

Abstract
mRNA vaccines have proven to be more stable, effective, and specific than protein/peptide-based vaccines in stimulating both humoral and cellular immune response. However, mRNA’s fast degradation rate and low-transfection efficiency in vivo impede its potential in vaccination. Recent research in gene delivery has focused on nonviral vaccine carriers and either implantable or injectable delivery systems to improve transgene expression in vivo. Here, an injectable chitosan-alginate gel scaffold for the local delivery of mRNA vaccines is reported. Gel scaffold biodegradation rates and biocompatibility are quantified. Scaffold-mediated mRNA in vivo transgene expression as well as ovalbumin antigen specific cellular and humoral immune responses are evaluated in vivo. Luciferase reporter protein expression resulting from mRNA lipoplex-loaded gel scaffolds is five times higher than systemic injection. Compared to systemic injections of naked mRNA or mRNA:lipoplexes, elevated levels of T cell proliferation and IFN-γ secretion are seen with in vivo scaffold-mediated mRNA lipoplex delivery. Furthermore, a humoral response (ovalbumin antigen specific IgG levels) is observed as early as week 1 for scaffold-mediated mRNA lipoplex delivery, while protein-based immunization did not elicit IgG production until 2 weeks post-injection. Results suggest that injectable scaffold mRNA vaccine delivery maybe a viable alternative to traditional nucleic acid immunization methods.
Keywords: biodegradable, injectable immunizing scaffold, mRNA vaccine delivery
1. Introduction
Protein/peptide-based vaccines and inactivated viruses are common immunization vectors, but they tend to induce humoral immunity rather than a robust cell mediated immune response. Nucleic acid vaccines (pDNA, mRNA) have several advantages compared to traditional protein antigen vaccines: a) nucleic vaccines can elicit CD8+ T cell responses; to date, there is no clinically acceptable protein format to do this and b) nucleic acid vaccines are not subject to neutralization by the host immune response, thus allowing repeat boosting.[1,2] Messenger RNA (mRNA) vaccines have gained attention recently due to their efficient and specific antigen production in transfected antigen presenting cells (APCs) without the risk of producing any other infectious progeny. By stimulation of toll-like receptors (TLR) 7/8 and 3, mRNA vaccines can also serve as a self-adjuvant,[3,4] which allows mRNA to trigger both the humoral and cellular immune response. Although mRNA has been considered more stable in the subcutaneous space, the major drawback of naked mRNA is its fast degradation in vivo and low-transfection efficiency by dendritic cells (DCs).[4] Thus, an efficient delivery system is critical to mRNA vaccine development.
Over the past decades, biomaterial-based nanoparticle platforms have been gradually applied to nucleic acid vaccine development and gene therapy.[5] Many polymer-based or lipid-based carrier systems have been developed to overcome the major barriers of DNA transfection: a) low uptake across the cell membrane, b) inadequate endosomal escape-release of nucleic acid molecules, and c) lack of nuclear entry.[6–8] Cationic polymers such as poly(ethyleneimine) (PEI),[9] poly(l-lysine) (PLL),[10] chitosan,[11] and dendrimers[12] have been widely reported as excellent nonviral carriers for plasmid DNA delivery. Meanwhile, liposomes and cationic lipids such as 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP)[13] and Lipofectamine[14] have also been widely investigated for DNA transfection both in vitro and in vivo. Nonviral carrier systems developed for DNA have also been applied for siRNA transfections due to their similar physiochemical properties. Besides PEI, poly(lactic-co-glycolic acid) PLGA is used most extensively to improve the stability and to provide sustained release of siRNA.[15] However, relative to the technologies developed for DNA and siRNA delivery, large single stranded mRNA delivery requires much more improvement. Some of the carriers established for DNA and siRNA have been extrapolated successfully to mRNA delivery; such as poly(β-amino ester) PBAE[16] and DOTAP.[17] The similar structure of liposomes to cell membranes promotes cellular endocytosis; while the weak binding with mRNA carriers facilitates “endosome escape” and release of mRNA into the cytoplasm. However, the development of relevant mRNA delivery systems is limited due to mRNAs perceived instability, susceptibility to degradation, insufficient transgene expression, and immunostimulatory effect.[18] Herein, we utilized a standard liposomal mRNA nanoparticle carrier system combined with an injectable biodegradable scaffold platform for mRNA vaccine delivery in vivo.
Native immune responses to cancers are limited, so many cell-based vaccination approaches attempt to enhance the response by differentiating and activating dendritic cells (DC) ex vivo and then introducing these programmed DCs back into the patient.[19–21] These ex vivo approaches require a) bone marrow isolation from the host, b) in vitro differentiation and modifications of DCs, then c) return of modified DC to the host; essentially two procedures involving the patient, which leads to high cost and significant regulatory concerns. More importantly, most (>90%) transplanted DCs die and few actually migrate to the lymph nodes (≈0.5–2.0%).[19,20,22] Further, ex vivo DC modifications are dependent on culture conditions and are transient, thus losing effectiveness upon in vivo transplantation. These limitations create a need,[19,20] to recruit and engineer DCs in vivo. 3D polymer scaffolds may be ideal for this purpose, due to their established success in controlled drug release and cell delivery,[23–31] which could be exploited to promote in vivo recruitment and manipulation of DCs. Development of a material system that can deliver an effective vaccine, eliminating the time, expense, and regulatory burden inherent to cell therapies will be a major advance, particularly if it can achieve this effect without the need for multiple, systemic injections and high-total drug loading.
Classical vaccine/adjuvant combinations are notoriously inefficient at transfecting DCs in vivo. Only recently, due to the needs of cancer immunotherapies, has the biomaterials/gene delivery community begun to develop new strategies for adjuvant engineering. Babensee and coworkers[32] incorporated the antigen, ovalbumin (OVA), into two different PLGA carriers; either in PLGA microparticles (diameter = 3.5 μm) or in PLGA porous scaffolds (0.7 cm dia. × 0.2 cm thick discs; pores ≈400 μm). Carriers with antigen were then injected (particles) or implanted (scaffolds) into mice and the resulting systemic immune response assessed. Total amount of polymer and antigen delivered were kept constant. OVA-specific IgG was significantly higher and sustained longer for antigen released from implanted scaffolds with associated tissue damage versus when the same amount of polymer/antigen was delivered by injected particles. Ali et al. reported similar findings for PLG scaffolds versus PLG microparticles, both releasing tumor extracted antigens.[33] Kim et al.[34] demonstrate that high-aspect ratio, mesoporous silica rods (MSRs) injected with a needle spontaneously assemble in vivo to form macroporous structures that provide a 3D cellular microenvironment for attraction of host immune cells. In mice, substantial numbers of dendritic cells are recruited to the pores between the scaffold rods. The recruitment of dendritic cells and their subsequent homing to lymph nodes can be modulated by sustained release of inflammatory signals and adjuvants from the scaffold. Injection of an MSR releasing OVA as antigen enhances systemic helper T cells TH1 and TH2 serum antibody and cytotoxic T-cell levels compared to bolus injections. Steinle et al.[35] evaluated similar degradable, injectable chitosan-alginate hybrid hydrogels for the delivery of mRNA to HEK293 cells that were both incorporated into the hydrogels; a simplistic in vitro assay showed enhanced expression of their reporter gene over 3 weeks; no in vivo studies were carried out.
