXA9, through an interaction between hDot1L and AF10 (Okada et al. 2005). Upregulation of HOXA9 expression results in defective hematopoiesis and leukemogenic transformation making regulation of H3K79 methylation a possible therapeutic target. Also, mammalian protein 53BP1 interacts with H3K79me3 through a tudor domain at sites of DNA damage (Huyen et al. 2004).
1.2.4.4. H3K27
In mammals, H3K27 methylation is a repressive mark catalyzed by the Polycomb Repressor Complex 2 (PRC2), which contains the SET-domain containing lysine HMT, Enhancer of Zester 2 (EZH2). H3K27me3 serves as a repressive mark at homeotic genes, the inactive X-chromosome, and imprinted genes while H3K27me1 is enriched at pericentric heterochromatin (Martin and Zhang 2005). PRC2 is made up of four core components: EZH2, embryonic ectoderm development (EED), suppressor of zeste 12 homolog (SUZ12), and histone-binding protein retinoblastoma-binding protein p48/46 (RbAp48/46). Both EED and SUZ12 are necessary for EZH2 HMT activity (Simon and Kingston 2009). EED contains repeats of WD40 domains that bind H3K27me3 and promote PRC2 propagation (Margueron et al. 2009) and SUZ12 contains C2-H2 zinc finger and VEFS domain. RbAp48/46 contains six WD40 domains and is a core histone binding subunit.
PCR2 also interacts with AEBP2, PCLs and JARID2. AEBP2 contains three zinc-fingers that may play a role in DNA binding (Kim, Kang, and Kim 2009). PCL1, PCL2 and PCL3 (also known as PHF1, MTF2 and PHF19, respectively) contain a tudor domain and two PHD finger proteins, a PCL extended domain and a carboxy-terminal domain tail (Wang, Robertson, and Zhu 2004). PCL proteins interact with PRC2 through EZH2, and to some extent through SUZ12 and the histone chaperones RbAp46/48 (Nekrasov et al. 2007). JARID2 is the founding member of the Jumonji family of proteins that catalyses the demethylation of histone proteins. However, it lacks demethylase activity. JARID2 contains JmjC and JmjN domains and two potential DNA binding domains, ARID and a zinc finger (Margueron and Reinberg 2011). The core components of PRC2 and its associated proteins discussed above are all necessary for EZH2 optimal function.
The targeting of PRC2 in D. melanogaster is a well understood mechanism compared to humans. In D. melanogaster, transcription factors, such as Pho and PhoL, bind to the Polycomb responsive element and recruit EZ of PRC2. Only now is the mammalian mechanism coming to light with the recent discovery of long non-coding RNA (lncRNA) dependent PRC2 recruitment. The lncRNA, HOTAIR, is transcribed from the HOXC locus, binds PRC2, and targets the complex to the HOXD locus where several genes are repressed (Rinn et al., 2007). Also, the lncRNA Xist and a short internal transcript RepA have been to shown target PRC2 to the inactivated female X-chromosome, which subsequently is repressed and enriched with H3K27me3. In contrast the lncRNA and antagonist to Xist, Tsix, also interacts with PRC2 suggesting an inhibitory mechanism to X-chromosome inactivation (Zhao et al. 2008).
1.2.4.5. H3K9
H3K9 methylation is one of the most intensely studied histone modifications to date. H3K9me1 is catalyzed by methyltransferases HMT1C/G9a or demethylases KDM3A/JMJD1A and KDM4D/JMJD2D (Shi and Whetstine 2007). The mark is enriched at the 5’ UTR and found minimally in non-genic regions (Barski et al. 2007; Rosenfeld et al. 2009). Although no function has been ascribed to H3K9me1, its proposed mechanism of action may be to act as an intermediary between gene activation and repression through rapid methylation or demethylation (Black and Whetstine 2011). Most studies have focused on H3K9me2/3 as a heterochromatin mark catalyzed by the lysine HMT SUV39H1/2 and recognized by the chromodomain of heterochromatin protein-1 (HP1), which dictates the compaction of heterochromatin. H3K9me2/3 is enriched in pericentromeric, subtelomeric, and gene desert regions. Gene deserts are megabase sized regions devoid of coding genes, and unlike H3K9me3, H3K9me2 is rarely found in at individual active or silenced genes (Rosenfeld et al 2009). In support of H3K9me2’s function as a repressive mark, it has been shown to associate with Lamin B1, a protein localized to the nuclear periphery and part of the nuclear lamina, which is commonly associated with inactive genes. Lamin B1 associated regions are also devoid of the activating mark, H3K4me3, and RNAPII further suggesting H3K9me2 is most likely a repressive mark that facilitates separation of active and inactive genes through chromosomal localization within the nuclear architecture (Guelen et al. 2008).
H3K9me3 is commonly found at heterochromatin and repressed promoters, and unlike H3K9me2, H3K9me3 is also localized to centromeres, subtelomeric regions, and in some cases, the coding region of genes (Vakoc et al. 2007; Mikkelsen et al. 2007). H3K9me3 is usually associated with H3K20me3 at heterochromatic locations such as pericentromeric chromatin, but this bivalent mark is absent at subtelomeric regions and gene deserts suggesting different silencing mechanisms at these different heterochromatic regions (Rosenfeld et al. 2009). In addition to its heterochromatin formation function, H3K9me2/3 is implicated in the silencing of euchromatic genes. RB and KAP1 corepressor complexes recruit lysine HMTs SUV39H1 and ESET/SETDB1 respectively to promoters of active genes. HP1 is recruited to sites of H3K9 methylation but is restricted to the promoter region of genes and does not spread (Kouzarides and Berger 2007). The role of H3K9me3 in the coding region of genes has not been elucidated, but enrichment of H3K9me3 at the 3’ ORF increases and co-localizes with the elongating form of RNAPII during active transcription. Moreover, despite the accepted dogma that HP1 is thought to always be repressive, a γ-isoform of HP1 has been found to also be enriched in the coding regions of active genes (Vakoc et al. 2005). During transcriptional activation, promoter repression by HP1β is replaced by HP1α, which seems to facilitate RNAPII processivity through the coding region of the gene in addition to an increase in H3K9me3 (Matteescu et al. 2008).
