

Abstract
Primary liver cancer (PLC) is heterogeneous and it is an aggressive malignancy with a poor prognostic outcome. Current evidence suggests that PLC tumorigenesis is driven by rare subpopulations of cancer stem cells (CSCs), which contribute to tumor initiation, progression, and therapy resistance through particular molecular mechanisms. Energy metabolism and mitochondrial function play an important role in the regulation of cancer stemness and stem cell specifications. Since the role of mitochondrial function as central hubs in cell growth and survival, studies on the critical physiological mechanisms of the liver underlying their therapy-resistant phenotype is important. In this review, we focus on liver CSC-related mitochondrial metabolism that contributes to the liver CSC features, in terms of enhanced drug-resistance and increased tumorigenicity, and to discuss their roles on potential therapies windows for PLC therapies.
Introduction
Primary liver cancer (PLC) is the sixth most common malignant cancer worldwide [57]. Moreover, liver cancer is among the most aggressive and difficult-to-treat malignancies, with a 5-year relative survival rate of less than 21% in the United States [34]. PLC mainly consists of two histologic types, i.e., hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA). HCC is the most common type of PLC, accounting for 90% of all liver cancer cases, followed by iCCA [71]. Potentially curative treatments, such as surgical resection, radiofrequency ablation, and liver transplantation, can only be applied in 30-40 percent of patients in the West, and even a smaller proportion of patients in Asia [19]. In addition, recurrence is quite frequent even after curative treatment; therefore, the long-term outcome for PLC treatment is still unsatisfactory [71]. The main challenge to overcome this issue is that PLC is clinically and biologically heterogeneous [82]. Due to the high recurrence, high mortality and resistance to conventional therapies, the development of new chemopreventive agents for precision management of PLC is an important research priority.
Emerging evidence supports the hierarchical model of cancer stem cells (CSCs) as the main driver of tumor progression, cancer recurrence and metastasis [4]. CSCs exhibit features of normal stem cells, e.g., self-renewal and multilineage differentiation capacity but also is responsible for tumor initiation. Therefore, eradicating CSCs may be a critical approach to cancer therapy. Previous studies have shown the existence of CSCs in human liver cancers [61, 70, 90]. Indeed, accumulating evidence supports that liver CSCs are histologically heterogeneous and contain a small fraction of cells with stem cell properties (e.g., self-renewal and differentiation) in PLC such as expressions of a variety of CSC markers. Currently, a number of cell surface markers have been identified as liver CSC markers including epithelial cell adhesion molecule (EpCAM), CD44, CD24, CD133, CD90, and CD13 (Table 1). In addition, other markers including oval cell marker OV6, Hoechst dye efflux, detoxifying enzymes aldehyde dehydrogenases (ALDH) are also frequently used to identify liver CSCs [67]. Indeed, our group has identified a novel HCC subtype defined by the liver CSC markers EpCAM and alpha-fetoprotein (AFP), which is associated with poor prognosis [89]. As cells expressing these markers may be functionally linked to CSC properties, studies on targeting CSC markers may help understanding therapeutic resistance of PLC.
Table 1:
Known liver CSC markers and their role in cellular metabolism
| Marker | Metabolic phenotype | Function in therapy | Reference |
|---|---|---|---|
| CD13 | Maintain lower levels of mitochondrial ROS | Combining a CD13 inhibitor with a chemo/radiation therapy inhibit tumor progression | 27,38 |
| CD133 | Decrease mitochondrial OXPHOS and promotes glycolysis | Targeting glycolytic enzymes represses stemness properties in CD133+ PLC cells | 32,72 |
| CD90 | Higher levels of the mitochondrial ROS | Actively proliferate and are sensitive to 5-FU therapy | 27 |
| CD44 | Maintaining low ROS levels through promoting glutathione synthesis | Combining a CD44 inhibitor with a Sulfasalazine inhibit tumor progression | 74 |
| EPCAM | Maintaining low ROS levels | Not reported in this paper | 16 |
| CD24 | Not reported | Not reported | |
| DLK1 | Not reported | Not reported | |
| OV6 | Not reported | Not reported |
The mitochondria of the liver, compared to other tissue types, have unique features since the liver plays a central role in a variety of critical biological metabolism functions including the homeostasis of carbohydrate, lipid, amino acids and protein synthesis [50]. In addition, the liver is one of the abundant tissues in terms of density and count of mitochondria [3]. The density of mitochondria is distinct depending on the demands of mitochondrial oxidative phosphorylation (OXPHOS) in different organs. Accumulation of damaged mitochondria is a crucial factor in chronic liver diseases [3]. Consequently, mitochondrial dysfunctions are frequently described in PLC [64], which have been reported to be associated with decreased ROS production, impaired apoptosis, increased anabolism rate, and proliferative potential, reduced autophagic degradation [78]. Interestingly, mitochondria have been demonstrated specifically affecting stem cell faith and differentiation potential, suggesting that modulation of mitochondrial activities contribute to the stem cell phenotype. However, there is no simple concept for the role of mitochondria in liver CSCs. Given the central role of mitochondria of the liver and stem cells in cell function and death decisions, we will focus on mitochondrial metabolism in liver CSC biology. This review will summarize functions of mitochondria, including mitochondrial metabolism, mitochondrial biogenesis, mitochondrial dynamics and mitophagy, cell death, oxidative stress, and mitochondrial bioenergetics, in the context of functional regulations of liver CSCs (Figure 1).
Figure 1. An integrative model of the roles of mitochondrial metabolism in liver cancer stem cells.