In this study, a biodegradable gel was made by chemical crosslinking via Schiff-base reaction between N-succinyl chitosan (S-CS) and oxidized alginate (O-Alg). A macroporous structure was introduced by lyophilization of the gel to improve its swelling property for loading. Single stranded mRNA was complexed into nanosized particles using a commercial liposomal carrier; resultant mRNA lipoplexes were loaded onto lyophilized chitosan-alginate scaffolds during the rehydration step. The rehydration-loading process transferred the scaffold from a dry state to gel state that could be injected through syringe (needle 25G × 5/8″) at room temperature. By either diffusion or degradation of the scaffold, mRNA lipoplexes are released from the gel scaffold and enter local cells by endocytosis. Physical properties and mRNA release kinetics of the gel were determined in vitro. Immunization efficiency (i.e., T cell response, IFN-γ secretion, and OVA-IgG production) of injectable gel:mRNA complexes were determined in an in vivo murine model using ovalbumin mRNA as a model antigen.
2. Experimental Section
2.1. Reagents and Materials
Chitosan (50 000–190 000 Da, 75–85% deacetylated), lactic acid (≈90%), succinic anhydride (≥99%), sodium periodate (≥99.8%), and methanol were supplied by Sigma-Aldrich (St. Louis, MO). Sodium alginate was purchased from Spectrum Chemical Manufacturing Corporation (New Brunswick, NJ). Ethylene glycol (≥99%) was supplied by J. T. Baker Chemicals (Avantor Performance Materials; Center Valley, PA). Bovine serum albumin (BSA) was purchased from Thermo Fisher Scientific Inc. (Waltham, MA). All chemicals were used as received.
2.2. Methods
2.2.1. Synthesis of N-Succinyl Chitosan
One gram chitosan was dissolved in 80 mL lactic acid (5% v/v) at room temperature. The chitosan solution was diluted with 320 mL methanol by magnetic stirring for 30 min. Succinic anhydride (2 g) was added gently into the solution and mixed by stirring at 600 rpm for 24 h at room temperature. After increasing the pH value to 8.0 by adding 0.1 m NaOH solution, a white precipitate was produced. The produced N-succinyl chitosan was centrifuged at 2500 rpm for 5 min and dissolved in deionized water. The solution was dialyzed against ultrapure water for 3 days for further purification. The purified solution was then lyophilized at −55 °C to obtain solid N-succinyl chitosan (S-CS).
2.2.2. Synthesis of Oxidized Alginate
Three grams sodium alginate were dissolved in 200 mL deionized water to obtain an alginate solution with a concentration of 1.5% (w/v). Prepared sodium periodate solution (15% w/v, 10 mL) was then added to the alginate solution dropwise. The oxidation reaction proceeded thoroughly in the dark by magnetic stirring at 600 rpm for 24 h at room temperature. 2 mL ethylene glycol was added for 2 h to stop the oxidization. The solid oxidized alginate (O-Alg) was purified and collected after a 3-day dialysis against ultrapure water and lyophilization at −55 °C.
2.2.3. Preparation of Gel Scaffold
To prepare an evenly crosslinked hydrogel (10CS/50Alg), S-CS and O-Alg were dissolved in PBS separately at a concentration of 10 mg mL−1 and 50 mg mL−1, respectively. Then, 100 μL O-Alg and 200 μL S-CS solution were mixed thoroughly in an injector or a 96-well plate for 10 min. The hydrogel was then lyophilized at −55 °C overnight to form a chitosan-alginate gel scaffold for further in vitro and in vivo study.
2.2.4. Scaffold Swelling, Degradation, and mRNA Release
To characterize the swelling behavior of the gel scaffolds as a function of time, cylindrical disc samples were immersed in PBS (pH = 7.4) at 37 °C for 2 h after the dry-state weight (Wd) was measured. The swollen hydrated gels were taken out of tubes at each time point (5, 10, 20, 30, 60, and 120 min post incubation) and the wet-state weight (Ws) was immediately recorded after the excess PBS on the surface were eliminated by filter paper. The swelling ratio (SR) was determined as (Ws−Wd)/Wd.
The kinetics of weight loss of the degradable gel scaffold in PBS and PBS-BSA (20% w/v) medium was monitored over 2 weeks. The gel scaffold was weighed initially (W0) after being immersed into 5 mL degradation medium for 2 h to reach equilibrium. Samples were then placed into an incubator shaking at 70 rpm at a constant 37 °C. The mass of gel scaffolds was weighted (Wt) after removing the residual medium on their surface at each time point. The suspending medium was refreshed every day. The fraction of gel scaffold mass remaining was defined as Wt/W0 × 100%. Experiments were repeated for scaffolds containing a) nothing, b) Cy5-stained naked mRNA 4 μg-mRNA total, and c) Stemfect:Cy5-stained mRNA lipoplexes 4 μg-mRNA total. Amount of mRNA loaded (naked or complexed) into the degradable scaffolds was determined by measuring the Cy5-fluorescence (ex/em 649 nm:666 nm) of the mRNA solution before and after scaffold loading and converting the difference into mRNA concentration by prior calibration. Stemfect complexed Cy5-stained mRNA did not affect the fluorescence intensity of Cy-5.
Concomitantly with gel degradation studies, the amount of mRNA released (naked or complexed) during degradation was quantified again by periodically sampling the liquid and measuring the Cy-5 fluorescence. Cumulative mRNA released was calculated from Equation (1),
| (1) |
where MACC (t) = total amount of cumulative mRNA released at any time t, [μg]; Vs = volume of elution medium (L3); and CmRNA (t) = concentration of mRNA at time t, [μg L−3]. The cumulative percent of mRNA released at time t is calculated from Equation (2),
| (2) |
where Mtotal = the total amount of mRNA in each sample.
2.2.5. Gel Scaffold Pore Structure
The microscopic pore structure of scaffolds was observed using a scanning electron microscope (FEI SEM XL Siron). The samples were imaged at 3 kV accelerating voltage with 5 mm working distance after being sliced and gold-coated for 90 s.
2.2.6. Cell Viability
All cell culture media and reagents were purchased from Gibco (Waltham, MA). BHK-21 hamster fibroblast cell line (ATCC; Manassas, VA) was maintained in Eagle’s Medium with 10% FBS and 1% penicillin-streptomycin. The DC 2.4 murine dendritic cell line (a gift from K. L. Rock, University of Massachusetts Medical School) was cultured in L-glutamine RPMI 1640 medium supplied with 0.1 mM non-essential amino acids, 10 mM HEPES, 55 μM 2-mercaptoethanol, 10% FBS, and 1% penicillin-streptomycin. The JAWsII murine dendritic cell line (ATCC) was incubated in alpha minimum essential medium with 2 mM L-glutamine, 1.5 g L−1 sodium bicarbonate, 5 ng mL−1 GM-CSF, 20% FBS, and 1% penicillin-streptomycin.