1.2.4.6. H3K20
In addition to H3K9me2/3, H4K20me3 is also indicative of silenced chromatin. H4K20 methylation is catalyzed by two SET-domain containing lysine HMTs, SUV4-20H1 and SUV4-20H2. Interestingly, both of these HMTs have been shown to interact with the repressive HP1 isoforms, α and β, indicating a possible upstream function for H3K9 methylation and subsequent H4K20 methylation (Schotta et al. 2004). This idea is further illustrated by the dual enrichment of H3K9me3 and H4K20me3 at constitutively repressed regions such as transposons, satellite and long terminal repeats (LTRs), and pericentromeric chromatin, a region rich with repetitive satellite elements and interspersed with long and short interspersed nuclear elements (LINEs and SINEs). As discussed in the previous section, gene deserts are enriched with H3K9me2/3 but not H4K20me3. Interestingly, neither mark is found at telomeric and subtelomeric regions, which suggests a different mechanism of repression mediates constitutive heterochromatin at telomeres (Rosenfeld et al. 2009).
In contrast to H4K20me3, H4K20me1 is associated with highly expressed genes and is enriched at the 5’ coding region along with H2BK5me1, H3K4me1/2/3, H3K9me1, H3K27me1, and H3K79me1/2/3 (Wang et al. 2008). As previously discussed, H3K36me3 is located at the 3’ end of the coding region and marks transcriptionally active genes. Studies have shown that H4K20me1, H3K36me3, and H3K79me1/2/3 facilitate transcriptional elongation as all three marks fluctuate in a similar temporal manner during gene activation and subsequent transcription (Vakoc et al. 2006). H4K20me2 also seems to be required for checkpoint function and cell survival after DNA damage through the recruitment of Tudor-domain containing protein Crb2 (Greeson et al. 2008).
1.2.5. Histone Demethylases
Reversal of histone methylation was thought to be impossible due to the stable nature of the modification until the discovery of lysine-specific demethylase 1 (LSD1). LSD1 is a FAD dependent amine oxidase that catalyzes lysine demethylation and releases the product hydrogen peroxide (Shi et al. 2004). Protein arginine deiminase 4 (PADI4) converts methyl-arginine to citrulline rather than an unmodified arginine. PADI4 does not complete full demethylation and therefore requires processing by histone replacement or aminotransferases for complete arginine demethylation (Bannister et al. 2002). Lastly, the JumonjiC-domain containing histone demethylases (JHDMs) are Fe2+ and α-ketoglutarate dependent histone demethylases that release the product formaldehyde (Tsukada et al. 2006). Specifics about individual enzymes, mechanisms, specificity, and transcriptional activity can be found in Table 1.2.
Table 1.2.
Histone demethylases
| Enzymatic Family | Subfamily | Enzymes | Specific residue activity | Transcriptional Activity | References |
|---|---|---|---|---|---|
| PADI | PAD4 | H3R2me1 H3R8me1 H3R17me1 H3R26me1 H4R3me1 |
Derepressors | Bannister et al. 2002; Wang et al. 2004; Cuthbert et al. 2004 | |
| Amine oxidase | LSD1 | H3K4me1/2 H3K9me1/2 |
Repressors: CoREST, NuRD Activator: AR/ERα |
Lee et al. 2005; Shi et al. 2005; Wang et al. 2009; Metzger et al. 2005; Garcia-Bassets et al. 2007 | |
| JmjC | JHDM1 | JHDM1A JHDM1B |
H3K36me1/2 | Tsukada et al. 2006 | |
| JHDM3/JMJD2 | JMJD2/JHDM3A JMJD2B JMJD2C/GASC1 JMJD2D |
H3K9me2/3 H3K36me2/3 |
Whetstine et al. 2006; Klose et al. 2006; Cloos et al. 2006; Fodor et al. 2006; | ||
| JARID | JARID1A JARID1B JARID1C JARID1D |
H3K4me2/3 | Repressor of growth inhibitors | Iwase et al. 2007; Klose et al. 2007; Lee et al. 2007; Yamane et al. 2007 | |
| UTX/UTY | JMJD3 UTX |
H3K27me2/3 | Activator: MLL | Agger et al. 2007; Issaeva et al. 2007 | |
| JHDM2 | JHDM2A JHDM2B JHDM2C |
H3K9me1/2 | Activator: AR | Yamane et al. 2006 |
Acronyms: Peptidyl arginine deiminase (PADI), Lysine specific demethylase (LSD), Jumonji C (JmjC), JmjC-domain-containing histone demethylase (JHDM), Androgen receptor (AR), Estrogen receptor (ER), Corepressor for RE1 silencing transcription factor/neural-restrictive silencing factor (CoREST), Nucleosome remodeling and histone deacetylase (NuRD)
1.2.6. Histone Proteolysis
In addition to demethylation and deacetylation, previous reports of H3 N-terminal tail proteolytic cleavage have also been described as a mechanism that facilitates the removal of HPTMs (Allis et al. 1980). Recently, H3 tail cleavage by Cathepsin L has been linked to transcriptional activation and induction of differentiation in embryonic stem cells. N-terminal tail cleavage is also regulated by the HPTMs present on the tail (Duncan et al. 2008). Studies have shown (Santos-Rosa et al. 2009) that cleavage is inhibited by the activation mark H3K4me3 and facilitated by the repressive mark H3R2me2 suggesting that tail clipping is a rapid way to void promoters of repressive marks and complexes during the regulation of gene expression. Moreover, tail clipping directly precedes histone eviction at promoters, which provides strong evidence that H3 tail cleavage is a gene activating event (Santos-Rosa et al. 2009). A major challenge in the chromatin field remains in understanding how patterns of modifications are generated and interpreted by nuclear machinery.
