

Abstract
ABSTRACTBackground:
Gain of function (GOF) mutations in PIK3CD cause a primary immunodeficiency characterized by recurrent respiratory tract infections, susceptibility to herpes virus infections and impaired antibody responses. Previous work revealed defects in CD8+ T and B cells that contribute to this clinical phenotype but less is understood about the role of CD4+ T cells in disease pathogenesis.
Objective:
To dissect the effects of increased phosphoinositide-3 kinase (PI3K) signaling on CD4+ T cell function.
Methods:
We performed detailed ex vivo, in vivo and in vitro phenotypic and functional analyses of patient CD4+ T cells and a novel murine disease model due to overactive PI3K signaling.
Results:
PI3K overactivation caused substantial increases in memory and T follicular helper (Tfh) cells, and dramatic changes in cytokine production in both patients and mice. Furthermore, PIK3CD GOF human Tfh cells had dysregulated phenotype and function, characterized by increased PD1, CXCR3, and IFNγ expression – the phenotype of a Tfh subset with impaired B-helper function. This was confirmed in vivo where Pik3cd GOF CD4+ T cells also acquired an aberrant Tfh-phenotype and provided poor help to support germinal center reactions and humoral immune responses by antigen-specific wild-type B cells. The increase in both memory and Tfh cells was largely CD4+ T cell extrinsic while changes in cytokine production and Tfh cell function were cell intrinsic.
Conclusion:
Our studies reveal that CD4+ T cells with overactive PI3K have aberrant activation and differentiation, thereby providing mechanistic insight into dysfunctional antibody responses in patients with PIK3CD GOF mutations.
Keywords: PI3 kinase, PIK3CD, activated PI3K delta syndrome (APDS), CD4+ T cell function, T follicular helper cells, humoral immunity, immune class regulation, humans, mouse models
Graphical Abstract
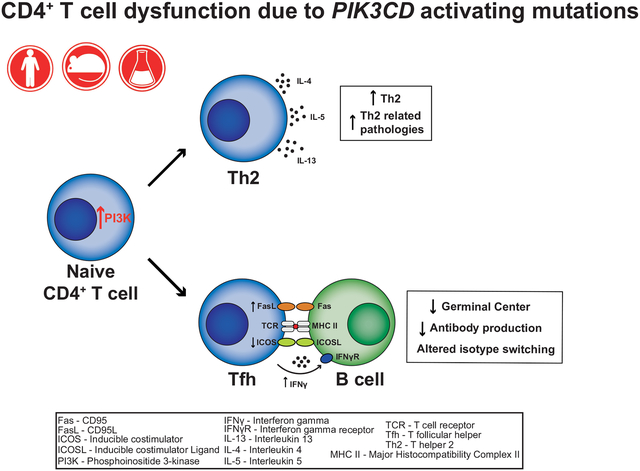
INTRODUCTION
Heterozygous germline gain-of-function (GOF) mutations in PIK3CD have recently been identified in individuals with recurrent respiratory tract infections, impaired Ab responses following natural infection and vaccination, hepatosplenomegaly, autoimmune cytopenias, increased susceptibility to infection with human herpes viruses, and an increased incidence of B-cell lymphoma1–3. PIK3CD encodes the p110δ catalytic subunit of phosphoinositide-3 kinase (PI3K). Class I PI3Ks (hereafter referred as PI3K) are heterodimeric enzymes composed of a catalytic (p110α, p110β or p110δ) and a regulatory (p85α, p85β, p50α or p55γ) subunit. The p110α and p110β subunits are ubiquitously expressed in mammalian tissues, whereas p110δ is mainly restricted to leukocytes. Thus, PIK3CD GOF mutations induce hyperactive PI3K signaling almost exclusively in immune cells, thereby resulting in a broad spectrum of clinical manifestations of immune dysregulation in affected patients2, 3, as well as defects in the differentiation and function of different lymphocyte populations4–10. This condition has been termed activated PI3Kδ syndrome (APDS)1, 2. Recent work has revealed intrinsic defects in B cells4–7 and CD8+ T cells8–10 that contribute to the clinical phenotype of APDS patients. While PIK3CD GOF mutations have been found to result in reduced thymic output and CD4+ T cell lymphopenia in most (>60%) of APDS patients2, the consequences of overactive PI3K on the differentiation and function of CD4+ T cells remains ill defined.
Following the delivery of signals from Ag presenting cells, naïve CD4+ T cells have the remarkable capacity of differentiating into a myriad of effector subsets with defined function, such as Th1, Th2, Th17, T follicular helper (Tfh) and regulatory T (Treg) cells11, 12. This provides a diverse and specialized array of effector CD4+ T cells capable of protecting the host against a broad range of pathogenic threats. Thus, Th1 cells play key roles in defense against intracellular bacterial and viral infections, Th2 cells are required for protection against parasites, and Th17 cells have a critical and non-redundant role in immunity against fungi13. Tfh cells regulate the differentiation of cognate B cells into long-lived memory and plasma cells (PC) in response to T-dependent antigens (Ags)12, 14, 15. In contrast to Th1, Th2 and Th17 subsets, Tfh cells are important for protective immunity against most, if not all, pathogens. This is evidenced by the success of most vaccines, which depends on establishing long-lived serological memory in the form of memory B cells, PC and high-affinity Ag-specific Abs12, 14, 15. Lastly, Tregs prevent the aberrant activation of immune cells, thereby maintaining immune homeostasis11.
In T cells, PI3K is activated downstream of several surface receptors, such as the T cell receptor (TCR), CD28, ICOS and various cytokine receptors. Several mouse models have been established to unravel the role of PI3K in CD4+ T cells. Gene-targeted mice that either lack p110δ or express catalytically-inactive p110δ have normal frequencies of CD4+ and CD8+ T cells but decreased proportions of effector and memory-type T cells in the lymph nodes16, 17. Furthermore, the generation of Th1, Th2 and Th17 cells in vitro, and of Tfh cells in vivo following immunization, were compromised in the absence of functional p110δ16, 18, 19. Mechanistically, p110δ functions downstream of ICOS for Tfh cell generation, evidenced by analysis of CD4+ T cells from mice that either lacked ICOS, or expressed an ICOS variant unable to activate PI3K20. In contrast to memory and Tfh cells, the consequences of p110δ deficiency on Tregs are complex. Thus, PI3K restrains the generation of thymic Tregs, consistent with PI3K/Akt signaling inhibiting FoxP3 expression, yet is required for the maintenance and function of Tregs in the periphery16, 17, 21. Collectively, these data establish that PI3K is important in regulating the development of specific populations of effector CD4+ T cells.
Given the hyperactive T cell phenotype and impaired humoral immune defects observed in APDS, we hypothesized that PIK3CD GOF mutations would perturb the function of CD4+ T cells, and these defects would contribute to the clinical manifestations of APDS. Thus, to elucidate the impact of p110δ hyper-activation on CD4+ T cell function and the potential contribution this makes to the pathophysiology of the disease, we analyzed a large cohort of individuals with PIK3CD GOF mutations, as well as a corresponding CRISPR/Cas9 gene edited mouse model harboring a mutation in Pik3cd found in >80% of individuals with APDS.
Methods
HUMAN STUDIES
Human samples
Buffy coats from healthy donors were purchased from the Australian Red Cross Blood Service. Peripheral blood was collected from patients with GOF mutations in PIK3CD4. All studies were approved by Institutional Human Research Ethics Committees (X16–0210 approved by the Sydney Local Health District Human Research Ethics Committee; 95-I-0066; approved by National Institute of Allergy and Infectious Diseases Institutional Review Board).
Antibodies and Reagents
The following were purchased from eBiosciences: anti-IL-21 eFluor660, anti-IL-10 Alexa Fluor 488, anti-IFNγ BV605, anti-CD45RA FITC, anti-ICOS PE, anti-PD-1 biotin. The following were purchased from Becton Dickinson: anti-CXCR5 Alexa Fluor 647, anti-CD4 APC-Cy7, anti-IL-2 BV711, anti-CD25 PE-Cy7, anti-CD127 PerCP-Cy5.5, SA-PerCpCy5.5, anti-IL-17F BV786, anti-TNFα BUV395, anti-IL-9 PerCP-Cy5.5, anti-IL-13 BV421. The following were purchased from Biolegend: anti-IL-22 PE, anti-IL-4 PE-Cy7, anti-IL-17A APC-Cy7. The TCR Vß repertoire kit was from Beckman Coulter. Recombinant human IL-12 was purchased from R&D Systems.
CD4+ T cell isolation and functional characterisation
PBMCs were incubated with mAbs to CD4, CD45RA, CCR7, CXCR5, CD127 and CD25. Naïve, memory and circulating Tfh (cTfh) CD4+ T cells were isolated by first excluding Tregs (CD25hiCD127lo) and then sorting CD4+CD45RA+CCR7+CXCR5−, CD4+CD45RA−CXCR5−CCR7± or CD4+CD45RACXCR5+CCR7± cells, respectively. Isolated naïve, memory and cTfh cells were then cultured in 96 well round bottomed well plates (30–40 ×103 cells/well) with T cell activation and expansion (TAE) beads (anti-CD2/CD3/CD28; Miltenyi Biotech) alone (Th0) or under Th1 (50 ng/mL IL-12) polarising conditions. After 5 days, supernatants were harvested and production of IL-4, IL-5, IL-9, IL-10, IL-13, IL-17A, IL-17F, and IFNγ determined by cytometric bead arrays (Becton Dickinson); IL-22 secretion was measured by ELISA (eBioscience). For cytokine expression, activated CD4+ T cells were re-stimulated with PMA (100 ng/ml)/ionomycin (750 ng/ml) for 6 hours, with Brefeldin A (10 μg/ml) being added after 2 hours. Cells were then fixed and intracellular expression of IL-4, IL-9, IL-13, IL-10, IL-17A, IL-17F, IL-22, IL-21 and IFNγ determined. In some experiments, naïve CD4+ T cells were first labeled with CFSE, and proliferation determined by assessing CFSE dilution.