Cells were seeded on tissue culture-treated 24-well plates at a concentration of 1.5 × 105 cells well−1. The cells were incubated at 37 °C for 4 h to allow adherence. After removal of culture medium, chitosan-alginate gels were injected into each well. Fresh cell medium was then replaced. At each time point (1, 3, and 5 days), after removal of the gel scaffolds, cells were stained using the LIVE/DEAD dual viability stain assay (Invitrogen L-3324); stained cells were imaged by a Zeiss Axio Observer Z1 fluorescence microscope (20 ×). The polyanionic dye calcein is well retained within live cells, producing an intense uniform green fluorescence in live cells (ex/em 495 nm/515 nm). EthD-1 enters cells with damaged membranes and undergoes a 40-fold enhancement of fluorescence upon binding to nucleic acids, thereby producing a bright red fluorescence in dead cells (ex/em 495 nm/635 nm). EthD-1 is excluded by the intact plasma membrane of live cells. The pH value of medium was also recorded at each time point.
2.2.7. mRNA Vector Construction
Plasmid DNA was prepared using the QIAprep Spin Miniprep Kit (Qiagen). Plasmid pGEM4Z-OVA-A64 encoding the gene for the ovalbumin (OVA) protein (model antigen) with a synthetic poly-A-tail has been previously described.[36] Briefly, the ovalbumin cDNA EcoRI fragment from plasmid pAc-neo-OVA was subcloned into the pVAX1 vector (Invitrogen) and amplified by PCR; XbaI and NotI sites were added to the 5′ and 3′ ends of the cDNA. The product was ligated into pGEM4Z-A64 to produce pGEM4Z-OVA-A64. pGEM4Z-OVA-A64 plasmids were linearized by SpeI and used as template for in vitro transcription using a mMESSAGE mMACHINE T7 Kit (Ambion). No modified nucleotides were used and a standard 7-methylguanosine(m7G) was linked to the 5′end of the mRNA through a 5′−5′-triphosphate bridge (ppp). The resultant mRNA was purified using a RNeasy MinElute Cleanup Kit (Qiagen). CleanCap FLuc mRNA encoding Luciferase (Luc mRNA) reporter gene (protein half-life = 5 h) was purchased from Tri-Link BioTechnologies (San Diego, CA).
To determine whether dendritic cells transfected with mRNA lipoplexes could successfully process and present the encoded antigen on MHC molecules, an in vitro MHC I antigen presentation assay was performed using the B3Z CD8-T cell hybridoma. The B3Z cell line produces β-galactosidase upon recognition of the ovalbumin CD8 epitope (SIINFEKL) presented in the context of MHC-I H-2Kb. DC2.4 cells were transfected with lipoplexes containing ovalbumin mRNA for 4 h, then cocultured with B3Z cells for 24 h, and the overall T cell response determined by assaying for β-galactosidase activity. Results of the B3Z T cell assay have been previously published.[36]
2.2.8. mRNA Lipoplex Formulation and Loading Strategy
Luc mRNA and OVA mRNA were utilized in the in vivo gene expression and ovalbumin immunizations, respectively. mRNA lipoplexes were formed by combining equal volumes of mRNA and Stemfect to a final mRNA concentration of 20 μg mL−1. To load naked mRNA or mRNA lipoplexes onto a gel scaffold, 200 μL mRNA (4 μg) was placed into a 1 mL syringe containing lyophilized gel scaffold and was absorbed thoroughly upon swelling of the gel scaffold. The scaffold in mRNA solution became liquid after 20 min and could then be injected through a (25G × 5/8″) needle.
2.2.9. In vivo mRNA Transgene Expression
C578L/6J mice (6–8 weeks old) were purchased from Jackson Laboratory and housed under normal conditions. All animal experiments were approved by the University of Washington Animal Care and Use Committee.
Mice were anesthetized by 2% isoflurane inhalation followed by subcutaneous injection in the dorsal right flank of the liquid gel scaffold (≈200 μL) loaded with naked Luc mRNA or loaded with mRNA nanoparticles. Naked mRNA in PBS and fresh-made mRNA nanoparticles in PBS were injected subcutaneously in the dorsal right flank of a separate cohort of mice. Each dose had the same volume of 200 μL and contained 4 μg Luc mRNA. A separate cohort of mice were injected 200 μL PBS alone by the same delivery route, as a negative control. The in vivo mRNA transgene expression was monitored periodically for 2 days. Bioluminescence imaging was performed by collecting emitted photons for 60 s using Xenogen IVIS200 Spectrum Imager after intraperitoneal injection of 200 μL D-luciferin (Gold Biotechnology, St. Louis, MO) at a concentration of 15 mg mL−1 (≈150 mg kg−1 of mouse body weight). The total luminescence intensity was quantified by Living Image software (Caliper).
2.2.10. T Cell Response to mRNA-OVA Vaccine
Individual groups of C578L/6J mice (six mice per group; 1:1 male:female) were subcutaneously injected with either 200 μL of naked OVA mRNA, Stemfect/OVA mRNA nanoparticles, liquid gel scaffolds releasing naked OVA mRNA, liquid gel scaffolds releasing OVA mRNA nanoparticles, ovalbumin protein solution (100 μg mL−1 in PBS), or PBS as negative controls. mRNA dose (4 μg) was the same as the in vivo mRNA transgene expression tests. Lymphocytes were isolated by homogenizing the lymph nodes 5 days post immunization, collecting cells through a 100-μm cell strainer, and incubating with ACK lysis buffer for 5 min to lyse red blood cells. T-cell antigen response was evaluated as: a) IFN-γ secretion and b) T-cell proliferation.
To quantify IFN-γ secretion, isolated lymphocytes were seeded on U-bottomed 96-well plates at 2 × 106 cells well−1 in 100 μL culture medium. Cells were stimulated with 100 μg mL−1 OVA protein for 5 days at 37 °C in 5% CO2. Culture supernatant was collected after 5 days and soluble protein IFN- γ expression level was measured with ELISA assay (BioLegend, San Diego, CA).
T cell population increase in response to an antigen stimulus is determined by measuring the time-dependent decrease in initial T cell fluorescence. Recovered T cells were stained at time equal zero with carboxyfluorescein succinimidyl ester (CFSE) and as they multiply the per-cell fluorescence decreases; which is measured by flow cytometry. To label cells with CFSE, the isolated lymphocytes were resuspended in 1 mL medium and placed carefully in the bottom of a fresh (non-wetted) 10 mL conical tube. The tube was placed horizontally and 110 μL PBS added at the top ensuring it did not make contact with the cell suspension. 1.1 μL of a 5 mM stock of carboxyfluorescein diacetate (CFDA) and succinimidyl ester (SE) solutions were added in the 110 μL PBS. The tube was quickly capped, inverted and vortexed gently for 1 min to uniformly label T-lymphocytes with CFSE. Cell suspensions (200 μL well−1) were transferred into U-bottomed 96-well plates and cultured for another 5 days at 37 °C in an incubator. Samples were collected after 5 days and flow cytometry used to analyze 10 000 events per sample gated on viable propidium iodide negative cells using a BD FACScan flow cytometer (BD Biosciences) and FlowJo software (TreeStar).
