

Abstract
Myofibroblasts are central mediators of fibrosis. Typically derived from resident fibroblasts, myofibroblasts represent a heterogeneous population of cells that are principally defined by acquired contractile function and high synthetic ability to produce extracellular matrix (ECM). Current literature sheds new light on the critical role of ECM signaling coupled with mechanotransduction in driving myofibroblastic activation. In particular, transforming growth factor β1 (TGF-β) and extra domain A containing fibronectin (EDA-FN) are thought to be the primary ECM signaling mediators that form and also induce positive feedback loops. The outsidein and inside-out signaling circuits are transmitted and integrated by TGF-β receptors and integrins at the cell membrane, ultimately perpetuating the abundance and activities of TGF-β and EDA-FN in the ECM. In this review, we highlight these conceptual advances in understanding myofibroblastic activation, in hope of revealing its therapeutic anti-fibrotic implications.
Keywords: myofibroblast, extracellular matrix, TGF-β1, EDA-FN, positive feedback loop
1. Introduction
Principally characterized by accumulation of disorganized extracellular matrix (ECM), fibrosis is the endstage hallmark of a broad range of diseases. The central cellular mediator of fibrosis is myofibroblast with a prominent contractile/synthetic phenotype. While myofibroblasts are heterogeneous [1, 2], they commonly express smooth muscle proteins such as a smooth muscle actin (α-SMA) that forms contractile stress fibers. Moreover, myofibroblasts produce large amounts of ECM proteins, including collagen I, extra-domain A containing fibronectin (EDA-FN), and matrix metalloproteinases (MMPs). In recent lineage tracing studies, de novo expressed periostin was identified as a robust marker of myofibroblasts [3]. Many terms have been used for the process that generates myofibroblasts; e.g. myofibroblast (trans)differentiation or activation or transformation, fibroblast activation, fibroblast-to-myofibroblast transition or conversion, etc. Current literature reveals that rather than terminally differentiated, myofibroblasts represent a range of highly plastic cell states. Thus, in this review we opt to use the term “myofibroblastic activation” [4–6] attempting to reconcile the deviatory terminologies with updated understanding of myofibroblasts.
A classical view is that myofibroblasts derive from fibroblasts for wound repair, where they produce and contract granulation tissue to fill and close a lesion. Recent studies have revealed that myofibroblasts derive from a wide variety of cell types [7, 8], maybe triggered by signaling cues beyond injury [9]. Early-stage myofibroblastic activation is deemed protective. However, in fibrotic diseases it fails to terminate, resulting in progressive and chronic damage through continuous synthesis of fibrotic scar tissue [10]. The biological basis for the occurrence of fibrosis instead of resolution remains unclear. Myofibroblast-mediated fibrotic tissue remodeling occurs in most organs, especially at the end stage of major diseases ranging from scleroderma, atherosclerosis, heart failure, liver cirrhosis, renal fibrosis, and many cancers [11]. As such, a better understanding of signaling mechanisms underlying myofibroblastic activation is imperative for producing knowledge that could be broadly applied in anti-fibrotic therapeutic developments.
Extensive research has been done to uncover many signaling pathways responsible for myofibroblastic activation and fibrosis, for which a large number of reviews have been published, mostly in specific disease contexts. With new investigative tools available, ECM remodeling and mechanotransduction have recently received refreshed enthusiasm [12, 13], likely for their reciprocal actions that profoundly influence myofibroblastic activation [14, 15]. Of particular note, the duet of transforming growth factor β1 (TGF-β) and the alternatively spliced EDA-FN constitute a potent myofibroblastic activator [16]. They form a positive feedback loop by coupling with TGF-β receptors and mechanotranducer integrins on the cell surface (see Figure 1). This circuit transmits mechanical and biochemical signals from the ECM into the cell, triggering transcriptional reprograming (outsidein). Consequently, elevated production of α-SMA stress fibers and EDA-FN in turn mediates cell contraction and ECM alteration and further TGF-β activation (inside-out). To highlight this integrated concept, the current review focuses on TGF-β1/EDA-FN and associated integrin and intracellular signaling that orchestrate myofibroblastic activation.
Figure 1. Outside-in and inside-out signal transduction in myofibroblastic activation.
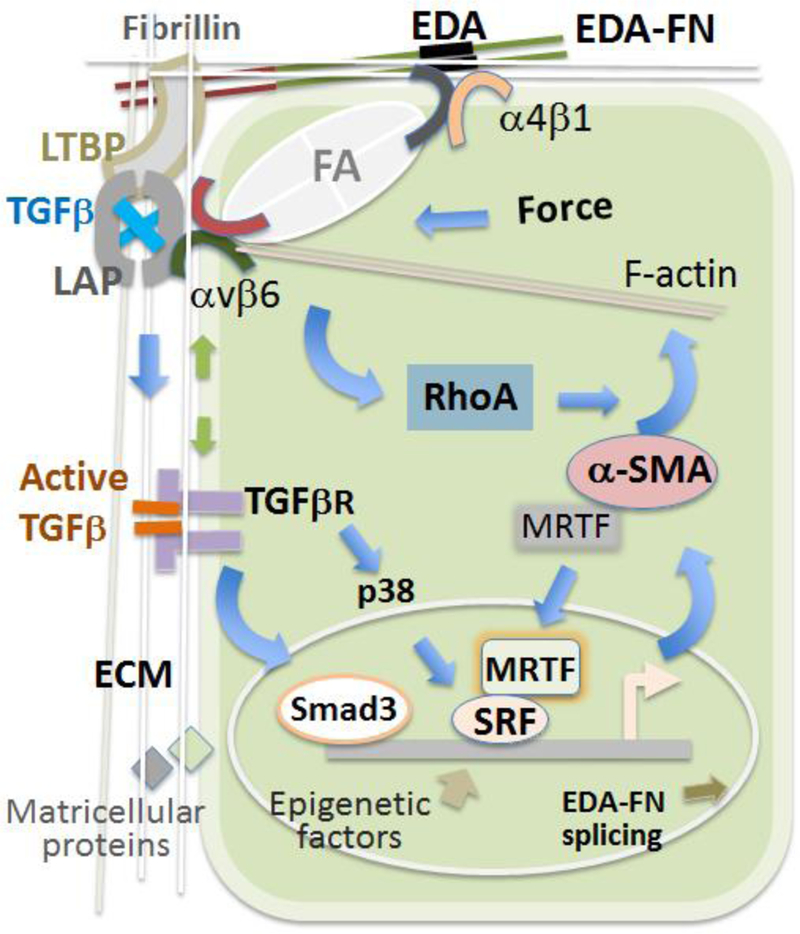
Outside-in: Activated TGF-β1 (freed from LAP) binds to the TGF-βR complex stimulating intracellular signaling that promotes α-SMA production. In parallel, ECM-to-cell mechanical transduction through the integrin/focal adhesion (FA) pathway activates RhoA, leading to assembly of α-SMA stress fibers.
Inside-out: TGF-β1-activated EDA-FN splicing increases the EDA-FN protein in the ECM; cell contraction executed by α-SMA stress fibers alters the LAP (gray) conformation via the integrin/LAP interaction releasing TGF-β1 (blue) from LAP.
Positive feedback loops: 1. TGF-β1 stimulates the production of EDA-FN which in turn facilitates the ECM incorporation and activation of TGF-β1 via the LTBP-1/EDA-FN/integrin interaction network. 2. ECM-integrin mechano-transduction activates RhoA which enhances the assembly of α-SMA stress fibers that execute cell contraction and integrin mechano-transduction, further augmenting RhoA activation. 3. TGF-β1-stimulated stress fiber production ultimately enhances TGF-β1 release from the ECM via mechano-activation through the integrin/LAP interaction. 4. The assembly of G-actin (monomer α-SMA) into F-actin (stress fiber) allows for the escape of MRTF from G-actin sequestration in the cytosol and its nuclear translocation and SRF activation, which in turn propagates α-SMA and stress fiber production.
Notes:
1. Latent TGF-β forms SLC with the LAP “straight jacket”, where it is trapped; SLC and LTBP forms LLC.
2. Green arrows indicate TGF-βR and integrin/FA mutual modulations.
3. Activated RhoA promotes MRTF nuclear translocation by enhancing F-actin assembly while reducing G-actin in the cytosol.
2. Precursors of Myofibroblasts
Origins of myofibroblasts, particularly in vivo, have been hotly debated [17]. Recent lineage tracing studies overall support the assertion that the main sources of myofibroblasts are resident mesenchymal cells such as fibroblasts and pericytes [18, 19]. It has been shown that periostin-expressing myofibroblasts in the heart are derived from resident fibroblasts of the TCF21 lineage, but not from endothelial, immune/myeloid or smooth muscle cells [3]. Fibroblasts are systemically ubiquitous ECM-producing cells that assume a spindle-shaped morphology in cell culture [20–22]. Myofibroblasts are distinguished from fibroblasts by highly upregulated α-SMA stress fibers and contractility, de novo expression of periostin and EDA-FN, increase of smooth muscle myosin heavy chain, vimentin, focal adhesion proteins, and elevated collagen secretion [23]. Lineage tracing experiments also identified that resident pericytes in the kidney [24] and those in the liver (hepatic stellate cells, HSCs) [18, 25] account for the major sources of fibrogenic myofibroblasts. Moreover, a new report indicates that Gli1+ mesenchymal stem cell (MSC)-like cells, which reside in the perivascular niche of many organs, substantially contribute to myofibroblasts and injury-induced organ fibrosis [26]. Interestingly, a novel population of Lin-integrin-α7-Sca1+PDGFRα+ multipotent mesenchymal progenitor cells, termed fibro/ adipogenic progenitors (FAPs), were recently identified [27]. These are the main source of myofibroblasts in injured skeletal muscle [28, 29], but are also found in multiple other tissues [30, 31].
Other cell types have also been proposed as precursors of myofibroblasts. Earlier reports suggested that epithelial cells undergoing EMT [21] or endothelial cells following EndoMT [32, 33] generate major populations of myofibroblasts. However, recent lineage tracing analyses refuted those assessments [1, 34]. While the origin of myofibroblasts from vascular SMCs has not been extensively studied, this paradigm has been suggested as an explanation for the pathogenesis of artery stiffness [35]. Fibrocytes, the circulating blood-borne cells similar to fibroblasts but expressing α-SMA and CD45, were reported to give rise to myofibroblasts [36]. However, using Collagen1a1-GFP as a robust cardiac fibroblast marker, a new report demonstrated that the majority of infarct fibroblasts are of epicardial origin but not derived from bone marrow lineages, EndoMT, or blood [37].