MURINE STUDIES
Mice
Pik3cd GOF mice were generated by introducing the E1020K mutation into Pik3cd by CRISPR/Cas9 as previously described4. For some experiments WT or Pik3cd GOF mice were crossed with OT-II22 mice to generate OTII WT or OTII Pik3cd GOF mice. Donor cells from either lineage were adoptively transferred into C57Bl/6 (CD45.1 congenic) or Sh2d1a deficient23 (CD45.1 congenic) mice (hereafter referred to as SAP KO mice). SWHEL mice expressing a BCR specific for hen egg lysozyme (HEL) have been previously described24. Fas-deficient were produced by the Mouse Engineering Garvan/ABR (MEGA) Facility (Moss Vale and Sydney, Australia) by CRISPR/Cas9 gene targeting in C57BL/6J mouse embryos following established molecular and animal husbandry techniques25. A single guide RNA (sgRNA) was designed to target within Exon 2 (target with protospacer-associated motif underlined = TCTCCGAGAGTTTAAAGCTGAGG) and injected with polyadenylated Cas9 mRNA into C57BL/6J zygotes. Microinjected embryos were cultured overnight and introduced into pseudo-pregnant foster mothers. Pups were screened by PCR and Sanger sequencing of ear-punch DNA and a founder mouse identified that carried a 14bp deletion that caused a frame shift after the E29 codon that generated an in frame stop codon 15bp downstream. All experiments were approved by the Garvan Institute-St Vincent’s Animal Ethics Committee (approval 16/29). Mice were bred and housed in specific pathogen-free conditions in the Garvan Institute Biological Testing Facility or Australian BioResources.
Mixed bone marrow chimeras
Pik3cd GOF or WT (CD45.2) and congenic C57BL/6 WT (CD45.1) bone marrow (BM) from donor mice were harvested and processed. Each recipient C57BL/6 WT (CD45.1) mouse was irradiated twice with 475 rad 6 hours apart. A 50:50 mix of either WT:WT or WT:Pik3cd GOF cells were injected intravenously (i.v.) into recipient mice (5×106 cells/mouse). Spleens from recipient mice were harvested at a young and old age (12 or 36 weeks after reconstitution, respectively) and processed for flow cytometry analysis.
mAb and reagents for flow cytometry
The following were purchased from BD Biosciences: anti-CD44 FITC, APC, BV605 (IM7); anti-CD45R/B220 FITC, BV786 (RA3–6B2); Anti CD16/CD32 Fc block (2.4G2), Streptavidin-BV605, Streptavidin-BUV395; Streptavidin-PerCP-Cy5.5; anti-CD45.1 BV421 (A20); anti-CD45.2 BUV395 (104); anti-CD62L APC, PE (MEL-14); anti-IL-4 BV786 (11B11); anti-mouse CD25 (PC61); anti-CD95 PE (Jo2); anti-CD8a FITC, PB (53–6.7); anti-CD4 APC (RM4–5); anti-CD3 PerCP-Cy5.5 (17A2); anti-CD279 PE (J43); anti-CD185 purified (2G8); anti-CD184 BV421, biotinylated (2B11/CXCR4); anti-IgG1 biotinylated (A85–1); anti-IFNγ PE-Cy7 (XMG1.2);. The following were purchased from eBiosciences: anti-Vα2 FITC (B20.1); anti-CD45.1 FITC, PeCy7, biotinylated (A20); anti-CD45.2 APC-eFluor 780, PerCp Cy5.5 (104), anti- CD45.1 PeCy7 (A20); anti-CD62L FITC (MEL-14); Streptavidin-PE-Cy7; anti-mouse CD25 (PC61.5); anti-CD4 APC-eFluor 780 (RM4–5); anti-CD39 PerCP-eFluor 710 (24DMS1); anti-CD278 PE, Biotinylated (C398.4A); anti-CD279 PE-Cy7 (J43); anti-CD152 APC (UC10–4B9); anti-CD197 APC (4B12); anti-FoxP3 PE-Cy7 (FJK-16s); anti-Vβ5.1/5.2 TCR PerCP-eFluor 710 (MR9–4); The following was purchased from Invitrogen: Streptavidin-PB. The following were purchased from Biolegend: anti-IgM(b) FITC (AF6–78); anti-CD45.1 PerCP Cy5.5 (A20); anti-CD62L BV605 (MEL-14); anti-CD86 BV650 (GL-1); Streptavidin-BV605; Streptavidin-BV421; anti-CD38 Pe-Cy7 (90); anti-CD127 APC (A7R34); anti-CD127 Alexa Fluor 647 (SB/199); anti-γδ TCR PECy7 (GL3); anti-IFNγ PerCP-Cy5.5 (XMG1.2); anti-IL-2 Alexa Fluor 647 (JES6–5H4); anti-IL-5 BV421 (TRFK5); anti-TNFα PE (MP6-XT22); anti-IL17A PE-Cy7 (TC11–1810.1); anti-IgD Alexa Fluor 647 (11–26c.2a); Zombie Aqua Fixable Viability Kit. The following was purchased from Jackson ImmunoResearch: Donkey Anti-Rat IgG (H+L) Biotinylated.
Flow cytometry
Spleens of WT, Pik3cd GOF, or recipient mice in adoptive transfer studies were harvested at the indicated times, prepared and stained for flow cytometry. Approximately 2 × 106 events were collected per sample. Data was acquired on an LSRII SORP or FORTESSA (Becton Dickinson) and analyzed using FlowJo software (Tree Star). All data are representative of 2 or more experiments as indicated.
Cell sorting
Spleens were prepared and stained for flow cytometry. For in vitro cultures naïve CD4+ T cells were identified using anti-CD3 PerCP-Cy5.5, anti-B220 BV786, anti-CD44 FITC, anti-CD62L PE, anti-CD8a Pacific Blue, anti-CD4 APCe780, anti-γδ TCR PE-Cy7. For adoptive transfers sorts, spleens were enriched with MACS CD4+ T cell isolation kit (Miltenyi Biotec) and separated using AutoMACS Pro separator. Recovered cells were identified as OT II or endogenous cells based on their expression of anti-CD4 APCe780, anti-Vα2 FITC, anti-Vβ5.1/5.2 PerCP-eFluor710, anti-B220 BV786, anti-CD45.1 Pacific Blue and anti-CD45.2 PeCy7. Pure populations were sorted using FACSAria (BD Biosciences).
In vitro cultures
Sorted Pik3cd GOF or WT naïve CD4+ T cells were cultured in flat bottom 96 well plates previously coated overnight with anti-CD3 (BioLegend) (4 μg/mL) in RPMI1640 (Life technologies) supplemented with 10% heat inactivated FCS (Life technologies), 5×10−5 M 2-ME, 0.1mM non essential amino acids, 1mM sodium pyruvate, 10mM HEPES, 100u/mL penicillin, 100ug/mL Streptomycin, 100ug/mL Noromycin (all from Sigma) at a density of 0.5 ×106 cells/mL. CD4+ naïve cells were cultured for 4 days in the following conditions: Th0 - anti-CD28 1 μg/mL + anti-TGFβ 5 μg/mL + anti-IL-4 5 μg/mL + anti-IFNγ 5 μg/mL; Th1 - anti-CD28 1 μg/mL + anti-TGFβ 5 μg/mL + anti-IL-4 5 μg/mL + IL-12 10ng/mL; Th2 - IL-4 10ng/mL + anti-CD28 1 μg/mL + anti-TGFβ 5 μg/mL + anti-IFNγ 5 μg/m; Th17 - IL-6 20ng/mL + human TGFβ 1ng/mL + anti-IFNγ 5 μg/m + anti-IL-4 5 μg/mL + anti-CD28 1 μg/mL. After 4 days of culture, cells were stimulated with PMA (50ng/mL) and ionomycin (375ng/mL) for 6 hours. Brefeldin A (10 μg/mL) was added to each well at 2 hours of culture. Cells were then harvested, washed and stained with Zombie Aqua Viability dye (BioLegend). CD4+ T cells were fixed with 2% formalin, permeabilised with saponin (0.1%), and stained intracellularly with mAbs directed against TNFα, IFNγ, IL17A, IL-2, IL5, IL-4 and analysed by flow cytometry.
OT-II adoptive transfers
Donor OT-II Pik3cd GOF or OT-II WT spleen cells containing 3 ×104 OT-II cells were injected i.v. into CD45.1 or CD45.2 congenic C57BL/6 or SAP KO recipient mice. Recipient mice were immunized intraperitoneally with ovalbumin in Alum (OVA-Alum; 1:1, OVA 1mg/ml: Imject® Alum Thermo Scientific). Recipient spleens were harvested at day 5 and day 7 and sera harvested at day 7. Cells (4 × 106 cells in a 48 well flat bottom plate) were stimulated with PMA (50ng/mL) and ionomycin (375ng/mL) for 6 hours. Brefeldin A (10 μg/mL) was added to each well at 2 hours of culture. Cells were then harvested, washed and stained with Zombie Aqua Viability dye (BioLegend) followed by CD4, CD45.1 and CD45.2 surface stain. Cells were fixed with 2% formalin and stained for intracellular cytokines as above. Anti-OVA antibody levels of the various Ig subclasses in the sera of recipient mice were analyzed by ELISA. 96 or 384-well ELISA plates were coated with OVA and bound serum Ig was detected using Ig heavy chain isotype specific Ab (BD Biosciences; biotinylated anti-IgG1 [A85–1], biotinylated anti-IgG2a/c [R19–15], biotinylated anti-IgM [R6–60.2]). Ig levels for each class were normalized to pooled sera from mice previously immunized with OVA.
