

Summary
Fluorinated organic compounds are produced in abundance by the pharmaceutical and agrochemical industry, making such compounds attractive as building blocks for further functionalization. Unfortunately, activation of C(sp3)-F bond in saturated fluorocarbons, especially for aliphatic gem-difluoroalkanes, remains challenging. Here we describe the selective activation of inert C(sp3)-F bonds catalyzed by B(C6F5)3. In hexafluoro-2-propanol (HFIP), chemically robust aliphatic gem-difluorides are converted in high yields to the corresponding substituted 2,2′,3,3′-tetrahydro-1,1′-spirobiindenes via a B(C6F5)3-catalyzed intramolecular cascade Friedel-Crafts cyclization, not requiring a silicon-based trapping reagent. However, in the absence of a hydrogen-bonding donor solvent such as HFIP, the aliphatic gem-difluorides preferentially engage in a defluorination/elimination process that provides monofluorinated alkenes in good yields. Furthermore, a series of substituted 1-alkyl-2,3-dihydro-1H-indenes was obtained in high yield from the B(C6F5)3-catalyzed defluorinative cyclization of aliphatic secondary monofluorides in HFIP. The protocol could inspire development of a new class of main-group Lewis acid-catalyzed C(sp3)-F bond activation in general unactivated fluorocarbons.
Subject Areas: Chemistry, Organic Chemistry, Organic Synthesis
Graphical Abstract
Highlights
-
•
C(sp3)-F bond activation in general unactivated fluorocarbons
-
•
The activation of C(sp3)-F bonds in aliphatic gem-difluoroalkanes
-
•
The selective activation of inert C(sp3)-F bonds catalyzed by B(C6F5)3
-
•
An intramolecular cascade defluorinative Friedel-Crafts cyclization
Chemistry; Organic Chemistry; Organic Synthesis
Introduction
The demand for the selective activation of C-F bonds is growing as a result of the increased availability of fluorinated compounds in the pharmaceutical and agrochemical industries (Amii and Uneyama, 2009, Ahrens et al., 2015, Kuehnel et al., 2013, Hamel and Paquin, 2018, Klare, 2017). Recently, remarkable progress has been made in the transition-metal-mediated heterolysis of C(sp2)-F bonds in aromatic and vinylic fluorocarbons (Ahrens et al., 2015, Kuehnel et al., 2013, Pike et al., 2017, Guo et al., 2015, Luo et al., 2018). However, the defluorinative functionalization of C(sp3)-F bonds in unactivated aliphatic fluorides is less frequently reported and still a challenging issue in synthetic organic chemistry (Stahl et al., 2013, Shen et al., 2015). Indeed, the notorious chemical robustness of C-F bonds not only stems from their thermodynamic stability—the C-F bond is among the strongest covalent single bonds that carbon can form—but also from kinetic factors because the fluoride moiety is neither a good leaving group nor a good Lewis base (O'Hagan, 2008, Nolte et al., 2012).
The direct abstraction of the fluoride moiety in inert C(sp3)-F bonds by p-block-based Lewis acids that exhibit high fluoride affinity has emerged as a promising strategy for the degradation of saturated fluorocarbons (Stahl et al., 2013, Shen et al., 2015), because of that the formation of covalent bonds between fluorine and main-group elements (e.g., B, Al, Si, and P), which are more stable than C-F bonds, may offer a thermodynamic driving force for the scission of the C-F bonds (Stahl et al., 2013). In addition, the stronger Lewis acidity of fluorophilic electrophiles is essential for the direct heterolytic cleavage of C(sp3)-F bonds, given the high activation barrier. The substitution of the fluoride in C(sp3)-F bonds to form C-H, C-C, and C-heteroatom bonds has been initiated by neutral, strong aluminum- and boron-based Lewis acids (Stahl et al., 2013, Greb, 2018, Morgan et al., 2013, Koerte et al., 2017, Ahrens et al., 2013, Jaiswal et al., 2017) or cationic species such as [CPh3]+, [SiEt3]+, [iBu2Al]+, [(C6F5)3FP]+, and even P(III) dications such as [(bipy)PPh]2+ bearing weakly coordinating counter anions such as [B(C6F5)4]- (Stahl et al., 2013, Klahn et al., 2007, Gu et al., 2009, Forster et al., 2017, Douvris and Ozerov, 2008, Scott et al., 2005, Großekappenberg et al., 2015, Zhu et al., 2016, Chitnis et al., 2018).
In their seminal reports, Olah and co-workers described the cleavage of unactivated C(sp3)-F bonds initiated by boron-based Lewis acids, specifically the preferential abstraction of fluorides from aliphatic fluorohaloalkanes by boron trifluoride to generate the stable BF4- anion and Friedel-Crafts-type alkylation products with excess arenes (Olah et al., 1957, Olah and Kuhn, 1964). Consistent with the greater stability of tertiary carbocations derived from tertiary aliphatic fluorides, Oshima and co-workers have reported that BF3·OEt2 catalyzes C-C bond couplings between silicon enolates and tertiary fluorides (Hirano et al., 2004). Subsequently, Stephan et al. have reported the splitting of unactivated C(sp3)-F bonds using stoichiometric amounts of B(C6F5)3 and a phosphine as frustrated Lewis pairs (FLPs) to produce [R3PR’][FB(C6F5)3] salts (Caputo and Stephan, 2012). Alternatively, using catalytic amounts of B(C6F5)3 with an excess of Et3SiH, a C-H bond is formed at the expense of the corresponding C(sp3)-F bond (Caputo and Stephan, 2012). Furthermore, HB(C6F5)2 has been used to induce the direct C(sp3)-F borylation of secondary and primary aliphatic fluoroalkanes via an initial dehydrofluorination and a subsequent borylation of the resulting olefin intermediates (Bamford et al., 2018). Recently, Moran and co-workers have reported the Friedel-Crafts reactions of tertiary fluoroalkanes with an excess of arenes (3.0–5.0 equiv.) catalyzed by B(C6F5)3 in MeNO2 under ambient atmosphere; interestingly, in this case the Lewis acid B(C6F5)3 absorbs water to generate [(C6F5)3B(OH2)n], which then acts as a Brønsted acid (Dryzhakov and Moran, 2016, Dryzhakov et al., 2017, Beringhelli et al., 2001). Despite the general progress in this area, the development of alternative catalytic methods based on boron-based Lewis acids as fluorophilic electrophiles for the activation of inert C(sp3)-F bonds in saturated fluorocarbons remains highly desirable.