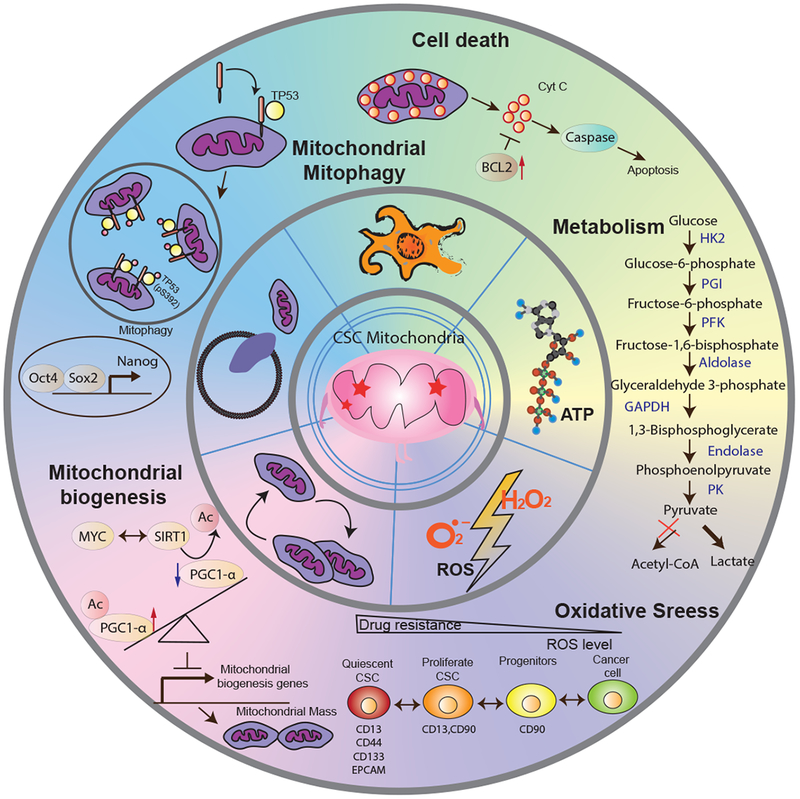
This illustration encompasses five key features of mitochondrial dysfunction needed for the maintenance of liver CSCs. In particular, Liver CSCs may preferentially (1) rely on glycolytic pathways to increase ATP production for biosynthesis; (2) reduce ROS levels to acquire a quiescent state in response to drug resistance; (3) decrease mitochondrial biogenesis through increased expression of acetylated PGC-1α to reduce mitochondrial respiration; (4) enhance mitophagy to block p53 mitochondrial translocation, which may bind to the NANOG promoter to inhibit the expression of NANOG, resulting in reduced stemness and self-renewal ability of liver CSCs; (5) acquire the ability for evading mitochondria-mediated death pathway by overexpressing BCL-2 family proteins thereby resistance to anticancer treatments. Inner circle: a dysfunctional CSC mitochondrium. Middle circle: five features of mitochondrial dysfunction. Outer circle: molecular signaling pathways of mitochondria, including mitochondrial metabolism, oxidative stress, mitochondrial biogenesis, mitochondrial mitophagy, and cell death, in the context of functional regulations of liver CSCs. Abbreviation: CSC: cancer stem cell; Cyt C: Cytochrome C; ROS: Reactive Oxygen Species; HK2: Hexokinase 2; PGI: Phosphoglucoisomerase; PFK: Phosphofructokinase; PK: Pyruvate kinase.
Mitochondrial metabolism in liver CSCs
One of the most striking characteristics of CSCs is their ability to form a specialized niche to adapt to changing microenvironmental conditions for their own benefit. This specialized niche is termed the CSC microenvironment [65]. This specific microenvironment maintains the principal properties of CSCs, protects them from multiple drug transporters and immune surveillance, and acquires resistance to DNA damage and mitochondria-mediated cell death mechanisms to facilitate tumor progression. The CSC microenvironment is dependent on activation of certain signaling pathways including altered tumor’s metabolic activities, which is conducive to CSC resistance to anticancer treatments. Due to distinct microenvironmental conditions they survive in, CSCs are considered highly heterogeneous and exhibit a distinct metabolic phenotype in different tumor types in terms of stemness features [86]. Indeed, a number of studies suggest that CSCs preferentially rely on glycolytic pathways, which presents low or absent rates of OXPHOS and high lactate production [10, 86]. In fact, there are several metabolic benefits to use “Warburg effect” for CSC, including increased ATP production rates while glycolytic signaling produces intermediates for biosynthesis, reduced ROS production in response to stressful environmental conditions characterized by low oxygen (hypoxia) [10, 86]. Interestingly, some studies indicate that mitochondrial oxidative metabolism may be a prevalent source of energy for CSC, suggesting a possible function for metabolic plasticity in CSCs [18].
Immune escape plays an important role in the initiation and progression of a malignant tumor. CSCs have the ability to evade immune surveillance as well as promote immunosuppression to maintain stem-like features and resistance to therapy through a variety of niche-specific mechanisms [66]. Thus, disruption of the interactions between tumor cells and infiltrating immune cells that drives a CSC microenvironment will be crucial for effective treatment. In addition, a number of studies suggest that the “Warburg effect” also is critical for instigating immunosuppressive response [1, 24]. On the one hand, glycolysis of cancer cells has been implicated in the inhibition of the function of anticancer immune cells [24]. Glucose utilization is required for the functional activation of T cells; however, rapidly dividing tumor cells may compete with T-cells for limited resources thereby disrupting their activity. Specifically, cancer cells may increase lactate production via activated glycolytic metabolism to maintain an acidic, low-pH tumor microenvironment, which is a product of tumor glycolysis to suppress antitumor immune cells, such as T effector cells and NK cells [24]. On the other hand, mitochondria, as the master regulators of many stress-induced signals, may trigger signaling mechanisms that are critical for the activation of antitumor immune responses [22]. Collectively, existing data convincingly demonstrate that metabolic reprogramming from mitochondrial respiration to glycolysis is a key mechanism to block immune surveillance during tumorigenesis.
