

Summary
Background
Lumacaftor/ivacaftor combination therapy demonstrated clinical benefits in patients with cystic fibrosis homozygous for the Phe508del CFTR mutation. Pretreatment lung function is a confounding factor that potentially impacts the efficacy and safety of lumacaftor/ivacaftor therapy.
Methods
Two multinational, randomised, double-blind, placebo-controlled, parallel-group Phase 3 studies randomised patients to receive placebo or lumacaftor (600 mg once daily [qd] or 400 mg every 12 hours [q12h]) in combination with ivacaftor (250 mg q12h) for 24 weeks. Prespecified analyses of pooled efficacy and safety data by lung function, as measured by percent predicted forced expiratory volume in 1 second (ppFEV1), were performed for patients with baseline ppFEV1 <40 (n=81) and ≥40 (n=1016) and screening ppFEV1 <70 (n=730) and ≥70 (n=342). These studies were registered with ClinicalTrials.gov ( NCT01807923 and NCT01807949).
Findings
The studies were conducted from April 2013 through April 2014. Improvements in the primary endpoint, absolute change from baseline at week 24 in ppFEV1, were observed with both lumacaftor/ivacaftor doses in the subgroup with baseline ppFEV1 <40 (least-squares mean difference versus placebo was 3∙7 and 3.3 percentage points for lumacaftor 600 mg qd/ivacaftor 250 mg q12h and lumacaftor 400 mg q12h/ivacaftor 250 mg q12h, respectively [p<0∙05] and in the subgroup with baseline ppFEV1 ≥40 (3∙3 and 2∙8 percentage points, respectively [p<0∙001]). Similar absolute improvements versus placebo in ppFEV1 were observed in subgroups with screening ppFEV1 <70 (3∙3 and 3∙3 percentage points for lumacaftor 600 mg qd/ivacaftor 250 mg q12h and lumacaftor 400 mg q12h/ivacaftor 250 mg q12h, respectively [p<0∙001]) and ≥70 (3∙3 and 1∙9 percentage points, respectively [p=0.002] and [p=0∙079]). Increases in BMI and reduction in number of pulmonary exacerbation events were observed in both LUM/IVA dose groups vs placebo across all lung function subgroups. Treatment was generally well tolerated, although the incidence of some respiratory adverse events was higher with active treatment than with placebo.
Interpretation
Lumacaftor/ivacaftor combination therapy benefits patients homozygous for Phe508del CFTR who have varying degrees of lung function impairment.
Keywords: cystic fibrosis, lumacaftor, ivacaftor, Phe508del, lung function, cystic fibrosis transmembrane conductance regulator
Introduction
The most common cystic fibrosis (CF)-causing mutation, Phe508del CF transmembrane conductance regulator (CFTR), leads to a variety of defects, including reduced folding and trafficking of the CFTR protein to the epithelial cell surface and defective channel gating, among others.1–4 Therefore, restoring the chloride transport activity of the Phe508del CFTR channel is complex. Lumacaftor (LUM) is a CFTR corrector, which selectively increases the processing and trafficking of Phe508del CFTR to the cell surface and enhances CFTR–mediated chloride transport in vitro.5 Ivacaftor (IVA) is a CFTR potentiator, which facilitates chloride transport by increasing the channel-open probability of CFTR on the cell surface.6 Monotherapy with either LUM or IVA was not shown to be clinically beneficial in patients with CF homozygous for the Phe508del CFTR mutation.7,8 In contrast, clinically meaningful benefits were observed with combination therapy in patients with CF homozygous for the Phe508del CFTR mutation in a Phase 29 and in two Phase 3, randomised, double-blind, placebo-controlled studies, TRAFFIC and TRANSPORT.10
Significant improvements in lung function were observed with LUM 600 mg once daily (qd)/IVA 250 mg every 12 hours (q12h) and LUM 400 mg q12h/IVA 250 mg q12h in the TRAFFIC and TRANSPORT studies; the mean absolute change in percent predicted forced expiratory volume in 1 second (ppFEV1) at week 24 versus placebo ranged from 2∙8 to 3∙3 percentage points in the pooled analysis (p<0∙001).10 Improvements were also observed in nutritional status and rate of pulmonary exacerbations (PEx). These data orted the approval of LUM/IVA combination therapy (Orkambi; Vertex Pharmaceuticals Incorporated; Boston, MA, USA) in patients aged 12 and older with CF homozygous for the Phe508del CFTR mutation in the United States, the European Union, and Canada.
Patients with CF whose ppFEV1 is in the severe range have a greater burden of disease associated with a higher rate of PEx and worse nutritional status.11,12 The safety and efficacy of new treatments in patients with severe lung dysfunction may not be the same as in patients with milder dysfunction. The TRAFFIC and TRANSPORT studies enrolled patients with ppFEV1 values of 40 to 90 percentage points at screening, reflecting a range of lung function impairment from mild (ppFEV1 ≥70 to ≤90) to moderate (ppFEV1 40 to 69). Some patients had a ppFEV1 value that decreased to below 40 between screening and baseline, providing an opportunity to assess treatment response in this clinically important subgroup.10 Prospective evaluation of the safety and efficacy of LUM/IVA in patients with severe lung dysfunction is ongoing. Here, we describe a prespecified pooled analysis of data from the TRAFFIC and TRANSPORT studies performed to determine the efficacy and safety of LUM/IVA combination therapy in patients with CF homozygous for the Phe508del CFTR mutation, defined by specific categories of lung function, including those with severe lung dysfunction (ppFEV1 <40 at baseline).
Methods
Study design and patients
The TRAFFIC and TRANSPORT trials were multinational, randomised, double-blind, placebo-controlled, parallel-group, 24-week Phase 3 studies conducted from April 2013 through April 2014. Both studies were conducted in accordance with the principles of the Declaration of Helsinki and in compliance with Good Clinical Practice guidelines and all applicable local and national regulations. The study protocol was approved by ethics committees, and all patients provided written informed consent.
