

Abstract
Idiopathic pulmonary fibrosis (IPF) is characterised by unpredictable disease course and poor survival. After the introduction of novel antifibrotic drugs, the prognosis of patients with IPF is probably changing.
FinnishIPF, a nationwide registry of carefully characterised patients, was initiated in Finland in 2011. For the data analysis, we included 453 incident IPF patients diagnosed during 2011–2015. In this study, we describe the demographics and prognosis of these real-life patients.
The median overall survival time of registered IPF patients was 4.5 years. The transplant-free survival at 1, 2, 3, 4 and 5 years was 95%, 83%, 70%, 58% and 45%, respectively. Smoking did not have any effect on survival. 117 (26%) patients received pirfenidone or nintedanib. Patients who received ≥6 months of treatment had better survival compared with those who did not receive treatment but this difference disappeared after age adjustment. The transplantation rate was 3%.
Although IPF is diagnosed in Finland at a older age, the prognosis is better than expected due to a relatively well preserved lung function at diagnosis. Age and pulmonary function were identified as independent predictors of survival in the entire IPF patient population as well as in patients who had received antifibrotic treatment.
Short abstract
Survival in idiopathic pulmonary fibrosis is better than previously reported. Independent predictors of survival are age and pulmonary function. Patients treated with antifibrotics have better prognosis but this is explained by younger age. http://bit.ly/2HWwCrH
Introduction
Idiopathic pulmonary fibrosis (IPF) is a disease with an unpredictable disease course and poor prognosis. The usually quoted median survival of 2–3 years is based on studies from the 1990s and the early 2000s [1], before the era of the new American Thoracic Society (ATS)/European Respiratory Society (ERS) classification and the introduction of antifibrotic medication. More recent studies suggest that IPF patients might live longer [2]: mortality rates are relatively low at a stage when lung function is still quite well preserved [3]. Nevertheless, a recent study states that IPF mortality is increasing in Europe, especially in Finland and in the UK [4].
Estimation of prognosis is important when choosing patients for lung transplantation and when considering different therapies. Clinical, biochemical and radiological variables have been proposed for estimating prognosis. Pulmonary function tests (PFTs), i.e. forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DLCO), at diagnosis are most commonly associated with prognosis [5]. However, a decrease in PFTs within 6 months after diagnosis may have a superior predictive power [6].
Two drugs, pirfenidone and nintedanib, have now been approved for the prevention of progression of IPF. Results of clinical trials for up to 1 year with pirfenidone [7] and nintedanib [8] have been published, but the results may not be applicable to all patients with IPF because of strict inclusion and exclusion criteria. These trials were not powered to assess the impact on survival. However, pooled analyses and meta-analyses have suggested a survival benefit [9, 10]. As data on survival of patients with IPF in the 2010s are sparse and the long-term survival benefits of the antifibrotic therapies remain unknown, observational studies in real-life scenarios are required to facilitate our understanding of the natural history of IPF and the efficacy of these medications.
Several IPF registries [11–16] have been founded around the world to enhance clinical research and to enable real-world studies. The Finnish population has had an exceptional history due to geographical and cultural isolation. The unique genetic background offers special opportunities for research and a national IPF registry is therefore justified [17]. This IPF registry (FinnishIPF) was initiated in 2011 [18] and it is currently including patients from all Finnish hospitals that have a pulmonary unit. At present, 700 patients have been included in the registry and to our knowledge, it is one of the most extensive IPF registries in the world in relation to population (5.5 million). Despite the numerous IPF registries being built around the world, the publications are still sparse and more data are needed by clinicians, patients and the medical industry.
We defined the transplant-free survival and explored the effects of demographic factors, physiological impairment, antifibrotic therapies and different GAP stages on survival.
Material and methods
Data for this study were collected from the national FinnishIPF registry. This is a multicentre, observational registry of incident and prevalent IPF patients in Finland. Currently, the registry covers all Finnish pulmonary clinics in 28 different hospitals.