2.2.11. Antibody Response to mRNA-OVA Vaccine
Mice were immunized as described above. Blood samples were collected on day 7, 14, 21, and 28 by submandibular bleeding; Mice were then euthanized by compressed CO2. Plasma aliquots were collected by centrifuging for 5 min at 2500 g and were stored at −80 °C. To prepare ovalbumin-coated 96-well plates, 100 μL of 1 μg mL−1 ovalbumin stock was added in each well and incubated overnight at 4 °C. Plates were then washed and blocked with 1% BSA in DPBS for an hour at 37 °C. Plates were washed twice with 200 μL PBS at room temperature. Then, diluted plasma samples were added to the plates and incubated for an hour at 37 °C. The secondary antibody, horseradish peroxidase-conjugated goat anti-mouse IgG (Biolegend, San Diego, CA) was added at a 1:2 000 dilution and incubated for 30 min at 37 °C. Plates were developed for 10 min in dark using TMB 2-component peroxidase substrate (Thermo Scientific), with 2 M H2SO4 as the stop solution, and analyzed for absorbance at 450 nm on the spectrophotometer.
2.3. Statistical Analysis
All in vivo samples comprised n = 6 mice for each independent variable unless otherwise stated. Data are displayed as mean ± standard error of the mean. Statistical outliers were removed using Grubbs’ outlier test at alpha = 0.05 using GraphPad Prism v6 Software (GraphPad Software Inc., La Jolla, CA). Two-way ANOVAs were performed (GraphPad Prism v6), with statistical significance set at p ≤ 0.05. For multiple comparisons, Tukey or Dunnet post-test corrections were applied.
3. Results
3.1. Scaffold Formation
Injectable degradable hydrogels were prepared by crosslinking N-succinyl chitosan (S-CS) with oxidized alginate (O-Alg) upon mixing (Figure 1A). By introducing hydrophilic side groups (succinic anhydride) onto the chitosan backbone, the water solubility of chitosan was increased from 1 mg mL−1 to 20 mg mL−1. Oxidization of alginate not only introduced aldehyde groups on its polymer chain but also reduced the molecular weight by 50% approximately. As a result, an oxidized alginate with a high solubility (100 mg mL−1) was produced. Spontaneous crosslinking of the two constituents into a hydrogel was achieved by C = N bond formation in a Schiff-base reaction between the amino groups on S-CS and the aldehyde groups on O-Alg. Once the two constituents crosslinked for 10 min and the formed gel was lyophilized; the resultant dried gel structure demonstrated an even and interconnected pore architecture (100–200 μm) (Figure 1B). The resultant porous structure of the dried gel scaffold aided in mRNA loading upon hydration. The Schiff-base reaction exhibited a controlled reasonably slow sol-to-gel phase transition behavior that allowed for even crosslinking throughout the hydrogel. Therefore, the rehydrated gel scaffold could be easily injected through a (25G × 5/8″) needle without resistance (Figure 1C). Our preliminary studies found that an optimized hydrogel composition of 10 mg mL−1 S-CS and 50 mg mL−1 O-Alg exhibited desired injection/degradation profiles. All subsequent in vitro and in vivo studies were carried out using these 10CS/50Alg gel scaffolds.
Figure 1.
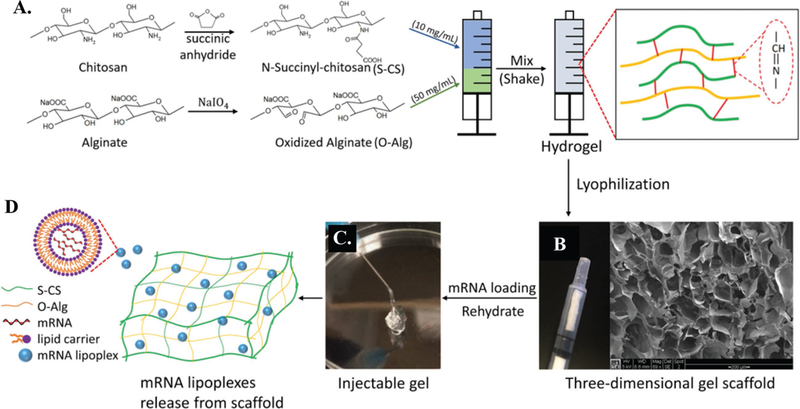
Schematic illustration of chitosan-alginate 3D gel scaffold formation. Hydrogel (10CS/50Alg) was first prepared via Schiff-base reaction in an injector by mixing N-succinyl chitosan (S-CS) and oxidized alginate (O-Alg) solutions A). Scaffold was formed by lyophilization of hydrogel overnight B). mRNA lipoplexes were loaded by rehydration of scaffold, which transferred to gel state and was able to be injected through needle (25G × 5/8″) at room temperature C). By either diffusion or degradation of scaffold, mRNA lipoplexes released from gel and were taken by cells in surrounding environment D).
3.2. Swelling, Degradation, and mRNA Release
Swelling ability of the gel scaffolds is of great significance for substance exchange in tissue regeneration and drug release. The swelling ratio directly reflects the efficiency of substance absorption and release. The swelling kinetics of a formed gel scaffold was measured in PBS at 37 °C for 2 h (Figure 2A). Results showed that the water absorption of the gel scaffolds equilibrated after 20 min and approached ≈20 times the gel’s initial dry weight.
Figure 2.
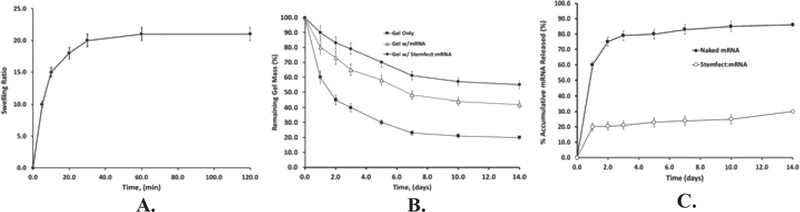
A) Swelling ratio of a chitosan:alginate gel scaffold (10CS/50Alg) over 2 h in PBS at 37 °C, B) in vitro degradation of gel scaffold (with or without mRNA), in PBS-BSA (20% w/v) at 37 °C. C) Concomitant mRNA released (either naked mRNA or Stemfect:mRNA) during gel degradation in PBS-BSA (20% w/v) at 37 °C. Values reported are an average n = 3, ± standard deviation.