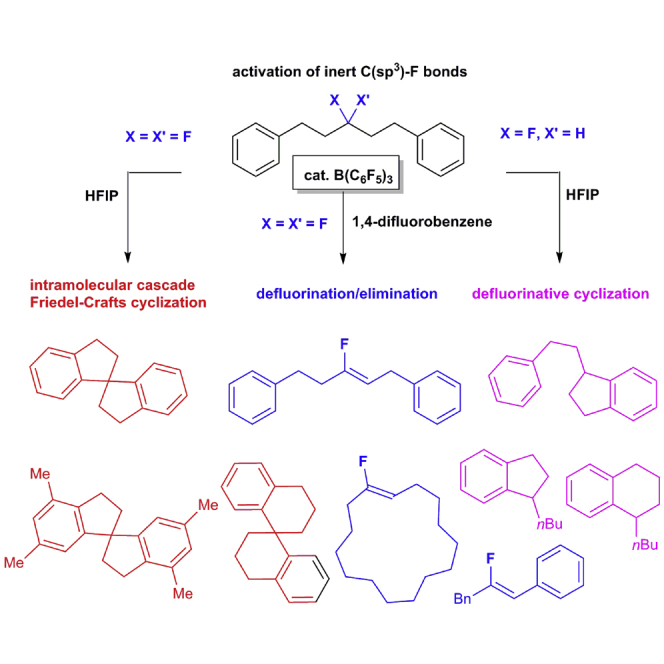
The modification of C(sp3)-F bonds in aliphatic gem-difluoroalkanes is much more difficult than in the corresponding saturated monofluorocarbons because the strength of C-F bonds increases with the number of geminal fluorine atoms (Hamel and Paquin, 2018, O'Hagan, 2008). Indeed, in most cases, the fluorine moiety in gem-difluorides is found at activated benzylic, allylic, or propargylic positions (Figure 1A), as well as at the α-position of a carbonyl group or in gem-difluorocyclopropanes (Hamel and Paquin, 2018, Song et al., 2017). In a representative example of unactivated aliphatic gem-difluoroalkanes from Ozerov and co-workers, the ethylation of 1,1-difluorocyclopentane was observed together with the reduction side product cyclopentane (67:33) by gas chromatography-mass spectrometry analysis as one special case (Figure 1B) by using catalytic amounts of [Et2Al][HCB11H5Br6] in the presence of an excess amount of AlEt3 (Gu et al., 2009). In 2018, the group of Young reported two examples for the monodefluorination of an acyclic aliphatic gem-difluoromethyl group in 1,1-difluoroethane and 1,1-difluorodecane: using FLPs obtained from Al(C6F5)3 and P(o-Tol)3, the α-fluoroalkylphosphonium salts were generated in moderate yield (Figure 1C) (Mandal et al., 2018). Building upon our long-standing interest in the activation of inert C(sp3)-F bonds (Haufe et al., 2012, Tanaka et al., 2016, Cui et al., 2018), we discovered in this study that a catalytic amount of the Lewis acid B(C6F5)3 activated the C(sp3)-F bond in aliphatic gem-difluoroalkanes of type 1 to selectively generate substituted 2,2′,3,3′-tetrahydro-1,1′-spirobiindenes (2) and monofluorinated alkenes (3) in good yield (Figure 1D). Moreover, this method was also used for the functionalization of the C(sp3)-F bond in secondary monofluoroalkanes to C(sp3)-C(sp3) bonds in good yield. The use of hydrogen-bonding hexafluoro-2-propanol (HFIP) as the solvent was essential to induce the catalyst turnover for the defluorinative Friedel-Crafts alkylation.
Figure 1.

Activation of C(sp3)-F Bonds in gem-Difluoroalkanes
(A) Activated benzylic, allylic, and propargylic gem-difluoroalkanes.
(B) [Et2Al][HCB11H5Br6]-induced C(sp3)-F functionalization of 1,1-difluorocyclopentane.
(C) Monodefluorination of a gem-difluoromethyl group initiated by the FLP Al(C6F5)3/P(o-Tol)3.
(D) Synthesis of spirobiindanes, monofluoroalkenes, and 1-alkyl-2,3-dihydro-1H-indenes via a B(C6F5)3-catalyzed activation of C(sp3)-F bonds (this work).
Results and Discussion
Optimization Study
Initially, based on the pioneering work of Olah and Stephan on the activation of C(sp3)-F bonds in saturated monofluoroalkanes initiated by boron-based Lewis acids (Olah et al., 1957, Olah and Kuhn, 1964, Caputo and Stephan, 2012), we attempted to use stoichiometric amounts of BF3·OEt2 and B(C6F5)3 (2.2 equiv.) to induce cleavage of the C(sp3)-F bond in unactivated gem-difluoroalkane 1a (Table 1, entries 1 and 2). Although no reaction was detected upon using BF3·OEt2, the use of a stoichiometric amount of B(C6F5)3 afforded 2,2′,3,3′-tetrahydro-1,1′-spirobi[indene] 2a in 85% yield. This result was very encouraging, considering that examples of the activation of C(sp3)-F bonds in inert aliphatic gem-difluoroalkanes are extremely rare (Figure 1) (Gu et al., 2009, Mandal et al., 2018). Subsequently, we turned our attention to the development of a catalytic B(C6F5)3-induced cascade intramolecular Friedel-Crafts cyclization (Lan et al., 2006, Birman et al., 1999, Li et al., 2016, Zheng et al., 2018).
Table 1.