Patient recruitment to the registry is implemented by screening patients from hospital registries with International Classification of Diseases (10th Revision) codes J84.1 and J84.9. Diagnoses of IPF are confirmed by pulmonary specialists and/or multidisciplinary panels. IPF is diagnosed according to ATS/ERS 2011 criteria [1]. This includes at least exclusion of other known causes of interstitial lung disease by taking a detailed history of medication and environmental exposures, usually serological testing to exclude connective tissue disease, measuring spirometry and DLCO, and high-resolution computed tomography. Bronchoalveolar lavage and surgical biopsies are performed in otherwise unclear cases. At both baseline and follow-up visits, all examinations and treatment decisions occur as part of routine clinical care without additional visits or investigations mandated for FinnishIPF.
In the present study, we included patients diagnosed during 2011–2015, resulting in a total of 453 patients. PFTs were performed according to ERS/ATS standards [19] and repeated when clinically indicated. Finnish reference values for PFTs were used [20]. In the Finnish population, the predicted values for lung volumes are higher than those obtained by the 2012 Global Lung Function Initiative predictions for the Caucasian subgroup for FVC [21]. GAP index was calculated based on gender, age and pulmonary function [22]. All these data were gathered from the date of diagnosis. A 6-month treatment cut-off was used to categorise patients as treated or not treated with antifibrotic medicines [23]. The date of death was obtained from the Population Register Centre in the end of April 2017, and the cause and place of death were collected from the death certificates. Lung transplantation was considered as an endpoint equal to death.
Ethical considerations
A positive statement was received from the Helsinki University Central Hospital Ethical Committee and all four other university hospitals in Finland. The National Institute of Health and Welfare has given the authorisation for patient screening from the hospital databases from all Finnish hospitals. All patients have given their written informed consent. Data are stored in a secure electronic database. Only responsible doctors and educated study nurses can access the registry with electronic identity verification.
Data analysis
Data analysis was conducted with SAS System for Windows, version 9.4 (SAS Institute Inc., Cary, NC, USA). The differences in age, FVC % predicted and DLCO % predicted between sexes were tested with two-sample t-tests. Mean ages between smokers, ex-smokers and nonsmokers were compared using a one-way ANOVA with Tukey's test in pairwise comparisons. Transplant-free survival was measured from diagnosis until death or to the date of transplantation. Survival was evaluated using the Kaplan–Meier method and differences in survival curves were evaluated using the log-rank test. The associations of variables with survival were analysed using univariate Cox regression analysis. The significant variables in the univariate analysis were included into a multivariate Cox regression model. GAP stage was excluded from multivariate model due to high correlation with FVC % predicted and DLCO % predicted to avoid collinearity problems.
Results
Altogether 28 centres (five university hospitals, 15 central hospitals and eight regional hospitals) entered data for 453 patients. The baseline characteristics are presented in table 1. 65% of the patients were male. At diagnosis, the mean age of the patients was 73.0 years and the age varied from 44 to 91 years. Men were somewhat younger than women (mean±sd 72.3±8.8 and 74.3±9.3 years, p=0.026). The smokers were younger at diagnosis than ex-smokers and non-smokers (63.9±9.5, 71.8±8.6 and 75.1±8.3 years, respectively; one-way ANOVA, p<0.0001). The mean duration of symptoms before diagnosis was 575±799 days and the median duration was 284 days. 56% of the patients were diagnosed within a year from their first symptoms. At diagnosis, women had better FVC (83.3±18.9% predicted for women, 78.6±17.7% predicted for men; p=0.013) and a trend towards better DLCO (57.9±16.6% predicted for women, 54.5±16.2% predicted for men; t-test, p=0.063). 22 (5%) patients reported a family history of pulmonary fibrosis. 64% of patients had two or more comorbidities; 43% had coronary artery disease, 60% had other cardiovascular diseases, 38% had hypertension, 38% had diabetes, 10% had asthma, 4% had COPD and 5% had sleep apnoea.
TABLE 1.