OT-II and SWHEL co-transfers
Donor WT SWHEL or FAS-deficient SWHEL spleen cells were enriched with MACS B cell isolation kit (Miltenyi Biotec) and separated using an AutoMACS Pro separator. Recovered cells were identified as HEL-binding B cells after being stained with saturating levels of HEL (Sigma) at 200ng/ml followed by HyHEL9-Alexa Fluor 647 and B220 BV786 (BD Biosciences). 2 ×105 donor OT-II Pik3cd GOF or OT-II WT spleen cells were injected i.v. together with 2 ×105 WT SWHEL or FAS-deficient SWHEL B cells into CD45.1+ SAP KO recipient mice. On the following day of the transfer, recipient mice were immunised sub-cutaneously with 20 g HEL-OVA in Sigma Adjuvant System (SAS; Sigma) in the lower flank and base of the tail. Inguinal lymph nodes were harvested at day 7.
Real time PCR
Sorted OT-II Pik3cd GOF or WT cells and recipient WT cells had their RNA isolated using the QIAGEN RNeasy Mini Prep. First strand cDNA synthesis kit (Invitrogen) was used for each sample and standards. Light cycle probes, master mix and system were used for the real time PCR reaction. Target genes primers (all purchased from IDT integrated DNA technologies): Il21 5’ GACATTCATCATTGACCTCGTG 3’ TCACAGGAAGGGCATTTAGC; Cd40lg ACGTTGTAAGCGAAGCCAAC 3’ TATCCTTTCTTGGCCCACTG; Il4 5’CCTGCTCTTCTTTCTCGAATG 3’CACATCCATCTCCGTGCA; Fasl 5’ACCGGTGGTATTTTTCATGG 3’AGGCTTTGGTTGGTGAACTC; House keeping genes primers: UBC 5’GACCAGCAGAGGCTGATCTT 3’CCTCTGAGGCGAAGGACTAA (IDT integrated DNA technologies); c-alpha based 5’ TAGGGATAACAGCGCAATCC 3’ GACTTTAATCGTTGAACAAACGAAC (Roche). ACTB (β actin) UPL purchased from Sigma-Aldrich. Light Cycle 480 machine was used to run the assay. Data analysis was performed using the Light Cycle 480 SW 1.5.
Statistical analysis.
Significant differences were determined using Prism (GraphPad Software).
RESULTS
cTfh cells are increased in individuals with PIK3CD GOF mutations
To assess the impact of aberrant PI3K signaling on CD4+ T cells, we examined the CD4+ T cell compartment in peripheral blood of patients with PIK3CD GOF mutations (n=37; mean age 20 ± 1 years). Clinical and laboratory features of these patients, where available, are listed in Supplementary Table 1. Consistent with previous findings2, 3, 50% (12/24) of patients had CD4+ T cell lymphopenia, defined as cell counts below the lower end of the normal range; most patients had brochiecstasis and/or respiratory infections, 62% (16/26) had hyper-IgM, 50% (13/26) had hypogammaglobulinemia, and 27% (3/11) had reduced levels of serum IgA. Furthermore, Ab responses against pneumococcal vaccines/polysaccharide Ags were reduced in 95% (20/21) of patients, responses to Haemophilus influenzae type B vaccine were reduced in 75% of patients, while responses to tetanus or diphtheria vaccines were largely intact.
Flow cytometric analysis revealed that the proportions of CD4+ T cells within lymphocytes from PIK3CD GOF patients were significantly reduced compared to healthy donors (31.0 ± 2.1% vs 44.7 ± 1.8%; p<0.001). We also quantified naïve (CD45RA+CCR7+), central memory (TCM; D45RA−CCR7+), and effector memory (TEM; CD45RA−CCR7−) CD4+ T cells. Naïve CD4+ T cells were significantly reduced while those of TCM and TEM CD4+ T cells were significantly increased in patients compared to healthy donors (Figure 1A, B). In contrast to these subsets, proportions of Tregs CD4+CD127loCD25hi) were similar in PIK3CD GOF patients and controls (Figure 1C).
Figure 1: PIK3CD GOF patients have a selective expansion of cTfh cells.

(A-E) PBMCs from healthy donors (n=40–55) and patients with PIK3CD GOF mutations (n=22–34) were labeled with Abs against CD4, CD45RA, CCR7, CXCR5, CD127 and CD25 and different populations identified by flow cytometric analysis. (A, B) Percentages of naïve (CD4+CD45RA+CCR7+), central memory (T CD4+CD45RA−CCR7dimCM;), effector memory (TEM; CD4+CD45RA−CCR7−) cells and (C) Tregs (CD4+CD127loCD25hi) within the CD4+ T cell population. (D, E) Proportions of non-cTfh memory (CD4+CD45RA−CCR7±CXCR5−) and cTfh cells (CD4+CD45RA−CCR7±CXCR5+) cells within the CD4+ T cell population. (F) Frequency of cTfh cells as a proportion of total memory CD4+ T cells. Contour plots in (A) and (D) show representative staining for healthy donors and PIK3CD GOF patients. Graphs give mean ± SEM. Significant differences were determined by unpaired Students t-tests. * p<0.05; ** p<0.01; **** p<0.0001.
(G) Percentage of cTfh cells (of all CD4+ T cells) in healthy donors (black) and PIK3CD GOF patients (red) were determined as a function of age.
(H) The TCR repertoire of cTfh cells present in PBMC of healthy donors (n=7) and PIK3CD GOF patients (n=3) was determined using flow cytometry by quantifying the proportion of CD4+CD45RACXCR5+ T cells expressing the different Vβ chains. Graphs show mean ± SEM.
To investigate further the nature of the expanded population of memory CD4+ T cells in PIK3CD GOF patients, we examined CD4+CD45RA−CXCR5+ T cells, which have been established to be the circulating counterpart of tissue resident Tfh cells and thus are termed circulating Tfh (cTfh) cells26–29. This revealed that the increase in memory CD4+ T cells was due to a selective increase in cTfh cells, as the proportion of CD45RA−CXCR5− memory CD4+ T cells in patients and healthy donors was comparable (Figure 1D, E). Thus, the pool of cTfh cells in PIK3CD GOF individuals was increased >3-fold (Figure 1E). Consequently, the cTfh population comprised approximately one third of the total memory CD4+ T cell population in PIK3CD GOF patients (Figure 1F). Thus, activating mutations in PIK3CD disproportionately expand cTfh cells.
The numbers and proportions of cTfh cells are very low/undetectable in cord blood, but rapidly increase over the first 12 months of life, reaching stable levels from ~2 years of age30. These levels then remain relatively unchanged during adolescence and adulthood30. Thus, even though the average age of the cohort of PIK3CD GOF patients was less than the healthy donors used in this study (ie 20 vs ~33 yrs), the increased proportions of cTfh cells in PIK3CD GOF patients should not be influenced by age. To formally establish this, we determined proportions of cTfh cells according to age. This confirmed that the numbers of cTfh cells in healthy donors remains constant from ~15 to 65 years of age (Figure 1G). In PIK3CD GOF patients, proportions of cTfh cells generally exceeded those in healthy donors, irrespective of age (Figure 1G). Indeed, the proportions of cTfh cells in many of the younger patients exceeded 20% of all CD4+ T cells, which was markedly increased compared to healthy donors. Thus, PIK3CD GOF mutations result in a profoundly expanded population of cTfh cells from a young age.
One possible explanation for the increased proportion of cTfh cells in PIK3CD GOF patients is the preferential expansion of particular clonotypes. To test this, we examined the TCR repertoire of cTfh cells using mAbs that recognize 24 specific TCR Vβ chains. While one PIK3CD GOF patient exhibited an expansion of cTfh cells expressing Vβ8 or Vβ11 TCRs, the overall proportions of cTfh cells expressing specific TCR Vβ chains in PIK3CD GOF patients did not differ significantly from those in healthy controls (Figure 1H). A limitation of this approach is that the TCR Vβ chain-specific mAbs collectively only detect TCRs expressed by ~70% of CD4+ T cells. However, preliminary analysis of the TCR repertoire of PIK3CD GOF patients and healthy donors by deep sequencing confirmed the polyclonality of the TCR expressed by cTfh cells (data not shown). Thus, it is unlikely that the expansion of cTfh cells in PIK3CD GOF is due to selective proliferation of clones with defined specificities. Collectively, these findings establish that PIK3CD GOF mutations dramatically influence differentiation of CD4+ T cells, particularly cTfh cells.
PIK3CD GOF mutations alter the phenotype of CD4+ T cell subsets
Tfh cells in secondary lymphoid tissues and peripheral blood express lower levels of CCR7 and elevated levels of ICOS and PD-1 than naïve CD4+ T cells, however the levels of these latter receptors on cTfh cells are less than on lymphoid tissue Tfh cells27, 31–33. To explore the consequences of constitutive PI3K activation on CD4+ T cell differentiation, we further analyzed the phenotype of naïve, memory and cTfh cells.