Optimization of the Selective Cleavage of C(sp3)-F Bonds in Aliphatic gem-Difluoroalkanes
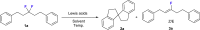 | ||||||
|---|---|---|---|---|---|---|
| Entry | Lewis Acids (Equiv.) | Solvent (0.1M) | T (°C) | t (h) | Yields (%) |
|
| 2a | 3aa | |||||
| 1 | BF3·OEt2 (2.2) | CH2Cl2 | RT | 30 | NR | NR |
| 2 | B(C6F5)3 (2.2) | CH2Cl2 | RT | 30 | 85 | 0 |
| 3 | B(C6F5)3 (0.2) | CH2Cl2 | RT | 30 | Trace | 0 |
| 4 | B(C6F5)3 (0.2) | (CF3)2CHOH | RT | 17 | 28 | 0 |
| 5 | B(C6F5)3 (0.2) | (CF3)2CHOH | 50 | 2 | 75 | 0 |
| 6 | B(C6F5)3 (0.1) | (CF3)2CHOH | 50 | 20 | 16 | 0 |
| 7 | B(C6F5)3 (0.2) | (CF3)2CHOHb | 50 | 2 | 27 | 0 |
| 8 | B(C6F5)3 (0.2) | (CF3)2CHOHc | 50 | 2 | 84 | 0 |
| 9 | B(C6F5)3 (0.2) | (CF3)2CHOHc H2O (2.2 equiv.) |
50 | 2 | 0d | 0d |
| 10 | B(C6F5)3 (0.2) | iPrOHc | 50 | 2 | NR | NR |
| 11 | B(C6F5)3 (0.2) | CF3CH2OHc | 50 | 2 | NR | NR |
| 12 | B(C6F5)3 (0.2) | (CF3)2PhOHc | 50 | 2 | NR | NR |
| 13 | – | (CF3)2CHOHc | 50 | 2 | NR | NR |
| 14 | B(C6F5)3 (0.2) | o-C6H4Cl2 | 160 | 3 | 0 | 64 |
| 15 | B(C6F5)3 (0.1) | o-C6H4Cl2 | 160 | 3 | 0 | 30 |
| 16 | – | o-C6H4Cl2 | 160 | 3 | NR | NR |
| 17 | B(C6F5)3 (0.2) | o-C6H4Cl2 | 220 | 3 | 0 | 81e |
| 18 | B(C6F5)3 (0.2) | p-C6H4F2 | reflux | 3 | 0 | 75 |
| 19 | B(C6F5)3 (0.2) | p-C6H4F2 | reflux | 24 | 0 | 87f |
RT, room temperature; NR, no reaction.
Determined by19F NMR analysis using PhCF3 as the internal standard.
(CF3)2CHOH was used as purchased without any precaution to exclude moisture. The hydrolysis product 1,5-diphenylpentan-3-one was observed as the major product.
Concentration: 0.05 M.
The hydrolysis product 1,5-diphenylpentan-3-one was obtained in quantitative yield after 12 h at 50°C.
Z/E = 7.3:1.
Z/E = 7.1:1.
Recently, HFIP has attracted considerable attention as a solvent to promote Friedel-Crafts acylations and alkylations (Motiwala et al., 2015, Vekariya and Aube, 2016, Tang et al., 2018) owing to its unique properties, which include reduced nucleophilicity, a strong propensity to engage as a hydrogen-bonding donor, and the ability to stabilize cationic intermediates (Colomer et al., 2017). Indeed, intermolecular Friedel-Crafts alkylations by hydrogen-bonding interactions between activated benzylic C-F bonds and HFIP in the absence of any Lewis or Brønsted acids has been reported by Paquin and co-workers (Champagne et al., 2014, Champagne et al., 2015). Using a combination of the hydrogen-bonding-donor solvent HFIP and B(C6F5)3 (20 mol %) in the absence of any silicon-based trapping reagent, afforded the defluorinative Friedel-Crafts-type product 2a in 75% yield (Table 1, entry 5). It is worth noting here that moisture was strictly excluded in our method, owing to the hydrolysis of 1a under acidic conditions. When using “wet” HFIP, i.e., HFIP that was used as purchased under an atmosphere of argon without any precaution to exclude moisture, the corresponding hydrolysis product (1,5-diphenylpentan-3-one) was observed as the major product and 2a was obtained in only 27% yield (Table 1, entry 7). Upon adding H2O (2.2 equiv.), the intramolecular Friedel-Crafts transformation was completely suppressed, and the quantitative hydrolysis into a ketone was confirmed instead when prolonging the reaction time (Table 1, entry 9). Other reaction parameters, such as solvents, concentration, and temperature, were also investigated (for more details, see also Table S1). Finally, we were able to identify the optimal reaction conditions for the synthesis of spirobiindanes 2: B(C6F5)3 (20 mol%) in HFIP (0.05 M) at 50°C for 2 h (Table 1, entry 8). In the absence of B(C6F5)3, or when using other hydrogen-bonding solvents such as iPrOH, CF3CH2OH, or (CF3)2PhOH, the reaction did not proceed (Table 1, entries 10–13). Unexpected results were obtained using 1,2-dichlorobenzene as the solvent. Indeed, the formation of monofluoroalkene 3a, derived from the defluorination/elimination sequence of aliphatic gem-difluoroalkane 1a (Yanai et al., 2011, Yang et al., 2013, Surmont et al., 2009, Li et al., 2018, Drouin et al., 2018), was observed in good yield when conducting the reaction at very high temperatures (Table 1, entries 14, 17). Specifically, when the temperature was increased from 160°C to 220°C in a sealed tube, the yield of the elimination product 3a increased from 64% to 81% with good stereoselectivity (Z/E = 7.3:1), whereas no reaction was detected in the absence of B(C6F5)3 (entry 16). These results indicate that the increased reaction temperature is beneficial for the defluorination/elimination. Furthermore, after screening the reaction conditions to find an acceptable reaction temperature (for further details, see also Table S2) in the presence of B(C6F5)3 (20 mol%), we discovered that stirring the reaction mixture in refluxing 1,4-difluorobenzene (boiling point 88–89°C) instead of using harsher reaction conditions (220°C) afforded 3a in 87% yield with good stereoselectivity (Z/E = 7.1:1).