The liver is an exquisitely dynamic organ, being able to change metabolic shift in response to body is conditions during fasting and feeding. Recent studies demonstrate the importance of metabolic reprograming in liver CSCs (Table 1). The CSC metabolism in PLC can be functionally identified by the expression of liver CSC markers, including CD133, CD44, and Nanog, and in general, liver CSCs have been found to favor glycolysis and suppress OXPHOS to promote stemness and resistance to treatment [12, 32, 72, 74]. Indeed, CD133+ tumors display higher expression of glycolytic enzymes and glycolytic capacity, coupled with a decrease in oxygen consumption rate (OCR) in order to promote glycolysis in liver CSCs [72]. Further, knockdown of glycolytic enzymes, LDHA and PDK4, in CD133+ PLC displays increased protein levels of several stemness markers (NANOG, OCT4, and SOX4), suggesting that a metabolic shift is important factor in sustaining stemness [72]. Similarly, global metabolic analysis demonstrates that CD133+ HCC exhibits an increased proliferation through enhancing glycolytic metabolism compared with CD133− HCC [32]. Furthermore, Thanee et al. found that CD44+ iCCA is advantageous for maintaining low ROS levels through promoting glutathione synthesis, resulting in increasing chemotherapy resistance [74]. In addition, knockdown of NANOG in HCC impaires glycolytic activity as demonstrated by the glycolytic flux (extracellular acidification rate or ECAR) [12]. Importantly, NANOG directly represses OXPHOS and maintains low intracellular ROS levels of CSCs required for the maintenance of CSCs properties [12]. Restoration of OXPHOS activity renders liver CSCs more susceptible to chemotherapy drugs [12]. Overall, these studies show that liver CSCs have specific metabolism pathways to maintain stemness properties of CSCs and resistance to chemotherapeutic drugs.
Mitochondrial biogenesis in liver CSCs
Mitochondrial dysfunction is often detected as an early alteration of liver diseases such as insulin resistance, non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD) and HCC, suggesting a causative effect [3, 20]. Cells have developed different mechanisms, including mitochondrial biogenesis, mitophagy, fusion and fission, to maintain mitochondrial functions or to block the effects of mitochondrial damage in response to metabolic demands and stressors. Mitochondrial biogenesis plays an important role in keeping mitochondrial mass to balance energy homeostasis during energy deprivation or to adapt mitochondrial insults. Interestingly, stem cells with increased mitochondrial biogenesis are associated with various stem cell differentiation, suggesting that loss of mitochondrial function is required to maintain the stemness properties [13, 88]. In addition, the hepatogenic differentiation of stem cells is accompanied by an increase in the mitochondrial biogenesis [79, 93].
There is a difference in the metabolic stage from the development and progression of PLC (Figure 2. In a healthy liver, the metabolic energy consumption relies primarily on mitochondrial OXPHOS, which is efficient to generate ATP than glycolysis pathway [94]. Once the liver starts to accumulate lipids, it enhances the liver susceptibility to subsequent damage induced by inflammation, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress and fibrinogenesis, which may lead to nonalcoholic fatty liver disease (NAFLD) [7, 56]. Liver tissues from patients with early-stage NAFLD have increased mitochondrial respiratory rates and mitochondrial biogenesis to mitigate disease progression. However, mitochondrial adaptations are lost in the early stages of nonalcoholic steatohepatitis (NASH), which has a low mitochondrial respiratory function despite an increased mitochondrial mass [39]. In PLC, there is a metabolic shift from mitochondrial OXPHOS to glycolysis accompanied by a decrease in mitochondrial mass, which makes PLC more reliant on the glycolysis pathway than on mitochondrial metabolism [53, 77]. These findings indicate that liver adapts to stress conditions by acquiring a mitochondrial flexibility at early stages of NAFLD, which is subsequently lost as a liver tumor progresses. Therefore, increasing mitochondrial biogenesis while preserving intact antioxidant defenses would benefit future treatment for PLC.
Figure 2. Principal metabolic alterations during hepatocarcinogenesis.
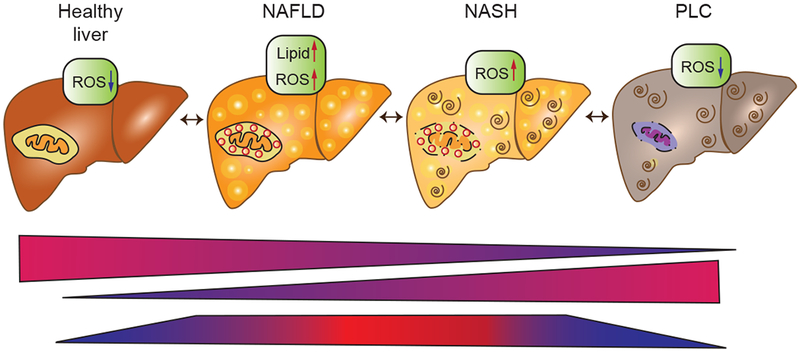
The metabolic energy consumption relies primarily on mitochondrial OXPHOS in a healthy liver. Once the liver starts to subsequent damage induced by oxidative stress, and lipids accumulation, which may lead to NAFLD. In the early stages of NASH, mitochondrial adaptations are lost, which includes a low mitochondrial respiratory function. Finally, a metabolic shift from mitochondrial OXPHOS to glycolysis accompanied by a decrease in mitochondrial mass occurs during malignant transformation to PLC. Abbreviation: NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PLC, Primary liver cancer.
Mitochondrial biogenesis is a highly regulated multi-step process involving the coordinated transcription and translation of both mitochondria and transcripts of nuclear origin [60]. Mitochondrial biogenesis is known to be regulated by peroxisome proliferator-activated receptor-γ coactivators (PGC1s), adenosine monophosphate (AMP)-activated protein kinase (AMPK), eNOS, NRFs, SIRT1, SIRT3, and mitochondrial transcription factor A (TFAM). The PGC1a, including PGC1a and PGC1p, is a family of PGC-1 transcriptional coactivators that are master regulators of mitochondrial biogenesis as well as involve different liver cellular energy metabolic processes including OXPHOS and fatty acid β-oxidation and gluconeogenesis [64]. PGC1s co-activate a variety of transcription factors and nuclear receptors, such as the peroxisome proliferator-activated receptors (PPARs), estrogen-related receptors (ERR), NRF1 and NRF2, to activate expression of genes implicated in mitochondrial metabolism [56, 64]. PGC1a and PGC1p have distinct roles in the regulation of liver metabolism depending on the different environmental conditions. During energy starvation, PGC-1α could upregulate gluconeogenesis, which contributes to de novo glucose synthesis in the liver, to adapt to the fasting environment. Conversely, PGC1p could upregulate the hepatic de novo lipogenesis by co-activation of LXRα and the sterol response element binding protein (SREBP1) due to the reduction of saturated fatty acid consumption [48, 60]. During PLC, cancer cells may upregulate glycolysis by negative regulation of gluconeogenesis, thereby favoring cell survival in the hypoxic and nutrient-deprived tumor microenvironment that characterizes early stages of tumorigenesis [26, 85]. Indeed, the expression levels of PGC1a and its target genes involved in mitochondrial metabolism and gluconeogenesis are significantly decreased in late stages of PLC and in a mouse model of HCC [37, 80]. Interestingly, liver CSCs tend to have lower levels of mitochondrial metabolism compared with non-Liver CSCs through increased expression of acetylated PGC-1α [32], which leads to its inactivation and decreases mitochondrial biogenesis [2]. However, other studies demonstrate that cancer cells shift to more efficient energy production to support their migrating phenotype by PGC1a-mediated promotion of mitochondrial biogenesis and respiration, during the metastatic cascade in PLC [42, 47]. Thus, given that the role of PGC1a in regulating mitochondrial biogenesis and gluconeogenesis, it can be hypothesized that altered mitochondrial biogenesis could be related to the liver CSC microenvironment.