The design of these nearly identical studies has been described previously and is briefly reviewed in the supplemental appendix.10 The studies included patients aged 12 years or older with a confirmed diagnosis of CF, homozygous for the Phe508del CFTR mutation, and a ppFEV1 of 40 to 90 at the time of screening. Some patients had ppFEV1 levels that decreased to below 40 between the screening and baseline visits (≤4 weeks). In the pooled analysis, data from the two studies were pooled by dosing regimens.
Outcomes
For the pooled TRAFFIC and TRANSPORT study data, preplanned subgroup analyses of ppFEV1 <40 versus ≥40 at baseline and ppFEV1 <70 versus ≥70 at screening were performed for the primary endpoint and key secondary endpoints in a manner similar to that reported previously for the entire study cohort.10 The primary endpoint was the absolute change from baseline in ppFEV1 at week 24, calculated by averaging the mean absolute change at week 16 and the mean absolute change at week 24. Key secondary endpoints were: the relative change from baseline in ppFEV1 at week 24 (calculated by averaging the mean values for weeks 16 and 24); the percentage of patients with at least a 5% relative increase from baseline in ppFEV1 (response derived using average relative change at weeks 16 and 24); the absolute change from baseline in body mass index (BMI) at week 24; the absolute change from baseline in the Cystic Fibrosis Questionnaire-Revised (CFQ-R) respiratory domain score at week 24; and the number of PEx through week 24 (expressed as a rate over 48 weeks). In addition, post hoc subgroup analyses were performed for the absolute change from baseline in ppFEV1 at each study visit, the percentage of patients with at least a 10% relative increase from baseline in ppFEV1 (response derived using average relative change at weeks 16 and 24), the number of PEx requiring intravenous (IV) antibiotics, and the number of PEx requiring hospitalisation. Safety and tolerability were assessed by reports of adverse events (AEs) and by clinical laboratory parameters.
Statistical analyses
The efficacy population included all patients who were randomised and received at least one dose of study drug; patients were analysed according to the study group to which they were randomised. Pooled data were analysed for each subgroup separately, defined according to ppFEV1 <40 and ≥40 at baseline and ppFEV1 <70 and ≥70 at screening. The least squares (LS) means for the subgroup analysis of the absolute and relative changes from baseline in ppFEV1 were calculated using a mixed-effects model for repeated measures (MMRM) that included study, sex, age (<18 vs ≥18 years), treatment, visit, and treatment-by-visit interaction. The odds ratio versus placebo for the percentage of patients with at least a 5% and at least 10% relative increase from baseline in ppFEV1 for each subgroup was estimated using the Cochran-Mantel-Haenszel test, stratified by study, baseline age (<18 vs ≥18 years), and sex. The LS means for the subgroup analysis of absolute change in BMI and CFQ-R respiratory domain were calculated using an MMRM model that included study, sex, age, treatment, visit, and treatment-by-visit interaction, plus the corresponding baseline as a covariate. The rate ratio of PEx events for each subgroup (ie, event rate per year for the treatment group vs that for the placebo group) was calculated using a negative binomial regression model that included study, treatment, sex, and age, with log(time on study in years) as an offset; 48 weeks was considered equivalent to 1 year for the analysis.
The safety analysis included all patients who received any amount of study drug and was based on actual treatment received. Patients who received medication from more than one treatment group during the studies were considered to be in the lower dose of the active treatment group.
Statistical analyses were performed using Statistical Analysis Software version 9∙2 or higher; p values <0∙05 were considered statistically significant and were not adjusted for multiplicity. The studies were registered with ClinicalTrials.gov (NCT01807923 and NCT01807949).
Role of the funding source
The funder participated in the design of the protocol, performed the statistical analysis, and was involved in data interpretation. Medical writing as well as editorial support and coordination were provided by the funder. All authors had full access to the study data. JSE contributed to data interpretation and manuscript conception, writing and revision, and made the final decision to submit for publication.
Results
Of the 1108 patients who were randomised and received at least one dose of study treatment in the pooled TRAFFIC and TRANSPORT studies, 342 patients (30∙9%) had a ppFEV1 of ≥70 at screening. Eighty-one patients (7∙3%) had a ppFEV1 level that decreased to <40 between the screening and baseline visits (range: 31∙1–39∙9). In the pooled data, treatment groups were well balanced across demographic and baseline characteristics, as reported previously.10 Characteristics of the subgroups at baseline classified by ppFEV1 <40 versus ≥40 and by ppFEV1 <70 versus ≥70 are shown in Table 1. A high percentage of patients in each subgroup reported maintenance use of bronchodilators and multiple other CF treatments. The majority of patients in each subgroup completed 24 weeks of study treatment, including 78 of the 81 patients (96∙3%) with severe lung dysfunction at baseline (ppFEV1 <40). With respect to patients who received the LUM 400 mg q12h/IVA 250 mg q12h dose, there were 29 in the subgroup with ppFEV1 <40 at baseline, 336 in the subgroup with ppFEV1 ≥40 at baseline, 245 in the subgroup with ppFEV1 <70 at screening, and 114 in the subgroup with ppFEV1 ≥70 at screening.