Despite many publications, the relative contributions of different cell sources to myofibroblasts and fibrosis have yet to be clearly defined in various organs or tissues [2]. Application of unambiguous lineage tracing technologies to distinguish the origins of myofibroblasts is an active direction of current research, and may successfully address this question [8, 38]. The answers will likely be contextually dependent on tissue and pathology [25], and precisely targeting a defined cell population for effective anti-fibrotic therapy may motivate extensive studies to delineate myofibroblastic mechanisms.
3. Myofibroblast-activating ECM Signaling
Defining the signal transduction that leads to myofibroblastic activation is inherently difficult due to the involvement of nearly all the major signaling pathways. Recent literature highlights a previously underappreciated role of ECM signaling as a primary mediator of myofibroblastic activation. While many signaling molecules are involved as myofibroblast activators, e.g. platelet derived growth factor and connective tissue growth factor [39, 40], TGF-β1 has been identified as mediating predominant impacts in most of the studies [41–43]. Therefore, we will focus on the TGF-β1 interactions with the ECM and its critical influence on myofibroblastic activation. Immune-mediated myofibroblastic activation involving TGF-β1 has been extensively investigated [44] but will not be included in the scope of this review.
3.1. Latent TGF-β1 “caged” in the ECM
Before maturation and activation, TGF-β is stored as a latent protein in the ECM, where it is bound to a latency associated peptide (LAP) that is conjugated to a latent TGF-β binding protein (LTBP) [45]. TGF-β1 and its pro-protein LAP-1 are translated from the same transcript. This pro-TGF-β1 dimerizes via disulfide bonding [46]. Subsequently, the linkage between LAP-1 and TGF-β1 is cleaved by a furin-like convertase and the LAP-1 dimer folds into a “straight jacket” that sterically trap the TGF-β1 dimer (Figure 1), resulting in the heterotetrameric small latent complex (SLC) [46]. The SLC is typically disulfide bonded with LTBP-1 at both LAP-1 monomers, forming the large latent complex (LLC), which is secreted and incorporated into the ECM. Four LTBPs exist in various tissues but with consensus high expression in elastic tissues such as heart, lung, and skeletal muscle [47]. They have different affinities to each of the three TGF-β proteins, and LTBP-1 is thought to sequester the most TGF-β1 in the ECM [48]. LTBP-2 is not known to conjugate to the TGF-β1 SLC; LTBP-3 and LTBP-4 have not been extensively studied for this function [49]. Aside from tethering the SLC to the ECM to mechanically secure TGF-β1’s latency [50], LTBP-1 may also serve a chaperone-like function to assist the folding and secretion of pro-TGF-β1 [51]. The LLC is conjugated to the ECM at LTBP-1 by transglutaminase [52]. The N-terminus of LTBP-1 is known to bind to FN [47, 49]. A recent study suggests that Fibrillin-1 binds to the C-terminal LTBP-1 at a site adjacent to the LAP-binding motif [53]. The Fibrillin-1 assembly into the ECM is FN dependent [48, 54]. The functional significance of TGF-β1 sequestration in the ECM is still not clear [45]. Studies provide interesting clues from Marfan syndrome, where Fibrillin-1 mutation leads to excessive TGF-β1 activation. This may be explained by insufficient Fibrillin-1 to keep TGF-β1 latent in the LLC, but the mechanisms can be far more complex [55].
3.2. Activation of TGF-β1
Compelling evidence indicates that the activation or “uncaging” of TGF-β1 from the ECM is a critical early step in driving myofibroblastic activation [42]. The release of mature TGF-β1 from the SLC can be triggered by a plethora of biological, physical, and physico-chemical factors (e.g. pH) [56], with integrins and proteases being well-established mediators [45]. The LAP protein contains an Arg-Gly-Asp (RGD) sequence which binds a number of integrins. All av integrins are known to bind RGD, with αvβ6 and αvβ8 as the best studied TGF-β1 activators [45]. A recent report demonstrates αvβ1 as another important TGF-β1 activator that binds LAP-1 [57]. The critical role of αv in myofibroblastic activation has been confirmed in several mouse models of organ fibrosis [57–59]. The literature to date on TGF-β1 activation involving αvβ6 is mostly focused on a traction-based mechanism [60]. As LAP-1 is anchored to the ECM via LTBP-1 and also bound to the cell surface through the binding of its RGD motif to αvβ6, a traction force between ECM and cells can deform the LAP-1 “straight jacket” thus liberating TGF-β1 [61] (Figure 1). Atomic structural information for this model has lately been updated [62]. This mechanism is also extrapolated to the “pulling” force generated by myofibroblast stress fiber contraction [63], or from ECM alone in a cell-free system [50]. There is evidence for an in vivo scenario where mechanical straining of the ECM primes the SLC (or LAP) for TGF-β1 activation triggered by cell contraction [64]. How αvβ8 activates ECM-caged TGF-β1 is less understood. Unlike αvβ6, the mechanism seems to involve recruitment of MMPs and protease activities independent of traction [65, 66].
Several proteinases are known to activate TGF-β1 without necessarily involving traction. MMP14 (aka: MT1-MMP) has been shown to degrade LAP-1 but is dependent on simultaneous cell-surface binding of the αvβ8 integrin to the RGD motif of LAP [67]. MMP-2 has been shown to directly cleave LAP-1 and LTBP-1 [68–70]. The role of MMP-9 is more complex, because it cleaves the soluble form but not ECM-bound form of LTBP-1 [71]. BMP1 has been shown to cleave LTBP-1 at two specific sites, liberating the LLC from ECM and resulting in activation of TGFβ1 after cleavage of LAP by non-BMP proteinases [69]. Serine proteinases, specifically plasmin, thrombin, elastase, chymase, trypsin, and kallikrein have been known for decades to cleave LTBP-1 and stimulate TGF-β1 activation [68, 72, 73]. However, the TGF-β1-activating mechanisms of these proteases are still not well understood in vivo in the context of myofibroblastic fibrosis. Thrombospondin-1, a non-protease protein, is well-documented in vitro and in vivo for its critical role in activating TGF-β1 and promoting fibrosis [74]. Its action likely involves an interaction with the N-terminal region of LAP-1 that alters the SLC conformation [75]. The other factors that turn TGF-β1 from latency to an active form have been updated by informative reviews [23, 45, 60].
3.3. The FN splice variant containing EDA (EDA-FN)
Based on studies using different precursor cells, three basic conditions are considered necessary for myofibroblastic activation [16], including strained ECM [76] and activated TGF-β1 [42, 77] as discussed above, and EDA-FN. Cellular but not plasma FN contains splice variants A (EDA or EIIIA) and B (EDB or EIIIB). EDA-FN is not expressed under normal conditions but is highly upregulated in myofibroblasts. While EDA-FN is recognized as necessary for myofibroblastic activation in various different precursors and fibrotic conditions in vitro and in vivo [78–82], EDB-FN is known as a hallmark of tumor angiogenesis [83]. It is worth noting that EDA-FN is reported to be dispensable for transformation of hepatic stellate cells and portal fibroblasts into myofibroblasts in culture and in mouse models of liver fibrosis [84], suggesting that the role of EDA could be compensated for by other factors.
It has been suggested that the insertion of EDA-FN into bulk fibronectin ECM may at a micro-level change overall material stress-strain properties [85]. The EDGIHEL motif at the EDA C’-C region binds to fibroblast-expressed integrins α4β1 and α9β1 [80, 86, 87]; the α4 and α9 subunits are considered the essential EDA-binding components [80, 88]. Another EDA-binding integrin, α4β7, has been found to be important in mouse lung myofibroblastic activation [89, 90], but the α4β7-binding EDA sequence has yet to be identified [89]. Of note, a recent report demonstrates that EDA-FN forms a signaling axis with Toll like receptor 4 (TLR4) driving fibrosis in scleroderma [81].
In addition to EDA and EDB, FN type III domains also contain RGD and PHSRN motifs. They have been shown to bind the canonical FN-recognition integrin α5β1 [91], as well as allbpi and α3β1 [92]. Interestingly, while the binding of LAP-1’s RGD with integrins facilitates TGF-β1 release from latency, as discussed above, the binding of FN’s RGD and its synergy site (the PHSRN motif) with integrins is thought to activate the integrin signaling that promotes FN filament assembly [92, 93]. However, whether this synergistic integrin-binding property of the RGD and PHSRN motifs plays an important role in TGF-β1 activation is not clear.
3.4. The TGF-β1/EDA-FN positive feedback
Activated TGF-β1 and EDA-FN positively affect and are affected by the cellular-mediated changes they induce in the ECM, essentially linking their functionality together in an ECM-cell feedback loop. It has been suggested that EDA-FN plays a positive role in activating TGF-β1signaling, possibly in multiple ways. First, there is direct evidence indicating that EDA plays a supporting role for the interaction of LTBP-1 with FN and its incorporation into the ECM [16, 64, 94]; blocking the EDA-FN/LTBP-1 interaction impairs TGF-β1 activation [16, 95]. The detailed molecular mechanism has yet to be revealed. Second, the EDA/integrin interaction has been reported to enhance the interaction of the LAP-1 RGD with integrins [96], implicating a role in facilitating the release of TGF-β1 from the ECM. In addition, in view of a network formed by protein-protein interactions in the LAP-1/LTBP-1/EDA-FN/integrin complex, it is an intriguing question as to whether perturbation of ECM may modify the LAP-1 conformation and hence the TGF-β1 activation status via the EDA-FN/integrin binding. Given the complexity of this ECM network and technical challenges, it will take careful future studies to delineate this potential mechanism. Moreover, further studies are also required to elucidate whether the EDA-FN binding with integrins assists with the incorporation of latent TGF-β1 into the ECM.
Interestingly, it has been shown that TGF-β1 directly regulates EDA-FN expression during myofibroblastic activation. For example, TGF-β1 has been identified as a modulating factor in EDA-FN splicing. Exposure of lung fibroblasts to TGF-β1 modulates serine-arginine-rich splicing factors (SRps), which influence the splicing of EDA into the cellular FN transcript. One study showed that active SRp40, SRp55, and SRp75 increased threefold and SRp20 increased eightfold after 24-hour TGF-β1 exposure. Additionally, several U5 proteins and helicases were modified. These changes modified EDA inclusion into the FN transcript [97]. This effect was further explored in a recent publication, where it was shown that TGF-β1 stimulates SRp40 via the PI3K-Akt pathway, which will be discussed later in this review. Akt was shown to directly bind SRp40, which bound the EDA-FN exon splicing enhancer on pre-mRNA and significantly increased the proportion of total cFN containing an EDA domain [98]. The observation in both of these studies that TGF-β1 mediates cellular redistribution of SRp proteins is intriguing and potentially worth further study, along with the signaling pathways governing this TGF-β1 guided induction of EDA-FN.