Baseline characteristics
| Variable | Patients | Mean±sd |
| Age at diagnosis years | 453 (100.0%) | 73.0±9.0 |
| Male | 295 (65.1%) | 72.3±8.8 |
| Female | 158 (34.9%) | 74.3±9.3 |
| BMI kg·m−2 | 355 (78.4%) | 28.4±5.2 |
| <25 | 93 (26.2%) | |
| 25–29.9 | 147 (41.4%) | |
| >30 | 115 (32.4%) | |
| Smoking | 438 (96.7%) | |
| Never smoker | 197 (45.0%) | |
| Ex-smoker | 210 (48.0%) | |
| Current smoker | 31 (7.0%) | |
| DLCO % predicted | 367 (81.0%) | 55.6±16.5 |
| FVC L | 407 (89.8%) | 2.8±0.8 |
| FVC % predicted | 407 (89.8%) | 80.2±18.2 |
| GAP stage | 370 (81.7%) | |
| GAP 1 | 200 (54.1%) | |
| GAP 2 | 151 (40.8%) | |
| GAP 3 | 19 (5.1%) |
BMI: body mass index; DLCO: diffusing capacity of the lung for carbon monoxide; FVC: forced vital capacity.
Altogether, 117 (26%) patients received antifibrotic treatment. The mean age and lung function in the treatment group were lower than those who did not receive medication and men were relatively overrepresented in the medication group (table 2). The mean duration between diagnosis and the start of treatment was 380±416 days. 95 (21%) patients were treated with antifibrotics for >6 months, which we considered to be a significant enough period to change the progression of the disease. 18 patients had stopped the medication and four patients had started treatment so late that they were not able to fulfil the 6 months criteria until April 28, 2017. 82 (18%) patients received pirfenidone and 13 (3%) patients received nintedanib. The mean duration of treatment was 804±416 days. As this was not a clinical trial, drug compliance was not measured. 13 (3%) patients underwent lung transplantation.
TABLE 2.
Characteristics of patients who received antifibrotic treatment and those who did not
| Patients with >6 months treatment | Patients without treatment or <6 months treatment |
Difference between groups p-value |
|
| Patients | 95 | 358 | |
| Age at diagnosis years | 67.4±7.8 | 74.5±8.8 | <0.0001 |
| Sex | 0.003 | ||
| Male | 74 (77.9%) | 221 (61.7%) | |
| Female | 21 (22.1%) | 137 (38.3%) | |
| BMI kg·m−2 | 29.0±5.3 | 28.2±5.1 | 0.207 |
| Smoking | 0.015 | ||
| Never smoker | 31 (33.3%) | 166 (48.1%) | |
| Ex-smoker | 57 (61.3%) | 153 (44.4%) | |
| Current smoker | 5 (5.4%) | 26 (7.5%) | |
| DLCO % predicted | 53.2±14.1 | 56.5±16.9 | 0.111 |
| FVC L | 2.8±0.7 | 2.7±0.9 | 0.338 |
| FVC % predicted | 72.4±13.6 | 82.4±18.7 | <0.0001 |
| GAP stage | 0.006 | ||
| GAP 1 | 33 (40.2%) | 167 (58.0%) | |
| GAP 2 | 46 (56.1%) | 105 (36.5%) | |
| GAP 3 | 3 (3.7%) | 16 (5.6%) | |
| Transplantation | 7 (7.3%) | 6 (1.7%) | |
| Deaths | 25 (26.3%) | 154 (43.0%) | |
| IPF-related deaths | 21 (84.0%) | 93 (60.8%) |
Data are presented as mean±sd unless otherwise stated. BMI: body mass index; DLCO: diffusing capacity of the lung for carbon monoxide; FVC: forced vital capacity; IPF: idiopathic pulmonary fibrosis. Bold indicates statistically significant p-values.