Mitochondrial mitophagy in liver CSCs
Mitophagy is a selective autophagic process to eliminatie dysfunctional mitochondria. This process is important for the maintenance of overall mitochondrial integrity to defective mitochondria following damage or stress [6]. Moreover, mitophagy/autophagy regulates cellular homeostasis and prevents cell death by keeping mitochondrial bioenergetics and decreasing oxidative stress and its alteration has been linked to various liver diseases including PLC [11, 45, 84]. Increasing evidence supports that dysregulation of mitophagy could be an etiological key factor in tumorigenesis of PLC [11]. In addition, an imbalance in the dysregulation of mitophagy could induce mitochondrial degradation process, which in turn results in alteration of cellular homeostasis and promotes tumor initiation and progression. Mitophagy/autophagy acts as a double-edged sword in the tumorigenesis, depending on the different environmental conditions [76, 91]. Whereas tumorigenesis of PLC relies on suppression of mitophagy, tumor progression possiblly relies on the bearing of functional mitophagy. Parkin RBR E3 Ubiquitin Protein Ligas (PARK2) is an E3 ubiquitin ligase that is responsible for ubiquitination of damaged mitochondria [87]. Homozygous mutations in PARK2 are commonly found in various tumors, which lead to repression of mitophagy, as well as an accumulation of dysfunctional mitochondria [11]. In addition, knockout of the PARK2 gene in mice develops spontaneous liver tumors [21]. Moreover, hepatocyte-specific knockout of Becn1, a gene essential for autophagy, can develop spontaneous tumors, while knockout of other important autophagic genes such as Atg5 or Atg7 can develop only benign hepatic tumors [75]. Collectively, these results suggest that an ability to increase mitochondrial mitophagy may be an effective strategy for treatment to prevent PLC.
Interestingly, tumors cells isolated from atg5-knockout mice have a significantly reduced population of CD133 and ITGA6/CD49f double-positive cells [44]. Recent studies found that mitophagy/autophagy plays an important role in the maintenance of stemness properties in liver CSCs. Liu et al. found that when mitophagy is inhibited, serine-392 of p53 is phosphorylated to modulate p53 mitochondrial translocation, which may lead to its activation and translocation into the nucleus, thereby binding to the NANOG promoter to inhibit the expression of NANOG, which results in the decrease of stemness and self-renewal ability of liver CSCs [49]. However, when mitophagy/autophagy is enhanced, p53 is recruited to mitochondria and subsequently is removed through mitophagy/autophagy mechanism [49]. Thus, mitophagy plays a critical role in the quality control of mitochondria, it may be possible to target tumor suppressor to mediate the stemness of liver CSCs [44]. Together, it may be possible to target mitophagy to deplete liver CSCs, which is dependent on wild-type p53 status.
Mitochondrial induced cell death in liver CSC homeostasis
CSCs is resistance to treatment and is associated with other mitochondria-related function, such as impaired cell death [17, 25]. In fact, several studies suggest that CSC-resistant features can be impaired by targeting components of the anti-cell death machinery [68]. Therefore, unique metabolism in liver CSCs can be associated with abnormalities in mitochondrial function, which affect cell death programs.
Apoptotic death is an energy-dependent cell death program whose regulatory pathways are important in CSCs. Apoptotic processes are regulated via two signaling pathways: extrinsic pathways (death receptor pathway) and the intrinsic pathway (mitochondrial pathway) [29]. Moreover, apoptosis is also regulated by the inhibitors of apoptosis proteins (IAPs), such as survivin, which can inhibit the initial activation of caspases-8 and caspases-10. Extracellular stimuli, including cytokines, growth factors, nitric oxide or toxins, may trigger the extrinsic apoptotic pathway through the binding of extracellular death receptors to cell surface receptors, such as Fas ligand (FasL), nerve growth factor receptor (NGFR), TNF-α and TNF-related apoptosis-inducing ligand (TRAIL) receptors. The intrinsic pathway of apoptosis, also known as the mitochondria-mediated death pathway, refers to cell death by a variety of mitochondrial stress signals, which lead to the activation of BH3-only proapoptotic B-cell leukemia/lymphoma 2 (Bcl-2) family protein. The BCL-2 family proteins could inhibit (anti-apoptotic members) or induce (pro-apoptotic members) mitochondrial outer membrane permeabilization (MOMP) that releases the apoptosis-triggering factors, such as cytochrome c and Smac, from the mitochondrial intermembrane space into the cytoplasm, resulting in activated caspase-induced apoptosis. Mitochondrial dysfunction could trigger intrinsic apoptotic pathways, which is associated with overexpression of BCL-2 family protein in cancer [5]. Both intrinsic and extrinsic apoptosis pathways may be linked to therapy evasion of liver CSCs. For instance, the Bcl-2 family proteins consist of anti-apoptotic proteins (Bcl-2, Bcl-XL, and Mcl-1) and pro-apoptotic proteins (Bax, Bak, Bid et. al), which regulate the intrinsic pathway of apoptosis in liver CSCs. Recent studies show that antiapoptotic genes (BCL-2, and BCL-xl), as well as IAP family of proteins (survivin) are up-regulated in liver CSCs [36], which is correlated with enhanced chemotherapy resistance to sorafenib [23]. Furthermore, TRAIL is a promising anticancer agent, which preferentially kills tumor cells without significant cytotoxicity toward normal cells [58]. It can bind to the death receptors TRAIL-RI (DR4) and/or TRAIL-RII (DR5) and activate caspase-8 to promote extrinsic apoptotic pathway. In addition, liver CSCs show up-regulation of DR4 and DR5 [46], which may lead to cancer cells and CSCs to have a differential sensitivity to TRAIL apoptosis induction. Moreover, liver CSCs treated with a recombinant human soluble TRAIL show increased cell death significantly [46]. These data suggest that one of the reasons for the failure of existing therapies is that CSCs may have the ability to evade cell death through anti-apoptotic mechanisms.