Substrate Scope
With the optimized reaction conditions in hand, we explored the substrate scope (Figure 2). First, we examined intramolecular Friedel-Crafts reactions as shown in Figure 2A. For aliphatic gem-difluoroalkanes substituted with alkyl groups (1a-h), good to high yields were observed; in particular, gem-difluoroalkane 1f, bearing a methyl group at the C2 position of the benzene ring, generated the desired product (2f) in high yield (up to 90%). However, for methoxy-substituted gem-difluorides 1c and 1g, substantially lower yields were observed due to the presence of the electron-rich heteroatom acting as a Lewis base that is able to provide lone electron pairs to interact with the Lewis acid catalyst. This unexpected donor-acceptor interaction between the oxygen atom and the electron-deficient boron moiety hampered the fluoride abstraction via the C(sp3)-F→B(C6F5)3 interaction, leading to decreased yields. In contrast, gem-difluoroalkanes (1i-m) with a halogen (F, Cl, Br) group at the C2 or C4 position afforded acceptable yields (57–79%), whereas dialkyl-substituted substrates 1n and 1o furnished good to excellent product yields (up to 95%). Naphthyl-type 2,2′,3,3′-tetrahydro-1,1′-spirobi[cyclopenta[b]naphthalene] 2p was also prepared in good yield (84%). Moreover, 4,6-dimethyl-2,2′,3,3′-tetrahydro-1,1′-spirobi[indene] 2q and 4-bromo-4′-methyl-2,2′,3,3′-tetrahydro-1,1′-spirobi[indene] 2r were generated in moderate yields (42% and 59%, respectively). Six-membered spiro-compound 2s and five- or six-membered spiro-compound 2t were also prepared in high yields (up to 90%). As shown in Figure 2B, the substrate scope for the defluorination/elimination process was explored. When using symmetric substrates, the desired acyclic monofluoroalkenes (3a, 3e-g, 3j-k, 3n-o, 3u, and 3aa) were prepared in moderate to good yields (up to 84%) with good Z/E stereocontrol. Specifically, substrates with electron-donating substituents such as methyl, methoxy, or dialkyl groups on the benzene ring gave the desired products (3e-g and 3n-o) in moderate to good yields (51%–70%) in refluxing 1,4-difluorobenzene. Halogen groups were well tolerated in the elimination transformation, although an unexpected decrease in yield (38%) was observed for the preparation of bromo-substituted 3j, albeit that the stereocontrol was high (Z/E = 25:1). In addition, benzylic gem-difluoroalkanes afforded 3bb and 3cc in merely low to moderate yields (41% and 25%, respectively), and only the Z-isomer is formed, even though the fluorinated moiety is located at the activated position and was thus expected to be removed more easily. For cyclic gem-difluoroalkanes (Strobach and Boswell, 1971), the formation of six-membered substrates was favored, i.e., 3dd and 3ee were prepared in 80% and 64% yield, respectively. Furthermore, the defluorination of large-ring-type gem-difluoroalkanes proceeded smoothly to afford the corresponding cyclic monofluoroalkenes 3gg and 3hh in good yields, albeit that the Z/E selectivity was low.
Figure 2.

Substrate Scope of the Defluorination of Aliphatic gem-Difluoroalkanes to Afford Spirobiindanes 2 and Monofluoroalkenes 3
(A) Intramolecular Friedel-Crafts reactions.
(B) Defluorination/elimination reactions.
B(C6F5)3 Catalyzed Friedel-Crafts Reactions of Secondary Aliphatic Fluorides
Although the cleavage of the C(sp3)-F bond in unactivated aliphatic monofluorides was expected to be easier than in the corresponding saturated gem-difluoroalkanes, the Friedel-Crafts alkylation of secondary monofluorinated alkanes was less successful (Hamel and Paquin, 2018, Stahl et al., 2013). Under the previously established optimal reaction conditions for the synthesis of spirobiindanes 2, using 20 mol % B(C6F5)3 in HFIP, an intramolecular defluorinative cyclization of secondary fluorocarbon 4a was observed in good yield (90%; Figure 3A, entry 1). Subsequently, upon decreasing the catalyst loading to 2 mol %, the desired 1-phenethyl-2,3-dihydro-1H-indene (5a) was smoothly prepared (91%; Figure 3A, entry 5). However, in the absence of a Lewis-acidic catalyst, a reaction was not observed (Figure 3A, entry 6), which demonstrates the crucial importance of B(C6F5)3 for abstraction of fluoride. Subsequently, we explored the substrate scope (Figure 3B) of this reaction. With long-chain symmetric substrates with electron-donating groups (4a-f), good to high yields were observed (70%–93%). Conversely, yields for the intramolecular Friedel-Crafts transformation of halogen-substituted monofluoroalkanes 5g-j were a bit lower (68%–80%). Similarly, 2,3-dimethyl- and 2,4-dimethyl-substituted 4k and 4l were converted into the cyclic products 5k and 5l in 50% and 67% yield, respectively, whereas the naphthyl-type product 5m was obtained in 79% yield. Miscellaneous monofluorides 4n-p furnished the desired alkyl-substituted indanes in moderate to good yields (44%–91%). Six-membered ring products 5r and 5s were also prepared in 82%–85% yield. However, benzylic secondary monofluoride 4q furnished 1-phenyl-2,3-dihydro-1H-indene (5q) in merely 29% yield. It should be noted that an increased yield (46%) for the synthesis of 5q was observed in the absence of a Lewis acid catalyst. Although intermolecular Friedel-Crafts alkylations of primary benzylic monofluoride using excess amounts of electrophiles in HFIP in the absence of acids have already been reported (Champagne et al., 2014), we have observed the first example of the functionalization of a secondary benzylic monofluoride such as 4q in the absence of any catalyst or additive (Figure 3B).
Figure 3.

Intramolecular Friedel-Crafts Cyclization of Secondary Monofluoroalkanes 4
(A) Optimization of reaction conditions.
(B) Substrate Scope.
Mechanistic Investigations
We found that the donor-acceptor interactions between the fluorine moiety of C(sp3)-F bonds in unactivated aliphatic gem-difluoroalkanes, a weak Lewis base, and the strong Lewis acid B(C6F5)3, is of vital importance; this is emphasized by the overwhelming chemoselectivity for the Friedel-Crafts cyclization of C(sp3)-F bonds rather than the cleavage of weaker C-halogen bonds or the removal of other good leaving groups (Table 2). Specifically, when using a stoichiometric amount of B(C6F5)3 (2.2 equiv.) in CH2Cl2 at room temperature for 30 h, gem-difluoroalkane 1a afforded only the desired 2,2′,3,3′-tetrahydro-1,1′-spirobi[indene] 2a in 85% yield, whereas the formation of elimination product 3a was not observed. In contrast, the intramolecular Friedel-Crafts cyclization was not observed when using 1,5-diphenylpentan-3-one (1v), (3,3-dimethoxypentane-1,5-diyl)dibenzene (1w), (3,3-dichloropentane-1,5-diyl)dibenzene (1x), and (3,3-dibromopentane-1,5-diyl)dibenzene (1y) (Table 2, entries 1–5). Although the cleavage of C-OMe bonds of substrate 1w by B(C6F5)3 (2.2 equiv.) was observed, as the electron-rich heteroatom is a good Lewis base, only a complex mixture was found, and the formation of 2a was not observed (Table 2, entry 3). For the gem-dibromoalkane 1y, the formation of an unexpected elimination product in 29% yield was detected, which was ascribed to the ability of the bromine to act as a good leaving group (Table 2, entry 5). In addition, under optimized conditions of HFIP, the formation of C(sp3)-C(sp3) bonds was only detected for gem-difluoroalkane 1a, but not for the relatively weaker C(sp3)-OMe, C(sp3)-Cl, and C(sp3)-Br bonds (Table 2, entries 6–12). However, the unexpected elimination products (monochloroalkene 3x and monobromoalkene 3y) were formed in of 63% and 81% yield, respectively (Table 2, entry 9 and 11). Interestingly, in the absence of B(C6F5)3 but still using HFIP as solvent, the yields of elimination products 3x and 3y remained essentially unchanged (61% and 86%, respectively). In other words, it is the hydrogen-bonding interaction between HFIP and either the C(sp3)-Cl or C(sp3)-Br bonds rather than the interaction with Lewis acids B(C6F5)3 that governs the elimination process. Similarly, under the standard reaction conditions for the defluorinative elimination of 1a, i.e., treatment with B(C6F5)3 (20 mol %) in refluxing 1,4-difluorobenzene for 24 h, gem-difluoride 1a afforded the desired monofluorinated olefin 3a in 87% yield, whereas a reaction was not observed for the corresponding aliphatic halides and ketals, with the exception of (3,3-dibromopentane-1,5-diyl)dibenzene, which afforded the monobromoalkene elimination product in 9% yield (Table 2, entries 13–16). Therefore the synthesis of spirobiindanes and monofluoroalkenes from aliphatic gem-difluoroalkanes 1 catalyzed by B(C6F5)3 proceeds from a C-F bond activation process.