ICOS was similarly expressed on CD4+ T cell subsets in patients and healthy donors (not shown). In contrast, CCR7 was significantly reduced on PIK3CD GOF naïve, memory and cTfh cells compared to healthy donors (Figure 2A). This was largely due to a decrease in expression on CCR7+ cells rather than an increase in the percentage of CCR7− cells. PD-1 was expressed at low levels on naïve CD4+ T cells, and induced on memory and cTfh cells from both healthy donors and PIK3CD GOF patients (Figure 2B). However, it was significantly upregulated on memory and cTfh cells from PIK3CD GOF patients, being 2–4-fold greater than controls (Figure 2B). Expression of additional markers of Tfh cells (BCL-6, CD200, CD57) did not differ between controls and PIK3CD GOF patients (not shown).
Figure 2: Patients with PIK3CD GOF mutations have altered cTfh cell differentiation.

PBMCs from healthy donors (n=37–50) and patients with PIK3CD GOF mutations (n=22–34) were labeled with Abs against CD4, CD45RA, CCR7, CXCR5, CD127, CD25, PD-1, CXCR3 and CCR6 and flow cytometric analysis performed. (A, B) Expression of (A) CCR7 and (B) PD-1 on naïve, non-Tfh memory and cTfh cells. Histogram plots show representative staining of healthy donors and PIK3CD GOF patients. Graphs display the expression (mean ± SEM) of CCR7 or PD-1 on naïve, non-Tfh memory and cTfh cells relative to naïve cells (normalized to 1.0). (C, D) Percentages of Th1 (CXCR3+CCR6−), Th17 (CXCR3−CCR6+), Th1/17 (CXCR3+CCR6+) and CXCR3−CCR6− cells within the (C) non-Tfh memory and (D) cTfh subsets were determined. Contour plots show representative staining of healthy donors and PIK3C GOF patients. Graphs display mean ± SEM of the indicated CD4+ T cell subsets. Significan differences were determined by unpaired Students t-tests. * p<0.05; ** p<0.01; **** p<0.0001.
Next, we quantified functional subsets of memory and cTfh cells based on differential expression of CXCR3 and CCR6. CXCR3 expression identifies Th1 cells, while CCR6 expression correlates with Th17 cells13. Expression of these chemokine receptors also defines functionally-distinct subsets of cTfh cells, with CXCR3+ cTfh cells being enriched for IFNγ production and poor inducers of B-cell differentiation in vitro, while CCR6+ cTfh cells produce Th17 cytokines (IL-17A, IL-17F, IL-22) and are potent B-cell helpers28, 29. Within the population of non-Tfh memory CD4+ T cells, proportions of cells with a Th1 (CXCR3+ CCR6−), Th17 (CXCR3−CCR6+) or Th1/17 (CXCR3+CCR6+) phenotype were comparable for healthy donors and PIK3CD GOF patients (Figure 2C). CXCR3−CCR6− memory CD4+ T cells, which comprise a mixed population of Th2 and non-differentiated memory cells28, 34, were also present at comparable frequencies in patients and healthy donors (Figure 2C). In marked contrast, there were significant increases in proportions of CXCR3+ Tfh1 cells, and concomitant reductions in CCR6+ Tfh17 and CXCR3−CCR6− cTfh cells, in PIK3CD GOF individuals compared to healthy donors (Figure 2D). The ratio of Th1/Th17-type Tfh cells in healthy donors was 1.8, while in PIK3CD GOF patients it was 6.4, equating to a 3.5-fold increase. Thus, hyper-active PI3K signaling has a selective effect on the differentiation of cTfh cells, skewing them to a Th1 and away from a Th17 phenotype.
Cytokine production by memory CD4+ T cells is differentially impacted by PIK3CD GOF mutations
To correlate changes in surface phenotype with function, we sorted naïve and total memory CD4+ T cells from healthy donors and PIK3CD GOF patients and examined production of different cytokines following in vitro culture. Expression (Figure 3A) and secretion (Supplementary Figure 1A) of IFNγ as well as of the Th17 cytokines IL-17A, IL-17F and IL-22 by memory CD4+ T cells from healthy donors and PIK3CD GOF patients were comparable. This is consistent with the proportions of CXCR3+ Th1 and CCR6+ Th17 memory CD4+ T cells in healthy donors and PIK3CD GOF patients being similar (Figure 2C). In contrast, the Th2 cytokines IL-4, IL-5, IL-9 and IL-13 were expressed and secreted in much greater quantities by PIK3CD GOF memory CD4+ T cells compared to corresponding cells from healthy donors (Figure 3A; Supplementary Figure 1A).
Figure 3: CD4+ T cells from patients with PIK3CD GOF mutations show altered cytokine production.

(A) Naïve and total memory CD4+ T cells (n=4–9) or (B) cTfh (n=8) cells were sort-purified from the peripheral blood of healthy donors and patients with PIK3CD GOF mutations and then cultured with beads coated with anti-CD2/CD3/CD28 mAbs. After 5 days, expression of the indicated cytokines was determined by flow cytometric analysis of intracellular immunofluorescent staining.
Sort-purified (C) naïve or (D) cTfh CD4+ T cells from healthy donors or patients with PIK3CD GOF mutations (n=8) were cultured under Th1 polarizing conditions (+IL-12). After 5 days, expression of IL-21 or IFNγ by naïve CD4+ T cells, or secretion of IFNγ by naïve CD4+ T cells or cTfh cells were determined. Data depict mean ± SEM of the experiments performed using CD4+ T cells from the indicated numbers of healthy controls or PIK3CD GOF patients. Significant differences were determined by unpaired Students t-tests, * p<0.05, **p<0.01, ****p<0.0001.
Consistent with skewing of the cTfh population to a CXCR3+ Tfh1 and away from a CCR6+ Tfh17 phenotype (Figure 2D), isolated cTfh cells from PIK3CD GOF patients had greater expression and secretion of IFNγ, and expression of TBX21, encoding Tbet, and corresponding reductions in IL-17A, IL-17F and IL-22 production compared to cTfh cells from controls (Figure 3B; Supplementary Figure 1B; not shown). Furthermore, production of Th2 cytokines by PIK3CD GOF cTfh cells was markedly increased compared to controls (Figure 3B; Supplementary Figure 1B). Since we observed that PIK3CD GOF patients have a modest decrease in CD4+CXCR5+CXCR3−CCR6− T cells (Figure 2D), which are enriched for production of Th2 cytokines28, 34, our finding that Th2 cytokine production by cTfh cells was increased suggests either that the relative decrease in CXCR3−CCR6− cells was largely due to a decrease in the undifferentiated memory cells that also comprise this population28 or that all subsets of cTfh cells exhibit aberrant production of Th2 cytokines. Thus, PIK3CD GOF mutations not only generate more memory CD4+ T cells, particularly cTfh cells, compared to healthy donors, they also promote production of Th2 cytokines by all memory CD4+ T cells, and selectively promote cTfh cells to acquire a Th1 fate. Importantly, these cells are poorly equipped to promote B-cell differentiation28, 29, 35.
Altered differentiation of naïve PIK3CD GOF CD4+ T cells in vitro
To investigate whether these changes reflected a defect in the differentiation of PIK3CD GOF naïve CD4+ T cells in response to polarizing conditions, we cultured sort-purified naïve CD4+ T cells with IL-12 in vitro, which induces both Th1 (IFNγ production) or Tfh-type (IL-21 expression) fates32, 34. PIK3CD GOF naïve CD4+ T cells exhibited a 2–3-fold reduction in the acquisition of IL-21 expression compared to control naïve CD4+ T cells (Figure 3C, left panel). This did not result from a generalized inability to respond to IL-12, as PIK3CD GOF naïve CD4+ T cells exhibited intact, or modestly enhanced, differentiation into IFNγ-producing cells under these same cultures (Figure 3C, middle and right panels). PIK3CD GOF cTfh cells continued to secrete significantly higher amounts of IFNγ than control cells when cultured under Th1 conditions (Figure 3D).
Differentiation of naïve CD4+ T cells into cytokine-expressing cells often requires cell division36. To determine whether the defect in PIK3CD GOF naïve CD4+ T cells in differentiating into IL-21+ cells resulted from impaired proliferation, we labeled sorted naïve CD4+ T cells with CFSE and determined cell division and cytokine expression. Naïve CD4+ T cells from healthy controls and PIK3CD GOF patients exhibited similar levels of proliferation, determined by CFSE dilution, when cultured under Th0 conditions; this was comparably enhanced in the presence of IL-12 (Supplementary Figure 1C, left panel). Despite intact proliferation, under Th1 polarizing conditions there was a reduced frequency of PIK3CD GOF naïve CD4+ T cells expressing IL-21 in each division compared to naïve cells from healthy donors (Supplementary Figure 1C, middle panel). In contrast, the division-linked acquisition of IFNγ by PIK3CD GOF naïve CD4+ T cells was comparable to that from healthy donors (Supplementary Figure 1C, right panel). Thus, impaired generation of IL-21+ cells from naïve PIK3CD GOF CD4+ T cells resulted from a defect in differentiation, rather than reduced proliferation. Collectively, the impaired ability of naïve PIK3CD GOF CD4+ T cells to differentiate into IL-21-expressing cells in vitro, coupled with a skewed Th1-type phenotype and cytokine profile of PIK3CD GOF cTfh cells ex vivo, are consistent with a major functional defect in Tfh cells in these patients.