1.2.7. Histone Crosstalk
Given all the histone modifications discussed in the previous sections, regulation of chromatin structure and transcriptional activity can be tightly controlled through the use of combinatorial modifications. Histone modifications can affect the stimulation or inhibition of multiple cellular processes, which subsequently affects the capacity for the creation or erasure of other HPTMs (Figure 1.3) (adapted from Bannister and Kouzarides 2011). Some modifications can inhibit the targeting of other modifications as seen with H3K27, which can be exclusively methylated or acetylated. Various modifications are also dependent on one another. For example, H2B120 ubiquitylation is necessary for H3K79 methylation in both yeast and humans (Lee et al. 2007, Kim et al. 2009). Modifications can also prevent the binding of certain effector proteins as is the case with the inhibition of HP1’s targeting to H3K9me2/3 by H3S10 phosphorylation (Fischle et al. 2005). Some marks can also facilitate the binding of effector proteins that in turn perform other modifications. As mentioned above, ING2 contains a PHD finger domain that binds H3K4me3 (Champagne and Kutateladze 2009) in response to DNA damage. Once Sin3a is recruited, HDAC1 activity is stimulated to deacetylate histones and reduce transcriptional activity of genes that promote cell growth and division (Shi et al. 2006).
1.3. Epigenetics and Human Disease
The human body is comprised of trillions of cells, each of which concurrently performs a specific function in order to form a functional human being. The function that one cell serves may be drastically different from another, yet each cell contains identical genetic information. Such phenotypic diversity is a result of a cell’s distinctive gene expression profile. Gene expression is directly influenced by various factors including histone modifications, DNA methylation, histone variants, and availability of functional chromatin modifying complexes. Occasionally, DNA sequences targeted for modifications are expanded or contracted, or the enzymes that catalyze the addition or removal of modifications are lost or mutated. Respectively, these events cause a redistribution of DNA methylation and histone modification patterns. Alteration in the localization of these marks at sites such as promoters, repeat elements, and constitutive heterochromatin ultimately result in diseased states due to dysregulated gene expression (Kaufman and Rando 2010).
The idea that influences beyond the genetic code could determine phenotype is not by any means novel. In 1942, C.H. Waddington coined the phrase “epigenetic landscape” to denote changes in phenotype during development despite an identical genotype (Waddington et al. 1957). To date, the epigenetic landscape portrayed by Waddington could be described by two important areas of chromatin research: the elaborate patterns of histone modifications and histone variant substitutions coined, “the histone code” (Jenuwein et al. 2001) and DNA methylation patterns (Bird and Wolffe 1999). Through its direct effects on transcriptional regulation, histone modifications and DNA methylation affect many essential cellular processes such as embryogenesis, genomic imprinting, DNA replication, microRNA expression, and X-chromosomal inactivation.
Evidence that some human diseases are caused by something other than just the genes you possess is seen in cancer (Estellar et al. 2007), autoimmune disorders (Javierre et al. 2010), and health related issues such as type 2 diabetes (Miao et al. 2008), coronary artery disease (Ordovas and Smith 2010), and obesity (Campion et al. 2009), to name a few. The role of epigenetics in the development of disease is further illustrated by the discordance of disease and trait development in monozyotic twins. Based on this study, environmental factors seem to play a significant role in disease susceptibility and dictating an individual’s epigenetic landscape (Fraga et al. 2005). Ultimately, an increase in disease susceptibility can be attributed to environmentally influenced differences in DNA methylation and histone modification patterns that affect levels of gene expression.
With so many new advents in biomedical research, using human epigenetic profiling for understanding disease and even developing medical treatments has never seemed so tangible. Genome-wide association studies (GWAS) and high-throughput sequencing has allowed for high resolution comparison of modifications and gene expression in various organisms. With a future understanding of the basic functional roles these modifications play as transcriptional regulators in the cell, development of targeted treatments resulting in artificial epigenetic landscaping can potentially be established.
1.4. Summary
Despite the rapid progression of discoveries in the epigenetics fields, there still remain many obstacles and questions left unanswered. Several HPTMs have been discovered without finding the enzyme or complex that performs the covalent addition onto histones or its’ removal. Some chromatin modifications are scarce enough that studying them would be impossible without new nanotechnologies such as ChIP-seq, RNA-seq, and MeDIP-seq. However, a problem that many GWAS run into is that many modifications are context dependent. Frequently, experiments performed to locate modifications and their effects on transcription produce results that represent a static state for a specific cell type. Both the cell type and time point at which the data was collected also affects what genes are expressed. Moreover, as in the case of H3K9 methylation, some modifications have the ability to alter a gene’s 3D spatial positioning within the nucleus (Guelen et al. 2008). Therefore, in addition to the direct effects that chromatin modifying complexes and covalent histone modifications have on promoters, another layer of complexity is added to transcriptional regulation by the way of a gene’s spatiotemporal positioning and organization within the nuclear architecture.