Table 2.
Control Experiments to Probe Reaction Mechanism
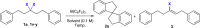 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Xa | Lewis Acids (Equiv.) | Solvent (0.1M) | T (oC) | t (h) | Yields (%) |
|
| 2a | 3ab | ||||||
| 1 | F | B(C6F5)3 (2.2) | CH2Cl2 | RT | 30 | 85 | 0 |
| 2 | 1v (C=O) | B(C6F5)3 (2.2) | CH2Cl2 | RT | 30 | NR | NR |
| 3 | MeO | B(C6F5)3 (2.2) | CH2Cl2 | RT | 30 | NDc | NDc |
| 4 | Cl | B(C6F5)3 (2.2) | CH2Cl2 | RT | 30 | NR | NR |
| 5 | Br | B(C6F5)3 (2.2) | CH2Cl2 | RT | 30 | 0 | 29 |
| 6 | F | B(C6F5)3 (0.2) | (CF3)2CHOHd | 50 | 2 | 84 | 0 |
| 7 | 1v (C=O) | B(C6F5)3 (0.2) | (CF3)2CHOHd | 50 | 2 | NR | NR |
| 8 | MeO | B(C6F5)3 (0.2) | (CF3)2CHOHd | 50 | 2 | NR | NR |
| 9 | Cl | B(C6F5)3 (0.2) | (CF3)2CHOHd | 50 | 2 | 0 | 63 |
| 10 | Cl | – | (CF3)2CHOHd | 50 | 2 | 0 | 61 |
| 11 | Br | B(C6F5)3 (0.2) | (CF3)2CHOHd | 50 | 2 | 0 | 81 |
| 12 | Br | – | (CF3)2CHOHd | 50 | 2 | 0 | 86 |
| 13 | F | B(C6F5)3 (0.2) | p-C6H4F2 | reflux | 24 | 0 | 87 |
| 14 | MeO | B(C6F5)3 (0.2) | p-C6H4F2 | reflux | 24 | 0 | 0 |
| 15 | Cl | B(C6F5)3 (0.2) | p-C6H4F2 | reflux | 24 | 0 | Trace |
| 16 | Br | B(C6F5)3 (0.2) | p-C6H4F2 | reflux | 24 | 0 | 9 |
NR, no reaction; RT, room temperature; ND, not detected.
Substrates: 1,5-diphenylpentan-3-one (1v), (3,3-dimethoxypentane-1,5-diyl)dibenzene (1w), (3,3-dichloropentane-1,5-diyl)dibenzene (1x), and (3,3-dibromopentane-1,5-diyl)dibenzene (1y).
NMR yields.
Complex mixture.
Concentration (0.05 M).
Based on the results discussed above, a reaction mechanism of C-F bond cleavage induced by the C(sp3)-F→B(C6F5)3 interaction is proposed in Figure 4A. In our opinion, two effects of HFIP favor the intramolecular Friedel-Crafts process. (1) The strong hydrogen-bonding interaction between the hydrogen-bonding donor solvent HFIP and the fluoride anion in [FB(C6F5)3]- reduces the Brønsted basicity of the fluoride anion (cf. intermediate III) (Lee et al., 2016, Liang et al., 2017), which would result in the suppression of the E1-type elimination. Indeed, it has already been reported that the Lewis basicity of the fluoride anion of CsF or tetrabutylammonium fluoride (TBAF) is decreased in tertiary alcohols or urea (Kim et al., 2006, Kim et al., 2008a, Kim et al., 2008b, Pfeifer et al., 2016). For instance, relative to anhydrous TBAF, the TBAF(t-BuOH)4 complex significantly favors nucleophilic substitution over elimination pathways (Kim et al., 2008a, Kim et al., 2008b). (2) HFIP, with its high dielectric constant (ε = 15.7) and low nucleophilicity (Colomer et al., 2017), provides additional stabilization for several carbocation intermediates in the intramolecular Friedel-Crafts alkylation (e.g., Figure 4B, II, IV, and VI). In addition, the alternative and probable reaction pathway via further defluorinative cyclization of monofluoroalkene 3 with a C(sp2)-F bond was ruled out as shown in Figure 4B. This result also indicates that the selective C-F bond activation by B(C6F5)3 is limited to C(sp3)-F bonds. For benzylic secondary monofluoride (1-fluoropropane-1,3-diyl)dibenzene (4q), the hydrogen bonding between the benzylic C(sp3)-F bond and HFIP could enable the heterolytic cleavage of the C-F bond to generate carbonium ion VIII in Figure 4C, followed by the formation of the C(sp3)-C(sp3) bond. Accordingly, it is reasonable to extrapolate that in the intramolecular cyclization of aliphatic gem-difluoroalkanes 1, the hydrogen-bond interaction enhances the ability of the fluoride to act as a leaving group, thus promoting the generation of carbonium ion II via the removal of a fluoride anion at relative low reaction temperatures. Indeed, without HFIP, higher temperatures were beneficial for the defluorinative elimination to generate monofluoroalkene 3; the yield of 3a increases from 64% to 81% when the temperature is increased from 160°C to 220°C in 1,2-dichlorobenznene in a sealed tube (Table 1, entries 14 and 17). Therefore, a combination of the hydrogen-bonding donor solvent HFIP and a catalytic amount of B(C6F5)3 promotes the cascade intramolecular Friedel-Crafts reactions of gem-difluorides 1. It also should be pointed out that the HF generated in situ from Friedel-Crafts cyclization might enhance the hydrogen-bonding interaction with C(sp3)-F bonds to improve further the ability of fluoride moiety to act as a leaving group, which would benefit the heterolytic cleavage of C(sp3)-F bonds induced by B(C6F5)3. Indeed, the intermolecular Friedel-Crafts reaction of primary benzylic monofluoride was controlled only by hydrogen-bonding effect initiated by HFIP and HF generated in situ (Champagne et al., 2014).