Interestingly, increased cell death occurs in a majority of human liver diseases, which could serve as a sensitive parameter for the detection of chronic and acute liver diseases due to toxic, viral, metabolic, or autoimmune origin-related insults [51]. Clinical data and animal models suggest that mitochondria is a crucial organelle in the trigger of liver disease progression characterized by increasing hepatocyte death, which is correlated with the subsequent development of inflammation, fibrosis, cirrhosis and PLC [20, 55]. Distinct modes of cell death including apoptosis (programmed cell death), necrosis (unprogrammed cell death, in response to injury) and necroptosis (programmed form of necrotic cell death) trigger specific cell death responses and the development of liver disease, such as I/ R injury, NASH, PLC [51]. Hepatocyte necrosis is a largely unregulated consequence of environmental stress, characterized by mitochondrial dysfunction and consequent rapid ATP depletion. This consequence results in rapid swelling of cells and ultimately cellular rupture, which then elicits significant inflammatory responses. Sakurai et al. found hepatocyte necrosis acts as a crucial mediator of carcinogen-induced HCC development [69]. The tumor-promoting effects of apoptosis of hepatocyte have been demonstrated by the antiapoptotic proteins Mcl-1 or Bcl-xl liver knockout model, which shows an increased rate of apoptosis of hepatocyte and an increased spontaneous development of HCC [83]. Interestingly, tumor development could be inhibited by hepatocyte-specific deletion of pro-apoptotic proteins Bak, which provides a direct connection between apoptosis and tumor development of PLC [30]. In contrast to the role of tumor-promoting effects of cell death in normal liver tissue, cell death in liver cancer represents the role of tumor suppressing effects. Accordingly, liver tumor, especially CSCs, may have undergone a selection process, such as mitochondrial dysfunction, which enables cells to evade apoptosis. Therefore, one needs to carefully distinguish the role of cell death between normal liver tissue and liver tumor for treatment of liver diseases.
Mitochondrial control of redox balance in liver CSC homeostasis
Mitochondria are the major contributors to the production of reactive oxygen species (ROS) [31]. ROS are chemically reactive molecules that have been implicated as a major contributor to stress and diseases, including cancer [33]. It is evident that intracellular ROS in redox homeostasis also play prominent roles in normal stem cells and CSCs including maintenance of stem cell self-renewal, differentiation and survival. To maintain the steady state of cellular conditions, ROS can be scavenged by antioxidant enzymes, such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (Gpx) or peroxiredoxins (Prx). The superoxide dismutase converts superoxide anion radicals into hydrogen peroxide, then be further detoxified into the water by catalase [81]. Indeed, both stem cells and CSCs possess critical mechanisms by which to cope with the mitochondrial ROS accumulation through elevation of antioxidant defenses. Thus these defense mechanisms act as an important redox regulator on self-renewal and stemness. For instance, hematopoietic stem cells maintain a low intracellular level of ROS to acquire a quiescent state, whereas a high level of ROS confers potent capacity for cell proliferation and differentiation that abolishes self-renewal of stem cell [35]. Proliferative neural stem cells (NSCs) display a high ROS status, which could act as the second messengers [41]. In addition, ROS can cause mitochondrial DNA damage, which is associated with the development of PLC [62]. The accumulation of mitochondrial DNA mutations in PLC suggests its contribution to tumorigenesis [62]. Further, ROS production and ChREBP activation trigger advanced glycation end products (AGEs)–mediated cell proliferation in liver cancers [14]. Despite this, it was suggested that maintaining a low level of intracellular ROS within CSCs may be a crucial property of the self-renewal process. Haraguchi et al. have reported that CD13+ liver CSCs are relatively resistant to chemo/radiation therapy, perhaps due to the low intracellular ROS, including lower levels of mitochondrial ROS (MitoSOX) [27, 38]. The enriched CD13+ liver CSCs might actually be a “G0-like” subpopulation by keeping a lower level of ROS to survive from chemotherapy [27]. Pharmacologic depletion of CD13 expression in liver CSCs by ubenimex treatment inhibits stemness properties and reduces tumorigenicity [27]. Interestingly, CD13 inhibition exhibits high ROS level and proliferation status [27]. On the other hand, CD13+/N-cadherin+ cells display high ROS level in liver cancer, suggesting that the EMT process may be associated with an increase of ROS level [38]. Similarly, CD133+ liver CSCs are also more radioresistance that is associated with low ROS levels [63]. In addition, EpCAM+ liver CSCs have been shown to contain lower ROS levels to maintain long-term self-renewal and survival. However, Disulfiram (DSF), a CSC marker aldehyde dehydrogenase inhibitor, suppresses self-renewal capability of liver CSCs mainly through increases in mitochondrial ROS production rather than reducing the scavenging of ROS [16]. Further, NANOG decreases OXPHOS function to prevent mitochondrial ROS production, which maintains stemness characteristics of liver CSCs [12]. Liver CSC treated with the mitochondrial ROS inducer, Paraquat, represses the self-renewal capacity, indicating that induction of ROS inhibits self-renewal ability of liver CSCs [12, 54]. Additionally, we recently report that, in solid tumor, cancer cells harboring low ROS display enhanced stemness whereas cancer cells containing higher ROS are more proliferative [9]. Given these findings, CSCs may be partially differentiated due to the imbalance of redox homeostasis in the light of tumor microenvironment changes, which leads to heterogeneous tumor cell population including CSCs and non-CSCs.