In this chapter, several classes of chromatin modifications and their subsequent effects on transcription have been described. There are many other mechanisms of transcriptional regulation that were mentioned but not discussed including arginine methylation, ubiquitylation, deiminination, and sumoylation. Although the enzymes that catalyze many of these and previously discussed reactions have been discovered, the mechanisms by which they control transcription, are established during development, and are stably maintained in somatic cells are still unclear. Albeit, many modifications have been characterized by their individual effects on gene transcription, developing a more complete picture of the complex orchestration between the enzymes that catalyze the reactions of chromatin modifications will lead to a better understanding of transcriptional regulation. Elucidating the code behind the interplay between chromatin modifying complexes and HPTMs provides exciting new prospects for development of medical treatments in the future that will target chromatin modifying enzymes.
References
- Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. (2007). UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature, 449(7163), 731–734. [DOI] [PubMed] [Google Scholar]
- Allis CD, Bowen JK, Abraham GN, Glover CV, & Gorovsky MA (1980). Proteolytic processing of histone H3 in chromatin: A physiologically regulated event in tetrahymena micronuclei. Cell, 20(1), 55–64. [DOI] [PubMed] [Google Scholar]
- Antequera F, & Bird A (1993). Number of CpG islands and genes in human and mouse. Proceedings of the National Academy of Sciences of the United States of America, 90(24), 11995–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SP, Phillips J, Anderson S, Qiu Q, Shabanowitz J, Smith MM, et al. (2010). Histone H3 thr 45 phosphorylation is a replication-associated post-translational modification in S. cerevisiae. Nature Cell Biology, 12(3), 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, & Kouzarides T (2011). Regulation of chromatin by histone modifications. Cell Research, 21(3), 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Schneider R, & Kouzarides T (2002). Histone methylation: Dynamic or static? Cell, 109(7), 801–806. [DOI] [PubMed] [Google Scholar]
- Bardwell VJ, & Treisman R (1994). The POZ domain: A conserved protein-protein interaction motif. Genes & Development, 5(14), 1664–1677. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell, 129(4), 823–837. [DOI] [PubMed] [Google Scholar]
- Bell AC, & Felsenfeld G (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature, 405(6785), 482–485. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell, 125(2), 315–326. [DOI] [PubMed] [Google Scholar]
- Bestor T, Laudano A, Mattaliano R, & Ingram V (1988). Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. the carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. Journal of Molecular Biology, 203(4), 971–983. [DOI] [PubMed] [Google Scholar]
- Bird A (2002). DNA methylation patterns and epigenetic memory. Genes & Development, 16(1), 6–21. [DOI] [PubMed] [Google Scholar]
- Bird AP, & Wolffe AP (1999). Methylation-induced repression--belts, braces, and chromatin. Cell, 99(5), 451–454. [DOI] [PubMed] [Google Scholar]
- Black JC, & Whetstine JR (2011). Chromatin landscape: Methylation beyond transcription. Epigenetics : Official Journal of the DNA Methylation Society, 6(1), 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, & Jacobsen SE (2007). UHRF1 plays a role in maintaining DNA methylation in mammalian cells Science (New York, N.Y.), 317(5845), 1760–1764. [DOI] [PubMed] [Google Scholar]
- Brenner C, Deplus R, Didelot C, Loriot A, Vire E, De Smet C, et al. (2005). Myc represses transcription through recruitment of DNA methyltransferase corepressor. The EMBO Journal, 24(2), 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Howe L, Sousa K, Alley SC, Carrozza MJ, Tan S, et al. (2001). Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit Science (New York, N.Y.), 292(5525), 2333–2337. [DOI] [PubMed] [Google Scholar]
- Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, et al. (1996). Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell, 84(6), 843–851. [DOI] [PubMed] [Google Scholar]
- Campion J, Milagro FI, & Martinez JA (2009). Individuality and epigenetics in obesity. Obesity Reviews : An Official Journal of the International Association for the Study of Obesity, 10(4), 383–392. [DOI] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, et al. (2005). Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell, 123(4), 581–592. [DOI] [PubMed] [Google Scholar]
- Champagne KS, & Kutateladze TG (2009). Structural insight into histone recognition by the ING PHD fingers. Current Drug Targets, 10(5), 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Nott TJ, Jin J, & Pawson T (2011). Deciphering arginine methylation: Tudor tells the tale. Nature ReviewsMolecular Cell Biology, 12(10):629–642 [DOI] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, & Frommer M (1995). CpNpG methylation in mammalian cells. Nature Genetics, 10(1), 20–27. [DOI] [PubMed] [Google Scholar]
- Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. (2006). The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature, 442(7100), 307–311. [DOI] [PubMed] [Google Scholar]
- Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, et al. (2009). Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell, 4(1), 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, et al. (2004). Histone deimination antagonizes arginine methylation. Cell, 118(5), 545–553. [DOI] [PubMed] [Google Scholar]
- Delaval K, Wagschal A, & Feil R (2006). Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. BioEssays : News and Reviews in Molecular, Cellular and Developmental Biology, 28(5), 453–459. [DOI] [PubMed] [Google Scholar]
- Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, et al. (2002). Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor Science (New York, N.Y.), 295(5557), 1079–1082. [DOI] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. (2009). Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature Genetics, 41(12), 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan EM, Muratore-Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF, et al. (2008). Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell, 135(2), 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, et al. (1982). Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Research, 10(8), 2709–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, & Kellis M (2010). Discovery and characterization of chromatin states for systematic annotation of the human genome. Nature Biotechnology, 28(8), 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M (2007). Cancer epigenomics: DNA methylomes and histone-modification maps. Nature Reviews.Genetics, 8(4), 286–298. [DOI] [PubMed] [Google Scholar]
- Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, et al. (2002). Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Current Biology : CB, 12(12), 1052–1058. [DOI] [PubMed] [Google Scholar]
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, et al. (2011). Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature, 473(7347), 398–402. [DOI] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, et al. (2005). Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature, 438(7071), 1116–1122. [DOI] [PubMed] [Google Scholar]
- Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, et al. (2006). Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes & Development, 20(12), 1557–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America, 102(30), 10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, et al. (2007). Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell, 128(3), 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, & Seto E (2005). Acetylation and deacetylation of non-histone proteins. Gene, 363, 15–23. [DOI] [PubMed] [Google Scholar]
- Govind CK, Qiu H, Ginsburg DS, Ruan C, Hofmeyer K, Hu C, et al. (2010). Phosphorylated pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Molecular Cell, 39(2), 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant PA, Duggan L, Cote J, Roberts SM, Brownell JE, Candau R, et al. (1997). Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: Characterization of an ada complex and the SAGA (Spt/Ada) complex. Genes & Development, 11(13), 1640–1650. [DOI] [PubMed] [Google Scholar]
- Grant PA, Schieltz D, Pray-Grant MG, Steger DJ, Reese JC, Yates JR 3rd, et al. (1998). A subset of TAF(II)s are integral components of the SAGA complex required for nucleosome acetylation and transcriptional stimulation. Cell, 94(1), 45–53. [DOI] [PubMed] [Google Scholar]
- Greeson NT, Sengupta R, Arida AR, Jenuwein T, & Sanders SL (2008). Di-methyl H4 lysine 20 targets the checkpoint protein Crb2 to sites of DNA damage. The Journal of Biological Chemistry, 283(48), 33168–33174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, et al. (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature, 453(7197), 948–951. [DOI] [PubMed] [Google Scholar]
- Hampsey M, & Reinberg D (2003). Tails of intrigue: Phosphorylation of RNA polymerase II mediates histone methylation. Cell, 113(4), 429–432. [DOI] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, & Tilghman SM (2000). CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature, 405(6785), 486–489. [DOI] [PubMed] [Google Scholar]
- Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, et al. (2002). Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell, 111(3), 369–379. [DOI] [PubMed] [Google Scholar]
- Hatada I, Fukasawa M, Kimura M, Morita S, Yamada K, Yoshikawa T, et al. (2006). Genome-wide profiling of promoter methylation in human. Oncogene, 25(21), 3059–3064. [DOI] [PubMed] [Google Scholar]
- Hebbes TR, Thorne AW, & Crane-Robinson C (1988). A direct link between core histone acetylation and transcriptionally active chromatin. The EMBO Journal, 7(5), 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz E (1929). Heterochromatin, Chromocentren, Chromomenen. Berichte der Deutschen Botanischen Gesellschaft, 47, 274–284. [Google Scholar]
- Hendrich B, Hardeland U, Ng HH, Jiricny J, & Bird A (1999). The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature, 401(6750), 301–304. [DOI] [PubMed] [Google Scholar]
- Hendrich B, & Tweedie S (2003). The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends in Genetics : TIG, 19(5), 269–277. [DOI] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, Ditullio RA,Jr, Gorgoulis, V. G., Zacharatos, P., Petty, T. J., et al. (2004). Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature, 432(7015), 406–411. [DOI] [PubMed] [Google Scholar]
- Iioka H, Doerner SK, & Tamai K (2009). Kaiso is a bimodal modulator for Wnt/beta-catenin signaling. FEBSLetters, 583(4), 627–632. [DOI] [PubMed] [Google Scholar]
- Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, et al. (2008). A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biology, 6(1), e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Jin SG, Pfeifer GP, & Szabo PE (2011). Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proceedings of the National Academy of Sciences of the United States of America, 108(9), 3642–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP (2000). CpG-island methylation in aging and cancer. Current Topics in Microbiology and Immunology, 249, 101–118. [DOI] [PubMed] [Google Scholar]
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, & Zhang Y (2010). Role of tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature, 466(7310), 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, et al. (2007). The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell, 128(6), 1077–1088. [DOI] [PubMed] [Google Scholar]
- Jacobson RH, Ladurner AG, King DS, & Tjian R (2000). Structure and function of a human TAFII250 double bromodomain module Science (New York, N.Y.), 288(5470), 1422–1425. [DOI] [PubMed] [Google Scholar]
- Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. (2010). Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Research, 20(2), 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, & Allis CD (2001). Translating the histone code Science (New York, N.Y.), 293(5532), 1074–1080. [DOI] [PubMed] [Google Scholar]
- Jones PA, Wolkowicz MJ, Rideout WM 3rd, Gonzales FA, Marziasz CM, Coetzee GA, et al. (1990). De novo methylation of the MyoD1 CpG island during the establishment of immortal cell lines. Proceedings of the National Academy of Sciences of the United States of America, 57(16), 6117–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen HF, Ben-Porath I, & Bird AP (2004). Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Molecular and Cellular Biology, 24(8), 3387–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AA, & Struhl K (2005). Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to pol II elongation. Molecular Cell, 20(6), 971–978. [DOI] [PubMed] [Google Scholar]
- JOSSE J, KAISER AD, & KORNBERG A (1961). Enzymatic synthesis of deoxyribonucleic acid. VIII. frequencies of nearest neighbor base sequences in deoxyribonucleic acid. The Journal of Biological Chemistry, 236, 864–875. [PubMed] [Google Scholar]
- Kacem S, & Feil R (2009). Chromatin mechanisms in genomic imprinting. Mammalian Genome : Official Journal of the International Mammalian Genome Society, 20(9–10), 544–556. [DOI] [PubMed] [Google Scholar]
- Kafri T, Ariel M, Brandeis M, Shemer R, Urven L, McCarrey J, et al. (1992). Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes & Development, 6(5), 705–714. [DOI] [PubMed] [Google Scholar]
- Kanduri C, Pant V, Loukinov D, Pugacheva E, Qi CF, Wolffe A, et al. (2000). Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Current Biology : CB, 10(14), 853–856. [DOI] [PubMed] [Google Scholar]