Figure 4.

Proposed Reaction Mechanism
(A) Plausible reaction pathway for the defluorinative Friedel-Crafts cyclization and defluorinative elimination of gem-difluoroalkanes (1).
(B) To rule out the possibility of affording spirobiindanes 2 via the intermediate of monofluoroalkene 3.
(C) Proposed reaction mechanism for the hydrogen-bonding-induced intramolecular Friedel-Crafts reaction of benzylic monofluoride 4q.
In conclusion, the selective cleavage of C(sp3)-F bonds in unactivated aliphatic gem-difluoroalkanes 1 afforded substituted spirobiindanes 2 and monofluoroalkenes 3 in good yields. In addition, the intramolecular Friedel-Crafts cyclization of aliphatic secondary monofluoroalkanes 4 was also described. The C(sp3)-F→B(C6F5)3 interaction was probed by control experiments by the use of the corresponding ketone, ketal, and other halide-substituted derivatives. Accordingly, the combination of the hydrogen-bonding donor solvent HFIP and a catalytic amount of the Lewis acid B(C6F5)3 enables the selective functionalization of inert C(sp3)-F bonds into C(sp3)-C(sp3) bonds.
Limitations of the Study
The substrates with electron-withdrawing groups such as CF3 and nitro groups are not suitable, which is to support the Friedel-Crafts cyclization mechanism in Figure 4A. We also examined more reactive iodo-substituted substrates, but complex mixtures were obtained. Although the corresponding F-, Cl-, and Br-substituted substrates are acceptable (2i, 2j, 2k, 2l, and 2m, Figure 2A), these results also show some limitation of this method.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by JSPS KAKENHI grants JP 18H02553 (KIBAN B) and JP 18H04401 (Middle Molecular Strategy).
Author Contributions
N.S. conceived the concept. J.W. conducted and analyzed the experiments and synthesized compounds. Y.O. prepared the starting materials. N.S. designed and directed the project, and N.S. and J.W. wrote the manuscript. All authors contributed to discussions.
Declaration of Interests
The authors declare no competing interests.
Published: July 26, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.06.018.
Supplemental Information
References
- Ahrens M., Scholz G., Braun T., Kemnitz E. Catalytic hydrodefluorination of fluoromethanes at room temperature by silylium-ion-like surface species. Angew. Chem. Int. Ed. 2013;52:5328–5332. doi: 10.1002/anie.201300608. [DOI] [PubMed] [Google Scholar]
- Ahrens T., Kohlmann J., Ahrens M., Braun T. Functionalization of fluorinated molecules by transition-metal-mediated c-f bond activation to access fluorinated building blocks. Chem. Rev. 2015;115:931–972. doi: 10.1021/cr500257c. [DOI] [PubMed] [Google Scholar]
- Amii H., Uneyama K. C−F bond activation in organic synthesis. Chem. Rev. 2009;109:2119–2183. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- Bamford K.L., Chitnis S.S., Qu Z.-w., Stephan D.W. Interactions of C−F bonds with hydridoboranes: reduction, borylation and Friedel–Crafts Alkylation. Chem. Eur. J. 2018;24:16014–16018. doi: 10.1002/chem.201804705. [DOI] [PubMed] [Google Scholar]
- Beringhelli T., Maggioni D., D'Alfonso G. 1H and 19F NMR investigation of the reaction of B(C6F5)3 with water in toluene solution. Organometallics. 2001;20:4927–4938. [Google Scholar]
- Birman V.B., Rheingold A.L., Lam K.-C. 1,1′-Spirobiindane-7,7′-diol: a novel, C2-symmetric chiral ligand. Tetrahedron Asymmetry. 1999;10:125–131. [Google Scholar]
- Caputo C.B., Stephan D.W. Activation of alkyl C-F bonds by B(C6F5)3: stoichiometric and catalytic transformations. Organometallics. 2012;31:27–30. [Google Scholar]
- Champagne P.A., Benhassine Y., Desroches J., Paquin J.-F. Friedel–Crafts reaction of benzyl fluorides: selective activation of C−F bonds as enabled by hydrogen bonding. Angew. Chem. Int. Ed. 2014;53:13835–13839. doi: 10.1002/anie.201406088. [DOI] [PubMed] [Google Scholar]
- Champagne P.A., Desroches J., Paquin J.-F. Organic fluorine as a hydrogen-bond acceptor: recent examples and applications. Synthesis. 2015;47:306–322. [Google Scholar]
- Chitnis S.S., LaFortune J.H.W., Cummings H., Liu L.L., Andrews R., Stephan D.W. Phosphorus coordination chemistry in catalysis: air stable P(III)-dications as lewis acid catalysts for the allylation of C–F bonds. Organometallics. 2018;37:4540–4544. [Google Scholar]
- Colomer I., Chamberlain A.E.R., Haughey M.B., Donohoe T.J. Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem. 2017;1:0088. [Google Scholar]
- Cui B., Jia S., Tokunaga E., Shibata N. Defluorosilylation of fluoroarenes and fluoroalkanes. Nat. Commun. 2018;9:4393. doi: 10.1038/s41467-018-06830-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douvris C., Ozerov O.V. Hydrodefluorination of perfluoroalkyl groups using silylium-carborane catalysts. Science. 2008;321:1188–1190. doi: 10.1126/science.1159979. [DOI] [PubMed] [Google Scholar]
- Drouin M., Hamel J.-D., Paquin J.-F. Synthesis of monofluoroalkenes: a leap forward. Synthesis. 2018;50:881–955. [Google Scholar]