Table 1:
Pooled patient demographic and baseline characteristics
| Characteristics | Placebo overall (n=371) |
LUM/IVA overall | |||
|---|---|---|---|---|---|
| ppFEV1 <40* (n=53) |
ppFEV1 ≥40 (n=678) |
ppFEV1 <70 (n=527) |
ppFEV1 ≥70 (n=204) |
||
| Female, n (%) | 181 (48∙8) | 31 (58∙5) | 331 (48∙8) | 269 (51∙0) | 93 (45∙6) |
| Age, mean (range), years | 25∙4 (12–64) | 27∙3 (13–44) | 24∙7 (12–57) | 26∙3 (12–57) | 21∙0 (12–53) |
| ppFEV1 at baseline, mean (range) | 60·4 (33·9–99·8) | 37·2 (31·1–39·9) | 62·5 (40·0–96·5) | 54·0 (31·1–69·8) | 77·9 (70·0–96·5) |
| Body mass index (mg/kg2), mean (range) | 21·0 (14·1–32·2) | 20·9 (16·1–31·4) | 21·3 (14·2–35·1) | 21·2 (14·2–35·1) | 21·4 (14·6–29·8) |
| Chronic CF therapy use at baseline, n (%) | |||||
| Bronchodilators (any) | 342 (92·2) | 50 (94·3) | 631 (93·1) | 496 (94·1) | 185 (90·7) |
| Dornase alfa | 281 (75·7) | 41 (77·4) | 517 (76·3) | 407 (77·2) | 151 (74·0) |
| Inhaled antibiotic | 258 (69·5) | 33 (62·3) | 421 (62·1) | 351 (66·6) | 103 (50·5) |
| Inhaled hypertonic saline | 220 (59·3) | 34 (64·2) | 386 (56·9) | 294 (55·8) | 126 (61·8) |
| Inhaled corticosteroids | 220 (59·3) | 35 (66·0) | 386 (56·9) | 311 (59·0) | 110 (53·9) |
Eighty-one patients (placebo, n=28; LUM/IVA, n=53) had ppFEV1 that decreased to <40 between screening and baseline.
CF=cystic fibrosis; IVA=ivacaftor; LUM=lumacaftor; ppFEV1=percent predicted forced expiratory volume in 1 second.
Significant improvements in the primary efficacy endpoint, absolute change from baseline in ppFEV1 at week 24, were observed with both doses of LUM/IVA (LUM 600 mg qd/IVA 250 mg q12h and LUM 400 mg q12h/IVA 250 mg q12h) in the subgroup with ppFEV1 <40 at baseline (LS mean difference versus placebo was 3∙7 and 3.3 percentage points, respectively [p<0∙05]) and in the subgroup with ppFEV1 ≥40 at baseline (3∙3 and 2∙8 percentage points, respectively [p<0∙001]) (Table 2). Generally similar results favoring LUM/IVA over placebo were observed in subgroups with ppFEV1 <70 and ≥70 at screening, although statistical significance was not reached in the ≥70 subgroup receiving LUM 400 mg q12h/IVA 250 mg q12h (Table 3). The absolute change versus placebo across all lung function subgroups ranged from 1∙9–3∙7 percentage points, consistent with differences observed in the overall population pooled from the two studies by dosing regimen (2∙8–3∙3 percentage points).10 Figure 1 shows the absolute change from baseline in ppFEV1 at each study visit throughout 24 weeks of treatment in subgroups defined by ppFEV1. Improvements in ppFEV1 were observed as early as day 15 and were sustained through week 24 with both LUM/IVA doses in these subgroups.
Table 2:
Efficacy results after treatment with LUM/IVA for 24 weeks in patients with ppFEV1 <40 vs ≥40 at baseline
| Parameter | Placebo | LUM 600 mg qd/IVA 250 mg q12h | LUM 400 mg q12h/IVA 250 mg q12h | |||
|---|---|---|---|---|---|---|
| ppFEV1 at baseline* | ||||||
| <40 (n=28) |
≥40 (n=338) |
<40 (n=24) |
≥40 (n=342) |
<40 (n=29) |
≥40 (n=336) |
|
| Absolute change in ppFEV1 | ||||||
| Within group LS mean (SE) | 0∙4 (1∙3) | −0∙4 (0∙4) | – | – | – | – |
| LS mean vs placebo (95% CI), percentage points† | – | – | 3·7 (0·5–6·9) | 3·3 (2·3–4·4) | 3·3 (0·2–6·4) | 2·8 (1·7–3·8) |
| p value | – | – | 0·024 | <0·001 | 0·036 | <0·001 |
| Relative change in ppFEV1 | ||||||
| Within group LS mean (SE) | 1·5 (3·4) | −0·2 (0·7) | – | – | – | – |
| LS mean vs placebo (95% CI), %† | – | – | 9·9 (1·2–18·5) | 5·3 (3·5–7·1) | 9·1 (0·7–17·4) | 4·5 (2·7–6·3) |
| p value | – | – | 0·026 | <0·001 | 0·034 | <0·001 |
| Relative increase of ≥5% from baseline in ppFEV1‡ | ||||||
| Odds ratio vs placebo (95% CI) | – | – | 2·4 (0·8–7·2) | 3·1 (2·2–4·3) | 1·7 (0·6–5·2) | 2·3 (1·6–3·2) |
| p value | – | – | 0·113 | <0·001 | 0·331 | <0·001 |
| Body mass index | ||||||
| Within group LS mean (SE) | 0·1 (0·2) | 0·1 (0·1) | – | – | – | – |
| LS mean vs placebo (95% CI), kg/m2 | – | – | 0·6 (0·1–1·2) | 0·3 (0·1–0·4) | 0·3 (−0·2–0·8) | 0·2 (0·1–0·4) |
| p value | – | – | 0·023 | <0·001 | 0·261 | 0·001 |
| CFQ-R respiratory domain | ||||||
| Within group LS mean (SE) | 5·8 (3·2) | 0·9 (0·9) | – | – | – | – |
| LS mean vs placebo (95% CI), points | – | – | 3·3 (−5·2–11·7) | 3·3 (1·0–5·7) | −4·2 (−12·0–3·7) | 2·9 (0·5–5·3) |
| p value | – | – | 0·446 | 0·006 | 0·298 | 0·017 |
Eighty-one patients had ppFEV1 levels that decreased to <40 between screening and baseline.