Generation of a mouse model of Pik3cd GOF
It was not clear whether the changes we observed in human PIK3CD GOF CD4+ T cells were intrinsic or extrinsic, resulting from PI3K over activation in other cells or recurrent infections. Thus, to dissect the intrinsic and extrinsic effects of overactive PI3K signaling on CD4+T cell development and function, we used CRISPR/Cas9 to introduce a heterozygous germline activating mutation (E1020K; orthologue of the most common mutation in human patients [E1021K]2) in murine Pik3cd (hereafter referred to as Pik3cd GOF)4. Lymphocytes from these mice displayed increased phosphorylation of S6 and Akt, consistent with elevated PI3K signalling4. Analysis of thymi of Pik3cd GOF mice revealed no major disruption to T cell development (Supplementary Figure 2).
Pik3cd GOF mice accumulate increased proportions of memory CD4+ T cells
Consistent with the lymphoproliferation and splenomegaly in APDS patients, spleens of Pik3cd GOF mice had increased total cellularity compared to WT spleens (data not shown)4. In aged mice this was accompanied by an increase in the percentage of CD4+ T cells (Figure 4A). We also observed a decrease in frequency of naïve CD4+ T and corresponding increase in memory cells (Figure 4B). This phenotype was particularly pronounced in aged mice (Figure 4B). Similarly, young Pik3cd GOF mice had increased numbers of Tfh cells and this became much more prominent as the mice aged (Figure 4C, D). We also observed an increase in FoxP3+ Tregs (Figure 4E, F). In young mice this increase in Treg frequency was mainly due to an expansion of the CD25+ compartment (Figure 4F). In contrast, the expanded FoxP3+ Treg compartment in aged mice was largely due to an increase in the percentage of CD25−FoxP3+ cells (Figure 4E, F).
Figure 4: Pik3cd GOF mutations enhance the formation of memory, Tfh and Treg cells.

Spleens from young (8–12 weeks) and aged (30–40 weeks) WT and Pik3cd GOF mice were stained to identify different CD4+ T cell populations by flow cytometric analysis. (A) Frequency of total CD4+ T cells. (B) Percentages of naïve (CD44loCD62Lhi), central memory (CD44hiCD62Lhi) or effector memory (CD44hiCD62Llo) CD4+ T cells. (C) Contour plots show representative staining of CXCR5 versus PD-1 on CD4+ T cells in aged mice. (D) Tfh cell (CXCR5hiPD-1hi) frequencies. (E) Contour plots show representative staining of FoxP3 versus CD25 on CD4+ T cells in aged mice. (F) The percentage of FoxP3+ T cells (CD25+ or CD25−). All graphs show mean ± SEM, n=7–11. Significant differences were determined by unpaired Students t-tests, **p<0.01; ****p<0.0001.
Cell extrinsic factors underlie aberrant differentiation of CD4+ T cells in Pik3cd GOF mice
To determine whether the perturbations we observed for murine Pik3cd GOF CD4+ T cells were cell intrinsic or extrinsic, we generated BM chimeras reconstituted with a 50:50 mix of WT(CD45.1):WT(CD45.2) or WT(CD45.1):Pik3cd GOF(CD45.2) BM. Approximately 12 weeks after reconstitution we analysed CD4+ T cell populations in these mice. Consistent with the data from intact mice, we observed a decrease in the percentage of naïve and an increase in effector memory CD4+ T cells derived from Pik3cd GOF BM (Figure 5A–D). Surprisingly, however, we observed even more pronounced changes in WT CD4+ T cells that developed in the presence of Pik3cd GOF cells including reduced naïve cells and increased memory cells (Figure 5A–D). We saw a similar pattern for Tfh and Treg cells with WT cells in the mixed BM chimeras displaying an expansion of these populations compared to CD4+ T cell populations in control WT:WT chimeras (Figure 5E, F). A similar pattern was observed in aged mixed BM chimeras, except we also observed a significant increase in Pik3cd GOF Tfh and Tregs cells that was not seen in young mice (Supplementary Figure 3). These significant changes induced in the WT T cells in the presence of Pik3cd GOF hematopoietic cells suggests that the numerical changes observed in Pik3cd GOF CD4+ T cell populations may not be strictly cell intrinsic, but rather result from extrinsic signals provided by other Pik3cd GOF cells.
Figure 5: Extrinsic factors contribute to CD4+ T cells activation in Pik3cd GOF mice.

Spleens from WT:WT and WT:Pik3cd GOF mixed BM chimeras 12 weeks after reconstitution were stained to identify different CD4+ T cell populations within the CD45.1+ or CD45.2+ compartments. (A) Schematics show the generation of mixed bone marrow chimeras. Representative plots show gating for naïve (CD44loCD62Lhi), central memory (CD44hiCD62Lhi), and effector memory (CD44hiCD62Llo) CD4+ T cells. CD45.1 plots show WT cells in the WT:WT chimera (left) and WT cells in the WT:Pik3cdGOF chimeras (right). CD45.2 plots show WT cells in the WT:WT chimera (left) and Pik3cdGOF cells in the WT:Pik3cdGOF chimeras (right). (B-F) The percentages of CD45.1+CD4+ or CD45.2+CD4+ T cells that were (B) naïve (CD44loCD62Lhi), (C) central memory (CD44hiCD62Lhi), (D) effector memory (CD44hiCD62Llo), (E) Tfh and (F) Treg CD4+ T cells were determined (mean ± SEM, n=5). Significant differences were determined by 2-way ANOVA, **p<0.01; ***p<0.001; ****p<0.0001.
PI3K overactivation results in altered T helper differentiation and cytokine production
Human PIK3CD GOF CD4+ T cells showed altered cytokine secretion, with memory cells showing increased production of Th2 cytokines and cTfh cells secreting more IFNγ and Th2 cytokines but less Th17 cytokines (Figure 3, Supplementary Figure 1). However, the small numbers of naïve CD4+ T cells in these patients (Figure 1) precluded detailed analysis of the ability of their naïve CD4+ T cells to differentiate into various T helper cell populations in vitro. Thus, we utilized our novel mouse model to ascertain whether these defects were cell intrinsic. Naïve WT or Pik3cd GOF murine CD4+ T cells were cultured in vitro under polarizing conditions to induce Th1, Th2 or Th17 fates. IL-2 production was equivalent between WT and Pik3cd GOF cells CD4+ T cells irrespective of the culture conditions used (Figure 6A, B). However, Pik3cd GOF augmented induction of Th1 and Th2 responses, evidenced by the generation of increased percentages of IFN-γ and IL-5-expressing cells, respectively (Figure 6C–F). In contrast, Th17 differentiation was impaired by Pik3cd GOF (Figure 6G, H).
Figure 6: Pik3cd GOF naïve CD4+ T cells have altered cytokine production in vitro.

Naïve CD4+ T cells (CD4+CD44loCD62Lhi) were sorted from spleens of WT or Pik3cd GOF mice, stimulated with anti-CD3 and anti-CD28 mAbs under (A, B) Th0, (C, D) Th1, (E, F) Th2 or (G, H) Th17 conditions for 4 days and then restimulated with PMA/ionomycin. Cells were harvested and stained for intracellular expression of IL-2, IL-5, IL-17A, IFN-γ and TNFα. Flow cytometry plots show representative cytokine expression under various conditions. Graphs depict summary data of cytokine production under each condition respectively (mean ± SEM, n=4–5). Significant differences were determined by 2-way ANOVA, ***p<0.001.
Impact of Pik3cd GOF mutations on Tfh development in vivo
The ability of CD4+ T cells to differentiate into Tfh cells and provide help to B cells for Ab production and formation of a germinal center (GC) is essential for an effective humoral response12, 14, 15. Even though PIK3CD GOF patients have an increased frequency of cTfh cells (Figure 1), these cells exhibit a dysregulated phenotype and altered cytokine profiles (Figures 2, 3; Supplementary Figure 1), with skewing towards a PD-1hi Th1-type Tfh subset which is predictive of poor B-cell help28, 29, 35. This is consistent with multiple clinical symptoms characteristic of these patients that point to defective Ab responses. Pik3cd GOF mice also have an increased number of Tfh cells (Figure 4), however analysis of mixed BM chimeras suggested this effect was not strictly cell intrinsic (Figure 5). To dissect the intrinsic capacity of Pik3cd GOF CD4+ T cells to differentiate into functional Tfh cell and provide help to B cells in the context of an Ag-specific response, we utilized an in vivo adoptive transfer model. WT or Pik3cd GOF TCR transgenic OT-II cells (expressing a TCR specific for ovalbumin [OVA]) were transferred into WT recipients and immunized intraperitoneally with alum/OVA. Seven days after immunization we observed ~30% fewer Pik3cd GOF OT-II cells within the spleen compared to WT OT-II (Figure 7A). However similar percentages of Tfh cells were generated from WT and Pik3cd GOF CD4+ OT-II cells (Figure 7B, C). In this adoptive transfer system, endogenous WT CD4+ T cells can also provide help to B cells. Thus, to directly assess the helper function of OT-II cells, we transferred Pik3cd GOF or WT OT-II cells into SAP-deficient mice, in which recipient CD4+ T cell are unable to provide B-cell help37. In this system we also observed decreased frequencies of OT-II cells following transfer (data not shown). Despite this, there was a trend towards an increased Tfh frequency (Figure 7D). Surprisingly this increase was not associated with an increase in GC cells (Figure 7E). Rather, significantly fewer GC B cells were present in SAP-deficient mice that received Pik3cd GOF OT-II cells (Figure 7E). GC numbers have been found to strongly correlate with the number of Tfh cells19, 38. Thus, to eliminate changes in Tfh numbers as a confounding factor and determine whether this decrease in GC B cells reflected decreased helper function of Tfh cells on a per cell basis, we plotted percentages of Tfh cells against GC B cells. This confirmed that Pik3cd GOF Tfh cells were less efficient than WT Tfh cells at providing help to B cells and forming an equivalent GC (Figure 7F).