The follow-up time was 3.2±1.4 years after diagnosis, accounting for 1470 person-years. In April 2017, 179 patients had died and 13 underwent a lung transplantation (of whom two died after transplantation) totalling a transplant-free survival of 58% (261 out of 453) (figure 1). The annualised mortality rate was 13.1%. The cause of death was known in 99% of the cases. The underlying cause of death was IPF in 66% of patients. The second most common cause of death was cardiovascular disease (in 16% of patients) and the third most common cause of death was malignant tumour (in 11% of patients) (table 2). 58% of cancer deaths were due to lung cancer. The median survival time was 4.5 years. Transplant-free survival at 1, 2, 3, 4 and 5 years was 95%, 83%, 70%, 58% and 45%, respectively (figure 1). There was no difference in survival between men and women (p=0.795), or between smokers, nonsmokers and ex-smokers (p=0.167). In addition, survival in patients who did not receive antifibrotic medication was similar in both sexes and in the different smoking groups (data not shown).
FIGURE 1.

Kaplan–Meier curve for transplant-free overall survival of FinnishIPF registry patients.
In 81% of patients with adequate data for calculations of GAP stage at baseline, it was a predictor of mortality. In GAP 1 stage, the median survival was not reached; in GAP 2 stage, the median survival was 4.5 years; and in GAP 3 stage, it was 2.2 years (p<0.0001) (figure 2 and table 3). The patients with >6 months antifibrotic treatment had better survival compared with those who did not receive antifibrotic treatment (p=0.035) (figure 3). When comparing patients with medication to patients who fulfilled the lung function criteria for reimbursement (FVC 50–90% predicted) but who had no antifibrotic medication (n=187), the survival difference remained in benefit of antifibrotic therapy (p=0.031) (figure 4).
FIGURE 2.
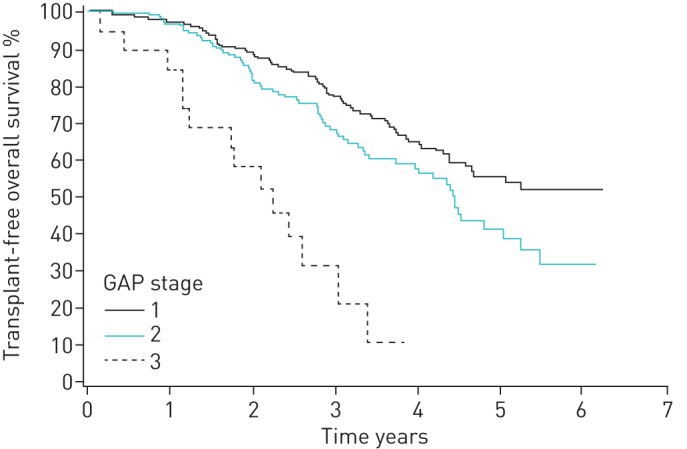
Kaplan–Meier analysis for transplant-free survival according to GAP stages (log-rank, p<0.0001).
TABLE 3.
Transplant-free survival according to GAP stage
| Survival | |||||
| 1 year | 2 years | 3 years | 4 years | 5 years | |
| All GAP stages | 95% | 85% | 72% | 60% | 48% |
| GAP 1 | 96% | 88% | 80% | 66% | 56% |
| GAP 2 | 96% | 85% | 69% | 61% | 42% |
| GAP 3 | 84% | 63% | 28% | 9% | 0% |
FIGURE 3.

Kaplan–Meier survival curve for patients treated with antifibrotics for >6 months versus patients without antifibrotic treatment (log-rank, p=0.035).
FIGURE 4.

Kaplan–Meier survival curve for patients treated with antifibrotics for >6 months versus patients without antifibrotic treatment, among those with forced vital capacity 50–90% of predicted (log-rank, p=0.031).
In the Cox univariate analysis, age and PFTs at diagnosis were predictors of survival (table 4). However, sex and smoking status did not predict survival. In the multivariate Cox regression analysis, age, FVC % predicted and DLCO % predicted were independent factors for survival (table 5). Although patients with antifibrotic medication seemed to have better prognosis than those without, the difference was not significant after age adjustment in the multivariate Cox regression analysis.
TABLE 4.