- Karlic R, Chung HR, Lasserre J, Vlahovicek K, & Vingron M (2010). Histone modification levels are predictive for gene expression. Proceedings of the National Academy of Sciences of the United States of America, 107(7), 2926–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman PD, & Rando OJ (2010). Chromatin as a potential carrier of heritable information. Current Opinion in Cell Biology, 22(3), 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, et al. (2005). Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell, 123(4), 593–605. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Podolny V, & Buratowski S (2003). Bur1 kinase is required for efficient transcription elongation by RNA polymerase II. Molecular and Cellular Biology, 23(19), 7005–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kang K, & Kim J (2009). AEBP2 as a potential targeting protein for polycomb repression complex PRC2. Nucleic Acids Research, 37(9), 2940–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, Milne TA, et al. (2009). RAD6-mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell, 137(3), 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, & Bird AP (2006). Genomic DNA methylation: The mark and its mediators. Trends in Biochemical Sciences, 31(2), 89–97. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, et al. (2006). The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature, 442(7100), 312–316. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P, et al. (2007). The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell, 128(5), 889–900. [DOI] [PubMed] [Google Scholar]
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, et al. (2011). Tetl and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell, 8(2), 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, & Ahringer J (2009). Differential chromatin marking of introns and expressed exons by H3K36me3. Nature Genetics, 41(3), 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T, & Berger S (2007). Chromatin modifications and their mechanisms of action In Allis CD, Jenuwein T & Reinberg D (Eds.), Epigenetics (pp. 191–206). Plainview, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Kouzarides T (2007). Chromatin modifications and their function. Cell, 128(4), 693–705. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, & Heintz N (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain Science (New York, N.Y.), 324(5929), 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, et al. (2003). Methylation of histone H3 by Set2 in saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Molecular and Cellular Biology, 23(12), 4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange M, Kaynak B, Forster UB, Tonjes M, Fischer JJ, Grimm C, et al. (2008). Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes & Development, 22(17), 2370–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, & Winston F (2001). The S. cerevisiae SAGA complex functions in vivo as a coactivator for transcriptional activation by Gal4. Genes & Development, 15(15), 1946–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen F, Gundersen G, Lopez R, & Prydz H (1992). CpG islands as gene markers in the human genome. Genomics, 13(4), 1095–1107. [DOI] [PubMed] [Google Scholar]
- Lee J, Jang SJ, Benoit N, Hoque MO, Califano JA, Trink B, et al. (2010). Presence of 5-methylcytosine in CpNpG trinucleotides in the human genome. Genomics, 96(2), 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, et al. (2007). Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell, 131(6), 1084–1096. [DOI] [PubMed] [Google Scholar]
- Lee MG, Norman J, Shilatifard A, & Shiekhattar R (2007). Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell, 128(5), 877–887. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, & Shiekhattar R (2005). An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature, 437(7057), 432–435. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, & Workman JL (2007). The role of chromatin during transcription. Cell, 128(4), 707–719. [DOI] [PubMed] [Google Scholar]
- Li B, Gogol M, Carey M, Lee D, Seidel C, & Workman JL (2007). Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin Science (New York, N.Y.), 316(5827), 1050–1054. [DOI] [PubMed] [Google Scholar]
- Li B, Howe L, Anderson S, Yates JR 3rd, & Workman JL (2003). The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. The Journal of Biological Chemistry, 278(11), 8897–8903. [DOI] [PubMed] [Google Scholar]
- Li W, & Liu M (2011). Distribution of 5-hydroxymethylcytosine in different human tissues. Journal of Nucleic Acids, 2011, 870726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature, 462(7271), 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Tesfai J, Evrard YA, Dent SY, & Martinez E (2003). c-myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. The Journal of Biological Chemistry, 278(22), 20405–20412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, & Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature, 389(6648), 251–260. [DOI] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, et al. (2009). Role of the polycomb protein EED in the propagation of repressive histone marks. Nature, 461(7265), 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, & Reinberg D (2011). The polycomb complex PRC2 and its mark in life. Nature, 469(7330), 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, & Zhang Y (2005). The diverse functions of histone lysine methylation. Nature Reviews.Molecular Cell Biology, 6(11), 838–849. [DOI] [PubMed] [Google Scholar]
- Martinez E, Kundu TK, Fu J, & Roeder RG (1998). A human SPT3-TAFII31-GCN5-L acetylase complex distinct from transcription factor IID. The Journal of Biological Chemistry, 273(37), 23781–23785. [DOI] [PubMed] [Google Scholar]
- Mateescu B, Bourachot B, Rachez C, Ogryzko V, & Muchardt C (2008). Regulation of an inducible promoter by an HP1beta-HP1gamma switch. EMBO Reports, 9(3), 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin PJ, & Allshire RC (2005). Methylation: Lost in hydroxylation? EMBO Reports, 6(4), 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, & Cole MD (2000). The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-myc. Molecular and Cellular Biology, 20(2), 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature, 437(7057), 436–439. [DOI] [PubMed] [Google Scholar]
- Miao F, Smith DD, Zhang L, Min A, Feng W, & Natarajan R (2008). Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: An epigenetic study in diabetes. Diabetes, 57(12), 3189–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature, 448(7153), 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk M, Boubelik M, & Lehnert S (1987). Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development Development (Cambridge, England), 99(3), 371–382. [DOI] [PubMed] [Google Scholar]