The subsequent loss of gel scaffold weight upon prolonged hydration was monitored in PBS-BSA (20% w/v) medium at 37 °C as a function of time (Figure 2B) for gel scaffolds alone, gel plus naked mRNA, and gel plus Stemfect:mRNA nanoparticles. For bare scaffolds, degradation rate was rapid over the first 3 days (from 100% to 40% remaining), then continued at a much slower rate over the next 9 days at 21%. By day 14, the gel macro-structure started to deteriorate, with large sections crumbling away suggesting degradation followed a bulk erosion mechanism.[37] Loading scaffold with either naked mRNA or Stemfect:mRNA nanoparticles provided a slower degradation compared to empty gels mainly due to the extra crosslinking between the mRNA and the hydrogel polymers. mRNA release kinetics (Figure 2C) showed that while both naked mRNA and Stemfect:mRNA nanoparticles reduced gel degradation rates, naked mRNA rapidly released from the gel, with ≈80% dispensed in just 3 days thus providing only a short window for potential cell transfection. Conversely, about 30% mRNA in nanoparticle form was released from the gel over the course of 2 weeks.
The in vivo degradation products of the scaffold are another critical aspect to consider. It is commonly agreed that chitosan is enzymatically degraded by lysozyme in vivo by breaking glucosamine–glucosamine bonds that produce chitosan oligosaccharides.[38] The chitosan oligosaccharides are then incorporated into glycoprotein pathways, metabolic pathways, or excreted.[38,39] For alginate, it is known that the in vivo degradation of pure alginate is slow (> 6 months) and unpredictable due to lack of natural enzymes in human organisms. Research has shown that oxidized alginate can be hydrolyzed within 2 months in vivo and is easily eliminated by the kidneys.[40]
3.3. Cell Viability
Cell viability studies (Figure 3) were performed to ascertain the toxicity, if any, of scaffold to mammalian cells. To test the cytocompatibility of gel scaffold, a Live/Dead assay was used to stain both fibroblast and dendritic cells exposed to hydrated scaffolds. The images were taken at 1 day, 3 days, and 5 days (Figure 3A–C).
Figure 3.
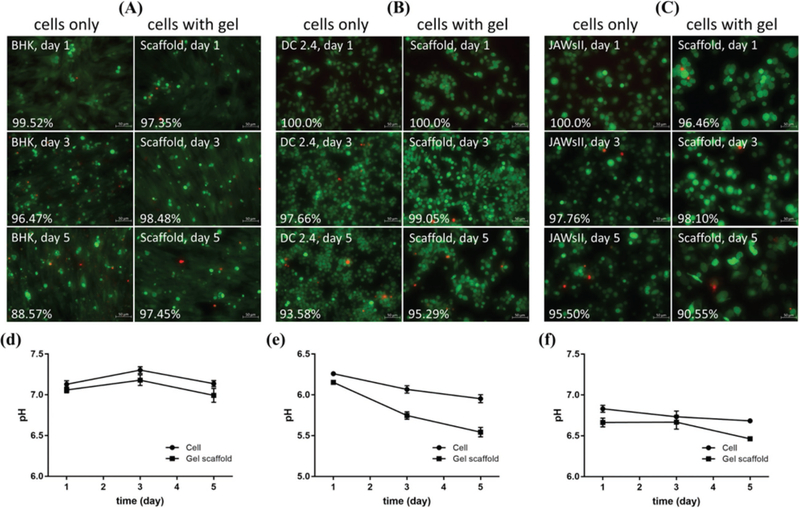
A) BHK, B) DC 2.4, and C) JAWsII cell viability over time as indicated by Live/Dead assay. Cells were seeded at 150 000 cells well−1 and 50 μL of 10CS/50Alg hydrogel was injected. Pictures of cells only (left) and cells with gel (right) were taken by inverted microscope (20×) at 1 day, 3 days, and 5 days. The number of live and dead cells were counted by ImageJ software; and cell viability were quantified by calculating the percentage of live cells. pH value of BHK D), DC 2.4 E) and JAWsII F) cell culture medium was also monitored at each time point. Scale bar is 50 μm. Results were averages of three independent experiments carried out in triplicate.
In general, chitosan/alginate gel scaffolds proved very bio-compatible to cells since there were no significant differences in cell viability when compared to control groups without scaffold. By counting the number of live cells by ImageJ, it was found that the majority of cells (over 90%) cultured with scaffolds were viable (green) and demonstrated no clear difference in viability between samples. Data (Figure 3D–F) demonstrate that the scaffold degradation did subtly lower the pH but that it apparently had no effect on cell viability.
3.4. In Vivo mRNA Mediated Protein Expression
To compare mRNA mediated protein expression in vivo for a) lipid-based mRNA-luc lipoplex alone versus b) an injected gel scaffold containing mRNA-luc:lipoplexes, expression of a luciferase reporter gene was measured over 2 days post-subcutaneous injection. As shown in Figure 4A, only mRNA-luc lipoplexes delivered by the injectable gel scaffold demonstrated local transfection in vivo, whereas no signal was detected from any other delivery system. It is also noted that the transfection mediated by the combined liposome:mRNA and gel scaffold was transient, with luciferase expression peaking at approximately 8 h (Figure 4B) post-injection and becoming undetectable by 48 h, probably due to mRNA degradation within the cells.
Figure 4.
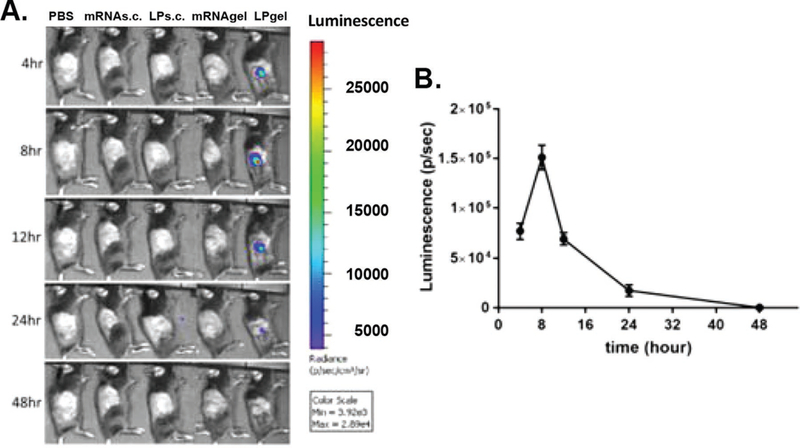
In vivo mRNA protein expression. Individual mice were injected subcutaneously in the back with either 200 μL PBS, naked mRNA (mRNA s.c.), Stemfect:mRNA lipoplexes (LP s.c.), naked mRNA loaded gel (mRNA gel) or Stemfect:mRNA lipoplex-loaded gel (LPgel). A) The mRNA-Luc reporter expression was imaged over 2 days by IVIS imaging. B) The luminescence for LP gel injections were quantified at each time point by Xenogen IVIS Living Image software.