Univariate Cox regression analysis for mortality
| Variable | Patients | HR (95% CI) | p-value |
| Age per year | 453 | 1.03 (1.01–1.05) | 0.001 |
| Female versus male | 453 | 1.04 (0.78–1.39) | 0.794 |
| BMI per kg·m−2 | 355 | 0.98 (0.95–1.02) | 0.353 |
| Ex/current smoker versus never-smoker | 438 | 1.17 (0.88–1.57) | 0.284 |
| DLCO per % predicted | 367 | 0.96 (0.95–0.97) | <0.0001 |
| FVC per % predicted | 407 | 0.98 (0.97–0.99) | <0.0001 |
| GAP stage | 370 | 1.71 (1.24–2.36) | 0.0010 |
| >6 months antifibrotic treatment | 453 | 0.67 (0.46–0.98) | 0.037 |
HR: hazard ratio; BMI: body mass index; DLCO: diffusing capacity of the lung for carbon monoxide; FVC: forced vital capacity. Bold indicates statistically significant p-values.
TABLE 5.
Multivariate Cox regression analysis for mortality
| Variable | Adjusted HR (95% CI) | p-value |
| Age per year | 1.02 (1.00–1.04) | 0.046 |
| FVC per % predicted | 0.99 (0.97–1.00) | 0.006 |
| DLCO per % predicted | 0.97 (0.96–0.98) | <0.0001 |
| >6 months antifibrotic treatment | 0.67 (0.43–1.05) | 0.078 |
HR: hazard ratio; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide. Bold indicates statistically significant p-values.
Discussion
We present the first data on prognosis, treatment and survival of the incident cases (n=453) in the FinnishIPF registry. The follow-up accounts for 1476 person-years. The diagnosis of IPF took almost 1.5 years from the first symptoms. During 2011–2017, only a quarter of the patients had received antifibrotic medicines and the transplantation rate was very low. The 5-year survival rate was 45%. The independent predictors of survival were age and lung function at diagnosis.
Compared with other registries and clinical trial cohorts [8, 12, 13, 15], our patients were older, and a greater proportion were men and nonsmokers. Using the Finnish lung function reference values, patients in the FinnishIPF registry had a milder disease than patients in earlier registry studies. Patients in our study had better conserved DLCO, which might be explained by the smaller number of smokers or earlier diagnosis. Compared with the Australian and German registries, the GAP stage was also lower in our study [12, 15]. As to comorbidities, in our cohort, coronary artery disease and diabetes were more common while asthma and COPD were less common than in the Australian study [12]. In our study, gastro-oesophageal reflux was not reported. The differences might be explained by differences in data collection.
Although registries are not the appropriate tool to demonstrate the efficacy of treatment, they provide the opportunity to monitor disease progression in patients with and without therapy in the real world [21]. The number of patients that received antifibrotic medicines was 23% in the Australian registry, 44% in the German study and 26% in the present study [12, 15]. In the Australian study, both antifibrotic medicines were available only during clinical trials, patient access programmes or through private purchase, which may give rise to bias. During our study, these medicines became available in Finland and they were reimbursed by the national health insurance. However, in Finland, the prevalence of patients on antifibrotic medication is much lower than, for example, in Sweden, where the medication is 100% reimbursed [24]. In Finland, patients pay an annual fee of €572 to receive antifibrotics. As the antifibrotic medicines have several side-effects, we acknowledge that some patients stop the medication before it can markedly modify the disease progression. In the integrated analysis of clinical studies with pirfenidone, 38.1% patients discontinued pirfenidone due to an adverse event, compared with 14.6% in the placebo group [25]. Previous real-world studies with pirfenidone or nintedanib reported discontinuation rates of 21–29% [26, 27]. One-sixth of our patients stopped the medication within 6 months. The 6 months treatment cut-off to categorise patients as treated or not treated was chosen because among the treated, there were several patients who received antifibrotics only for a short time and inclusion of them into the treatment group would have resulted to underestimation of the effect of medication. A limit of 1 year would have resulted in an even greater difference in survival between the groups but would raise questions about including patients with 1 year treatment in the nontreated group.