Assessed by averaging the mean values from weeks 16 and 24, as prespecified in the statistical analysis plan.
Average relative increase from baseline at weeks 16 and 24.
CFQ-R=Cystic Fibrosis Questionnaire-Revised; CI=confidence interval; IVA=ivacaftor; LUM=lumacaftor; LS=least squares; ppFEV1=percent predicted forced expiratory volume in 1 second; SE=standard error; q12h=every 12 hours; qd=every day.
Table 3:
Efficacy results after treatment with LUM/IVA for 24 weeks in patients with ppFEV1 <70 vs ≥70 at screening
| Parameter | Placebo | LUM 600 mg qd/IVA 250 mg q12h | LUM 400 mg q12h/IVA 250 mg q12h | |||
|---|---|---|---|---|---|---|
| ppFEV1 at screening* | ||||||
| <70 (n=244) |
≥70 (n=109) |
<70 (n=241) |
≥70 (n=119) |
<70 (n=245) |
≥70 (n=114) |
|
| Absolute change in ppFEV1 | ||||||
| Within group LS mean (SE) | −0∙5 (0∙4) | 0∙1 (0∙8) | – | – | – | – |
| LS mean vs placebo (95% CI), percentage points† | – | – | 3∙3 (2∙1–4∙4) | 3∙3 (1∙3–5∙4) | 3∙3 (2∙1–4∙4) | 1∙9 (−0∙2–4∙0) |
| p value | – | – | <0∙001 | 0∙002 | <0∙001 | 0∙079 |
| Relative change in ppFEV1 | ||||||
| Within group LS mean (SE) | −0∙3 (0∙9) | 0∙7 (1∙1) | – | – | – | – |
| LS mean vs placebo (95% CI), %† | – | – | 6∙0 (3∙7–8∙2) | 4∙4 (1∙5–7∙4) | 5∙9 (3∙6–8∙2) | 2∙5 (−0∙5–5∙5) |
| p value | – | – | <0∙001 | 0∙003 | <0∙001 | 0∙103 |
| Relative increase of ≥5% from baseline in ppFEV1‡ | ||||||
| Odds ratio vs placebo (95% CI) | – | – | 2∙5 (1∙7–3∙7) | 3∙8 (2∙1–6∙8) | 2∙4 (1∙6–3∙5) | 1∙9 (1∙0–3∙4) |
| p value | – | – | <0∙001 | <0∙001 | <0∙001 | 0∙045 |
| Body mass index | ||||||
| Within group LS mean (SE) | 0∙1 (0∙1) | 0∙1 (0∙1) | – | – | – | – |
| LS mean vs placebo (95% CI), kg/m2 | – | – | 0∙2 (0∙0–0∙4) | 0∙4 (0∙2–0∙7) | 0∙2 (0∙0–0∙3) | 0∙3 (0∙1–0∙6) |
| p value | – | – | 0∙017 | <0∙001 | 0∙041 | 0∙006 |
| CFQ-R respiratory domain | ||||||
| Within group LS mean (SE) | 1∙5 (1∙1) | 1∙7 (1∙4) | – | – | – | – |
| LS mean vs placebo (95% CI), points | – | – | 4∙1 (1∙3–6∙9) | 1∙9 (−1∙9–5∙7) | 1∙9 (−0∙9–4∙7) | 3∙6 (−0∙3–7∙4) |
| p value | – | – | 0∙005 | 0∙326 | 0∙184 | 0∙071 |
Eighty-one patients had ppFEV1 that decreased to <40 between screening and baseline.
Assessed by averaging the mean values from weeks 16 and 24, according to the prespecified statistical analysis plan.
Average relative increase from baseline at weeks 16 and 24.
CFQ-R=Cystic Fibrosis Questionnaire-Revised; CI=confidence interval; IVA=ivacaftor; LUM=lumacaftor; LS=least squares; ppFEV1=percent predicted forced expiratory volume in 1 second; SE=standard error; q12h=every 12 hours; qd=every day.
Figure 1:
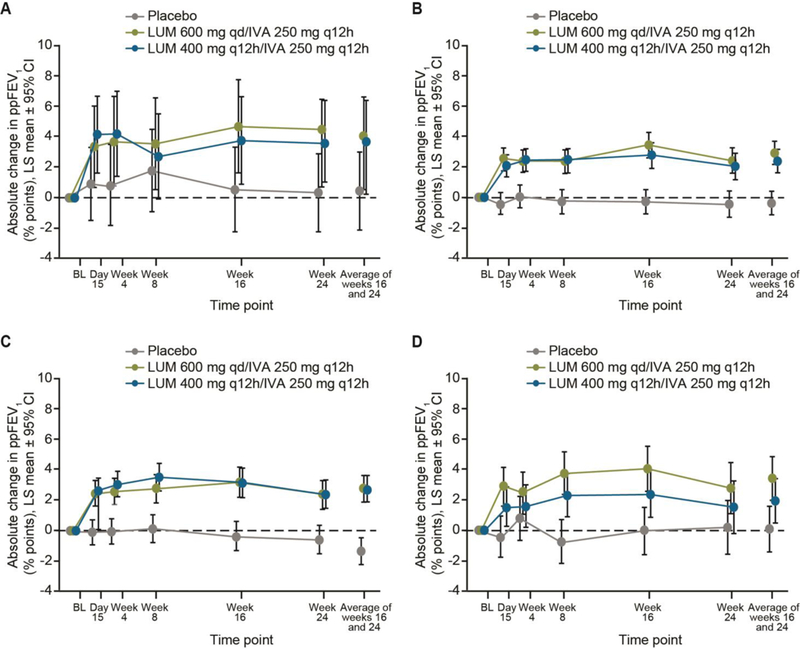
Absolute change from baseline in ppFEV1 at each study visit for patients with baseline ppFEV1 <40 (A) or ≥40 (B), and for patients with screening ppFEV1 <70 (C) or ≥70 (D)
BL=baseline; CI=confidence interval; IVA=ivacaftor; LUM=lumacaftor; LS=least squares; ppFEV1=percent predicted forced expiratory volume in 1 second; q12h=once every 12 hours; qd=once daily.