3.5. Ovalbumin Immunization and Immune Response
Based on the in vivo mRNA-luc transfection results, ovalbumin mRNA was then used to determine whether an improved immune response could be mediated by lipid-based carriers and gel scaffold in vivo. Mice were immunized with the following: naked mRNA expressing the antigen ovalbumin, mRNA-OVA nanoparticles alone, or within gel scaffolds as above. Humoral response was assessed weekly over a month, while cellular responses were evaluated at day 5 by both a T cell proliferation assay and an extracellular IFN-γ quantification.
Total ovalbumin-specific IgG serum levels for each immunization group were quantified by ELISA for relative antibody response (Figure 5). Among the four mRNA delivery groups, only gel-released mRNA lipoplexes led to an increased antibody production statistically different from the control group. Antibody production to the protein-based immunization produced over four times the antibody compared to gel-released mRNA lipoplexes after 2 weeks.
Figure 5.
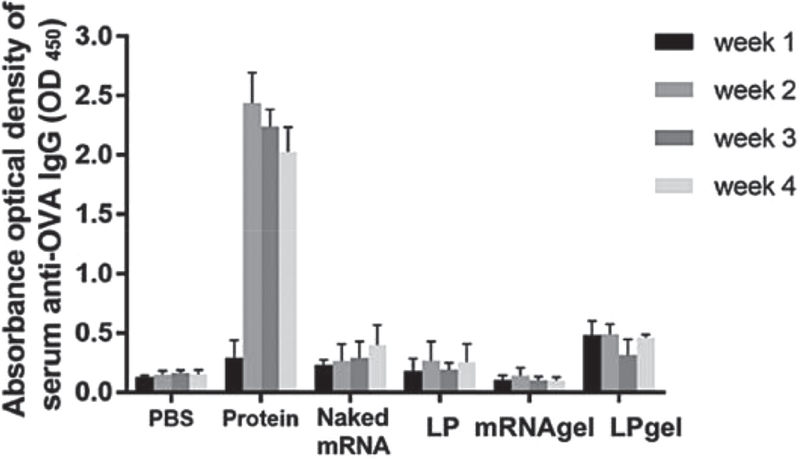
Ovalbumin-specific humoral IgG responses. Individual mice were injected once subcutaneously in the dorsal right flank with either 200 μL PBS, ovalbumin protein solution 100 μg mL−1(Protein), naked mRNA (naked mRNA), mRNA/Stemfect lipoplexes (LP), naked mRNA loaded gel scaffold (mRNA gel) or mRNA:lipoplex-loaded gel scaffold (LPgel). The amount of mRNA applied per mouse 4 μg. Serum samples were collected weekly for a month and analyzed for anti-ovalbumin IgG by ELISA.
To compare the cellular immune response by a traditional protein-based vaccine and mRNA vaccine, lymph nodes of mice were explanted, and cells were collected 5 days post immunization. A T cell proliferation test was carried out using an in vitro OVA protein stimulation assay based on CFSE-stained ex vivo collected lymphocytes; and analyzed by flow cytometry (Figure 6A). CFSE fluorescence decays upon cell division such that a broader distribution is observed by flow cytometry test if T cells proliferate (Figure 6B) in response to antigen stimulus. The quantitative results are shown in Figure 6C, where the NP-gel group exhibited the most significant level of T-cell proliferation (27.2%) over any other group. These results suggest that scaffold-released mRNA nanoparticles produced the highest level of cellular response versus alternative antigen delivery means. That T cells activated by both scaffold-released naked mRNA and NP-mRNA produced 3x more IFN-γ than either protein vaccine or systemically delivered mRNA vaccines (Figure 6 D) indicates the mRNA vaccines delivered by scaffold can significantly activate cytotoxic T cells in vivo.
Figure 6.
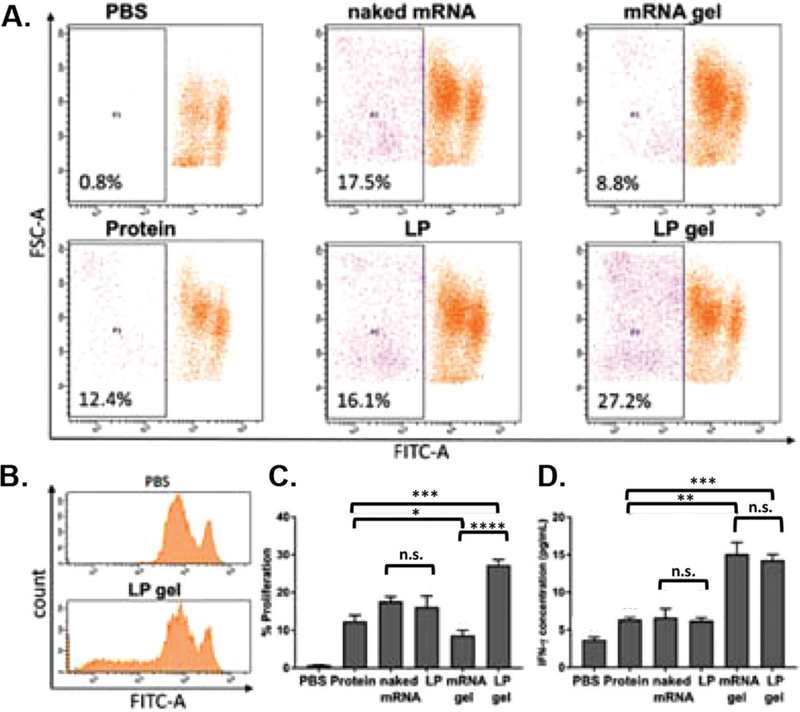
Ovalbumin immunization T cell response. Individual mice were injected once subcutaneously in the dorsal right flank with either 200 μL PBS, ovalbumin protein solution (Protein), 4 μg naked mRNA (naked mRNA), mRNA/Stemfect lipoplexes (LP), mRNA loaded gel scaffold (mRNA gel) or mRNA lipoplex-loaded gel scaffold (LP gel). Cells were isolated from recovered lymph nodes 5 days post immunization and analyzed for: T lymphocyte proliferation based on CFSE stain dilution A–C) and quantification of T cell IFN-γ secretion D) upon in vitro stimulation with OVA protein. (n.s., nonsignificant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
4. Discussion
Based on our ovalbumin immunization results, scaffold-mediated mRNA vaccines produced a robust T cell and humoral response in a short term compared to protein-based vaccines, which is attributed to the combination of lipid-based mRNA carriers and the injected gel scaffold delivery system.
In vivo Luciferase protein expression was only observed from the injected gel scaffold containing mRNA-luc:lipoplexes within the first 24 h post-injection. In vitro mRNA release kinetics (Figure 2C) showed that most release occurred during the first 2 days. This suggests that most protein expression resulted from transfection of mRNA released from the scaffold. Even though much less mRNA-luc:lipoplexes was released compared to naked mRNA (≈20% vs 80%, Figure 2C), the lipoplex formulation successfully retained mRNA bioactivity and led to higher protein expression. It is worth noting that after the initial burst release, the mRNA-luc:lipoplexes remained encapsulated in the scaffold did not result in detectable protein expression during the 24–48 h time frame. It is possible that as the gels completely crumble overtime, a second burst release of the mRNA lipoplexes would occur, providing a “booster” immunization. If more sustained protein expression is desired, it is also feasible to incorporate chemoattractant components to promote cell recruitment within the scaffold, where the mRNA lipoplexes are more accessible.