In pooled analyses of two multinational phase 3 trials [10] in thoroughly selected patients, the 1-year mortality was 3.5% in patients who received pirfenidone and 6.7% in the placebo group (p=0.01), and pirfenidone reduced the risk of death at 1 year by 48%. In separate studies, survival benefit was found neither with pirfenidone nor nintedanib [7–9]. 1 year is, however, quite a short time to estimate survival in a chronic illness like IPF. In our study, the patients with >6 months of medication had better overall survival compared with those who did not receive antifibrotic medicines, but the difference was not yet seen at 1-year follow-up. This might be explained by the mean delay of 380 days between diagnosis and the beginning of the treatment. Survival was not measured from the start of medication but from the day of diagnosis, as in the comparison group, which surely underestimates the effect of the treatment on survival. The number of lung transplant recipients in our study was low, which might be explained by the older age of the patients.
In studies from 1998–2005 [28], the 5-year survival rate ranged between 20% and 40%. Now, ∼15 years later, the prognosis of Finnish IPF patients seems markedly better. One reason for this might be earlier diagnosis and increased awareness of the disease. Well-preserved lung function implies this, but the 1.5-year time between first symptoms and diagnosis speaks against this. It is also possible that patients with slow progression are overrepresented in our registry, and patients with a rapid onset of symptoms and progressive disease may not be included in our study, which is based on screening from registries and informed consent. In a Finnish single-centre cohort (a registry study without informed consent) of 132 IPF patients from the years 2002–2012, the median survival time was almost equal to our study (3.75 years) [29].
In previous registry studies on IPF [12, 15, 16], both prevalent and incident cases were recruited simultaneously, which may affect survival results. It is known that slowly progressing diseases are overrepresented in prevalent patients and this induces bias in survival analysis. To avoid this survival bias, we excluded the prevalent patients from this study. Despite this, the survival rates at 1, 2, 3, 4 and 5 years of 95%, 83%, 70%, 58% and 45%, respectively, in the present study were quite close to the survival rates in the Australian registry with 1–4-year cumulative mortality ratea of 5%, 24%, 37% and 44%, although they also included prevalent patients.
A recent analysis from the US Medicare database [2] indicated that the median survival time in IPF was 3.8 years, with survival time decreasing sharply based on age at diagnosis. Patients between 66 and 69 years had a median survival of 8 years, compared with 4.5 years in those diagnosed between 75 and 79 years, and 2.5 years in those diagnosed at ≥80 years. In addition, in our study, age was an independent predictor of survival, and it was also an explanatory factor for the survival difference between the groups with and without medication. The effect of sex on survival has been variable; in our study, there was no survival advantage in women. Smoking has been associated with both increased and decreased mortality [30]. In our study, no difference was seen in survival between smokers and nonsmokers. In previous studies, high body mass index (BMI) has been protective against death in IPF [31, 32]. We did not find any correlation between BMI and survival. GAP stage at diagnosis seemed to work reasonably well in predicting survival. As our patients were older than in other studies, the better survival is thus explained by better lung function at diagnosis and lower GAP stage.
There are some limitations to this study. First, we were not able to capture all Finnish IPF patients to the registry. The patients were included only if they gave the informed consent. It is possible that the patients with the most aggressive diseases are missing from the registry. Second, we used Finnish reference values for lung function tests and therefore, the values may not be comparable with other countries. Third, quality of life was not measured and acute exacerbations were not registered. As in all real-life data, missing values may have some effect on the reliability of the results. Fourth, the number of patients who received antifibrotic medicines was low. There were 268 (59%) patients who fulfilled the criterion for reimbursement (FVC 50–90% of predicted) but only 30% of these patients received antifibrotics for >6 months. We do not know the reasons for low numbers of treated patients and this detail warrants further investigation. Antifibrotic medication was not available in Finland until 2013, which may make the inclusion of patients starting from 2011 to the same survival analysis somewhat artificial. However, this group can be viewed as a historical control group. There were 32 patients on medication whose diagnosis was made before 2013. Some of them had been included in a named patient programme starting from 2012. It must also be noted that overall, the median time between the diagnosis and beginning of the treatment was >1 year. This fact further minimises the error due to the inclusion of patients from the start of IPF registry in 2011. As a real-world database, the information available was based on clinical practice and therefore, we cannot make accurate comparisons between treatment groups like in randomised trials.