Figure 7: Effect of Pik3cd GOF mutation on Tfh cell differentiation.

(A-C) WT or Pik3cd GOF OT-II T cells were transferred into WT recipients, which were then immunized with OVA/Alum intraperitoneally. Spleens were harvested 7 days later and flow cytometry analysis performed (mean ± SEM, n=15–17). (A) Percentages of OT-II cells recovered 7 days post transfer. (B) Contour plots showing representative staining of CXCR5 versus PD-1 on OT-II cells. (C) Percentage of Tfh (CXCR5+PD-1+) cells in the OT-II cell population.
(D-F) WT or Pik3cd GOF OT-II cells were transferred into SAP-deficient hosts, which were then immunised with OVA/Alum. Spleens from recipient mice were harvested at day 7 and flow cytometry analysis performed. Percentage of (D) Tfh (CXCR5+PD-1+) cells (mean ± SEM) and (E) GC (B220+CD38loFashi) B cells in recipient mice (n=18–20). (F) Percentages of OT-II Tfh cells versus GC B cells in individual mice receiving either WT or Pik3cd GOF OT-II cells. Significant differences were determined by unpaired Students t-tests, **p<0.01. Linear regression analysis was performed and the slope of the lines compared in GraphPad Prism.
Pik3cd GOF qualitatively and quantitatively impacts the phenotype and function of Tfh cells in vivo
To gain insight into how Pik3cd GOF may intrinsically limit CD4+ T cell-mediated B-cell help, we examined expression of key genes important for Tfh function. While Cd40lg was expressed at similar levels by both WT and Pik3cd GOF OT-II cells (Figure 8A), Fasl was significantly higher in Pik3cd GOF than in WT OT-II cells (Figure 8B). Contrastingly, ICOS was significantly lower on Pik3cd GOF compared to WT OT-II cells (Figure 8C). We also examined expression of additional surface molecules associated with Tfh differentiation (Supplementary Figure 4A–E). Pik3cd GOF OT-II cells showed significantly greater downregulation of CD127 and CCR7 compared to WT cells (Supplementary Figure 4A–E).
Figure 8: Pik3cd GOF mutations result in altered Tfh function.
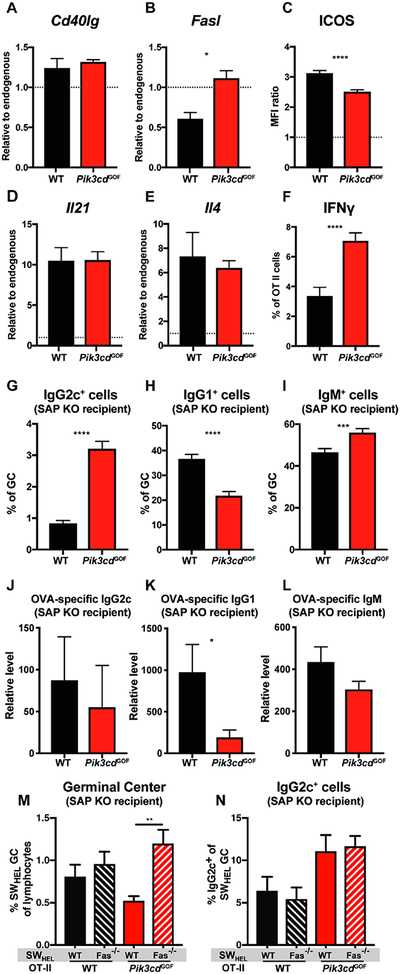
WT or Pik3cd GOF OT-II cells were transferred into WT (A-F) or SAP-deficient (G-I) recipients, which were then immunised with OVA/Alum. Splenic OT-II and recipient CD4+ T cell populations were sorted on day 5. (A) Cd40lg and (B) Fasl expression was determined by qPCR (mean ± SEM, n=4). (C) ICOS expression was assessed by flow cytometry (day 7). Graph depicts mean fluorescent intensity relative to recipient CD4+ T cells (dotted line; mean ± SEM, n=15–17). Expression of (D) Il21 and (E) Il4 were determined as in (A). (F) IFN-γ production by OT-II cells on day 7 was assessed by flow cytometry (mean ± SEM, n=15–17).
Expression of (G) IgG2c, (H) IgG1 and (I) IgM by GC B cells and serum levels of OVA-specific (J) IgG2c, (K) IgG1and (L) IgM (day 7; mean ± SEM, n=14–20).
(M-N) WT or Pik3cd GOF OT-II cells together with WT or Fas-deficient SWHEL B cells were transferred to SAP-deficient recipients which were then immunized with HEL-OVA. Lymph nodes were stained on day 7 to identify (M) GC and (N) IgG2c+ B cells (mean ± SEM; n=9–11). Significant differences were determined by unpaired Students t-tests or ANOVA, *p<0.05; **p<0.01, ***p<0.001; ****p<0.0001.
Given the dysregulated cytokine induction observed for human (Figure 3, Supplementary Figure 1) and murine (Figure 6) PI3K GOF CD4+ T cells in vitro, we also investigated cytokine production by OT-II CD4+ T cells in vivo following immunisation with OVA. Expression of Il21 and Il4 was similar between Pik3cd GOF and WT OT-II cells (Figure 8D, E). Consistent with data from the in vitro Th1 polarising cultures, we saw a large increase in the frequency of Pik3cd GOF CD4+ OT-II cells producing IFN-γ compared to WT OT-II cells (Figure 8F). While Pik3cd GOF OT-II cells produced equivalent amounts of IL-2, they exhibited significantly reduced production of TNFα and IL17A compared to WT OT-II (Supplementary Figure 4F–H). To determine the functional consequences of this altered cytokine profile of Pik3cd GOF OT-II cells we examined isotype switching in the GC. In mice IL-4 promotes class switching to IgG1, and IFNγ drives switching to IgG2a/c while inhibiting switching to IgG139, 40. Consistent with increased IFNγδproduction by Pik3cd GOF OT-II cells we saw an increase in switching to IgG2c (Figure 8G) and a concomitant decrease in switching to IgG1 (Figure 8H) in the GCs of mice that received Pik3cd GOF OT-II cells. Interestingly we also observed an increase in the percentage of IgM+ (i.e. unswitched) GC B cells (Figure 8I), establishing that Pik3cd GOF CD4+ T cells are overall less effective at inducing isotype switching than WT cells. To determine how the decreased help to B cells and altered isotype switching affected the production of specific antibodies we assessed the levels of anti-OVA antibody present in the serum of mice (Figure 8J–L). In mice in which T cell help was provided by Pik3cd GOF OT-II cells, the levels of serum IgG2c and IgM were relatively similar to that observed in mice that received WT OT-II cells (Figure 8J, L), despite the increased percentage of IgG2c+ and IgM+ GC B cells observed in the presence of Pik3cd GOF help (Figure 8G, I). In contrast, Pik3cd GOF CD4+ T cells failed to support production of anti-OVA IgG1 Abs (Figure 8K). Overall these results reveal that Pik3cd GOF CD4+ T cells have a significantly altered ability to support GCs and antibody secretion, and appropriately regulate Ig isotype switching.
To assess the contribution of dysregulated FasL expression on Pik3cd GOF Tfh cells (Figure 8B) to these altered B cells responses, WT or Pik3cd GOF OT-II cells were transferred together with WT or Fas-deficient SWHEL B cells into SAP-KO mice and immunized with HEL-OVA. Seven days later we assessed the GC response. The GC response induced by Pik3cd GOF OT-II cells was significantly larger from Fas-deficient B cells than from WT B cells (Fig 8M) indicating that increased expression of FasL on CD4+ T cells is partially responsible for the decreased B-cell help provided by these T cells. In contrast, the absence of Fas on B cells did not alter the increased switching to IgG2c that was seen in the presence of Pik3cd GOF T cells (Fig 8N).