The differences between LUM/IVA and placebo with respect to relative change from baseline at week 24 in ppFEV1 were consistent with results for the absolute change in ppFEV1. Relative improvements in ppFEV1 with LUM 600 mg qd/IVA 250 mg q12h and LUM 400 mg q12h/IVA 250 mg q12h versus placebo were 9∙9% and 9∙1%, respectively in the subgroup with baseline ppFEV1 <40 (p<0∙05) and 5∙3% and 4∙5%, respectively in the subgroup with baseline ppFEV1 ≥40 (p<0∙001) (Table 2). Relative improvements in ppFEV1 with both LUM/IVA doses versus placebo were also observed in the subgroups with screening ppFEV1 <70 (6∙0% and 5∙9%, respectively) and ≥70 (4∙4% and 2∙5%, respectively); once again, significance was not reached in the ≥70 subgroup receiving LUM 400 mg q12h/IVA 250 mg q12h (Table 3). The proportion of patients with ≥5% and ≥10% average relative increases from baseline at weeks 16 and 24 in ppFEV1 was significantly higher with both LUM/IVA doses than with placebo in subgroups with ppFEV1 ≥40 at baseline (p≤0∙002) and ppFEV1 <70 at screening (p<0∙001) (Figure 2). Similar trends favoring LUM/IVA doses were observed in the other subgroups, but statistical significance was not reached in most comparisons in the smaller subgroup with baseline ppFEV1 <40; significance was achieved for most comparisons in the subgroup with screening ppFEV1 ≥70 (Figure 2).
Figure 2:
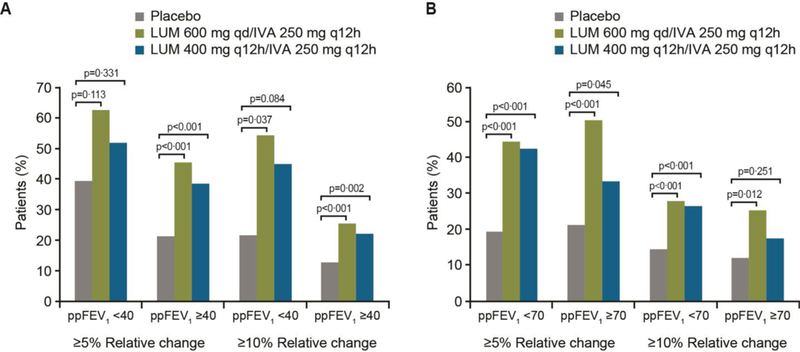
Percentage of patients with ≥5% and ≥10% average relative increases from baseline in ppFEV1 at weeks 16 and 24 in patients with ppFEV1 <40 or ≥40 at baseline (A) and ppFEV1 <70 or ≥70 at screening (B)
IVA=ivacaftor; LUM=lumacaftor; ppFEV1=percent predicted forced expiratory volume in 1 second; q12h=once every 12 hours; qd=once daily.
Although these subgroup analyses were not powered to detect statistical differences, improvements in lung function and other clinical parameters were observed. The absolute change in BMI was statistically significant in most subgroups (Tables 2–3). Improvements in the CFQ-R respiratory domain score favoring LUM/IVA over placebo were observed in some of the larger subgroups, including those with ppFEV1 ≥40 at both LUM/IVA doses (Tables 2–3), although variability on this measure was high, particularly in the subgroups with small patient numbers.
Treatment with LUM/IVA significantly reduced the number of PEx compared with placebo in most ppFEV1 subgroups (Table 4). Additionally, trends toward fewer PEx events requiring IV antibiotic therapy and hospitalisations were observed in both LUM/IVA dose groups versus placebo across all lung function subgroups (Table 4).
Table 4:
Pulmonary exacerbation events through week 24 by ppFEV1 subgroup and treatment group
| Rate ratio vs placebo (95% CI) | LUM 600 mg qd/IVA 250 mg q12h | LUM 400 mg q12h/IVA 250 mg q12h | ||
|---|---|---|---|---|
| ppFEV1 at baseline | ||||
| <40 (n=24)* |
≥40 (n=342) |
<40 (n=29)* |
≥40 (n=336) |
|
| Pulmonary exacerbation events | 0∙47 (0∙24–0∙93) | 0∙73 (0∙58–0∙92) | 0∙59 (0∙33–1∙05) | 0∙61 (0∙48–0∙77) |
| p value | 0∙030 | 0∙007 | 0∙074 | <0∙001 |
| Events requiring IV antibiotic therapy | 0∙41 (0∙17–0∙98) | 0∙57 (0∙43–0∙77) | 0∙56 (0∙27–1∙17) | 0∙42 (0∙30–0∙58) |
| p value | 0∙046 | <0∙001 | 0∙122 | <0∙001 |
| Events requiring hospitalisation | 0∙43 (0∙14–1∙33) | 0∙63 (0∙44–0∙89) | 0∙67 (0∙27–1∙65) | 0∙36 (0∙23–0∙54) |
| p value | 0∙142 | 0∙009 | 0∙382 | <0∙001 |
| ppFEV1 at screening | ||||
| <70 (n=241) |
≥70 (n=119) |
<70 (n=245) |
≥70 (n=114) |
|
| Pulmonary exacerbation events | 0∙74 (0∙57–0∙95) | 0∙55 (0∙35–0∙85) | 0∙65 (0∙50–0∙84) | 0∙51 (0∙32–0∙80) |
| p value | 0∙018 | 0∙007 | 0∙001 | 0∙003 |
| Events requiring IV antibiotic therapy | 0∙53 (0∙39–0∙73) | 0∙53 (0∙27–1∙01) | 0∙49 (0∙36–0∙68) | 0∙22 (0∙09–0∙55) |
| p value | <0∙001 | 0∙052 | <0∙001 | 0∙001 |
| Events requiring hospitalisation | 0∙59 (0∙40–0∙85) | 0∙53 (0∙27–1∙06) | 0∙48 (0∙32–0∙71) | 0∙09 (0∙02–0∙37) |
| p value | 0∙005 | 0∙072 | <0∙001 | 0∙001 |
Eighty-one patients had ppFEV1 that decreased to <40 between screening and baseline.