Due to the extracellular availability and high expression in fibrosis, EDA-FN has been used clinically as an indicator of fibrotic disease and tissue remodeling in humans [99]. Moreover, EDA function-blocking antibodies and competitive peptides inhibit activation of TGF-β1 and myofibroblasts [16, 100]. As such, the relationship of TGF-β1 and EDA-FN signaling pathways will continue to attract research interest directed toward the identification of new mechanisms for therapeutic targeting.
4. Signal Transmission and Integration at the Cell Membrane
At the plasma membrane, the TGF-β receptor complex is activated by TGF-β1 that is released from the ECM; Integrins interact with EDA-FN and the ECM in a mechanoreceptive manner. These two major events integrate to transmit ECM signals into the cell resulting in RhoA-stimulated stress fiber formation and transcriptional/ posttranscriptional reprograming that orchestrate myofibroblastic activation.
4.1. TGF-β1 signaling via TGF-β receptors
In the canonical pathway, TGF-β1 activates the TGF-β receptor complex which in turn phosphorylates Smad2 and Smad3. These two signaling proteins then complex with Smad4 and translocate to the nucleus to initiate transcription of many genes [46, 101]. The Smad2/3 signaling is known to elevate the expression of ECM-remodeling proteins such as collagen I and tissue inhibitor of metalloproteinases 2 (TIMP2)-an important regulator of MMP activity [102]. However, the role of this canonical TGF-β pathway in myofibroblastic activation was not confirmed in vivo until last year [103]. This study demonstrated that cardiac fibroblast-specific Smad2/3 deletion in mice inhibits the fibrogenic gene program and ECM remodeling [103]. Myofibroblastic transcription can also be activated through non-Smad pathways such as ERK and p38 [104, 105]. Revealing a crosstalk between canonical and non-canonical TGF-β pathways, a most recent report showed that TGF-β1 induced human lung fibroblast activation through the Raf1/ERK/Smad pathway [106]. Given the intricate TGF-β1 downstream signaling networks, the pathway(s) that mediates TGF-β1-induced myofibroblastic activation is likely tissue/signaling context dependent [107].
Since TGF-β1 activates intracellular signaling via TGF-β receptors, regulation of these receptors has profound influence on myofibroblastic activation [4]. TGF-βR endocytosis and degradation via clatherin-dependent or caveolin-1 dependent processes have been well established [108]. A relatively new thread of knowledge is that the TGF-βRI and TGF-βRII activities are regulated by their dissociation and association. In a recent report whereas TGF-βRI was found to be selectively enriched at focal adhesions, TGF-βRII was selectively excluded [109]. Reduction in Rho/ROCK signaling, as mediated by decrease in cellular tension, diminished this segregation and allows the formation of complete TGF-βRI/TGF-βRII complexes. Further, the av integrin subunit was shown to form a tension-mediated complex with TGF-βRI, TGF-βRII, and cofilin, although the significance of this regulation remains elusive. Taken together, these results suggest that the TGF-βR complex is modulated in part by mechanotransduction and that specific integrins, such αv -that are known to activate ECM-tethered TGF-β-may have multifaceted roles in the activation of TGF-β1 signaling.
4.2. Integrin signaling through focal adhesions
The ability for diverse isoforms of integrins to cluster and to mechanotransduce ECM mechanical, topographical, or motif-directed cues is primarily mediated by the formation of focal adhesions (FAs) [13, 110]. These structures were previously described as maturing by gaining increasing mechanical strength and α-SMA incorporation throughout the morphological development of myofibroblasts. Focal adhesions are dynamic structures known to consist of over 150 proteins including focal adhesion kinase (FAK) and Src, another tyrosine kinase. FAK activation is essential for mechanotransduction of integrin signaling during myofibroblastic activation. Activated integrins allow for autophosphorylation of FAK and its binding with Src, which results in mutual activation of the kinase domains of both molecules [111, 112]. They link clusters of integrins at the plasma membrane to the cytoskeleton and mechanoreceptive cytosolic signaling factors.
Focal adhesion biology is extremely complex. Focal adhesion mechanotransduction of myofibroblast pathways has been extensively investigated yet the inner workings of their subcomponents are not well understood [113]. The canonical integrin-FAK-Src signaling and downstream RhoA-activating pathways have been frequently reviewed [114–116]. Therefore, we will only highlight Hic-5, a paxillin family member that has recently garnered new attention as an essential component of myofibroblastic activation [117, 118]. Otherwise known as TGF-β1-induced transcript 1 (TGFB1H) [119], Hic-5 is induced by TGF-β1 and also induces TGF-β1 transcription [120] and regulates the coupling between focal adhesion activities and ECM degradation [121]. The Hic-5 protein shuttles between focal adhesions and the nucleus [122] in a process at least partially dependent on cellular mechanical stress [123, 124]. While knockdown of Hic-5 inhibits myofibroblastic activation, its precise regulatory role is only beginning to be unveiled. Recent studies show that Hic-5 transcription is directly regulated by and enhances myocardin-related transcription factor A (MRTF-A) nuclear accumulation [125], a downstream factor induced by non-canonical TGF-β1 signaling and mechanical stress (to be discussed later). Further, it appears that both MRTF-A and Hic-5 are required for the induction of α-SMA and formation of super-mature focal adhesions. MRTF-dependent Hic-5 expression is also regulated in a Smad3-dependent manner, although the exact mechanism is still unclear [117]. Signaling pathways connecting Hic-5 to α-SMA expression have yet to be elucidated, but are likely dominated by Hic-5 synergizing with extracellular stress to promote RhoA activation. Hic-5 may also affect myofibroblastic activation indirectly through modulating steroid receptor transcription [122, 126].
4.3. Crosstalk between focal adhesions and TGF-α signaling
The activities of integrins/focal adhesions have been shown to engage in crosstalk with TGF-β1 signaling. It is known that FAK-activated Src facilitates the activation of phosphoinositide 3-kinase (PI3K), a lipid inositol-modifying heterodimer that phosphorylates PIP2 into PIP3 [127] [128]. This further activates the downstream Akt1/mTOR pathway [129–131]. The phosphatase PTEN abolishes PI3K signaling by catalyzing the PIP3 conversion back to PIP2. TGF-β1-dependent alternative splicing of EDA-FN has been shown as dependent on the PI3K activation of Akt, which binds to the EDA-FN-inducing enzyme SRp40 [98]. As activated Akt1 potently induces EDA-FN production in addition to total FN [132], it appears possible that EDA-FN induces its own expression via FAK-PI3K-Akt activation mediated by the EDA/integrin interaction, thus resulting in a matrix-mediated positive feedback loop. While TGF-β1 modulates the PI3K-Akt pathway via FAK activation [133], its inhibition of PTEN has also been shown as necessary for both TGF-β1-induced α-SMA expression and EDA-FN production [134]. However, this interaction itself inhibits Smad-based TGF-β1 signaling, as decreased PTEN results in increased Smad7 which promotes TGF-βR degradation [22, 135].
The FAK/Src axis has also been linked to TGF-β1 signaling in stress fiber formation of myofibroblasts via TGF-βRs. Sequestering of Src by FAK decreases the activation of dynamin, a protein essential for endocytosis [136], and may hence reduce TGF-βR degradation [137]. Additionally, it has been shown that TGF-β1 signaling results in TGF-βR/Src-dependent association of RhoA and calveolin-1, activating RhoA, a necessary signaling step for TGF-β1-induced fibronectin synthesis [138]. Another recent study has shown that Src activation is an essential component for facilitating the profibrotic effects of TGF-β1, including the expression of α-SMA and the formation of stress fibers [139].
Interestingly, TGF-β1stimulation can robustly upregulate Pyk2 expression that leads to EMT [140]. Since Pyk2 is an FAK homolog that enhances RhoA activation [141], it is possible that TGF-β1 signaling may activate RhoA in an FAK-independent manner. Independent and compensatory regulations of downstream RhoA effectors by TGF-β1 and FAK signaling may provide an explanation for the ability of either TGF-β1 or EDA-FN to fully and independently induce myofibroblastic activation in vivo.
5. Intracellular Signaling
A cascade of signaling events downstream of integrin/FAK/Src involving ERK and MAPK pathways are known to induce myofibroblastic activation [142, 143]. Moreover, FAK and Src facilitate the RhoA GTPase activation by regulating guanine nucleotide exchange factors (GEFs) and GTPases-activating proteins (GAPs) in response to mechano-stimulation [144]. Updates on these pathways related to myofibroblasts have been covered by excellent recent reviews [143, 145, 146]. Therefore, we will focus on activated RhoA and downstream pathways responsible for α-SMA stress fiber production and associated nuclear regulations.
5.1. RhoA signaling in actin stress fiber formation
Rho GTPases have been shown to control the assembly of F-actin in all eukaryotic species examined to date. The best-characterized Rho-like proteins are RhoA, Rac1, and Cdc42. We will focus on RhoA because its activation is primarily mediated by mechanotransduction factors and is a specific requirement in the induction of α-SMA stress fiber formation in myofibroblastic activation [147]. RhoA activates the signaling molecules ROCK and mDia. ROCK phosphorylates and inactivates the myosin light chain phosphatase (MLCP) and can directly phosphorylate and activate the myosin light chain. Both of these ROCK actions facilitate the activation of non-muscle myosin II at focal adhesions [148, 149]. Additionally, ROCK further activates LIM kinases, which phosphorylate and inhibit the actin-cleaving enzyme cofilin, thereby decreasing actin degradation and while increasing the torsional rigidity of existing actin filaments [150]. While ROCK induces stability and myosin accumulation, mDia acts in a VASP-dependent manner to polymerize actin and allows the formation of lengthy filaments [151, 152]. These molecules balance the activity of one another as mDia induces a Src-dependent Cas/Crk/DOCK180 cascade resulting in decreased ROCK activity [153, 154]. Together, ROCK and mDia pathways interact to induce the formation of stress fibers via actin polymerization.
Several reports have shown that TGF-βRI phosphorylates and activates Par6. Subsequently, Par6 can dimerize with Smurf1, and the resulting complex can target RhoA at tight junctions for its ubiquitination and degradation. This process facilitates the dissolution of tight junctions and has been determined to be necessary for TGF-β1 induced EMT [155, 156]. As this process contradicts the necessity for RhoA-induced α-SMA stress fiber formation, the regulations of RhoA in EMT and in myofibroblastic activation are likely spatially and/or temporally separate processes.
5.2. MRTF-directed nuclear signaling
It has been well documented that the activation of serum response factor (SRF), a transcription factor, is essential for the transcription program that drives myofibroblastic activation [157]. RhoA activation facilitates the nuclear translocation of SRF co-activators, with MRTFs (A and B) being prominently important for instigating myofibroblastic phenotypes [158].