Conclusions and future perspectives
Most of the anticancer drugs such as cisplatin and 5-fluorouracil preferentially kill proliferating non-CSC tumor cells that, initially, causes the shrinkage of tumor size. However, due to mostly unharmed CSC populations, prolonged treatment with these drugs results in enriched CSCs, consequently contributing to therapy resistance. In this view, it is crucial to understand mitochondrial metabolism in the context of chemoresistance contributed by liver CSCs, with the purpose of improving the development of novel therapeutic strategies for targeting liver CSCs.
Accumulating evidence supports that liver CSCs preferentially relying on the glycolytic pathway and presents low or absent rates of OXPHOS. Due to the presence of unique metabolic activities of CSCs, drugs that suppress glycolysis have been studied as potential anticancer agents. Consistently, 2-deoxy-D-glucose (2-DG), a glucose analog by competitively inhibits glucose-uptake, has been found to induce apoptosis of liver CSCs in combination with Sorafenib [73]. ADI-PEG20 have been found to inhibit the Warburg Effect, which upregulates OXPHOS and targeting glutamine and glycolysis metabolism [40]. A randomized phase II study shows a beneficial effect of ADI-PEG20 in stabilizing the progression of pretreated advanced HCC in an Asian population [92]. Furthermore, the use of ADI-PEG20, in combination with other molecularly targeted or cytotoxic agents, should be investigated, which may improve the success of the therapeutic effect of PLC [28]. On the other hand, Metformin, which interferes with OXPHOS by repressing NADH-coenzyme Q oxidoreductase (complex I), has been shown to enhance tumor aggressiveness and resistance to Sorafenib treatment in diabetic patients with advanced HCC [8]. In addition, studies suggest that Sorafenib enhances glycolysis of liver CSCs [73]. Thus, co-treatment with glycolytic inhibitors or upregulation of OXPHOS to target CSCs in combination with chemotherapy might be more effective in the future treatment of PLC. Interestingly, Griffin et al. conducted a study with a large cohort of patients with diabetes [59]. They found a strong association between the use of metformin and a reduction of liver cancer. This study may support the hypothesis that enhancing mitochondrial function may be an effective strategy for treatment to prevent PLC.
Moreover, since mitochondrial function and liver CSC features seem to be closely linked, drugs that regulate mitochondrial function may be worth exploring as novel therapies. XIAP is the most effective inhibitor of caspases and has been identified as a major repressor of mitochondrial-mediated apoptosis [15]. AEG35156 is an antisense oligonucleotide to promote apoptosis by inhibiting the apoptosis protein XIAP. A randomized phase II study show that AEG35156 in combination with Sorafenib has a better effect in progression-free survival (PFS) of advanced HCC compared to sorafenib alone [43]. Furthermore, Vitamin C increased intracellular ROS in liver CSCs, leading to cell cycle arrest and apoptosis. In addition, intravenous Vitamin C use is associated with improved disease-free survival in HCC patients [52]. Together, the vision of PLC as a metabolic disease strengthens the clinical significance of mitochondria function, particularly the relevance of liver CSCs for cancer initiation, progression, recurrence, and therapy. Although the Warburg effect is linked to enhanced glycolysis, mitochondrial dysfunction, as the concept of cancer metabolism for the benefit survival of PLC, argues the importance of mitochondrial functions in promoting tumor progression. Furthermore, revealation of mitochondrial metabolism in chemoresistance of liver CSCs may have important therapeutic implications in the future.
Acknowledgements
This work was supported by the intramural research program of the Center for Cancer Research, National Cancer Institute of the United States, and by the Dragon-Gate Program of the Taiwan Ministry of Science and Technology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ahmed N, Escalona R, Leung D et al. (2018) Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Seminars in Cancer Biology 53:265–281 [DOI] [PubMed] [Google Scholar]
- 2.Audano M, Ferrari A, Fiorino E et al. (2014) Energizing Genetics and Epigenetics: Role in the Regulation of Mitochondrial Function. Curr Genomics 15:436–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auger C, Alhasawi A, Contavadoo M et al. (2015) Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front Cell Dev Biol 3:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batlle E, Clevers H (2017) Cancer stem cells revisited. Nat Med 23:1124–1134 [DOI] [PubMed] [Google Scholar]
- 5.Boland ML, Chourasia AH, Macleod KF (2013) Mitochondrial dysfunction in cancer. Front Oncol 3:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bordi M, Nazio F, Campello S (2017) The Close Interconnection between Mitochondrial Dynamics and Mitophagy in Cancer. Front Oncol 7:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buzzetti E, Pinzani M, Tsochatzis EA (2016) The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65:1038–1048 [DOI] [PubMed] [Google Scholar]
- 8.Casadei Gardini A, Marisi G, Scarpi E et al. (2015) Effects of metformin on clinical outcome in diabetic patients with advanced HCC receiving sorafenib. Expert Opin Pharmacother 16:2719–2725 [DOI] [PubMed] [Google Scholar]
- 9.Chang CW, Chen YS, Chou SH et al. (2014) Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res 74:6291–6305 [DOI] [PubMed] [Google Scholar]
- 10.Chang CW, Chen YS, Tsay YG et al. (2018) ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis 9:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang JY, Yi HS, Kim HW et al. (2017) Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim Biophys Acta Bioenerg 1858:633–640 [DOI] [PubMed] [Google Scholar]
- 12.Chen CL, Uthaya Kumar DB, Punj V et al. (2016) NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab 23:206–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CT, Hsu Sh, Wei YH (2012) Mitochondrial bioenergetic function and metabolic plasticity in stem cell differentiation and cellular reprogramming. Biochim Biophys Acta 1820:571–576 [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Li Y, Zhu Y et al. (2017) Advanced glycation end products promote ChREBP expression and cell proliferation in liver cancer cells by increasing reactive oxygen species. Medicine (Baltimore) 96:e7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung HH, Lacasse EC, Korneluk RG (2006) X-linked inhibitor of apoptosis antagonism: strategies in cancer treatment. Clin Cancer Res 12:3238–3242 [DOI] [PubMed] [Google Scholar]