CI=confidence interval; IV=intravenous; IVA=ivacaftor; LUM=lumacaftor; q12h=every 12 hours; qd=every day.
The overall incidence of AEs in both LUM/IVA groups and in the placebo group was similar among patients with ppFEV1 <40 and ≥40 at baseline and those with ppFEV1 <70 and ≥70 at screening (Table 5). Because the incidence of AEs was similar between the two LUM/IVA dose groups, the safety data of the two dosing regimens were pooled. The most commonly reported AEs across all treatment groups were infective PEx of CF and cough. The incidence of certain respiratory AEs was greater in the pooled LUM/IVA group than in the placebo group in all subgroups; in patients with baseline ppFEV1 <40, these AEs with higher incidence in the pooled LUM/IVA group than in placebo included cough (39∙6% vs 25∙0%), dyspnoea (26∙4% vs 14∙3%), and respiration abnormal (the Preferred Term for the verbatim term of chest tightness [7∙5% vs 3∙6%]). The incidence of dyspnoea and respiration abnormal was also greater in the pooled LUM/IVA group than in the placebo group in those with baseline ppFEV1 ≥40 (13∙0% vs 7∙4% and 10∙0% vs 6∙2%, respectively), as well as in those with screening ppFEV1 <70 and ≥70 (Table 5). Irrespective of lung function subgroup, respiratory AEs were associated with the initiation of treatment and usually resolved with continued treatment. The median time (min–max) to onset of the first AE of special interest of respiratory symptoms was 2 (1–170) days for the pooled LUM/IVA groups (n=738) and 43 (1–172) days for the placebo group (n=370).
Table 5:
Summary of treatment-emergent adverse events
| Variable, n (%) | Placebo | LUM/IVA* | Placebo | LUM/IVA* | ||||
|---|---|---|---|---|---|---|---|---|
| ppFEV1 at baseline | ppFEV1 at screening | |||||||
| <40 (n=28) |
≥40 (n=337) |
<40 (n=53) |
≥40 (n=679) |
<70 (n=243) |
≥70 (n=109) |
<70 (n=487) |
≥70 (n=233) |
|
| Patients who experienced any AE | 28 (100) | 322 (95·5) | 52 (98·1) | 649 (95·6) | 235 (96·7) | 102 (93·6) | 466 (95·7) | 224 (96·1) |
| AEs reported in ≥10% of patients in any subgroup of placebo or total LUM/IVA | ||||||||
| Infective PEx of CF | 20 (71·4) | 162 (48·1) | 27 (50·9) | 248 (36·5) | 125 (51·4) | 53 (48·6) | 211 (43·3) | 59 (25·3) |
| Cough | 7 (25·0) | 140 (41·5) | 21 (39·6) | 203 (29·9) | 94 (38·7) | 47 (43·1) | 153 (31·4) | 68 (29·2) |
| Dyspnoea | 4 (14·3) | 25 (7·4) | 14 (26·4) | 88 (13·0) | 26 (10·7) | 3 (2·8) | 83 (17·0) | 17 (7·3) |
| Sputum increased | 8 (28·6) | 62 (18·4) | 13 (24·5) | 94 (13·8) | 49 (20·2) | 18 (16·5) | 80 (16·4) | 25 (10·7) |
| Headache | 5 (17·9) | 52 (15·4) | 10 (18·9) | 103 (15·2) | 42 (17·3) | 14 (12·8) | 74 (15·2) | 36 (15·5) |
| Pyrexia | 5 (17·9) | 29 (8·6) | 8 (15·1) | 59 (8·7) | 28 (11·5) | 6 (5·5) | 51 (10·5) | 15 (6·4) |
| Diarrhoea | 2 (7·1) | 29 (8·6) | 7 (13·2) | 73 (10·8) | 19 (7·8) | 10 (9·2) | 62 (12·7) | 16 (6·9) |
| Nausea | 3 (10·7) | 25 (7·4) | 7 (13·2) | 67 (9·9) | 18 (7·4) | 9 (8·3) | 56 (11·5) | 17 (7·3) |
| Fatigue | 2 (7·1) | 27 (8·0) | 6 (11·3) | 57 (8·4) | 21 (8·6) | 7 (6·4) | 48 (9·9) | 15 (6·4) |
| Haemoptysis | 7 (25·0) | 43 (12·8) | 6 (11·3) | 95 (14·0) | 42 (17·3) | 8 (7·3) | 81 (16·6) | 18 (7·7) |
| Nasopharyngitis | 2 (7·1) | 37 (11·0) | 6 (11·3) | 65 (9·6) | 30 (12·3) | 8 (7·3) | 49 (10·1) | 20 (8·6) |
| Oropharyngeal pain | 1 (3·6) | 29 (8·6) | 6 (11·3) | 61 (9·0) | 17 (7·0) | 11 (10·1) | 43 (8·8) | 24 (10·3) |
| URTI | 0 (0) | 19 (5·6) | 6 (11·3) | 53 (7·8) | 12 (4·9) | 5 (4·6) | 39 (8·0) | 18 (7·7) |
| Nasal congestion | 1 (3·6) | 43 (12·8) | 5 (9·4) | 52 (7·7) | 22 (9·1) | 21 (19·3) | 34 (7·0) | 23 (9·9) |
| Respiration abnormal | 1 (3·6) | 21 (6·2) | 4 (7·5) | 68 (10·0) | 19 (7·8) | 2 (1·8) | 49 (10·1) | 22 (9·4) |
| Blood creatinine phosphokinase increased | 1 (3·6) | 19 (5·6) | 2 (3·8) | 39 (5·7) | 7 (2·9) | 12 (11·0) | 24 (4·9) | 16 (6·9) |
| Viral URTI | 4 (14·3) | 20 (5·9) | 2 (3·8) | 48 (7·1) | 15 (6·2) | 8 (7·3) | 34 (7·0) | 16 (6·9) |
Pooled data for the LUM 600 mg qd/IVA 250 mg q12h and LUM 400 mg q12h/IVA 250 mg q12h groups.