Factors known to directly interact with SRF include Myocardin (MyoCD), MRTFs, Elkl, β-catenin, YAP (Yes-activating protein)/TAZ (transcriptional co-activator with PDZ binding motif), and Smad3. MyoCD is an SMC and cardiac muscle specific SRF cofactor required for significant activation of SRF-directed transcription and is constitutively localized to the nucleus. Acting in a similar fashion as MyoCD, tissue ubiquitous MRTF is a cofactor for SRF activation. However, unlike MyoCD, MRTF is typically localized in the cytoplasm and bound to G-actin in an inactive state. RhoA stimulates the assembly of actin stress fibers thereby reducing the G-actin/F-actin ratio. Consequently, MRTF is freed from G-actin sequestration and is allowed to translocate to the nucleus, where it activates SRF-mediated transcription of myofibroblastic factors such as α-SMA (Figure 1). Elk1 acts as an inhibitor of MRTF, through direct antagonistic SRF binding or through operator binding adjacent to the SRF-binding CArG motif.
Recent research reveals an interplay between canonical TGF-β1 signaling and MRTF in mechanotransduction [147], e.g. nuclear Smad3 recruits GSK-3β to phosphorylate MRTF leading to subsequent ubiquitination and degradation of MRTF [159]. Regulations of MRTF stability also link the Wnt/β-catenin and YAP/TAZ pathways to TGF-β1 signaling in the nucleus, as discussed below.
5.3. MRTF-regulatory nuclear factors
Wnt cytokines act canonically through the Frizzled receptor complex and activate the scaffolding protein Disheveled, which degrades Axin thereby inhibiting constitutive destruction of β-catenin, which can then translocate to the nucleus [160]. Nuclear translocation of β-catenin has been linked to increased expression of TGF-β1 in addition to α-SMA [157, 161–164].
The YAP/TAZ pathway has been recognized as sensitive to mechanotransduction. Inhibition of YAP/TAZ signaling blocked fibroblast-to-myofibroblast transformation and renal fibrosis in mice [165]. Similar to β-catenin, inhibited YAP/TAZ degradation promotes their translocation to the nucleus where they complex with the transcription factor TEAD to co-activate transcription [166–168]. There is evidence that cellular tension increases YAP/TAZ nuclear concentrations [5]. Interestingly, while RhoA and the actin cytoskeleton are thought to be necessary to maintain YAP/TAZ activation, this complex does not seem to be directly regulated by G-actin/F-actin levels, implicating either upstream RhoA interaction or stress fiber interaction with an unknown mediator [169, 170].
Another pathway recently implicated in myofibroblastic activation involves zinc finger E-box-binding homeobox 2 (ZEB2) protein [171], which is a transcription factor and constitutive repressor of Meox½. Reduced expression of Meox½ is associated with increased expression of EDA-FN and α-SMA during myofibroblastic activation [172].
Studies suggest that these major nuclear factors may all crosstalk with the TGF-β1/Smad3 pathway at the resolution of nuclear MRTF stability, which is highly regulated. While nuclear Smad3 has been shown to promote MRTF-A degradation via GSK-3β-mediated phosphorylation followed by ubiquitination, this action is inhibited by nuclear β-catenin [159, 160, 173]. Moreover, nuclear-localized YAP/TAZ bind MRTF-A and enhance its activity while down-regulating Smad3 [163, 174]. The mechanisms by which these functions occur have yet to be clearly addressed. Despite Smad3 nuclear inhibition of MRTF, TGF-β1 has been shown to increase nuclear MRTF concentration as mentioned above. Whether this TGF-β1 action is executed via Smad or non-Smad pathways is unclear. The context-dependent role of TGF-β1 signaling in MRTF regulation warrants further investigation. In addition, the TGF-β1/Smad3 pathway inhibitor, Ski, has been found to bind and repress ZEB2 thereby derepressing Meox2 and suppressing myofibroblastic activation [175]. Though contradicting a preconceived inhibitory effect on myofibroblasts, this observation implicates an emerging significance of Ski in stimulating myofibroblasts. Whether Ski and ZEB2 directly regulate MRTF in the myofibroblastic context remains an open question.
5.4. Epigenetic regulation
Beyond aforementioned classical pathways, interest in epigenetic regulators, including DNA and histone modifiers and readers of the modifications [176, 177], microRNAs [178], and long noncoding RNAs [179], is rapidly growing. These chromatin-associated factors exert profound influence on transcriptomic dynamics and cell state transition without involving DNA sequence changes [14]. It has been demonstrated that both genetic and pharmacological inhibition of DNMT (DNA methyltransferase) mitigates mouse renal myofibroblasts and fibrosis [180, 181]. Inhibitors of HDACs have shown anti-fibrotic effects in several mouse models [182]. Whereas miR29a mitigates [183], miR21 aggravates [184] TGF-β1-induced myofibroblastic programs, both expected as potential interventional targets [182, 185]. Interestingly, there is also evidence suggesting that miR29a is a possible mediator of the EDA-FN-driven fibrosis in scleroderma [81]. More recently, the bromo and extraterminal domain (BET) family of epigenetic readers (binding to acetyl marks on histones) have attracted significant attention due to their potentially crucial role in regulating myofibroblastic activation [186–188]. It has been observed that pharmacological blockage of the BET family effectively inhibits fibrosis in the liver, kidney, and heart [187, 189–191].
How the epigenetic regulators cooperate with the aforementioned nuclear factors in fibrogenic pathways is an intriguing question that is soliciting growing research. As opposed to traditional perception that epigenetic factors regulate transcription globally, accumulating evidence suggests significant specificity of those regulations depending on cell type, extracellular cue and signaling context [192]. This feature may open a way for future epigenetic interventions to mitigate fibrotic progression with careful assessments of possible side effects [182, 190].
6. Other Myofibroblastic Regulators
Aside from SMA and EDA-FN, many other products resulting from myofibroblastic nuclear reprogramming in turn modulate the ECM architecture and signaling. They participate in the inside-out signal transmission that further accentuate myofibroblastic activation. Given a large number of those products, we will only highlight a few that have recently re-energized myofibroblast research.
6.1. Hyaluronic Acid
Evidence suggests that the ECM glycosaminoglycan hyaluronic acid (HA) and its receptor CD-44 are involved in myofibroblastic activation [193]. It has been shown that high molecular weight HA inhibits fibrotic pathways but low molecular weight HA appears to induce them [194]. Interestingly, naked mole rats express unique HA molecules that are five times the length of those in humans or mice, and they have seven-fold longer domestic lifespan than mice [195]. While the large HA confers cancer resistance in naked mole rats, the relationship between unique HA molecules and lifespan is not clear [196]. High molecular weight HA has been shown to induce CD-44-assisted translocation of activated TGF-βR to caveolin-1 lipid rafts, facilitating the previously discussed Smurf1/Smad7-mediated TGF-βR degradation [197]. However, inhibition of HA synthase (HAS) also antagonizes TGF-β1-induced expression of α-SMA [198]. Moreover, TGF-β1-mediated HAS2 activation results in some aspects of myofibroblastic activation [193]. These results seem to contradict the previously observed inhibition of myofibroblasts by the HA-CD-44 signaling [197]. Alternatively, as a viscous molecule, HA may also regulate myofibroblastic activation through its interactions with the ECM [199].
6.2. Matricellular proteins
Matricellular proteins reside in the ECM but do not serve a primary structural role [200]. They modulate the activities of proteases, cytokines/growth factors, and cell-surface receptors [201]. Regarded as a new myofibroblast marker in lineage-tracing studies [3], periostin prevents ventricular rupture but also contributes to myofibroblast activation and cardiac fibrosis [202, 203]. Secreted modular calcium-binding protein 2 (SMOC2) is a member of the secreted protein acidic and rich in cysteine (SPARC) family, and was recently found to promote myofibroblast-mediated fibrosis in the mouse retina [204]. Syndecans comprise a family of transmembrane proteins, their extracellular domains interacting with the ECM. Whereas beneficial effects of over-expressing Syndecan-4 have been found in preventing mouse lung [205] and cardiac [206] fibroblast-to-myofibroblast transition and subsequent fibrosis, opposite effects in mouse renal fibrosis have also been reported [207], likely involving its ectodomain shedding [208]. While more detailed mechanistic studies are required, these and other new findings exhibit an attractive potential of matricellular proteins as new interventional targets in the battle against fibrosis.
7. Conclusion
TGF-β1 signaling is generally regarded as the most important activator of myofibroblasts [6, 42, 43]. Latent TGF-β1 is stored in the ECM, and its release and binding to TGF-βRs transmit ECM signaling cues into the cell. This incites transcriptional and posttranscriptional reprograming (e.g. EDA-FN splicing) and cytosolic remodeling (e.g. RhoA activation and stress fiber assembly). These orchestrated events in turn lead to increased ECM accumulation and mechano-responsive activities, and ultimately, further TGF-β1 activation via the integrin/ECM interaction network. Therefore, TGF-β1-activated pathways in conjunction with mechanotransduction constitute outside-in and inside-out signaling loops that potently propagate myofibroblastic activation. As myofibroblasts mediate fibrosis in a broad range of pathologies, methods to break these vicious cycles may result in viable therapeutic options. We have therefore reviewed recent literature on myofibroblastic signaling mechanisms with a primary focus on TGF-β1 and associated pathways in the context of ECM/cell signaling integration. While studies have revealed several ECM-cell-ECM and intracellular positive feedback loops, antagonizing mechanisms have also been noted. In the myofibroblastic intracellular signaling network, RhoA is a cytosolic hub protein that perpetuates α-SMA stress fibers. This RhoA effect is converted into transcriptional reprogramming via nuclear translocation of MRTF-A, another hub protein that integrates the modulations from other nuclear factors including Smad3, β-catenin, and YAP/TAZ. Encouraging therapeutic progress has recently been achieved in preclinical models by targeting the major myofibroblast pathways to ameliorate fibrosis [10, 13, 17, 41, 57, 209]. More in-depth investigations into the myofibroblastogenic mechanisms in vitro and in vivo are required. New knowledge should help create effective anti-fibrotic therapies by precisely targeting the signaling vicious cycles that are propagated by defined myofibroblast or precursor populations.
Acknowledgments
Funding:
This work was supported by NIH grants HL133665, HL129785 and EY022678 (to L.-W. G.)
Abbreviations
- α-SMA
α-smooth muscle actin
- BET
bromo and extraterminal domain containing epigenetic readers
- ECM
extracellular matrix
- EDA-FN
extra domain A-containing fibronectin
- EMT
epithelial-to-mesenchymal transition
- FA
focal adhesion
- FAK
FA Kinase
- FN
fibronectin
- GEF
guanine nucleotide exchange factor
- HA
hyaluronic acid
- HAS
hyaluronic acid synthase
- LAP
latency associated peptide
- LLC
large latent complex
- LTBP
latent TGF-β binding protein
- MRTF
myocardin-related transcription factor
- MSC
mesenchymal stem cell
- MyoCD
myocardin
- RGD
the tripeptide sequence of Arg-Gly-Asp
- SLC
small latent complex
- SMC
smooth muscle cells
- SRF
serum response factor
- TGF-β
transforming growth factor
- TGF-βR
TGF-β receptor
- TAZ
transcriptional coactivator with PDZ binding motif
- YAP
Yes-activating protein
- ZEB2
zinc finger E-box-binding homeobox 2.