Discussion
Our parallel studies in humans and mice identified the necessity of balanced PI3K signaling in CD4+ T cells to maintain immune regulation in the setting of differentiation to effector cells with distinct fates. In particular we observed substantial dysregulation of the Tfh cell compartment – patients with PIK3CD GOF mutations displayed a significant increase in cTfh cells and this was mirrored by a significant increase in Tfh cells in the secondary lymphoid organs of mice carrying a Pik3cd GOF mutation. As the generation of Tfh cells in mice requires CD4+ T cell-intrinsic PI3K signaling19, this finding may have been predicted. Indeed a very recent study also reported increased proportions of cTfh cells in PIK3CD GOF patients, and in an independently-generated mouse model of Pik3cd GOF6. Based on adoptive transfer experiments, Schwartzberg and colleagues concluded that overactive PI3K caused a cell-intrinsic increase in Tfh cells due to PI3K GOF abrogating the need for ICOS signaling6. However, in our adoptive transfer experiments, while there was a slight trend towards increased Tfh cells this was not statistically significant. Further, our analysis of murine mixed BM chimeras revealed that WT CD4+ T cells also had an increased Tfh cell population in the presence of Pik3cd GOF immune cells, suggesting that the increased generation of Tfh cells was, to a large extent, cell extrinsic. Thus, while overactive PI3K in CD4+ T cells may be able to drive a slight increase in Tfh cells in certain situations where signaling is limited we suggest that the main driver of the Tfh expansion in these patients comes from dysregulation of other cells e.g. B cells. IL-6 can promote Tfh cell formation, and B cell-derived IL-6 can drive Tfh and subsequent cognate B cell responses both in infection41 and autoimmunity42, 43. Notably, PI3K is important for IL-6 production by B cells44. In addition, PI3K signaling downstream of IL-21/IL-21R upregulates CD86 expression on B cells which contributes to CD4+ T cell expansion45 and generation of Tfh cells46. Consistent with this we observed increased CD86 expression on B cells in our Pik3cd GOF mice (data not shown). Thus, B cells may contribute to heightened production of Tfh cells in Pik3cd GOF mice through dysregulated IL-6 and/or CD86 expression. We also noted an extrinsic increase in memory CD4+ T cells and loss of naïve CD4+ T cells suggesting that these mechanisms may contribute to general overactivation of CD4+ T cells.
Despite this expanded population of Tfh cells, patients with overactive PI3K have poor specific antibody responses and recurrent respiratory infections2 (see Supplementary Table 1) suggestive of defects in humoral immune defects. This raises the question of whether PIK3CD GOF Tfh cells can provide effective help to B cells in vivo, a question not addressed in previous studies6. Here, we provide several lines of evidence that overactive PI3K signaling in CD4+ T cells results in Tfh cells that are highly dysfunctional. PIK3CD GOF patients had increased proportions of cTfh cells expressing a Th1/CXCR3+ phenotype and concomitant loss of CCR6+ Tfh cells, and their cTfh cells exhibited enhanced production of IFNγ, and markedly elevated expression of PD-1. This exaggerated production of IFNγ, coupled with reductions in frequency of CCR6+ cTfh cells, would impede B cell differentiation, as established by (1) the intrinsically poor B-cell helper function of CXCR3+ cTfh cells in the settings of healthy donors27–29, other monogenic primary immunodeficiencies such as those resulting from STAT3 LOF or STAT1 GOF mutations28, common variable immunodeficiency35, 47 and HIV infection48, 49 and (2) previous findings of the inhibitory effect of IFNγ on human B-cell differentiation28, 35, 50–52. Further, enhanced engagement of PD-1, due to its heightened expression, would restrain TCR-induced proliferation and IL-21 production49. Thus, elevated expression of PD-1 on PIK3CD GOF cTfh would further compromise Tfh cell function. Data from our novel mouse model of Pik3cd GOF confirmed and extended these findings from patients. Our studies further revealed that, in contrast to the Tfh cell expansion, which was largely cell extrinsic, the altered functionality of the Tfh cells was cell intrinsic. Thus, Pik3cd GOF CD4+ T cells showed increased IFNγ production and generated smaller GC responses in vivo. Increased production of IFNγ by murine Pik3cd GOF CD4+ T cells also manifested as reduced and skewed Ig isotype switching, resulting in significantly fewer IgG1+ B cells and more IgG2c+ B cells and reduced Ag-specific IgG1 in the serum following immunization. This is consistent with the well-established roles of IFN-γ in inducing switching to IgG2a/c while inhibiting switching to IgG139, 40.
Importantly, we identified additional defects that may also contribute to less effective help from Pik3cd GOF Tfh cells in response to specific Ag. First, Pik3cd GOF OT-II cells expressed elevated levels of Fasl. Interestingly, a subset of Tfh-like cells has been identified in extrafollicular regions of human tonsils53. In contrast to GC Tfh cells, these extrafollicular Tfh-type cells were unable to efficiently induce the differentiation of autologous GC B cells into Ig-secreting cells. This was attributable to elevated FasL expression, as provision of B-cell help to GC B cells could be achieved by Fas/FasL blockade53. Here we showed that when Fas/FasL interactions were blocked Pik3cd GOF CD4+ T cells were able to support a greater GC response suggesting that the increased expression of FasL by Pik3cd GOF CD4+ Tfh cells at least partially underlies the impaired B-cell help in the setting of PI3K hyperactivation. It is likely that other factors may also contribute to the impaired help provided by the cells. For example, we also observed decreased ICOS expression on Pik3cd GOF CD4+ T cells following immunization. ICOS/ICOS-L interactions play an important role in the activation and function of Tfh cells54–57 as well as their migration to GCs58. Thus, decreased ICOS expression could contribute to the decreased function of Pik3cd GOF Tfh cells. Interestingly, ICOS expression is controlled by the transcription factor FOXO1, with loss of FOXO1 leading to decreased ICOS expression59. As PI3K/Akt signaling inactivates FOXO1, it is likely that PI3K overactivation in cells bearing Pik3cd GOF mutations leads to decreased FOXO1-induced ICOS expression. This is consistent with our finding of decreased expression of CCR7 and CD127, which are also induced by FOXO1, on Pik3cd GOF CD4+ T cells59, 60.
In addition to the alteration in Tfh cell numbers and function we observed increased production of Th2 cytokines by human PIK3CD GOF memory cells CD4+ T cells. Further, naïve Pik3cd GOF CD4+ T cells activated under Th2 polarizing conditions also produced increased IL-5 suggesting this defect was cell intrinsic. This skewing to production of IL-4, IL-5 and IL-13 by PIK3CD GOF CD4+ T cells may predict clinical features of Th2-type disease in PIK3CD GOF patients. While allergies have not previously been reported to occur at an increased frequency in these patients compared to the healthy population2, analysis of the clinical records or our cohort revealed that ~50% of patients displayed Th2 related pathologies61 such as asthma, eosinophilic esophagitis, eczema/atopic dermatitis, urticaria and rhinoconjunctivitis (Supplementary Table 1). Remarkably, very few of the patients reported increased IgE levels (Supplementary Table 1). The disconnect between increased Th2 cytokines and lack of hyper-IgE is most likely explained by the ability of PI3K to inhibit switching to IgE62, 63. Thus, by cataloguing the frequent occurrence of non-allergic Th2-type diseases in patients with PIK3CD GOF mutations, our study has not only substantially extended the clinical phenotype of this condition but importantly also provided a cellular basis for this aspect of the APDS.
Here, by examining in detail the phenotype, function and biology of human and mouse Tfh cells arising from PI3K GOF, we have substantially extended initial observations6 and importantly provided significant insight into the mechanisms underlying aberrant effector function of these Tfh cells. We find aberrant cytokine production with increased Th1 and Th2 cytokines as well as dysregulated expression of FasL which together impact the ability of PI3K GOF Tfh cells to provide optimal B cell help during T-dependent humoral immune responses. These defects, together with intrinsic impairments in the development and function of PIK3CD GOF B cells4–7, would contribute to the poor ability to induce memory B cells and Ab-secreting PCs in PIK3CD GOF patients and the resultant characteristically poor Ab responses observed in these patients following natural infection or vaccination with protein and polysaccharide2.
Supplementary Material
Key Messages:
PIK3CD GOF mutations result in aberrant differentiation and function of human and mouse CD4+ T cells
Increased PI3K activity leads to a cell extrinsic increase in memory and Tfh CD4+ T cells, cell intrinsic increases in IFNγ and IL-4 production, and poor Tfh helper function for B cell differentiation
These defects may contribute to antibody deficiencies and impaired humoral immune responses characteristic of patients with PIK3CD GOF mutations
Capsule Summary.
Gain of function mutations in PIK3CD cause multiple defects in CD4+ T cells including altered cytokine production and Tfh function which may contribute to the poor antibody responses in these patients.
Funding source:
This work was supported by project grants (1088215 to EKD and CSM; 1127157 to SGT, EKD and CSM), Program grants (1016953 and 1113904 to SGT, RB), and Principal Research Fellowships (1042925 to SGT; 1105877 to RB) from the National Health and Medical Research Council of Australia, the Susan and John Freeman Cancer Research Grant from Cancer Council NSW (Australia, to SGT), the Office of Health and Medical Research of the NSW Government, the Jeffrey Modell Foundation, the John Cook Brown Foundation, and a Bench to Bedside grant from the National Institute of Allergy and Infectious Diseases, NIH (to SGT, GU and LN). CSM is supported by an Early-Mid Career Research Fellowship from the New South Wales State Government; JB is supported by the Postgraduate Research Scholarship awarded by Fundação Estudar (Brazil); AL is supported by a Tuition Fee Scholarship from UNSW Sydney. SMH, LDN and GU are supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH.