We also want to highlight the strengths of the study: in Finland, there is a uniform healthcare system where practically all pulmonary physicians work in public health care. IPF is diagnosed and treated exclusively in public healthcare centres or hospitals. We have succeeded in recruiting IPF patients from all of these centres nationwide, resulting in a unique registry of a genetically homogenous population.
Conclusions
Although IPF is diagnosed in Finland at older age, the prognosis is better than expected due to well preserved lung function. The independent predictors of survival were age and PFTs. Younger age and better PFTs were also explanatory factors for better survival in patients treated with antifibrotics.
Acknowledgements
We especially thank all patients for participating in the FinnishIPF registry. We also want to thank coordinator Eva Sutinen (Helsinki University Hospital, Finland), study nurses and pulmonary colleagues in each Finnish hospital for collaboration. Finally, we thank Thomas Dean (University of Turku, Turku, Finland) for proofreading the manuscript.
Footnotes
Conflict of interest: J. Kaunisto reports lecture fees from Boehringer and Roche, and support to attend an international conference from Sanofi Genzyme, outside the submitted work.
Conflict of interest: E-R. Salomaa has nothing to disclose.
Conflict of interest: U. Hodgson has nothing to disclose.
Conflict of interest: R. Kaarteenaho reports grants from the Foundation of the Finnish Anti-Tuberculosis Association, the Research Foundation of the Pulmonary Diseases, Jalmari and Rauha Ahokas Foundation and the Research Foundation of North Finland, lecture fees and an advisory board fee from GlaxoSmithKline, Roche and Boehringer-Ingelheim, and conference travel costs from Orion Pharma, outside the submitted work.
Conflict of interest: H. Kankaanranta reports fees for lectures and consulting, costs for attending an international conference, and a research grant from AstraZeneca; fees for consulting from Chiesi Pharma AB; fees for lectures and consulting, and costs for attending and international conference from Boehringer-Ingelheim; fees for lectures and consulting from Novartis; fees for lectures from Mundipharma; fees for consulting from GlaxoSmithKline; fees for lectures and consulting from Orion Pharma; and fees for consulting from SanofiGenzyme, all outside the submitted work.
Conflict of interest: K. Koli has nothing to disclose.
Conflict of interest: T. Vahlberg has nothing to disclose.
Conflict of interest: M. Myllärniemi has nothing to disclose.
Support statement: We thank the Sigrid Jusélius Foundation, The Academy of Finland, The Research Foundation of the Pulmonary Diseases, Väinö and Laina Kivi Foundation, Jalmari and Rauha Ahokas Foundation, The Paulo Foundation and The Finnish Anti-Tuberculosis Association Foundation.
References
- 1.Raghu G, Collard HR, Egan JJ, et al. . An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raghu G, Chen SY, Yeh WS, et al. . Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med 2014; 2: 566–567. [DOI] [PubMed] [Google Scholar]
- 3.King TE Jr, Albera C, Bradford WZ, et al. . All-cause mortality rate in patients with idiopathic pulmonary fibrosis. Implications for the design and execution of clinical trials. Am J Respir Crit Care Med 2014; 189: 825–831. [DOI] [PubMed] [Google Scholar]
- 4.Marshall DC, Salciccioli JD, Shea BS, et al. . Trends in mortality from idiopathic pulmonary fibrosis in the European Union: an observational study of the WHO mortality database from 2001–2013. Eur Respir J 2018; 51: 1701603. [DOI] [PubMed] [Google Scholar]
- 5.Kim HJ, Perlman D, Tomic R. Natural history of idiopathic pulmonary fibrosis. Respir Med 2015; 109: 661–670. [DOI] [PubMed] [Google Scholar]
- 6.Reichmann WM, Yu YF, Macaulay D, et al. . Change in forced vital capacity and associated subsequent outcomes in patients with newly diagnosed idiopathic pulmonary fibrosis. BMC Pulm Med 2015; 15: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noble PW, Albera C, Bradford WZ, et al. . Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377: 1760–1769. [DOI] [PubMed] [Google Scholar]
- 8.Richeldi L, du Bois RM, Raghu G, et al. . Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. [DOI] [PubMed] [Google Scholar]