- Nagy Z, & Tora L (2007). Distinct GCN5/PCAF-containing complexes function as coactivators and are involved in transcription factor and global histone acetylation. Oncogene, 26(37), 5341–5357. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393(6683), 386–389. [DOI] [PubMed] [Google Scholar]
- Nekrasov M, Klymenko T, Fraterman S, Papp B, Oktaba K, Kocher T, et al. (2007). Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at polycomb target genes. The EMBO Journal, 26(18), 4078–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, & Struhl K (2003). Targeted recruitment of Set1 histone methylase by elongating pol II provides a localized mark and memory of recent transcriptional activity. Molecular Cell, 11(3), 709–719. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Kotani T, Zhang X, Schiltz RL, Howard T, Yang XJ, et al. (1998). Histone-like TAFs within the PCAF histone acetylase complex. Cell, 94(1), 35–44. [DOI] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, et al. (2005). hDOT1L links histone methylation to leukemogenesis. Cell, 121(2), 167–178. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, & Li E (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99(3), 247–257. [DOI] [PubMed] [Google Scholar]
- Ordovas JM, & Smith CE (2010). Epigenetics and cardiovascular disease. Nature Reviews.Cardiology, 7(9), 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatnani HP, & Greenleaf AL (2006). Phosphorylation and functions of the RNA polymerase II CTD. Genes & Development, 20(21), 2922–2936. [DOI] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, et al. (2005). Genome-wide map of nucleosome acetylation and methylation in yeast. Cell, 122(4), 517–527. [DOI] [PubMed] [Google Scholar]
- Portela A, & Esteller M (2010). Epigenetic modifications and human disease. Nature Biotechnology, 25(10), 1057–1068. [DOI] [PubMed] [Google Scholar]
- Pray-Grant MG, Daniel JA, Schieltz D, Yates JR 3rd, & Grant PA (2005). Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature, 433(7024), 434–438. [DOI] [PubMed] [Google Scholar]
- Prokhortchouk E, & Defossez PA (2008). The cell biology of DNA methylation in mammals. BiochimicaEtBiophysica Acta, 1783(11), 2167–2173. [DOI] [PubMed] [Google Scholar]
- Qiu H, Hu C, & Hinnebusch AG (2009). Phosphorylation of the pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Molecular Cell, 33(6), 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand E, Ben-Porath I, Keshet I, & Cedar H (2004). CTCF elements direct allele-specific undermethylation at the imprinted H19 locus. Current Biology : CB, 14(11), 1007–1012. [DOI] [PubMed] [Google Scholar]
- Reeves WM, & Hahn S (2005). Targets of the Gal4 transcription activator in functional transcription complexes. Molecular and Cellular Biology, 25(20), 9092–9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, & Lewis A (2005). Co-evolution of X-chromosome inactivation and imprinting in mammals. Nature Reviews.Genetics, 6(5), 403–410. [DOI] [PubMed] [Google Scholar]
- Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, et al. (2003). Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Molecular Cell, 12(6), 1591–1598. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell, 129(7), 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, & Zhang MQ (2009). Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics, 10, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Kirmizis A, Nelson C, Bartke T, Saksouk N, Cote J, et al. (2009). Histone H3 tail clipping regulates gene expression. Nature Structural &Molecular Biology, 16(1), 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, et al. (2002). Active genes are tri-methylated at K4 of histone H3. Nature, 419(6905), 407–411. [DOI] [PubMed] [Google Scholar]
- Saxonov S, Berg P, & Brutlag DL (2006). A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proceedings of the National Academy of Sciences of the United States of America, 103(5), 1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarano E, Iaccarino M, Grippo P, & Parisi E (1967). The heterogeneity of thymine methyl group origin in DNA pyrimidine isostichs of developing sea urchin embryos. Proceedings of the National Academy of Sciences of the United States of America, 57(5), 1394–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, et al. (2004). A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes & Development, 18(11), 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, et al. (2006). ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature, 442(7098), 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell, 119(7), 941–953. [DOI] [PubMed] [Google Scholar]
- Shi Y, & Whetstine JR (2007). Dynamic regulation of histone lysine methylation by demethylases. Molecular Cell, 25(1), 1–14. [DOI] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, & Shi Y (2005). Regulation of LSD1 histone demethylase activity by its associated factors. Molecular Cell, 19(6), 857–864. [DOI] [PubMed] [Google Scholar]
- Shiio Y, & Eisenman RN (2003). Histone sumoylation is associated with transcriptional repression. Proceedings of the National Academy of Sciences of the United States of America, 100(23), 13225–13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, & Peterson CL (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions Science (New York, N.Y.), 311(5762), 844–847. [DOI] [PubMed] [Google Scholar]
- Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, et al. (2003). Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. The EMBO Journal, 22(8), 1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, & Kingston RE (2009). Mechanisms of polycomb gene silencing: Knowns and unknowns. Nature Reviews.Molecular Cell Biology, 10(10), 697–708. [DOI] [PubMed] [Google Scholar]
- Sterner DE, & Berger SL (2000). Acetylation of histones and transcription-related factors. Microbiology and Molecular Biology Reviews : MMBR, 64(2), 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, & Allis CD (2000). The language of covalent histone modifications. Nature, 403(6765), 41–45. [DOI] [PubMed] [Google Scholar]
- Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, et al. (2009). Developmental programming of CpG island methylation profiles in the human genome. Nature Structural & Molecular Biology, 16(5), 564–571. [DOI] [PubMed] [Google Scholar]
- Swartz MN, Trautner TA, & Kornberg A (1962). Enzymatic synthesis of deoxyribonucleic acid. XI. further studies on nearest neighbor base sequences in deoxyribonucleic acids. The Journal of Biological Chemistry, 237, 1961–1967. [PubMed] [Google Scholar]