References
- 1.Habiel DM, Hogaboam CM: Heterogeneity of Fibroblasts and Myofibroblasts in Pulmonary Fibrosis. Curr Pathobiol Rep 2017;5:101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kisseleva T: The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017;65:1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, Molkentin JD: Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nature communications 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu C, Billadeau DD, Abdelhakim H, Leof E, Kaibuchi K, Bernabeu C, Bloom GS, Yang L, Boardman L, Shah VH, Kang N: IQGAP1 suppresses TbetaRII-mediated myofibroblastic activation and metastatic growth in liver. The Journal of clinical investigation 2013;123:1138–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma H, Killaars AR, DelRio FW, Yang C, Anseth KS: Myofibroblastic activation of valvular interstitial cells is modulated by spatial variations in matrix elasticity and its organization. Biomaterials 2017;131:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lagares D, Ghassemi-Kakroodi P, Tremblay C, Santos A, Probst CK, Franklin A, Santos DM, Grasberger P, Ahluwalia N, Montesi SB, Shea BS, Black KE, Knipe R, Blati M, Baron M, Wu B, Fahmi H, Gandhi R, Pardo A, Selman M, Wu J, Pelletier JP, Martel-Pelletier J, Tager AM, Kapoor M: ADAM10-mediated ephrin-B2 shedding promotes myofibroblast activation and organ fibrosis. Nature medicine 2017;23:1405–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El Agha E, Kramann R, Schneider RK, Li X, Seeger W, Humphreys BD, Bellusci S: Mesenchymal Stem Cells in Fibrotic Disease. Cell stem cell 2017;21:166–177. [DOI] [PubMed] [Google Scholar]
- 8.Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ: Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat Rev Nephrol 2015;11:233–244. [DOI] [PubMed] [Google Scholar]
- 9.Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, Tan J, Chothani SP, Ye L, Rackham OJL, Ko NSJ, Sahib NE, Pua CJ, Zhen NTG, Xie C, Wang M, Maatz H, Lim S, Saar K, Blachut S, Petretto E, Schmidt S, Putoczki T, Guimaraes-Camboa N, Wakimoto H, van Heesch S, Sigmundsson K, Lim SL, Soon JL, Chao VTT, Chua YL, Tan TE, Evans SM, Loh YJ, Jamal MH, Ong KK, Chua KC, Ong BH, Chakaramakkil MJ, Seidman JG, Seidman CE, Hubner N, Sin KYK, Cook SA: IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017;552:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jun JI, Lau LF: Resolution of organ fibrosis. The Journal of clinical investigation 2018;128:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, Sirven P, Magagna I, Fuhrmann L, Bernard C, Bonneau C, Kondratova M, Kuperstein I, Zinovyev A, Givel AM, Parrini MC, Soumelis V, Vincent-Salomon A, Mechta-Grigoriou F: Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer cell 2018;33:463–479 e410. [DOI] [PubMed] [Google Scholar]
- 12.Herrera J, Henke CA, Bitterman PB: Extracellular matrix as a driver of progressive fibrosis. The Journal of clinical investigation 2018;128:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lampi MC, Reinhart-King CA: Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci Transl Med 2018;10 [DOI] [PubMed] [Google Scholar]
- 14.Tschumperlin DJ, Ligresti G, Hilscher MB, Shah VH: Mechanosensing and fibrosis. The Journal of clinical investigation 2018;128:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pakshir P, Hinz B: The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol 2018 [DOI] [PubMed] [Google Scholar]
- 16.Klingberg F, Chau G, Walraven M, Boo S, Koehler A, Chow ML, Olsen AL, Im M, Lodyga M, Wells RG, White ES, Hinz B: The fibronectin ED-A domain enhances recruitment of latent TGF-beta-binding protein-1 to the fibroblast matrix. J Cell Sci 2018;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemos DR, Duffield JS: Tissue-resident mesenchymal stromal cells: Implications for tissue-specific antifibrotic therapies. Sci Transl Med 2018;10 [DOI] [PubMed] [Google Scholar]
- 18.Kramann R, DiRocco DP, Humphreys BD: Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J Pathol 2013;231:273–289. [DOI] [PubMed] [Google Scholar]
- 19.Di Carlo SE, Peduto L: The perivascular origin of pathological fibroblasts. The Journal of clinical investigation 2018;128:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phan SH: Biology of fibroblasts and myofibroblasts. Proceedings of the American Thoracic Society 2008;5:334–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalluri R, Weinberg RA: The basics of epithelial-mesenchymal transition. The Journal of clinical investigation 2009;119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White ES, Atrasz RG, Hu B, Phan SH, Stambolic V, Mak TW, Hogaboam CM, Flaherty KR, Martinez FJ, Kontos CD, Toews GB: Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10). American journal of respiratory and critical care medicine 2006;173:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinz B: Myofibroblasts. Experimental eye research 2016;142:56–70. [DOI] [PubMed] [Google Scholar]
- 24.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS: Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010;176:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, Liu X, Xu J, Wang P, Paik YH, Meng F, Asagiri M, Murray LA, Hofmann AF, Iida T, Glass CK, Brenner DA, Kisseleva T: Origin of myofibroblasts in the fibrotic liver in mice. Proceedings of the National Academy of Sciences of the United States of America 2014;111:E3297–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD: Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell stem cell 2015;16:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM: Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 2010;12:153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemos DR, Babaeijandaghi F, Low M, Chang CK, Lee ST, Fiore D, Zhang RH, Natarajan A, Nedospasov SA, Rossi FM: Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nature medicine 2015;21:786–794. [DOI] [PubMed] [Google Scholar]
- 29.Mueller AA, van Velthoven CT, Fukumoto KD, Cheung TH, Rando TA: Intronic polyadenylation of PDGFRalpha in resident stem cells attenuates muscle fibrosis. Nature 2016;540:276–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lees-Shepard JB, Yamamoto M, Biswas AA, Stoessel SJ, Nicholas SE, Cogswell CA, Devarakonda PM, Schneider MJ Jr., Cummins SM, Legendre NP, Yamamoto S, Kaartinen V, Hunter JW, Goldhamer DJ: Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nature communications 2018;9:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kopinke D, Roberson EC, Reiter JF: Ciliary Hedgehog Signaling Restricts Injury-Induced Adipogenesis. Cell 2017;170:340–351 e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R: Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature medicine 2007;13:952–961. [DOI] [PubMed] [Google Scholar]
- 33.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR: Conversion of vascular endothelial cells into multipotent stem-like cells. Nature medicine 2010;16:1400–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL: Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proceedings of the National Academy of Sciences of the United States of America 2011;108:E1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crosas-Molist E, Meirelles T, Lopez-Luque J, Serra-Peinado C, Selva J, Caja L, Gorbenko Del Blanco D, Uriarte JJ, Bertran E, Mendizabal Y, Hernandez V, Garcia-Calero C, Busnadiego O, Condom E, Toral D, Castella M, Forteza A, Navajas D, Sarri E, Rodriguez-Pascual F, Dietz HC, Fabregat I, Egea G: Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arteriosclerosis, thrombosis, and vascular biology 2015;35:960–972. [DOI] [PubMed] [Google Scholar]
- 36.Abu El-Asrar AM, De Hertogh G, van den Eynde K, Alam K, Van Raemdonck K, Opdenakker G, Van Damme J, Geboes K, Struyf S: Myofibroblasts in proliferative diabetic retinopathy can originate from infiltrating fibrocytes and through endothelial-to-mesenchymal transition (EndoMT). Experimental eye research 2015;132:179–189. [DOI] [PubMed] [Google Scholar]
- 37.Moore-Morris T, Cattaneo P, Guimaraes-Camboa N, Bogomolovas J, Cedenilla M, Banerjee I, Ricote M, Kisseleva T, Zhang L, Gu Y, Dalton ND, Peterson KL, Chen J, Puceat M, Evans SM: Infarct Fibroblasts Do Not Derive From Bone Marrow Lineages. Circulation research 2018;122:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greenhalgh SN, Iredale JP, Henderson NC: Origins of fibrosis: pericytes take centre stage. F1000Prime Rep 2013;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonner JC: Regulation of PDGF and its receptors in fibrotic diseases. Cytokine & growth factor reviews 2004;15:255–273. [DOI] [PubMed] [Google Scholar]
- 40.Gressner OA, Gressner AM: Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver international : official journal of the International Association for the Study of the Liver 2008;28:1065–1079. [DOI] [PubMed] [Google Scholar]
- 41.Kang JH, Jung MY, Yin X, Andrianifahanana M, Hernandez DM, Leof EB: Cell-penetrating peptides selectively targeting SMAD3 inhibit profibrotic TGF-beta signaling. The Journal of clinical investigation 2017;127:2541–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng XM, Nikolic-Paterson DJ, Lan HY: TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol 2016;12:325–338. [DOI] [PubMed] [Google Scholar]
- 43.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA: Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circulation research 2004;95:253–260. [DOI] [PubMed] [Google Scholar]
- 44.Velnar T, Bailey T, Smrkolj V: The wound healing process: an overview of the cellular and molecular mechanisms. Journal of International Medical Research 2009;37:1528–1542. [DOI] [PubMed] [Google Scholar]
- 45.Robertson IB, Rifkin DB: Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb Perspect Biol 2016;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA: Latent TGF-beta structure and activation. Nature 2011;474:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB: Latent TGF-beta-binding proteins. Matrix biology : journal of the International Society for Matrix Biology 2015;47:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Couroussé T, Sakai LY, Rifkin DB: Specificity of latent TGF -β binding protein (LTBP) incorporation into matrix: Role of fibrillins and fibronectin. Journal of cellular physiology 2012;227:3828–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Courousse T, Sakai LY, Rifkin DB: Specificity of latent TGF-beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. Journal of cellular physiology 2012;227:3828–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buscemi L, Ramonet D, Klingberg F, Formey A, Smith-Clerc J, Meister JJ, Hinz B: The single-molecule mechanics of the latent TGF-beta1 complex. Curr Biol 2011;21:2046–2054. [DOI] [PubMed] [Google Scholar]
- 51.Miyazono K, Olofsson A, Colosetti P, Heldin CH: A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO J 1991;10:1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rifkin DB: Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. The Journal of biological chemistry 2005;280:7409–7412. [DOI] [PubMed] [Google Scholar]
- 53.Robertson IB, Dias HF, Osuch IH, Lowe ED, Jensen SA, Redfield C, Handford PA: The N-Terminal Region of Fibrillin-1 Mediates a Bipartite Interaction with LTBP1. Structure 2017;25:1208–1221 e1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM: Fibronectin regulates latent transforming growth factor-β (TGFβ) by controlling matrix assembly of latent TGFβ-binding protein-1. Journal of Biological Chemistry 2005;280:18871–18880. [DOI] [PubMed] [Google Scholar]