AE=adverse event; CF=cystic fibrosis; IVA=ivacaftor; LUM=lumacaftor; PEx=pulmonary exacerbation; ppFEV1=percent predicted forced expiratory volume in 1 second; URTI=upper respiratory tract infection.
With respect to baseline ppFEV1 values, the incidence of dyspnoea was approximately two times higher in patients with ppFEV1 <40 versus ≥40 in both the placebo group (14∙3% vs 7∙4%) and active treatment group (26∙4% vs 13∙0%), consistent with what might be expected for a population of patients with more severe lung dysfunction. The proportion of patients who discontinued treatment because of AEs was small across all subgroups; such discontinuations occurred in 3∙6% of patients (n=1) who received placebo and 0% who received LUM/IVA in the <40 subgroup, and in 1∙5% of patients (n=5) who received placebo and 4∙6% of patients (n=31) who received LUM/IVA in the ≥40 subgroup.
Discussion
This pooled analysis of data from the TRAFFIC and TRANSPORT studies shows that the efficacy and safety of LUM/IVA in patients with CF homozygous for the Phe508del CFTR mutation was similar across lung function subgroups, including ppFEV1 <40 and ≥40 at baseline and ppFEV1 <70 and ≥70 at screening.
The data in the subgroup with ppFEV1 <40 at baseline were notable given the severity of lung function impairment in these patients (ppFEV1 range of 31∙1–39∙9 percentage points). In this subgroup, the absolute improvement in lung function, as measured by ppFEV1, from baseline at week 24 with both LUM/IVA doses compared with placebo ranged from 3∙3 to 3∙7 percentage points, which was similar to the improvement in lung function observed in those with ppFEV1 ≥40 (2∙8–3∙3 percentage points) and in the overall study population.10 Also notable were outcomes in patients whose ppFEV1 was ≥70 at screening; lung function improvements in this subgroup were also generally consistent with the overall study population.10
Clinical improvements in BMI were also seen with both LUM/IVA doses compared with placebo; these were generally similar in magnitude across lung function subgroups. Furthermore, clinically meaningful reductions in PEx events were observed across lung function subgroups, including those with ppFEV1 <40 at baseline and ≥70 at screening. Similarly, reductions in those events requiring the use of IV antibiotics and hospitalisation were observed across subgroups. The majority of these comparisons reached statistical significance, but the small sample size in some subgroups likely limited the ability to detect statistical differences. Using the respiratory domain of the CFQ-R, a CF-specific patient-reported outcome instrument,13 significant improvements were noted in some of the subgroups with larger patient numbers; however, variability was high, particularly in the smaller subgroups, which limited interpretation of these findings.
The side-effect profile of LUM/IVA therapy was acceptable in each lung function subgroup. The rates of discontinuation due to AEs were low across lung function subgroups. The incidence of certain respiratory AEs (such as dyspnoea) was higher in subgroups with more impaired lung function (eg, ppFEV1 <40 versus ≥40) in both the placebo and LUM/IVA groups. The increased incidence of certain respiratory AEs in those with ppFEV1 <40 versus ≥40 is consistent with the nature of CF in a population of patients with more severe lung dysfunction. The incidence of certain respiratory AEs was also higher in the active treatment groups versus placebo groups, notably in the subgroup with ppFEV1 <40 at baseline (eg, dyspnoea and respiration abnormal, or chest tightness); when respiratory AEs were present, they were generally associated with the initiation of treatment, irrespective of lung function impairment, and usually resolved with continued treatment.
It should be noted that these subgroup analyses were not powered statistically for efficacy comparisons between treatment groups. This is particularly important for subgroups with small numbers of patients, such as those with ppFEV1 <40 at baseline. Nevertheless, the outcomes in patients with severe lung dysfunction were consistent with improvements observed in patients with ppFEV1 ≥40 at baseline, suggesting a benefit of LUM/IVA combination therapy across a range of differing ppFEV1 values. The generalizability of these findings to patients with severe lung dysfunction should be approached cautiously, as these trials were not designed to recruit patients with ppFEV1 levels below 40. Prospective evaluation is needed to confirm these findings in this clinically important subgroup. It is also important to bear in mind that the subgroup of patients with severe lung dysfunction included in this analysis had ppFEV1 values ranging between 31∙1 to 39∙9 percentage points. Special attention may be needed in initiating patients with ppFEV1 below 30 until further results are available. An open-label Phase 3b trial to assess the safety and efficacy of LUM/IVA combination therapy in patients with severe lung dysfunction is currently ongoing (ClinicalTrials.gov number, NCT02390219).