- Dryzhakov M., Moran J. Autocatalytic Friedel–Crafts reactions of tertiary aliphatic fluorides initiated by B(C6F5)3·H2O. ACS Catal. 2016;6:3670–3673. [Google Scholar]
- Dryzhakov M., Richmond E., Li G., Moran J. Catalytic B(C6F5)3·H2O-promoted defluorinative functionalization of tertiary aliphatic fluorides. J. Fluor. Chem. 2017;193:45–51. [Google Scholar]
- Forster F., Metsänen T.T., Irran E., Hrobárik P., Oestreich M. Cooperative Al–H bond activation in DIBAL-H: catalytic generation of an alumenium-ion-like lewis acid for hydrodefluorinative Friedel–Crafts alkylation. J. Am. Chem. Soc. 2017;139:16334–16342. doi: 10.1021/jacs.7b09444. [DOI] [PubMed] [Google Scholar]
- Greb L. Lewis superacids: classifications, candidates, and applications. Chem. Eur. J. 2018;24:17881–17896. doi: 10.1002/chem.201802698. [DOI] [PubMed] [Google Scholar]
- Großekappenberg H., Reißmann M., Schmidtmann M., Müller T. Quantitative assessment of the lewis acidity of silylium ions. Organometallics. 2015;34:4952–4958. [Google Scholar]
- Gu W., Haneline M.R., Douvris C., Ozerov O.V. Carbon−carbon coupling of C(sp3)−F bonds using alumenium catalysis. J. Am. Chem. Soc. 2009;131:11203–11212. doi: 10.1021/ja903927c. [DOI] [PubMed] [Google Scholar]
- Guo W.-H., Min Q.-Q., Gu J.-W., Zhang X. Rhodium-catalyzed ortho-selective C-F bond borylation of polyfluoroarenes with Bpin-Bpin. Angew. Chem. Int. Ed. 2015;54:9075–9078. doi: 10.1002/anie.201500124. [DOI] [PubMed] [Google Scholar]
- Hamel J.-D., Paquin J.-F. Activation of C-F bonds α to C-C multiple bonds. Chem. Commun. (Camb.) 2018;54:10224–10239. doi: 10.1039/c8cc05108a. [DOI] [PubMed] [Google Scholar]
- Haufe G., Suzuki S., Yasui H., Terada C., Kitayama T., Shiro M., Shibata N. C-F bond activation of unactivated aliphatic fluorides: synthesis of fluoromethyl-3,5-diaryl-2-oxazolidinones by desymmetrization of 2-Aryl-1,3-difluoro-2-propanols. Angew. Chem. Int. Ed. 2012;51:12275–12279. doi: 10.1002/anie.201207304. [DOI] [PubMed] [Google Scholar]
- Hirano K., Fujita K., Yorimitsu H., Shinokubo H., Oshima K. Boron trifluoride-catalyzed reaction of alkyl fluoride with silyl enolate, allylsilane, and hydrosilane. Tetrahedron Lett. 2004;45:2555–2557. [Google Scholar]
- Jaiswal A.K., Goh K.K.K., Sung S., Young R.D. Aluminum-catalyzed cross-coupling of silylalkynes with aliphatic C–F bonds. Org. Lett. 2017;19:1934–1937. doi: 10.1021/acs.orglett.7b00712. [DOI] [PubMed] [Google Scholar]
- Kim D.W., Ahn D.-S., Oh Y.-H., Lee S., Kil H.S., Oh S.J., Lee S.J., Kim J.S., Ryu J.S., Moon D.H. A new class of SN2 reactions catalyzed by protic solvents: facile fluorination for isotopic labeling of diagnostic molecules. J. Am. Chem. Soc. 2006;128:16394–16397. doi: 10.1021/ja0646895. [DOI] [PubMed] [Google Scholar]
- Kim D.W., Jeong H.J., Lim S.T., Sohn M.-H., Katzenellenbogen J.A., Chi D.Y. Facile nucleophilic fluorination reactions using tert-alcohols as a reaction medium: significantly enhanced reactivity of alkali metal fluorides and improved selectivity. J. Org. Chem. 2008;73:957–962. doi: 10.1021/jo7021229. [DOI] [PubMed] [Google Scholar]
- Kim D.W., Jeong H.-J., Lim S.T., Sohn M.-H. Tetrabutylammonium Tetra(tert-Butyl Alcohol)-coordinated fluoride as a facile fluoride source. Angew. Chem. Int. Ed. 2008;47:8404–8406. doi: 10.1002/anie.200803150. [DOI] [PubMed] [Google Scholar]
- Klahn M., Fischer C., Spannenberg A., Rosenthal U., Krossing I. Hydrodefluorination of non-activated C–F bonds by diisobutyl-aluminiumhydride via the aluminium cation [i-Bu2Al]+ Tetrahedron Lett. 2007;48:8900–8903. [Google Scholar]
- Klare H.F.T. Catalytic C–H arylation of unactivated C–H bonds by silylium ion-promoted C(sp2)–F bond activation. ACS Catal. 2017;7:6999–7002. [Google Scholar]
- Koerte L.A., Schwabedissen J., Soffner M., Blomeyer S., Reuter C.G., Vishnevskiy Y.V., Neumann B., Stammler H.-G., Mitzel N.W. Tris(perfluorotolyl)borane-A boron lewis superacid. Angew. Chem. Int. Ed. 2017;56:8578–8582. doi: 10.1002/anie.201704097. [DOI] [PubMed] [Google Scholar]
- Kuehnel M.F., Lentz D., Braun T. Synthesis of fluorinated building blocks by transition-metal-mediated hydrodefluorination reactions. Angew. Chem. Int. Ed. 2013;52:3328–3348. doi: 10.1002/anie.201205260. [DOI] [PubMed] [Google Scholar]
- Lan K., Shan Z., Fan S. Synthesis of spirobiindanes via bis-cyclization reaction of the 1,5-diaryl-3-pentanones catalyzed by heteropoly acids. Tetrahedron Lett. 2006;47:4343–4345. [Google Scholar]
- Lee J.-W., Oliveira M.T., Jang H.B., Lee S., Chi D.Y., Kim D.W., Song C.E. Hydrogen-bond promoted nucleophilic fluorination: concept, mechanism and applications in positron emission tomography. Chem. Soc. Rev. 2016;45:4638–4650. doi: 10.1039/c6cs00286b. [DOI] [PubMed] [Google Scholar]
- Li S., Zhang J.-W., Li X.-L., Cheng D.-J., Tan B. Phosphoric acid-catalyzed asymmetric synthesis of SPINOL derivatives. J. Am. Chem. Soc. 2016;138:16561–16566. doi: 10.1021/jacs.6b11435. [DOI] [PubMed] [Google Scholar]
- Li Y., Liu J., Zhao S., Du X., Guo M., Zhao W., Tang X., Wang G. Copper-catalyzed fluoroolefination of silyl enol ethers and ketones toward the synthesis of β-fluoroenones. Org. Lett. 2018;20:917–920. doi: 10.1021/acs.orglett.7b03700. [DOI] [PubMed] [Google Scholar]