- 55.Zilberberg L, Phoon CK, Robertson I, Dabovic B, Ramirez F, Rifkin DB: Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proceedings of the National Academy of Sciences of the United States of America 2015;112:14012–14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang XX, Ostedgaard LS, Hoegger MJ, Moninger TO, Karp PH, McMenimen JD, Choudhury B, Varki A, Stoltz DA, Welsh MJ: Acidic pH increases airway surface liquid viscosity in cystic fibrosis. The Journal of clinical investigation 2016;126:879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reed NI, Jo H, Chen C, Tsujino K, Arnold TD, DeGrado WF, Sheppard D: The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci Transl Med 2015;7:288ra279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, Maher JJ, Iredale JP, Lacy-Hulbert A, Adams RH, Sheppard D: Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nature medicine 2013;19:1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murray IR, Gonzalez ZN, Baily J, Dobie R, Wallace RJ, Mackinnon AC, Smith JR, Greenhalgh SN, Thompson AI, Conroy KP, Griggs DW, Ruminski PG, Gray GA, Singh M, Campbell MA, Kendall TJ, Dai J, Li Y, Iredale JP, Simpson H, Huard J, Peault B, Henderson NC: alphav integrins on mesenchymal cells regulate skeletal and cardiac muscle fibrosis. Nature communications 2017;8:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hinz B: The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol 2015;47:54–65. [DOI] [PubMed] [Google Scholar]
- 61.Annes JP, Chen Y, Munger JS, Rifkin DB: Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. The Journal of cell biology 2004;165:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, Engen JR, Springer TA: Force interacts with macromolecular structure in activation of TGF-beta. Nature 2017;542:55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wipff PJ, Rifkin DB, Meister JJ, Hinz B: Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. The Journal of cell biology 2007;179:1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klingberg F, Chow ML, Koehler A, Boo S, Buscemi L, Quinn TM, Costell M, Alman BA, Genot E, Hinz B: Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. The Journal of cell biology 2014;207:283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL: The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. The Journal of cell biology 2002;157:493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang J, Dong X, Zhao B, Li J, Lu C, Springer TA: Atypical interactions of integrin alphaVbeta8 with pro-TGF-beta1. Proceedings of the National Academy of Sciences of the United States of America 2017;114:E4168–E4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL: The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-β1. The Journal of cell biology 2002;157:493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jenkins G: The role of proteases in transforming growth factor-β activation. The international journal of biochemistry & cell biology 2008;40:1068–1078. [DOI] [PubMed] [Google Scholar]
- 69.Ge G, Greenspan DS: BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. The Journal of cell biology 2006;175:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kobayashi T, Kim H, Liu X, Sugiura H, Kohyama T, Fang Q, Wen F-Q, Abe S, Wang X, Atkinson JJ: Matrix metalloproteinase-9 activates TGF-β and stimulates fibroblast contraction of collagen gels. American Journal of Physiology-Lung Cellular and Molecular Physiology 2014;306:L1006–L1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mott JD, Werb Z: Regulation of matrix biology by matrix metalloproteinases. Current opinion in cell biology 2004;16:558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dallas SL, Rosser JL, Mundy GR, Bonewald LF: Proteolysis of latent transforming growth factor-beta (TGF-beta )-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. The Journal of biological chemistry 2002;277:21352–21360. [DOI] [PubMed] [Google Scholar]
- 73.Saharinen J, Taipale J, Keski-Oja J: Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. The EMBO journal 1996;15:245. [PMC free article] [PubMed] [Google Scholar]
- 74.Kim CW, Pokutta-Paskaleva A, Kumar S, Timmins LH, Morris AD, Kang DW, Dalal S, Chadid T, Kuo KM, Raykin J, Li H, Yanagisawa H, Gleason RL, Jr., Jo H, Brewster LP: Disturbed Flow Promotes Arterial Stiffening Through Thrombospondin-1. Circulation 2017;136:1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumar R, Mickael C, Kassa B, Gebreab L, Robinson JC, Koyanagi DE, Sanders L, Barthel L, Meadows C, Fox D, Irwin D, Li M, McKeon BA, Riddle S, Dale Brown R, Morgan LE, Evans CM, Hernandez-Saavedra D, Bandeira A, Maloney JP, Bull TM, Janssen WJ, Stenmark KR, Tuder RM, Graham BB: TGF-beta activation by bone marrow-derived thrombospondin-1 causes Schistosoma-and hypoxia-induced pulmonary hypertension. Nature communications 2017;8:15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li CX, Talele NP, Boo S, Koehler A, Knee-Walden E, Balestrini JL, Speight P, Kapus A, Hinz B: MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat Mater 2017;16:379–389. [DOI] [PubMed] [Google Scholar]
- 77.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G: Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. The Journal of cell biology 1993;122:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kelsh RM, McKeown-Longo PJ, Clark RAF: EDA Fibronectin in Keloids Create a Vicious Cycle of Fibrotic Tumor Formation. J Invest Dermatol 2015;135:1714–1718. [DOI] [PubMed] [Google Scholar]
- 79.Booth AJ, Wood SC, Cornett AM, Dreffs AA, Lu G, Muro AF, White ES, Bishop DK: Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. J Pathol 2012;226:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shinde AV, Kelsh R, Peters JH, Sekiguchi K, Van De Water L, McKeown-Longo PJ: The alpha4beta1 integrin and the EDA domain of fibronectin regulate a profibrotic phenotype in dermal fibroblasts. Matrix Biol 2015;41:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, White ES, Varga J: FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med 2014;6:232ra250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kelsh-Lasher RM, Ambesi A, Bertram C, McKeown-Longo PJ: Integrin alpha4beta1 and TLR4 Cooperate to Induce Fibrotic Gene Expression in Response to Fibronectin’s EDA Domain. J Invest Dermatol 2017;137:2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bordeleau F, Califano JP, Negron Abril YL, Mason BN, LaValley DJ, Shin SJ, Weiss RS, Reinhart-King CA: Tissue stiffness regulates serine/arginine-rich protein-mediated splicing of the extra domain B-fibronectin isoform in tumors. Proceedings of the National Academy of Sciences of the United States of America 2015;112:8314–8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Olsen AL, Sackey BK, Marcinkiewicz C, Boettiger D, Wells RG: Fibronectin extra domain-A promotes hepatic stellate cell motility but not differentiation into myofibroblasts. Gastroenterology 2012;142:928–937 e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Niimi T, Osawa M, Yamaji N, Yasunaga K, Sakashita H, Mase T, T anaka A, Fujita S: Letter to the Editor: NMR structure of human fibronectin EDA. Journal of biomolecular NMR 2001;21:281–284. [DOI] [PubMed] [Google Scholar]
- 86.Leiss M, Beckmann K, Girós A, Costell M, Fässler R: The role of integrin binding sites in fibronectin matrix assembly in vivo. Current opinion in cell biology 2008;20:502–507. [DOI] [PubMed] [Google Scholar]
- 87.Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L: The EIIIA segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. The Journal of biological chemistry 2002;277:14467–14474. [DOI] [PubMed] [Google Scholar]
- 88.Sun X, Fa P, Cui Z, Xia Y, Sun L, Li Z, Tang A, Gui Y, Cai Z: The EDA-containing cellular fibronectin induces epithelial-mesenchymal transition in lung cancer cells through integrin alpha9beta1-mediated activation of PI3-K/AKT and Erk½. Carcinogenesis 2014;35:184–191. [DOI] [PubMed] [Google Scholar]
- 89.Kohan M, Muro AF, White ES, Berkman N: EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4beta7 integrin receptor and MAPK/Erk ½-dependent signaling. FASEB J 2010;24:4503–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kohan M, Muro AF, Bader R, Berkman N: The extra domain A of fibronectin is essential for allergen-induced airway fibrosis and hyperresponsiveness in mice. J Allergy Clin Immunol 2011;127:439–446 e431–435. [DOI] [PubMed] [Google Scholar]
- 91.Strohmeyer N, Bharadwaj M, Costell M, Fassler R, Muller DJ: Fibronectin-bound alpha5beta1 integrins sense load and signal to reinforce adhesion in less than a second. Nat Mater 2017;16:1262–1270. [DOI] [PubMed] [Google Scholar]
- 92.Benito-Jardon M, Klapproth S, Gimeno LI, Petzold T, Bharadwaj M, Muller DJ, Zuchtriegel G, Reichel CA, Costell M: The fibronectin synergy site re-enforces cell adhesion and mediates a crosstalk between integrin classes. eLife 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sechler JL, Corbett SA, Schwarzbauer JE: Modulatory roles for integrin activation and the synergy site of fibronectin during matrix assembly. Mol Biol Cell 1997;8:2563–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM: Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. The Journal of biological chemistry 2005;280:18871–18880. [DOI] [PubMed] [Google Scholar]
- 95.Klingberg F, Chow ML, Koehler A, Boo S, Buscemi L, Quinn TM, Costell M, Alman BA, Genot E, Hinz B: Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation. The Journal of cell biology 2014;207:283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.White ES, Muro AF: Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011;63:538–546. [DOI] [PubMed] [Google Scholar]
- 97.Hallgren O, Malmström J, Malmström L, Andersson-Sjöland A, Wildt M, Tufvesson E, Juhasz P, Marko-Varga G, Westergren-Thorsson G: Splicosomal and serine and arginine-rich splicing factors as targets for TGF-β. Fibrogenesis Tissue Repair 2012;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Phanish MK, Heidebrecht F, Nabi ME, Shah N, Niculescu-Duvaz I, Dockrell ME: The regulation of TGFbeta1 induced fibronectin EDA exon alternative splicing in human renal proximal tubule epithelial cells. Journal of cellular physiology 2014 [DOI] [PubMed] [Google Scholar]
- 99.Walraven M, Hinz B: Therapeutic approaches to control tissue repair and fibrosis: Extracellular matrix as a game changer. Matrix Biol 2018 [DOI] [PubMed] [Google Scholar]
- 100.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G: The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. The Journal of cell biology 1998;142:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kang JS, Liu C, Derynck R: New regulatory mechanisms of TGF-beta receptor function. Trends in cell biology 2009;19:385–394. [DOI] [PubMed] [Google Scholar]
- 102.Verrecchia F, Chu M-L, Mauviel A: Identification of novel TGF-β/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. Journal of Biological Chemistry 2001; 276:17058–17062. [DOI] [PubMed] [Google Scholar]
- 103.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD: Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. The Journal of clinical investigation 2017;127:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carthy JM, Sundqvist A, Heldin A, van Dam H, Kletsas D, Heldin CH, Moustakas A: Tamoxifen Inhibits TGF-beta-Mediated Activation of Myofibroblasts by Blocking Non-Smad Signaling Through ERK½. J Cell Physiol 2015;230:3084–3092. [DOI] [PubMed] [Google Scholar]