- Szabo P, Tang SH, Rentsendorj A, Pfeifer GP, & Mann JR (2000). Maternal-specific footprints at putative CTCF sites in the H19 imprinting control region give evidence for insulator function. Current Biology : CB, 10(10), 607–610. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1 Science (New York, N.Y.), 324(5929), 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, & Jones PA (2002). Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proceedings of the National Academy of Sciences of the United States of America, 99(6), 3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature, 439(7078), 811–816. [DOI] [PubMed] [Google Scholar]
- Turner BM, Birley AJ, & Lavender J (1992). Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in drosophila polytene nuclei. Cell, 69(2), 375–384. [DOI] [PubMed] [Google Scholar]
- Vakoc CR, Mandat SA, Olenchock BA, & Blobel GA (2005). Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Molecular Cell, 19(3), 381–391. [DOI] [PubMed] [Google Scholar]
- Vakoc CR, Sachdeva MM, Wang H, & Blobel GA (2006). Profile of histone lysine methylation across transcribed mammalian chromatin. Molecular and Cellular Biology, 26(24), 9185–9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valinluck V, & Sowers LC (2007). Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Research, 67(3), 946–950. [DOI] [PubMed] [Google Scholar]
- Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, & Sowers LC (2004). Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Research, 32(14), 4100–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, & Gottschling DE (2002). Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell, 109(6), 745–756. [DOI] [PubMed] [Google Scholar]
- Vettese-Dadey M, Grant PA, Hebbes TR, Crane-Robinson C, Allis CD, & Workman JL (1996). Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. The EMBO Journal, 15(10), 2508–2518. [PMC free article] [PubMed] [Google Scholar]
- Waddington CH (1957). The strategy of the genes; a discussion of some aspects of theoretical biology. London: Allen & Unwin. [Google Scholar]
- Walter J (2011). An epigenetic tet a tet with pluripotency. Cell Stem Cell, 8(2), 121–122. [DOI] [PubMed] [Google Scholar]
- Wang S, Robertson GP, & Zhu J (2004). A novel human homologue of drosophila polycomblike gene is up-regulated in multiple cancers. Gene, 343(1), 69–78. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, et al. (2004). Human PAD4 regulates histone arginine methylation levels via demethylimination Science (New York, N.Y.), 306(5694), 279–283. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, et al. (2009). LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell, 138(4), 660–672. [DOI] [PubMed] [Google Scholar]
- Wang YA, Kamarova Y, Shen KC, Jiang Z, Hahn MJ, Wang Y, et al. (2005). DNA methyltransferase-3a interacts with p53 and represses p53-mediated gene expression. Cancer Biology & Therapy, 4(10), 1138–1143. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. (2008). Combinatorial patterns of histone acetylations and methylations in the human genome. Nature Genetics, 40(7), 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RA (1980). Modified bases in bacteriophage DNAs. Annual Review of Microbiology, 34, 137–158. [DOI] [PubMed] [Google Scholar]
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. (2005). Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature Genetics, 37(8), 853–862. [DOI] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. (2006). Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell, 125(3), 467–481. [DOI] [PubMed] [Google Scholar]
- Wood A, Schneider J, Dover J, Johnston M, & Shilatifard A (2003). The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. The Journal of Biological Chemistry, 278(37), 34739–34742. [DOI] [PubMed] [Google Scholar]
- Workman JL, & Kingston RE (1998). Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annual Review of Biochemistry, 67, 545–579. [DOI] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, et al. (2011). 5-hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nature Communications, 2, 241. [DOI] [PubMed] [Google Scholar]
- Wyatt GR, & Cohen SS (1952). A new pyrimidine base from bacteriophage nucleic acids. Nature, 170(4338), 1072–1073. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, et al. (2005). WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell, 121(6), 859–872. [DOI] [PubMed] [Google Scholar]
- Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, et al. (2003). Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes & Development, 17(5), 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, et al. (2007). PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Molecular Cell, 25(6), 801–812. [DOI] [PubMed] [Google Scholar]
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, et al. (2006). JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell, 125(3), 483–495. [DOI] [PubMed] [Google Scholar]
- Yang XJ, & Seto E (2007). HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene, 26(37), 5310–5318. [DOI] [PubMed] [Google Scholar]
- Yang XJ, & Seto E (2008). The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nature Reviews.Molecular Cell Biology, 9(3), 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lu Y, Espejo A, Wu J, Xu W, Liang S, et al. (2010). TDRD3 is an effector molecule for arginine-methylated histone marks. Molecular Cell, 40(6), 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder JA, & Bestor TH (1998). A candidate mammalian DNA methyltransferase related to pmt1p of fission yeast. Human Molecular Genetics, 7(2), 279–284. [DOI] [PubMed] [Google Scholar]
- Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, & Zhou MM (2010). Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature, 466(7303), 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, et al. (2006). Genome wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell, 126(6), 1189–1201. [DOI] [PubMed] [Google Scholar]
- Zhang Y, & Reinberg D (2001). Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes & Development, 15(18), 2343–2360. [DOI] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, & Lee JT (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome Science (New York, N.Y.), 322(5902), 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, et al. (2009). PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nature Structural & Molecular Biology, 16(3), 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Wyrick JJ, & Reese JC (2010). Novel trans-tail regulation of H2B ubiquitylation and H3K4 methylation by the N terminus of histone H2A. Molecular and Cellular Biology, 30(14), 3635–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Zheng Y, Pham AD, Mandal SS, Erdjument-Bromage H, Tempst P, et al. (2005). Monoubiquitination of human histone H2B: The factors involved and their roles in HOX gene regulation. Molecular Cell, 20(4), 601–611. [DOI] [PubMed] [Google Scholar]
- Zollman S, Godt D, Prive GG, Couderc JL, & Laski FA (1994). The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in drosophila. Proceedings of the National Academy of Sciences of the United States of America, 91(22), 10717–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]