- Liang S., Hammond G.B., Xu B. Hydrogen bonding: regulator for nucleophilic fluorination. Chem. Eur. J. 2017;23:17850–17861. doi: 10.1002/chem.201702664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z.-J., Zhao H.-Y., Zhang X. Highly selective Pd-catalyzed direct C–F bond arylation of polyfluoroarenes. Org. Lett. 2018;20:2543–2546. doi: 10.1021/acs.orglett.8b00692. [DOI] [PubMed] [Google Scholar]
- Mandal D., Gupta R., Young R.D. Selective monodefluorination and wittig functionalization of gem-difluoromethyl groups to generate monofluoroalkenes. J. Am. Chem. Soc. 2018;140:10682–10686. doi: 10.1021/jacs.8b06770. [DOI] [PubMed] [Google Scholar]
- Morgan M.M., Marwitz A.J.V., Piers W.E., Parvez M. Comparative lewis acidity in fluoroarylboranes: B(o-HC6F4)3, B(p-HC6F4)3, and B(C6F5)3. Organometallics. 2013;32:317–322. [Google Scholar]
- Motiwala H.F., Vekariya R.H., Aube J. Intramolecular Friedel-Crafts acylation reaction promoted by 1,1,1,3,3,3-Hexafluoro-2-propanol. Org. Lett. 2015;17:5484–5487. doi: 10.1021/acs.orglett.5b02851. [DOI] [PubMed] [Google Scholar]
- Nolte C., Ammer J., Mayr H. Nucleofugality and nucleophilicity of fluoride in protic solvents. J. Org. Chem. 2012;77:3325–3335. doi: 10.1021/jo300141z. [DOI] [PubMed] [Google Scholar]
- O'Hagan D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- Olah G.A., Kuhn S.J. Selective Friedel-Crafts reactions. I. Boron halide catalyzed haloalkylation of benzene and alkylbenzenes with fluorohaloalkanes. J. Org. Chem. 1964;29:2317–2320. [Google Scholar]
- Olah G., Kuhn I., Olah J. Aromatic substitution. III. Alkylation of aromatic compounds by the boron trifluoride-catalyzed reaction of alkyl fluorides. J. Chem. Soc. 1957;1957:2174–2176. [Google Scholar]
- Pfeifer L., Engle K.M., Pidgeon G.W., Sparkes H.A., Thompson A.L., Brown J.M., Gouverneur V. Hydrogen-bonded homoleptic fluoride–diarylurea complexes: structure, reactivity, and coordinating power. J. Am. Chem. Soc. 2016;138:13314–13325. doi: 10.1021/jacs.6b07501. [DOI] [PubMed] [Google Scholar]
- Pike S.D., Crimmin M.R., Chaplin A.B. Organometallic chemistry using partially fluorinated benzenes. Chem. Commun. (Camb.) 2017;53:3615–3633. doi: 10.1039/c6cc09575e. [DOI] [PubMed] [Google Scholar]
- Scott V.J., Çelenligil-Çetin R., Ozerov O.V. Room-temperature catalytic hydrodefluorination of C(sp3)−F bonds. J. Am. Chem. Soc. 2005;127:2852–2853. doi: 10.1021/ja0426138. [DOI] [PubMed] [Google Scholar]
- Shen Q., Huang Y.-G., Liu C., Xiao J.-C., Chen Q.-Y., Guo Y. Review of recent advances in CF bond activation of aliphatic fluorides. J. Fluorine Chem. 2015;179:14–22. [Google Scholar]
- Song X., Xu C., Wang M. Transformations based on ring-opening of gem-difluorocyclopropanes. Tetrahedron Lett. 2017;58:1806–1816. [Google Scholar]
- Stahl T., Klare H.F.T., Oestreich M. Main-group lewis acids for C-F bond activation. ACS Catal. 2013;3:1578–1587. [Google Scholar]
- Strobach D.R., Boswell G.A., Jr. Synthesis of 1-fluorocycloalkenes. J. Org. Chem. 1971;36:818–820. [Google Scholar]
- Surmont R., Verniest G., Kimpe N.D. Gold-catalyzed synthesis of 2-Aryl-3-fluoropyrroles. Org. Lett. 2009;11:2920–2923. doi: 10.1021/ol900953n. [DOI] [PubMed] [Google Scholar]
- Tanaka J., Suzuki S., Tokunaga E., Haufe G., Shibata N. Asymmetric desymmetrization via metal-free C−F bond activation: synthesis of 3,5-Diaryl-5-fluoromethyloxazolidin-2-ones with quaternary carbon centers. Angew. Chem. Int. Ed. 2016;55:9432–9436. doi: 10.1002/anie.201603210. [DOI] [PubMed] [Google Scholar]
- Tang R.-J., Milcent T., Crousse B. Bisulfate salt-catalyzed Friedel-Crafts benzylation of arenes with benzylic alcohols. J. Org. Chem. 2018;83:14001–14009. doi: 10.1021/acs.joc.8b02361. [DOI] [PubMed] [Google Scholar]
- Vekariya R.H., Aube J. Hexafluoro-2-propanol-Promoted intermolecular Friedel-crafts acylation reaction. Org. Lett. 2016;18:3534–3537. doi: 10.1021/acs.orglett.6b01460. [DOI] [PubMed] [Google Scholar]
- Yanai H., Okada H., Sato A., Okada M., Taguchi T. Copper-free defluorinative alkylation of allylic difluorides through Lewis acid-mediated C–F bond activation. Tetrahedron Lett. 2011;52:2997–3000. [Google Scholar]
- Yang T.-P., Lin J.-H., Chen Q.-Y., Xiao J.-C. A novel reaction of gem-difluorocyclopropyl ketones with nitriles leading to 2-fluoropyrroles. Chem. Commun. (Camb.) 2013;49:9833–9835. doi: 10.1039/c3cc45456h. [DOI] [PubMed] [Google Scholar]
- Zheng Z., Cao Y., Chong Q., Han Z., Ding J., Luo C., Wang Z., Zhu D., Zhou Q.-L., Ding K. Chiral cyclohexyl-fused spirobiindanes: practical synthesis, ligand development, and asymmetric catalysis. J. Am. Chem. Soc. 2018;140:10374–10381. doi: 10.1021/jacs.8b07125. [DOI] [PubMed] [Google Scholar]
- Zhu J., Pérez M., Caputo C.B., Stephan D.W. Use of trifluoromethyl groups for catalytic benzylation and alkylation with subsequent hydrodefluorination. Angew. Chem. Int. Ed. 2016;55:1417–1421. doi: 10.1002/anie.201510494. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.