- 105.Dugina V, Fontao L, Chaponnier C, Vasiliev J, Gabbiani G: Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J Cell Sci 2001;114:3285–3296. [DOI] [PubMed] [Google Scholar]
- 106.Li S, Liu J, Tan J, Li L, Kaltreider MJ, Zhao J, Kass DJ, Shang D, Zhao Y: Inhibition of Raf1 ameliorates bleomycin-induced pulmonary fibrosis through attenuation of TGFbeta1 signaling. American journal of physiology Lung cellular and molecular physiology 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rosenbloom J, Mendoza FA, Jimenez SA: Strategies for anti-fibrotic therapies. Biochimica et biophysica acta 2013;1832:1088–1103. [DOI] [PubMed] [Google Scholar]
- 108.Clement CA, Ajbro KD, Koefoed K, Vestergaard ML, Veland IR, Henriques de Jesus MP, Pedersen LB, Benmerah A, Andersen CY, Larsen LA, Christensen ST: TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell reports 2013;3:1806–1814. [DOI] [PubMed] [Google Scholar]
- 109.Rys JP, DuFort CC, Monteiro DA, Baird MA, Oses-Prieto JA, Chand S, Burlingame AL, Davidson MW, Alliston TN: Discrete spatial organization of TGFβ receptors couples receptor multimerization and signaling to cellular tension. Elife 2015;4:e09300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oakes PW, Gardel ML: Stressing the limits of focal adhesion mechanosensitivity. Curr Opin Cell Biol 2014;30:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mitra SK, Hanson DA, Schlaepfer DD: Focal adhesion kinase: in command and control of cell motility. Nature reviews Molecular cell biology 2005;6:56–68. [DOI] [PubMed] [Google Scholar]
- 112.Parsons JT, Parsons SJ: Src family protein tyrosine kinases: cooperating with growth factor and adhesion signaling pathways. Current opinion in cell biology 1997;9:187–192. [DOI] [PubMed] [Google Scholar]
- 113.Goffin JM, Pittet P, Csucs G, Lussi JW, Meister J-J, Hinz B: Focal adhesion size controls tension-dependent recruitment of a-smooth muscle actin to stress fibers. The Journal of cell biology 2006;172:259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huveneers S, Danen EH: Adhesion signaling -crosstalk between integrins, Src and Rho. J Cell Sci 2009;122:1059–1069. [DOI] [PubMed] [Google Scholar]
- 115.Wehrle-Haller B: Structure and function of focal adhesions. Current opinion in cell biology 2012;24:116–124. [DOI] [PubMed] [Google Scholar]
- 116.Oakes PW, Gardel ML: Stressing the limits of focal adhesion mechanosensitivity. Current opinion in cell biology 2014;30:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Varney SD, Betts CB, Zheng R, Wu L, Hinz B, Zhou J, Van De Water L: Hic-5 is required for myofibroblast differentiation by regulating mechanically dependent MRTF-A nuclear accumulation. J Cell Sci 2016;129:774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leach DA, Need EF, Trotta AP, Grubisha MJ, DeFranco DB, Buchanan G: Hic-5 influences genomic and non-genomic actions of the androgen receptor in prostate myofibroblasts. Mol Cell Endocrinol 2014;384:185–199. [DOI] [PubMed] [Google Scholar]
- 119.Kim Y, Lim S, Ha T, Song YH, Sohn YI, Park DJ, Paik SS, Kim-Kaneyama JR, Song MR, Leung A, Levine EM, Kim IB, Goo YS, Lee SH, Kang KH, Kim JW: The LIM protein complex establishes a retinal circuitry of visual adaptation by regulating Pax6 alpha-enhancer activity. eLife 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dabiri G, Tumbarello DA, Turner CE, Van De Water L: Hic-5 promotes the hypertrophic scar myofibroblast phenotype by regulating the TGF-β1 autocrine loop. Journal of Investigative Dermatology 2008;128:2518–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Petropoulos C, Oddou C, Emadali A, Hiriart-Bryant E, Boyault C, Faurobert E, Vande Pol S, Kim-Kaneyama JR, Kraut A, Coute Y, Block M, Albiges-Rizo C, Destaing O: Roles of paxillin family members in adhesion and ECM degradation coupling at invadosomes. The Journal of cell biology 2016;213:585–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chodankar R, Wu DY, Schiller BJ, Yamamoto KR, Stallcup MR: Hic-5 is a transcription coregulator that acts before and/or after glucocorticoid receptor genome occupancy in a gene-selective manner. Proceedings of the National Academy of Sciences of the United States of America 2014;111:4007–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim-Kaneyama J-r, Suzuki W, Ichikawa K, Ohki T, Kohno Y, Sata M, Nose K, Shibanuma M: Uni-axial stretching regulates intracellular localization of Hic-5 expressed in smooth-muscle cells in vivo. J Cell Sci 2005;118:937–949. [DOI] [PubMed] [Google Scholar]
- 124.Shibanuma M, Kim-Kaneyama J-r, Ishino K, Sakamoto N, Hishiki T, Yamaguchi K, Mori K, Mashimo J-i, Nose K: Hic-5 communicates between focal adhesions and the nucleus through oxidant-sensitive nuclear export signal. Molecular biology of the cell 2003;14:1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Varney SD, Betts CB, Zheng R, Wu L, Hinz B, Zhou J, Van De Water L: Hic-5 is required for myofibroblast differentiation by regulating mechanically dependent MRTF-A nuclear accumulation. J Cell Sci 2016;129:774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee BH, Stallcup MR: Glucocorticoid receptor binding to chromatin is selectively controlled by the coregulator Hic-5 and chromatin remodeling enzymes. Journal of Biological Chemistry 2017;292:9320–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu C, You J, Fu J, Wang X, Zhang Y: Phosphatidylinositol 3-Kinase/Akt Mediates Integrin Signaling To Control RNA Polymerase I Transcriptional Activity. Mol Cell Biol 2016;36:1555–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim NG, Gumbiner BM: Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. The Journal of cell biology 2015;210:503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Laplante M, Sabatini DM: mTOR signaling in growth control and disease. Cell 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bertacchini J, Heidari N, Mediani L, Capitani S, Shahjahani M, Ahmadzadeh A, Saki N: Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci 2015;72:2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Inoki K, Li Y, Zhu T, Wu J, Guan KL: TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002;4:648–657. [DOI] [PubMed] [Google Scholar]
- 132.Abdalla M, Goc A, Segar L, Somanath PR: Akt1 mediates alpha-smooth muscle actin expression and myofibroblast differentiation via myocardin and serum response factor. The Journal of biological chemistry 2013;288:33483–33493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Horowitz JC, Rogers DS, Sharma V, Vittal R, White ES, Cui Z, Thannickal VJ: Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cellular signalling 2007;19:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.White ES, Thannickal VJ, Carskadon SL, Dickie EG, Livant DL, Markwart S, Toews GB, Arenberg DA: Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. American journal of respiratory and critical care medicine 2003;168:436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.White ES, Sagana RL, Booth AJ, Yan M, Cornett AM, Bloomheart CA, Tsui JL, Wilke CA, Moore BB, Ritzenthaler JD, Roman J, Muro AF: Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Experimental cell research 2010;316:2644–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD: Role of Src-induced dynamin-2 phosphorylation in caveolae-mediated endocytosis in endothelial cells. Journal of Biological Chemistry 2004;279:20392–20400. [DOI] [PubMed] [Google Scholar]
- 137.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL: Distinct endocytic pathways regulate TGF-ß receptor signalling and turnover. Nature cell biology 2003;5:410–421. [DOI] [PubMed] [Google Scholar]
- 138.Peng F, Zhang B, Wu D, Ingram AJ, Gao B, Krepinsky JC: TGFβ-induced RhoA activation and fibronectin production in mesangial cells require caveolae. American Journal of Physiology-Renal Physiology 2008;295:F153–F164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hu M, Che P, Han X, Cai G-Q, Liu G, Antony V, Luckhardt T, Siegal GP, Zhou Y, Liu R-m: Therapeutic Targeting of Src Kinase in Myofibroblast Differentiation and Pulmonary Fibrosis. Journal of Pharmacology and Experimental Therapeutics 2014;351:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wendt MK, Schiemann BJ, Parvani JG, Lee YH, Kang Y, Schiemann WP: TGF-beta stimulates Pyk2 expression as part of an epithelial-mesenchymal transition program required for metastatic outgrowth of breast cancer. Oncogene 2013;32:2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lim Y, Lim ST, Tomar A, Gardel M, Bernard-Trifilo JA, Chen XL, Uryu SA, Canete-Soler R, Zhai J, Lin H, Schlaepfer WW, Nalbant P, Bokoch G, Ilic D, Waterman-Storer C, Schlaepfer DD: PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. The Journal of cell biology 2008;180:187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis J: Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017;136:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Davis J, Molkentin JD: Myofibroblasts: trust your heart and let fate decide. Journal of molecular and cellular cardiology 2014;70:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guilluy C, Swaminathan V, Garcia-Mata R, O’Brien ET, Superfine R, Burridge K: The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol 2011;13:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stempien-Otero A, Kim DH, Davis J: Molecular networks underlying myofibroblast fate and fibrosis. Journal of molecular and cellular cardiology 2016;97:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lawson CD, Ridley AJ: Rho GTPase signaling complexes in cell migration and invasion. The Journal of cell biology 2018;217:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lighthouse JK, Small EM: T ranscriptional control of cardiac fibroblast plasticity. Journal of molecular and cellular cardiology 2016;91:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Somlyo AP, Somlyo AV: Signal transduction by G-proteins, Rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. The Journal of Physiology 2000;522:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Knipe RS, Tager AM, Liao JK: The Rho kinases: critical mediators of multiple profibrotic processes and rational targets for new therapies for pulmonary fibrosis. Pharmacological reviews 2015;67:103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J: Functions of cofilin in cell locomotion and invasion. Nature reviews Molecular cell biology 2013;14:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grosse R, Copeland JW, Newsome TP, Way M, Treisman R: A role for VASP in RhoA -Diaphanous signalling to actin dynamics and SRF activity. The EMBO journal 2003;22:3050–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zigmond SH: Formin-induced nucleation of actin filaments. Current opinion in cell biology 2004;16:99–105. [DOI] [PubMed] [Google Scholar]
- 153.Hanna S, El-Sibai M: Signaling networks of Rho GTPases in cell motility. Cellular signalling 2013;25:1955–1961. [DOI] [PubMed] [Google Scholar]
- 154.Narumiya S, Tanji M, Ishizaki T: Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer and Metastasis Reviews 2009;28:65–76. [DOI] [PubMed] [Google Scholar]
- 155.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL: Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science (New York, NY) 2005;307:1603–1609. [DOI] [PubMed] [Google Scholar]
- 156.Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL: Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science (New York, NY) 2003;302:1775–1779. [DOI] [PubMed] [Google Scholar]