- 16.Chiba T, Suzuki E, Yuki K et al. (2014) Disulfiram eradicates tumor-initiating hepatocellular carcinoma cells in ROS-p38 MAPK pathway-dependent and - independent manners. PLoS One 9:e84807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colak S, Zimberlin CD, Fessler E et al. (2014) Decreased mitochondrial priming determines chemoresistance of colon cancer stem cells. Cell Death Differ 21:1170–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Francesco EM, Sotgia F, Lisanti MP (2018) Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J 475:1611–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Lope CR, Tremosini S, Forner A et al. (2012) Management of HCC. J Hepatol 56 Suppl 1:S75–87 [DOI] [PubMed] [Google Scholar]
- 20.Degli Esposti D, Hamelin J, Bosselut N et al. (2012) Mitochondrial roles and cytoprotection in chronic liver injury. Biochem Res Int 2012:387626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujiwara M, Marusawa H, Wang HQ et al. (2008) Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27:6002–6011 [DOI] [PubMed] [Google Scholar]
- 22.Galluzzi L, Kepp O, Kroemer G (2012) Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 13:780–788 [DOI] [PubMed] [Google Scholar]
- 23.Galuppo R, Maynard E, Shah M et al. (2014) Synergistic inhibition of HCC and liver cancer stem cell proliferation by targeting RAS/RAF/MAPK and WNT/beta-catenin pathways. Anticancer Res 34:1709–1713 [PMC free article] [PubMed] [Google Scholar]
- 24.Ganapathy-Kanniappan S (2017) Linking tumor glycolysis and immune evasion in cancer: Emerging concepts and therapeutic opportunities. Biochim Biophys Acta Rev Cancer 1868:212–220 [DOI] [PubMed] [Google Scholar]
- 25.Guerra F, Arbini AA, Moro L (2017) Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg 1858:686–699 [DOI] [PubMed] [Google Scholar]
- 26.Gwak GY, Yoon JH, Kim KM et al. (2005) Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J Hepatol 42:358–364 [DOI] [PubMed] [Google Scholar]
- 27.Haraguchi N, Ishii H, Mimori K et al. (2010) CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest 120:3326–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harding JJ, Do RK, Dika IE et al. (2018) A phase 1 study of ADI-PEG 20 and modified FOLFOX6 in patients with advanced hepatocellular carcinoma and other gastrointestinal malignancies. Cancer Chemother Pharmacol 82:429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He YC, Zhou FL, Shen Y et al. (2014) Apoptotic death of cancer stem cells for cancer therapy. Int J Mol Sci 15:8335–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hikita H, Kodama T, Shimizu S et al. (2012) Bak deficiency inhibits liver carcinogenesis: a causal link between apoptosis and carcinogenesis. J Hepatol 57:92–100 [DOI] [PubMed] [Google Scholar]
- 31.Holmstrom KM, Finkel T (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature reviews. Molecular cell biology 15:411–421 [DOI] [PubMed] [Google Scholar]
- 32.Hur W, Ryu JY, Kim HU et al. (2017) Systems approach to characterize the metabolism of liver cancer stem cells expressing CD133. Sci Rep 7:45557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hussain SP, Hofseth LJ, Harris CC (2003) Radical causes of cancer. Nat Rev Cancer 3:276–285 [DOI] [PubMed] [Google Scholar]
- 34.Islami F, Miller KD, Siegel RL et al. (2017) Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA Cancer J Clin 67:273–289 [DOI] [PubMed] [Google Scholar]
- 35.Jang YY, Sharkis SJ (2007) A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 110:3056–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang N, Chen W, Zhang JW et al. (2015) Aberrantly regulated dysadherin and B-cell lymphoma 2/B-cell lymphoma 2-associated X enhances tumorigenesis and DNA targeting drug resistance of liver cancer stem cells. Mol Med Rep 12:7239–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin J, Iakova P, Jiang Y et al. (2013) Transcriptional and translational regulation of C/EBPbeta-HDAC1 protein complexes controls different levels of p53, SIRT1, and PGC1alpha proteins at the early and late stages of liver cancer. J Biol Chem 288:14451–14462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HM, Haraguchi N, Ishii H et al. (2012) Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial-mesenchymal transition-like phenomenon. Ann Surg Oncol 19 Suppl 3:S539–548 [DOI] [PubMed] [Google Scholar]
- 39.Koliaki C, Szendroedi J, Kaul K et al. (2015) Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 21:739–746 [DOI] [PubMed] [Google Scholar]
- 40.Kremer JC, Prudner BC, Lange SES et al. (2017) Arginine Deprivation Inhibits the Warburg Effect and Upregulates Glutamine Anaplerosis and Serine Biosynthesis in ASS1-Deficient Cancers. Cell Rep 18:991–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Belle JE, Orozco NM, Paucar AA et al. (2011) Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell stem cell 8:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lebleu VS, O’connell JT, Gonzalez Herrera KN et al. (2014) PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16:992–1003, 1001–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee FA, Zee BC, Cheung FY et al. (2016) Randomized Phase II Study of the X-linked Inhibitor of Apoptosis (XIAP) Antisense AEG35156 in Combination With Sorafenib in Patients With Advanced Hepatocellular Carcinoma (HCC). Am J Clin Oncol 39:609–613 [DOI] [PubMed] [Google Scholar]
- 44.Lee J, Liu K, Stiles B et al. (2018) Mitophagy and hepatic cancer stem cells. Autophagy 14:715–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee S, Kim JS (2014) Mitophagy: therapeutic potentials for liver disease and beyond. Toxicol Res 30:243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee SH, Hyun SK, Kim HB et al. (2016) Potential Role of CD133 Expression in the Susceptibility of Human Liver Cancer Stem-Like Cells to TRAIL. Oncol Res 24:495–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Xu S, Li J et al. (2016) SIRT1 facilitates hepatocellular carcinoma metastasis by promoting PGC-1alpha-mediated mitochondrial biogenesis. Oncotarget 7:29255–29274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin J, Yang R, Tarr PT et al. (2005) Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 120:261–273 [DOI] [PubMed] [Google Scholar]
- 49.Liu K, Lee J, Kim JY et al. (2017) Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol Cell 68:281–292 e285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loureiro R, Mesquita KA, Magalhaes-Novais S et al. (2017) Mitochondrial biology in cancer stem cells. Semin Cancer Biol 47:18–28 [DOI] [PubMed] [Google Scholar]
- 51.Luedde T, Kaplowitz N, Schwabe RF (2014) Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 147:765–783 e764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lv H, Wang C, Fang T et al. (2018) Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis Oncol 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma C, Kesarwala AH, Eggert T et al. (2016) NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531:253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Machida K (2017) Pluripotency transcription factors and Metabolic reprogramming of mitochondria in tumor-initiating stem-like cells. Antioxid Redox Signal [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malhi H, Guicciardi ME, Gores GJ (2010) Hepatocyte death: a clear and present danger. Physiol Rev 90:1165–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mansouri A, Gattolliat CH, Asselah T (2018) Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 155:629–647 [DOI] [PubMed] [Google Scholar]