In conclusion, the results of these subgroup analyses of the Phase 3 TRAFFIC and TRANSPORT studies revealed generally consistent improvements across lung function subgroups, including those with ppFEV1 <40 and ≥70, suggesting that LUM/IVA combination therapy was generally well-tolerated and benefits patients homozygous for the Phe508del CFTR mutation across a spectrum of lung function impairment.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed on April 12, 2016 for the terms “ivacaftor” or “VX-770”, “lumacaftor” or “VX-809”, and “clinical trial” with no restrictions on publication date or language and retrieved three relevant clinical studies. In Phase 2 studies, combination lumacaftor/ivacaftor therapy, but not monotherapy, improved lung function and had an acceptable side-effect profile in patients with cystic fibrosis (CF) homozygous for the Phe508del CFTR mutation. The Phase 3 TRAFFIC and TRANSPORT studies demonstrated a clinically meaningful benefit of lumacaftor/ivacaftor combination therapy in this population. To be eligible for these studies patients had to have a screening percent predicted forced expiratory volume 1 second (ppFEV1) of 40 to 90. Therefore, few data are available on which to base treatment decisions in patients whose ppFEV1 is below 40.
Added value of this study
We evaluated the response to lumacaftor/ivacaftor therapy in the Phase 3 TRAFFIC and TRANSPORT studies among patients with CF homozygous for the Phe508del CFTR mutation stratified by specific categories of lung function, including a subgroup of patients with severe lung dysfunction whose ppFEV1 declined to below 40 percentage points between screening and baseline. This provided an opportunity to assess the response in this group of patients that is often not studied. Results of this prespecified subgroup analysis provide evidence that lumacaftor/ivacaftor therapy improved ppFEV1 levels in patients across a spectrum of pretreatment lung function. The incidence of some respiratory adverse events (AEs) was higher among patients with baseline ppFEV1 <40 than those with baseline ppFEV1 ≥40. Across lung function subgroups, some respiratory AEs occurred more frequently in patients who received lumacaftor/ivacaftor therapy than placebo. These respiratory AEs were associated with the initiation of treatment, irrespective of lung function subgroup, and usually resolved with continued treatment. Discontinuations due to AEs were low and similar across subgroups.
Implications of all the available evidence
These data demonstrate that lumacaftor/ivacaftor combination therapy benefits patients with CF homozygous for the Phe508del CFTR mutation with varying degrees of lung function impairment, including those with moderate to severe dysfunction. Prospective evaluation is warranted in patients with ppFEV1 values below 40, in particular among those with ppFEV1 values below 30, in whom the safety and efficacy of lumacaftor/ivacaftor combination therapy are currently being evaluated.
Acknowledgments
Editorial coordination and support were provided by Dhrupad Patel, PharmD, an employee of Vertex Pharmaceuticals Incorporated who may own stock or stock options in that company. Medical writing and editorial support were provided by Michelle Yochum, PhD, and Dena McWain. MY and DM are employees of Infusion Communications, which received funding from Vertex Pharmaceuticals Incorporated.
Funding Vertex Pharmaceuticals Incorporated.
Footnotes
Declaration of interests
JSE reports speaker fees from Vertex Pharmaceuticals Incorporated, grants from Novartis and ProQR, and consultant fees from ProQR during the conduct of the study. BWR reports contract support from Aridis, Celtaxsys, Flatley Discover Lab LLV, KaloBios, Laurent Therapeutics, Nilvalis Therapeutics, Synedgen, and Vertex Pharmaceuticals Incorporated outside of the submitted work. MPB reports grants from Vertex Pharmaceuticals Incorporated during the conduct of the study. MWK reports grants, consultant fees, and travel support from Vertex Pharmaceuticals Incorporated during the conduct of the study; grants and travel support from the Cystic Fibrosis Foundation; consultant fees from Anthera, Chiesi, Digestive Care Inc, and Laurent; grants, consultant fees, and travel support from Genentech, Insmed, Novartis, PTC Therapeutics, and Vertex Pharmaceutical Incorporated; consultant fees and travel support from AbbVie, Celtaxsys, and Gilead; grants and consultant fees from Savara and KaloBios, outside of the submitted work. XH, GM, and DW are employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. CEW reports receiving grant income on a per patient basis for conducting studies, consultant fees, and travel support from Vertex Pharmaceuticals Incorporated during conduct of the study and outside of the submitted work; a research grant from Novo Nordisk and honoraria andtravel support from Novartis outside of the submitted work.
References
- 1.Bosch B, De Boeck K. Searching for a cure for cystic fibrosis. A 25-year quest in a nutshell. Eur J Pediatr 2016; 175: 1–8. [DOI] [PubMed] [Google Scholar]
- 2.O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet 2009; 373: 1891–904. [DOI] [PubMed] [Google Scholar]
- 3.Cystic Fibrosis Foundation Patient Registry 2013 Annual Data Report. Bethesda, MD: Cystic Fibrosis Foundation, 2014. [Google Scholar]
- 4.Cai ZW, Liu J, Li HY, Sheppard DN. Targeting F508del-CFTR to develop rational new therapies for cystic fibrosis. Acta Pharmacol Sin 2011; 32: 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011; 108: 18843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009; 106: 18825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012; 142: 718–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012; 67: 12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med 2014; 2: 527–38. [DOI] [PubMed] [Google Scholar]
- 10.Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015; 373: 220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goss CH, Burns JL. Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax 2007; 62: 360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinkamp G, Wiedemann, on behalf of the German CFQA Group. Relationship between nutritional status and lung function in cystic fibrosis: cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax 2002; 57: 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quittner AL, Modi AC, Wainright C, et al. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009; 135: 1610–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.