- 157.Miano JM, Long X, Fujiwara K: Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. American journal of physiology Cell physiology 2007;292:C70–81. [DOI] [PubMed] [Google Scholar]
- 158.Witt W, Buttner P, Jannasch A, Matschke K, Waldow T: Reversal of myofibroblastic activation by polyunsaturated fatty acids in valvular interstitial cells from aortic valves. Role of RhoA/G-actin/MRTF signalling. Journal of molecular and cellular cardiology 2014;74:127–138. [DOI] [PubMed] [Google Scholar]
- 159.Charbonney E, Speight P, Masszi A, Nakano H, Kapus A: beta-catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial-myofibroblast transition. Molecular biology of the cell 2011;22:4472–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Guo Y, Gupte M, Umbarkar P, Singh AP, Sui JY, Force T, Lal H: Entanglement of GSK-3beta, beta-catenin and TGF-beta1 signaling network to regulate myocardial fibrosis. Journal of molecular and cellular cardiology 2017;110:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Miao CG, Yang YY, He X, Huang C, Huang Y, Zhang L, Lv XW, Jin Y, Li J: Wnt signaling in liver fibrosis: progress, challenges and potential directions. Biochimie 2013;95:2326–2335. [DOI] [PubMed] [Google Scholar]
- 162.Zhou B, Liu Y, Kahn M, Ann DK, Han A, Wang H, Nguyen C, Flodby P, Zhong Q, Krishnaveni MS, Liebler JM, Minoo P, Crandall ED, Borok Z: Interactions between beta-catenin and transforming growth factor-beta signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). The Journal of biological chemistry 2012;287:7026–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Speight P, Nakano H, Kelley TJ, Hinz B, Kapus A: Differential topical susceptibility to TGFbeta in intact and injured regions of the epithelium: key role in myofibroblast transition. Mol Biol Cell 2013;24:3326–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J: Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nature communications 2012;3:735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liang M, Yu M, Xia R, Song K, Wang J, Luo J, Chen G, Cheng J: Yap/Taz Deletion in Gli(+) Cell-Derived Myofibroblasts Attenuates Fibrosis. J Am Soc Nephrol 2017;28:3278–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Harvey KF, Zhang X, Thomas DM: The Hippo pathway and human cancer. Nature reviews Cancer 2013;13:246–257. [DOI] [PubMed] [Google Scholar]
- 167.Ramos A, Camargo FD: The Hippo signaling pathway and stem cell biology. Trends in cell biology 2012;22:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Johnson R, Halder G: The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nature Reviews Drug Discovery 2014;13:63–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S: Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179–183. [DOI] [PubMed] [Google Scholar]
- 170.Dupont S: Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Experimental cell research 2016;343:42–53. [DOI] [PubMed] [Google Scholar]
- 171.Vandewalle C, Van Roy F, Berx G: The role of the ZEB family of transcription factors in development and disease. Cellular and molecular life sciences 2009;66:773–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cunnington RH, Northcott JM, Ghavami S, Filomeno KL, Jahan F, Kavosh MS, Davies JJ, Wigle JT, Dixon IM: The Ski-Zeb2-Meox2 pathway provides a novel mechanism for regulation of the cardiac myofibroblast phenotype. J Cell Sci 2014;127:40–49. [DOI] [PubMed] [Google Scholar]
- 173.Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E, Force T: Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014;130:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Piersma B, Bank RA, Boersema M: Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front Med (Lausanne) 2015;2:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cunnington RH, Northcott JM, Ghavami S, Filomeno KL, Jahan F, Kavosh MS, Davies JJ, Wigle JT, Dixon IM: The Ski-Zeb2-Meox2 pathway provides a novel mechanism for regulation of the cardiac myofibroblast phenotype. Journal of cell science 2014;127:40–49. [DOI] [PubMed] [Google Scholar]
- 176.Stratton MS, McKinsey TA: Epigenetic regulation of cardiac fibrosis. Journal of molecular and cellular cardiology 2016;92:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, Monzani V, Saripalli C, Mascagni P, Reece TB, Ambardekar AV, Granzier HL, Dinarello CA, McKinsey TA: Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med 2018;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang D, Zhang H, Li M, Frid MG, Flockton AR, McKeon BA, Yeager ME, Fini MA, Morrell NW, Pullamsetti SS, Velegala S, Seeger W, McKinsey TA, Sucharov CC, Stenmark KR: MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circulation research 2014;114:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting CC, Alexanian M, Maric D, Maison D, Nemir M, Young RA, Schroen B, Gonzalez A, Ounzain S, Pedrazzini T: The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, Muller CA, Kalluri R, Zeisberg M: Methylation determines fibroblast activation and fibrogenesis in the kidney. Nature medicine 2010;16:544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chang YT, Yang CC, Pan SY, Chou YH, Chang FC, Lai CF, Tsai MH, Hsu HL, Lin CH, Chiang WC, Wu MS, Chu TS, Chen YM, Lin SL: DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. The Journal of clinical investigation 2016;126:721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Breyer MD, Susztak K: The next generation of therapeutics for chronic kidney disease. Nature reviews Drug discovery 2016;15:568–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Montgomery RL, Yu G, Latimer PA, Stack C, Robinson K, Dalby CM, Kaminski N, van Rooij E: MicroRNA mimicry blocks pulmonary fibrosis. EMBO molecular medicine 2014;6:1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, Abraham E: miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med 2010;207:1589–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, Li J, Tran PT, Kaimal V, Huang X, Chang AN, Li S, Kalra A, Grafals M, Portilla D, MacKenna DA, Orkin SH, Duffield JS: MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med 2012;4:121ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ijaz T, Jamaluddin M, Zhao Y, Zhang Y, Jay J, Finnerty CC, Herndon DN, Tilton RG, Brasier AR: Coordinate activities of BRD4 and CDK9 in the transcriptional elongation complex are required for TGFbeta-induced Nox4 expression and myofibroblast transdifferentiation. Cell Death Dis 2017;8:e2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ding N, Hah N, Yu RT, Sherman MH, Benner C, Leblanc M, He M, Liddle C, Downes M, Evans RM: BRD4 is a novel therapeutic target for liver fibrosis. Proceedings of the National Academy of Sciences of the United States of America 2015;112:15713–15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wang B, Zhang M, T akayama T, Shi X, Roenneburg DA, Kent KC, Guo LW: BET Bromodomain Blockade Mitigates Intimal Hyperplasia in Rat Carotid Arteries. EBioMedicine 2015;2:1650–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhubanchaliyev A, Temirbekuly A, Kongrtay K, Wanshura LC, Kunz J: Targeting Mechanotransduction at the Transcriptional Level: YAP and BRD4 Are Novel Therapeutic Targets for the Reversal of Liver Fibrosis. Front Pharmacol 2016;7:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Stratton MS, Haldar SM, McKinsey TA: BRD4 inhibition for the treatment of pathological organ fibrosis. F1000Res 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA, Haldar SM: BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Shi J, Vakoc CR: The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Molecular cell 2014;54:728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, Steadman R: Transforming growth factor-beta1 (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. The Journal of biological chemistry 2013;288:14824–14838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ruppert SM, Hawn TR, Arrigoni A, Wight TN, Bollyky PL: Tissue integrity signals communicated by high-molecular weight hyaluronan and the resolution of inflammation. Immunol Res 2014;58:186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tian X, Azpurua J, Hine C, Vaidya A, Myakishev-Rempel M, Ablaeva J, Mao Z, Nevo E, Gorbunova V, Seluanov A: High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 2013;499:346–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gorbunova V, Seluanov A, Zhang Z, Gladyshev VN, Vijg J: Comparative genetics of longevity and cancer: insights from long-lived rodents. Nat Rev Genet 2014;15:531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ito T, Williams JD, Fraser DJ, Phillips AO: Hyaluronan regulates transforming growth factor-β1 receptor compartmentalization. Journal of Biological Chemistry 2004;279:25326–25332. [DOI] [PubMed] [Google Scholar]
- 198.Webber J, Jenkins RH, Meran S, Phillips A, Steadman R: Modulation of TGFβ1-dependent myofibroblast differentiation by hyaluronan. The American journal of pathology 2009;175:148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Evanko SP, Potter-Perigo S, Petty LJ, Workman GA, Wight TN: Hyaluronan Controls the Deposition of Fibronectin and Collagen and Modulates TGF-β1 Induction of Lung Myofibroblasts. Matrix Biology 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bornstein P: Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. The Journal of cell biology 1995;130:503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Li L, Zhao Q, Kong W: Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol 2018 [DOI] [PubMed] [Google Scholar]
- 202.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW 2nd, Conway SJ, Aronow BJ, Robbins J, Molkentin JD: Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circulation research 2007;101:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Elliott CG, Wang J, Guo X, Xu SW, Eastwood M, Guan J, Leask A, Conway SJ, Hamilton DW: Periostin modulates myofibroblast differentiation during full-thickness cutaneous wound repair. J Cell Sci 2012;125:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gerarduzzi C, Kumar RK, Trivedi P, Ajay AK, Iyer A, Boswell S, Hutchinson JN, Waikar SS, Vaidya VS: Silencing SMOC2 ameliorates kidney fibrosis by inhibiting fibroblast to myofibroblast transformation. JCI Insight 2017;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jiang D, Liang J, Campanella GS, Guo R, Yu S, Xie T, Liu N, Jung Y, Homer R, Meltzer EB, Li Y, Tager AM, Goetinck PF, Luster AD, Noble PW: Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. The Journal of clinical investigation 2010;120:2049–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Matsui Y, Ikesue M, Danzaki K, Morimoto J, Sato M, Tanaka S, Kojima T, Tsutsui H, Uede T: Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circulation research 2011;108:1328–1339. [DOI] [PubMed] [Google Scholar]
- 207.Herum KM, Lunde IG, McCulloch AD, Christensen G: The Soft-and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J Clin Med 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lipphardt M, Song JW, Ratliff BB, Dihazi H, Muller GA, Goligorsky MS: Endothelial dysfunction is a superinducer of syndecan-4: fibrogenic role of its ectodomain. American journal of physiology Heart and circulatory physiology 2018;314:H484–H496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M, Jackson JR, Le Saux CJ, Calhoun C, Peters J, Derynck R, Backes BJ, Chapman HA: Fibroblast-specific inhibition of TGF-beta1 signaling attenuates lung and tumor fibrosis. The Journal of clinical investigation 2017;127:3675–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]