- 57.Mcglynn KA, Petrick JL, London WT (2015) Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis 19:223–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meurette O, Fontaine A, Rebillard A et al. (2006) Cytotoxicity of TRAIL/anticancer drug combinations in human normal cells. Ann N Y Acad Sci 1090:209–216 [DOI] [PubMed] [Google Scholar]
- 59.Murff HJ, Roumie CL, Greevy RA et al. (2018) Metformin use and incidence cancer risk: evidence for a selective protective effect against liver cancer. Cancer Cause Control 29:823–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nassir F, Ibdah JA (2014) Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci 15:8713–8742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nio K, Yamashita T, Kaneko S (2017) The evolving concept of liver cancer stem cells. Mol Cancer 16:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishikawa M, Nishiguchi S, Shiomi S et al. (2001) Somatic mutation of mitochondrial DNA in cancerous and noncancerous liver tissue in individuals with hepatocellular carcinoma. Cancer Res 61:1843–1845 [PubMed] [Google Scholar]
- 63.Piao LS, Hur W, Kim TK et al. (2012) CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett 315:129–137 [DOI] [PubMed] [Google Scholar]
- 64.Piccinin E, Villani G, Moschetta A (2018) Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nat Rev Gastroenterol Hepatol [DOI] [PubMed] [Google Scholar]
- 65.Plaks V, Kong N, Werb Z (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16:225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prager BC, Xie Q, Bao S et al. (2019) Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 24:41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiu L, Li H, Fu S et al. (2018) Surface markers of liver cancer stem cells and innovative targeted-therapy strategies for HCC. Oncol Lett 15:2039–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Safa AR (2016) Resistance to Cell Death and Its Modulation in Cancer Stem Cells. Crit Rev Oncog 21:203–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sakurai T, He G, Matsuzawa A et al. (2008) Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 14:156–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sell S, Leffert HL (2008) Liver cancer stem cells. J Clin Oncol 26:2800–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sia D, Villanueva A, Friedman SL et al. (2017) Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 152:745–761 [DOI] [PubMed] [Google Scholar]
- 72.Song K, Kwon H, Han C et al. (2015) Active glycolytic metabolism in CD133(+) hepatocellular cancer stem cells: regulation by MIR-122. Oncotarget 6:40822–40835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tesori V, Piscaglia AC, Samengo D et al. (2015) The multikinase inhibitor Sorafenib enhances glycolysis and synergizes with glycolysis blockade for cancer cell killing. Sci Rep 5:9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thanee M, Loilome W, Techasen A et al. (2016) CD44 variant-dependent redox status regulation in liver fluke-associated cholangiocarcinoma: A target for cholangiocarcinoma treatment. Cancer Sci 107:991–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tian Y, Kuo CF, Sir D et al. (2015) Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ 22:1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Um JH, Yun J (2017) Emerging role of mitophagy in human diseases and physiology. BMB Rep 50:299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vivekanandan P, Daniel H, Yeh MM et al. (2010) Mitochondrial mutations in hepatocellular carcinomas and fibrolamellar carcinomas. Mod Pathol 23:790–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vyas S, Zaganjor E, Haigis MC (2016) Mitochondria and Cancer. Cell 166:555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wanet A, Remacle N, Najar M et al. (2014) Mitochondrial remodeling in hepatic differentiation and dedifferentiation. Int J Biochem Cell Biol 54:174–185 [DOI] [PubMed] [Google Scholar]
- 80.Wang B, Hsu SH, Frankel W et al. (2012) Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology 56:186–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang K, Zhang T, Dong Q et al. (2013) Redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell death & disease 4:e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang XW, Thorgeirsson SS (2014) The biological and clinical challenge of liver cancer heterogeneity. Hepat Oncol 1:349–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weber A, Boger R, Vick B et al. (2010) Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 51:1226–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Williams JA, Ni HM, Ding Y et al. (2015) Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol 309:G324–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilson GK, Tennant DA, Mckeating JA (2014) Hypoxia inducible factors in liver disease and hepatocellular carcinoma: current understanding and future directions. J Hepatol 61:1397–1406 [DOI] [PubMed] [Google Scholar]
- 86.Wong TL, Che N, Ma S (2017) Reprogramming of central carbon metabolism in cancer stem cells. Biochim Biophys Acta Mol Basis Dis 1863:1728–1738 [DOI] [PubMed] [Google Scholar]
- 87.Wong YC, Holzbaur EL (2015) Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 11:422–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu X, Duan S, Yi F et al. (2013) Mitochondrial regulation in pluripotent stem cells. Cell Metab 18:325–332 [DOI] [PubMed] [Google Scholar]
- 89.Yamashita T, Ji J, Budhu A et al. (2009) EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 136:1012–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamashita T, Wang XW (2013) Cancer stem cells in the development of liver cancer. J Clin Invest 123:1911–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yan C, Li TS (2018) Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res 38:617–621 [DOI] [PubMed] [Google Scholar]
- 92.Yang TS, Lu SN, Chao Y et al. (2010) A randomised phase II study of pegylated arginine deiminase (ADI-PEG 20) in Asian advanced hepatocellular carcinoma patients. Br J Cancer 103:954–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu Y, Liu H, Ikeda Y et al. (2012) Hepatocyte-like cells differentiated from human induced pluripotent stem cells: relevance to cellular therapies. Stem Cell Res 9:196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zheng J (2012) Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett 4:1151–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]