Non-standard abbreviations:
- TN
Naive T cells
- TCM
central memory T cells
- TEM
effector memory T cells
- Tfh
T follicular helper cells
- Tregs
regulatory T cells
- PC
plasma cells
- TAE
T cell activation and expansion
- PI3K
phosphoinositide-3 kinase
- BM
bone marrow
- OVA
ovalbumin
- HEL
hen egg lysozyme
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest: none
References
- 1.Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science 2013; 342:866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J Allergy Clin Immunol 2017; 139:597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol 2014; 15:88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Avery DT, Kane A, Nguyen T, Lau A, Nguyen A, Lenthall H, et al. Germline-activating mutations in PIK3CD compromise B cell development and function. J Exp Med 2018:jem.20180010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wray-Dutra MN, Al Qureshah F, Metzler G, Oukka M, James RG, Rawlings DJ. Activated PIK3CD drives innate B cell expansion yet limits B cell-intrinsic immune responses. J Exp Med 2018; 215:2485–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Preite S, Cannons JL, Radtke AJ, Vujkovic-Cvijin I, Gomez-Rodriguez J, Volpi S, et al. Hyperactivated PI3Kδ promotes self and commensal reactivity at the expense of optimal humoral immunity. Nat Immunol 2018; 19:986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stark A-K, Chandra A, Chakraborty K, Alam R, Carbonaro V, Clark J, et al. PI3Kδ hyper-activation promotes development of B cells that exacerbate Streptococcus pneumoniae infection in an antibody-independent manner. Nat Commun 2018; 9:3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cannons JL, Preite S, Kapnick SM, Uzel G, Schwartzberg PL. Genetic Defects in Phosphoinositide 3-Kinase δ Influence CD8+ T Cell Survival, Differentiation, and Function. Front Immunol 2018; 9:1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards ESJ, Bier J, Cole TS, Wong M, Hsu P, Berglund LJ, et al. Activating PIK3CD mutations impair human cytotoxic lymphocyte differentiation and function and EBV immunity. J Allergy Clin Immunol 2018. [DOI] [PubMed] [Google Scholar]
- 10.Wentink MWJ, Mueller YM, Dalm VASH, Driessen GJ, van Hagen PM, van Montfrans JM, et al. Exhaustion of the CD8+T Cell Compartment in Patients with Mutations in Phosphoinositide 3-Kinase Delta. Front Immunol 2018; 9:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu J, Paul WE. CD4+ T cell plasticity-Th2 cells join the crowd. Immunity 2010; 32:11–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly - TFH cells in human health and disease. Nat Rev Immunol 2013; 13:412–26. [DOI] [PubMed] [Google Scholar]
- 13.Sallusto F Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu Rev Immunol 2016; 34:317–34. [DOI] [PubMed] [Google Scholar]
- 14.Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med 2012; 209:1241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crotty S T follicular helper cell differentiation, function, and roles in disease. Immunity 2014; 41:529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okkenhaug K Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol 2013; 31:675–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han JM, Patterson SJ, Levings MK. The Role of the PI3K Signaling Pathway in CD4(+) T Cell Differentiation and Function. Front Immunol 2012; 3:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okkenhaug K, Patton DT, Bilancio A, Garçon F, Rowan WC, Vanhaesebroeck B. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol 2006; 177:5122–8. [DOI] [PubMed] [Google Scholar]
- 19.Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LMC, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol 2010; 185:4042–52. [DOI] [PubMed] [Google Scholar]
- 20.Gigoux M, Shang J, Pak Y, Xu M, Choe J, Mak TW, et al. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc Natl Acad Sci USA 2009; 106:20371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, et al. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol 2006; 177:6598–602. [DOI] [PubMed] [Google Scholar]
- 22.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 1998; 76:34–40. [DOI] [PubMed] [Google Scholar]
- 23.Czar MJ, Kersh EN, Mijares LA, Lanier G, Lewis J, Yap G, et al. Altered lymphocyte responses and cytokine production in mice deficient in the X-linked lymphoproliferative disease gene SH2D1A/DSHP/SAP. Proc Natl Acad Sci USA 2001; 98:7449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phan TG, Amesbury M, Gardam S, Crosbie J, Hasbold J, Hodgkin PD, et al. B cell receptor-independent stimuli trigger immunoglobulin (Ig) class switch recombination and production of IgG autoantibodies by anergic self-reactive B cells. J Exp Med 2003; 197:845–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 2014; 9:1956–68. [DOI] [PubMed] [Google Scholar]
- 26.He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5⁺ CD4⁺ CD4CXCR5⁺ CD4⁺ Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013; 39:770–81. [DOI] [PubMed] [Google Scholar]
- 27.Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-⁺ 1CXCR3⁻ CXCR5⁺ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 2013; 39:758–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol 2015; 136:993–1006.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011; 34:108–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schatorjé EJH, Gemen EFA, Driessen GJA, Leuvenink J, van Hout RWNM, De Vries E. Paediatric reference values for the peripheral T cell compartment. Scand. J. Immunol 2012; 75:436–44. [DOI] [PubMed] [Google Scholar]
- 31.Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med 2000; 192:1553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, et al. Early commitment of naïve human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol 2009; 87:590–600. [DOI] [PubMed] [Google Scholar]
- 33.Chevalier N, Jarrossay D, Ho E, Avery DT, Ma CS, Yu D, et al. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol 2011; 186:5556–68. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity 2009; 31:158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The TH1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol 2018; 141:730–40. [DOI] [PubMed] [Google Scholar]
- 36.Gett AV, Hodgkin PD. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc Natl Acad Sci USA 1998; 95:9488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crotty S, Kersh EN, Cannons JL, Schwartzberg PL, Ahmed R. SAP is required for generating long-term humoral immunity. Nature 2003; 421:282–7. [DOI] [PubMed] [Google Scholar]
- 38.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 2013; 38:596–605. [DOI] [PubMed] [Google Scholar]
- 39.Snapper CM, Paul WE. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science 1987; 236:944–7. [DOI] [PubMed] [Google Scholar]
- 40.Deenick EK, Hasbold J, Hodgkin PD. Decision criteria for resolving isotype switching conflicts by B cells. Eur J Immunol 2005; 35:2949–55. [DOI] [PubMed] [Google Scholar]
- 41.Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D'Costa K, et al. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med 2012; 209:2049–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsantikos E, Oracki SA, Quilici C, Anderson GP, Tarlinton DM, Hibbs ML. Autoimmune disease in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. J Immunol 2010; 184:1348–60. [DOI] [PubMed] [Google Scholar]
- 43.de Valle E, Grigoriadis G, O'Reilly LA, Willis SN, Maxwell MJ, Corcoran LM, et al. NFκB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J Exp Med 2016; 213:621–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dil N, Marshall AJ. Role of phosphoinositide 3-kinase p110 delta in TLR4- and TLR9-mediated B cell cytokine production and differentiation. Mol Immunol 2009; 46:1970–8. [DOI] [PubMed] [Google Scholar]
- 45.Attridge K, Kenefeck R, Wardzinski L, Qureshi OS, Wang CJ, Manzotti C, et al. IL-21 promotes CD4 T cell responses by phosphatidylinositol 3-kinase-dependent upregulation of CD86 on B cells. J Immunol 2014; 192:2195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salek-Ardakani S, Choi YS, Rafii-El-Idrissi Benhnia M, Flynn R, Arens R, Shoenberger S, et al. B cell-specific expression of B7–2 is required for follicular Th cell function in response to vaccinia virus. J Immunol 2011; 186:5294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cunill V, Clemente A, Lanio N, Barcelo C, Andreu V, Pons J, et al. Follicular T Cells from smB(−) Common Variable Immunodeficiency Patients Are Skewed Toward a Th1 Phenotype. Front Immunol 2017; 8:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boswell KL, Paris R, Boritz E, Ambrozak D, Yamamoto T, Darko S, et al. Loss of circulating CD4 T cells with B cell helper function during chronic HIV infection. PLoS Pathog 2014; 10:e1003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cubas RA, Mudd JC, Savoye A-L, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med 2013; 19:494–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.King CL, Gallin JI, Malech HL, Abramson SL, Nutman TB. Regulation of immunoglobulin production in hyperimmunoglobulin E recurrent-infection syndrome by interferon gamma. Proc Natl Acad Sci U S A 1989; 86:10085–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.King CL, Low CC, Nutman TB. IgE production in human helminth infection. Reciprocal interrelationship between IL-4 and IFN-gamma. J Immunol 1993; 150:1873–80. [PubMed] [Google Scholar]
- 52.Kim JR, Lim HW, Kang SG, Hillsamer P, Kim CH. Human CD57+ germinal center-T cells are the major helpers for GC-B cells and induce class switch recombination. BMC Immunol. 2005; 6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bentebibel SE, Schmitt N, Banchereau J, Ueno H. Human tonsil B-cell lymphoma 6 (BCL6)-expressing CD4+ T-cell subset specialized for B-cell help outside germinal centers. Proc Natl Acad Sci USA 2011; 108:E488–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McAdam AJ, Greenwald RJ, Levin MA, Chernova T, Malenkovich N, Ling V, et al. ICOS is critical for CD40-mediated antibody class switching. Nature 2001; 409:102–5. [DOI] [PubMed] [Google Scholar]
- 55.Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol 2005; 175:2340–8. [DOI] [PubMed] [Google Scholar]
- 56.Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol 2006; 177:4927–32. [DOI] [PubMed] [Google Scholar]
- 57.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011; 34:932–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 2013; 496:523–7. [DOI] [PubMed] [Google Scholar]
- 59.Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai C-Y, Emslie E, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity 2015; 42:239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol 2012; 12:649–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spergel J, Aceves SS. Allergic components of eosinophilic esophagitis. J Allergy Clin Immunol 2018; 142:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang T-T, Okkenhaug K, Nashed BF, Puri KD, Knight ZA, Shokat KM, et al. Genetic or pharmaceutical blockade of p110delta phosphoinositide 3-kinase enhances IgE production. J Allergy Clin Immunol 2008; 122:811–9.e2. [DOI] [PubMed] [Google Scholar]
- 63.Doi T, Obayashi K, Kadowaki T, Fujii H, Koyasu S. PI3K is a negative regulator of IgE production. Int Immunol 2008; 20:499–508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.