Significant T cell response (proliferation and INF-γ production) was observed in those groups delivered OVA mRNA, especially LP gel. This is not surprising since one of the advantages of nucleic acid-based vaccines is that they effectively engage both MHC-I and MHC-II pathways therefore allow for a more robust T cell response. By contrast, soluble antigens such as protein generally induces only antibody responses. We found that both carriers and scaffold served important roles in the improvement of local transfection in vivo 2 days post-injection. Previous results from scaffold-released mRNA vaccines demonstrated that dendritic cells are recruited into the scaffold, uptake mRNA lipoplexes, then migrate to draining lymph nodes for T cell stimulation to initiate an adaptive immune response.[41]
Various nanoparticle systems have been developed for maintaining mRNA stability and bioactivity. In this study, an injectable scaffold platform was designed specifically for mRNA loading and it was combined with the standard mRNA lipoplex nanoparticle technology for mRNA vaccine delivery in vivo. The current industrial approach of transfecting ex vivo extracted dendritic cells followed by re-infusion of the transfected DCs allows precise control of the cellular target and high-transfection efficiency by electroporation.[42] However, this approach is expensive and a labor-intensive endeavor fraught with numerous quality assurance issues. The porous structure of our injectable scaffold platform provides a local microenvironment for immune cell acquisition and modification,[33,43] effective loading and delivery mRNA vaccines.
In this study, we focused on subcutaneous delivery of mRNA vaccines, while most vaccines are given by intramuscular route since that option maximizes immunogenicity and limits adverse reactions at injection site.[44] The slow mobilization and processing of antigen in the subcutaneous space usually leads to vaccine failure,[45] although it has been shown that repeated booster immunizations by subcutaneous injection are more effective than other routes.[46] For nucleic acid vaccines, the subcutaneous route can induce greater immune response compared with those elicited by other routes of delivery.[1] However, a delivery strategy is necessary to administer vaccines through the layer of subcutaneous fat. Based on our positive results, the chitosan-alginate gel scaffold could be a suitable candidate for a subcutaneous delivery system of mRNA vaccines. Various scaffolds have been developed and widely used on for cell modification,[34,47,48] tissue regeneration,[39,49,50] and drug or gene delivery,[36,51,52] but none of them considered nucleic acid vaccine loading and delivery.
While scaffold-released mRNA produced a robust T cell response, the humoral response was much less than observed for the protein antigen. To improve humoral response, both protein-based and mRNA vaccine could be loaded and delivered by the gel scaffold. Furthermore, a long term immune response such as IFN-γ and memory B cell response after 6–12 months should be evaluated. It is possible that scaffold-released mRNA simply did not sufficiently stimulate B-cells to the level of that seen protein antigen. Memory B cells are generated during the primary immune response to foreign antigens. Initial antigen exposure and B cell proliferation produces short-lived immunoglobulin secreting plasmablasts and germinal center cells. After a repeat exposure to antigen, the memory cells proliferate rapidly and generate plasmablasts, which boost the amount of antigen-specific immunoglobulin in the serum to aid in antigen clearance. It is possible that the scaffold degradation and cessation of mRNA lipoplex release was insufficient in producing a secondary stimulus. This could be resolved by optimizing scaffold degradation rates or simply applying a secondary scaffold injection.
Besides antigen presentation resulting from mRNA transfection and protein production, sufficient APC activation is also crucial for a robust immune response. Sufficient B cell activation and antibody production require APCs such as activated DCs migrate to lymph nodes and/or spleen, where they will activate B and T cells. Therefore, DC transfection and protein expression in local tissue is not adequate for B and T cell activation. Additionally, antigen presentation to T cells by nonactivated/inadequately activated APCs may induce T cell tolerance. In this case, the immunostimulatory effect of both the biomaterial platform and the formulated mRNA should be taken into consideration. The biomaterials used in the scaffolds, chitosan and alginate, have the ability to activate DCs and promote DC costimulatory markers expression.[53] While mRNA can be used as self-adjuvant, its immunogenicity may be attenuated after lipoplex formulation. In future studies, adjuvants such as TLR agonists can be incorporated into the delivery system for a more controllable adjuvant effect.
5. Conclusion
In this study, we developed an injectable biodegradable chitosan-alginate gel scaffold using a Schiff-base reaction and evaluated its application in mRNA vaccine delivery in vitro and in vivo. The high-swelling ratio of the porous gel scaffold enhances the gel’s ability to load naked mRNA or Stemfect/mRNA lipoplexes. Degradable scaffolds also allow for the option to apply a booster injection of scaffold releasing vaccine in subsequent weeks. Biocompatibility was assessed by gel cell toxicity assays that show no adverse effects on either fibroblast or dendritic cells after 5 days. Both lipid-based mRNA carrier and the gel scaffold delivery combined were necessary for mRNA transgene expression in vivo. Cellular and humoral response to OVA immunization using our system was evaluated over 4 weeks. The elevated level of local gene expression by scaffold-released mRNA lipoplexes led to a stronger T cell response at 5 days post immunization as demonstrated by T cell proliferation and IFN-γ secretion. These positive results suggest that scaffold-mediated mRNA vaccine delivery may be a viable alternative to traditional nucleic acid immunization methods.