- 9.King TE, Bradford WZ, Castro-Bernanrdini S, et al. . A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083–2092. [DOI] [PubMed] [Google Scholar]
- 10.Nathan SD, Albera C, Bradford WZ, et al. . Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5: 33–41. [DOI] [PubMed] [Google Scholar]
- 11.Doubková M, Švancara J, Svoboda M, et al. . EMPIRE Registry, Czech Part: Impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J 2018; 12: 1526–1535. [DOI] [PubMed] [Google Scholar]
- 12.Jo HE, Glaspole I, Grainge C, et al. . Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J 2017; 49: 1601592. [DOI] [PubMed] [Google Scholar]
- 13.Ferrara G, Carlson L, Palm A, et al. . Idiopathic pulmonary fibrosis in Sweden: report from the first year of activity of the Swedish IPF-Registry. Eur Clin Respir J 2016; 3: 31090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Brien EC, Durheim MT, Gamerman V, et al. . Rationale for and design of the Idiopathic Pulmonary Fibrosis-Prospective Outcomes (IPF-PRO) registry. BMJ Open Respir Res 2016; 3: e000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behr J, Kreuter M, Hoeper MM, et al. . Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. Eur Respir J 2015; 46: 186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guenther A, Krauss E, Tello S, et al. . The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res 2018; 19: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax 2002; 57: 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaunisto J, Kelloniemi K, Sutinen E, et al. . Re-evaluation of diagnostic parameters is crucial for obtaining accurate data on idiopathic pulmonary fibrosis. BMC Pulm Med 2015; 15: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller MR, Hankinson J, Brusasco V, et al. . Standardisation of spirometry. Eur Respir J 2005; 26: 319–338. [DOI] [PubMed] [Google Scholar]
- 20.Viljanen AA, Halttunen PK, Kreus KE, et al. . Spirometric studies in non-smoking, healthy adults. Scand J Clin Lab Invest Suppl 1982; 159: 5–20. [PubMed] [Google Scholar]
- 21.Kainu A, Timonen KL, Toikka J, et al. . Reference values of spirometry for Finnish adults. Clin Physiol Funct Imaging 2016; 36: 346–358. [DOI] [PubMed] [Google Scholar]
- 22.Ley B, Ryerson CJ, Vittinghoff E, et al. . A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156: 684–689. [DOI] [PubMed] [Google Scholar]
- 23.Harari S, Caminati A, Poletti V, et al. . A real-life multicenter national study on nintedanib in severe idiopathic pulmonary fibrosis. Respiration 2018; 95: 433–440. [DOI] [PubMed] [Google Scholar]
- 24.Pesonen I, Carlson L, Murgia N, et al. . Delay and inequalities in the treatment of idiopathic pulmonary fibrosis: the case of two Nordic countries. Multidiscip Respir Med 2018; 13: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lancaster L, Albera C, Bradford WZ, et al. . Safety of pirfenidone in patients with idiopathic pulmonary fibrosis: integrated analysis of cumulative data from 5 clinical trials. BMJ Open Respir Res 2016; 3: e000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galli JA, Pandya A, Vega-Olivo M, et al. . Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: tolerability and adverse drug reactions. Respirology 2017; 22: 1171–1178. [DOI] [PubMed] [Google Scholar]
- 27.Hughes G, Toellner H, Morris H, et al. . Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med 2016; 5: E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006; 3: 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kärkkäinen M, Kettunen HP, Nurmi H, et al. . Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir Res 2017; 18: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ley B, Collard HR, King Jr TE. Clinical course and prediction of survival in idiopathic fibrosis. Am J Respir Crit Care Med 2011; 183: 431–440. [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Lee J, Ryu Y, et al. . Clinical predictors of survival in idiopathic pulmonary fibrosis. Tuberc Respir Dis 2012; 73: 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alakhras M, Decker PA, Nadrous HF, et al. . Body mass index and mortality in patients with idiopathic fibrosis. Chest 2007; 131: 1448–1453. [DOI] [PubMed] [Google Scholar]