Acknowledgements
J.D.B. designed and supervised the overall project conception and execution; and aided in revising the manuscript. J.Y. designed and executed the study, analyzed, and interpreted the data related to scaffold fabrication; and drafted and revised the manuscript. R.C. assisted in mRNA vaccine design and analysis of the data; and aided in revising the manuscript. H.Z. assisted in scaffold fabrication, characterization, and data interpretation; and aided in revising the manuscript. This work was funded by the National Institutes of Health/NIAID (5R01AI074661 and 2R56AI074661-06).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Ulmer JB, Mason PW, Geall A, Mandl CW, Vaccine 2012, 30, 4414. [DOI] [PubMed] [Google Scholar]
- [2].Siegrist C-A, Vaccine Immunology, in Vaccines 6th ed. (Eds: Plotkin SA, Orenstein WA, Offit PA), Elsevier; 2013, 14. [Google Scholar]
- [3].Geall AJ, Mandl CW, Ulmer JB, Semin. Immunol 2013, 25, 152. [DOI] [PubMed] [Google Scholar]
- [4].Leitner WW, Ying H, Restifo NP, Vaccine 1999, 18, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG, Nat. Rev. Genet 2014, 15, 541. [DOI] [PubMed] [Google Scholar]
- [6].Luo D, Saltzman WM, Nat. Biotechnol 2000, 18, 33. [DOI] [PubMed] [Google Scholar]
- [7].Al-Dosari MS, Gao X, AAPS J 2009, 11, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ramamoorth M, Narvekar A, J. Clin. Diagnostic Res 2015, 9, GE01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lungwitz U, Breunig M, Blunk T, Göpferich A, Eur. J. Pharm. Biopharm 2005, 60, 247. [DOI] [PubMed] [Google Scholar]
- [10].Lollo CP, Banaszczyk MG, Mullen PM, Coffin CC, Wu D, Carlo AT, Bassett DL, Gouveia EK, Carlo DJ, Methods Mol. Med 2001, 69, 1. [DOI] [PubMed] [Google Scholar]
- [11].Borchard G, Adv. Drug Delivery Rev 2001, 52, 145. [DOI] [PubMed] [Google Scholar]
- [12].Dufès C, Uchegbu IF, Schätzlein AG, Adv. Drug Delivery Rev 2005, 57, 2177. [DOI] [PubMed] [Google Scholar]
- [13].Lv H, Zhang S, Wang B, Cui S, Yan J, J. Controlled Release 2006, 114, 100. [DOI] [PubMed] [Google Scholar]
- [14].Goldman CK, Soroceanu L, Smith N, Gillespie GY, Shaw W, Burgess S, Bilbao G, Curiel DT, Nat. Biotechnol 1997, 15, 462. [DOI] [PubMed] [Google Scholar]
- [15].Khan A, Benboubetra M, Sayyed PZ, Ng KW, Fox S, Beck G, Benter IF, Akhtar S, J. Drug Targeting 2004, 12, 393. [DOI] [PubMed] [Google Scholar]
- [16].Su X, Fricke J, Kavanagh D, Irvine DJ, Chase C, Mol. Pharmaceutics 2011, 8, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rejman J, Tavernier G, Bavarsad N, Demeester J, De Smedt SC, J. Controlled Release 2010, 147, 385. [DOI] [PubMed] [Google Scholar]
- [18].Wu X, Brewer G, Gene 2012, 500, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Steinman RM, Banchereau J, Nature 2007, 449, 419. [DOI] [PubMed] [Google Scholar]
- [20].Gilboa E, J. Clin. Invest 2007, 117, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Banchereau J, Steinman RM, Nature 1998, 392, 245. [DOI] [PubMed] [Google Scholar]
- [22].Kleindienst P, Brocker T, Immunol J. 2003, 170, 2817. [DOI] [PubMed] [Google Scholar]
- [23].Freed LE, Guilak F, Guo XE, Gray ML, Tranquillo R, Holmes JW, Radisic M, Sefton MV, Kaplan D, Vunjak-Novakovic G, Tissue Eng 2006, 12, 3285. [DOI] [PubMed] [Google Scholar]
- [24].Discher DE, Mooney DJ, Zandstra PW, Science 2009, 324, 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Place ES, Evans ND, Stevens MM, Nat. Mater 2009, 8, 457. [DOI] [PubMed] [Google Scholar]
- [26].Dvir T, Timko BP, Kohane DS, Langer R, Nat. Nanotechnol 2011, 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huebsch N, Mooney DJ, Nature 2009, 462, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lutolf MP, Hubbell JA, Nat. Biotechnol 2005, 23, 47. [DOI] [PubMed] [Google Scholar]
- [29].Langer R, Vacanti JP, Science, 1993, 260, 920. [DOI] [PubMed] [Google Scholar]
- [30].Tsang KY, Cheung MCH, Chan D, Cheah KSE, Cell Tissue Res 2010, 339, 93. [DOI] [PubMed] [Google Scholar]
- [31].Walsh DP, Heise A, O’Brien FJ, Cryan S-A, Gene Ther 2017, 24, 681. [DOI] [PubMed] [Google Scholar]
- [32].Bennewitz NL, Babensee JE, Biomaterials 2005, 26, 2991. [DOI] [PubMed] [Google Scholar]
- [33].Ali O, Huebsch N, Cao L, Dranoff G, Mooney DJ, Nat. Mater 2009, 8, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G, Mooney DJ, Nat. Biotechnol 2014, 33, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Steinle H, Ionescu TM, Schenk S, Golombek S, Kunnakattu SJ, Özbek MT, Schlensak C, Wendel HP, Avci-Adali M, Int. J. Mol. Sci 2018, 19, 1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cheng C, Convertine AJ, Stayton PS, Bryers JD, Biomaterials 2012, 33, 6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lin CC, Metters AT, Adv. Drug Delivery Rev 2006, 58, 1379. [DOI] [PubMed] [Google Scholar]
- [38].Şenel S, McClure SJ, Adv. Drug Delivery Rev 2004, 56, 1467. [DOI] [PubMed] [Google Scholar]
- [39].Levengood SL, Zhang M, J. Mater. Chem. B 2014, 2, 3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bouhadir KH, Lee KY, Alsberg E, Damm KL, Anderson KW, Mooney DJ, Biotechnol. Prog 2001, 17, 945. [DOI] [PubMed] [Google Scholar]
- [41].Chen R, Zhang H, Yan J, Bryers JD, Gene Ther 2018, 10.1038/s41434-018-0040-9. [DOI] [PMC free article] [PubMed]
- [42].Benteyn D, Heirman C, Bonehill A, Thielemans K, Breckpot K, Expert Rev. Vaccines 2015, 14, 161. [DOI] [PubMed] [Google Scholar]
- [43].Kim J, Mooney DJ, Nano Today 2011, 6, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zuckerman JN, BMJ 2000, 321, 1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Anderson DM, Hall LL, Ayyalapu AR, Irion VR, Nantz MH, Hecker JG, Hum. Gene Ther 2003, 14, 191. [DOI] [PubMed] [Google Scholar]
- [46].Poland G, Borrud A, Jacobson R, McDermott K, Wollan P, Brakke D, Charboneau J, JAMA, J. Am. Med. Assoc 1997, 277, 1709. [PubMed] [Google Scholar]
- [47].Singh A, Peppas NA, Adv. Mater 2014, 26, 6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen R, Ma H, Zhang L, Bryers JD, Biotechnol. Bioeng 2018, 115, 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fan M, Ma Y, Tan H, Jia Y, Zou S, Guo S, Zhao M, Huang H, Ling Z, Chen Y, Hu X, Mater. Sci. Eng., C 2017, 71, 67. [DOI] [PubMed] [Google Scholar]
- [50].Lee KY, Mooney DJ, Chem. Rev 2001, 101, 1869. [DOI] [PubMed] [Google Scholar]
- [51].Li Z, Ning W, Wang J, Choi A, Lee PY, Tyagi P, Huang L, Pharm. Res 2003, 20, 884. [DOI] [PubMed] [Google Scholar]
- [52].Tan H, Fan M, Ma Y, Qiu J, Li X, Yan J, Adv. Healthcare Mater 2014, 3, 1769. [DOI] [PubMed] [Google Scholar]
- [53].Lei Y, Rahim M, Ng Q, Segura T, J. Controlled Release 2011, 153, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]