

Abstract
Motivation
Cell-free nucleic acid (cfNA) sequencing data require improvements to existing fusion detection methods along multiple axes: high depth of sequencing, low allele fractions, short fragment lengths and specialized barcodes, such as unique molecular identifiers.
Results
AF4 was developed to address these challenges. It uses a novel alignment-free kmer-based method to detect candidate fusion fragments with high sensitivity and orders of magnitude faster than existing tools. Candidate fragments are then filtered using a max-cover criterion that significantly reduces spurious matches while retaining authentic fusion fragments. This efficient first stage reduces the data sufficiently that commonly used criteria can process the remaining information, or sophisticated filtering policies that may not scale to the raw reads can be used. AF4 provides both targeted and de novo fusion detection modes. We demonstrate both modes in benchmark simulated and real RNA-seq data as well as clinical and cell-line cfNA data.
Availability and implementation
AF4 is open sourced, licensed under Apache License 2.0, and is available at: https://github.com/grailbio/bio/tree/master/fusion.
1 Introduction
Due to its clinical relevance in cancer, many fusion detection tools have been developed and applied to tissue RNA-seq data (Kumar et al., 2016; Liu et al., 2016). Fusion detection algorithms usually run in two stages: identification of candidate readpairs or fragments (The term ‘fragment’ refers to physical RNA or DNA molecule; whereas the term ‘readpair’ refers to the sequencing result of a fragment. A single fragment may produce multiple readpairs due to duplicates caused by polymerase chain reaction (PCR) amplification. These terms are used interchangeably in this text.) corresponding to candidate gene fusions and application of a list of empirically defined filtering criteria. The first stage typically makes use of a sequence alignment tool (Haas et al., 2017; Jia et al., 2013; Kim and Salzberg, 2011), which can identify discordant and unmapped readpairs. Some methods further assemble reads into longer contigs for more accurate mapping (Chen et al., 2012). This step dominates the runtime and memory usage of the fusion detection process. However, this process can be brittle as readpairs can be misaligned or misidentified as soft-clipping events. Because of this, the aligners must be configured to reduce misclassification due to such alignment artifacts (Haas et al., 2017). The second stage for fusion detection typically involves a variety of empirically defined criteria to eliminate spurious fusion events. Such criteria may include discarding fusions involving homologous genes, requiring a minimum number of supporting fragments, and a constraint on minimum genomic distance between genes. Because these rules are not universally applicable, there is no dominant method: instead, criteria are chosen to best match a particular application (Kumar et al., 2016).
Cell-free nucleic acid (cfNA) data serve as an important biosource for non-invasive cancer diagnoses and monitoring (Donaldson and Park, 2018). Fusion detection for cfNA sequencing data poses distinct challenges compared to tissue sequencing data. To capture the small amount of nucleic acid fragments from tumor cells, cfNA analyses must sequence fragments orders of magnitude more than those for tissue sequencing. This problem prevents many existing fusion callers from running in reasonable time frame (Haas et al., 2017, Fig. 4). Meanwhile, curtailing the loss of fusion-supporting fragments is critical to maintain sufficient evidence. PCR duplicates become prevalent with high sequencing depth, so fragments are typically tagged with unique molecular identifiers (UMIs), and the system must deduplicate them properly. As many cfNA fragments are shorter than the intended sequencing read length, barcode or primer sequences are incorporated into the 3’ end of reads. Reads that incorporate these extra sequences may be discarded by alignment-based methods; the high abundance of primer or barcode kmers may confound alignment-free methods.
Fig. 4.
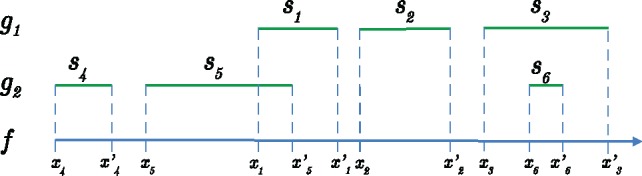
Computing maximum coverage of fragment f for a gene pair (g1, g2). g1 and g2 are two genes inferred to cover regions of f. g1 covers regions , and g2 cover regions . are start and end positions of f for region si
AF4 addresses these challenges with a two staged approach. In the first stage, this tool conservatively identifies discordant readpairs without relying on alignments. It finds kmers shared with candidate genes, ignoring positions of these kmers in the reference transcriptome altogether. This approach improves upon the sensitivity of alignment-based approaches by avoiding the drawbacks highlighted earlier: split reads are not treated as soft-clipping events and discordant readpairs are not misaligned. AF4 then identifies fusion-supporting readpairs by inferring the most likely gene or pair of genes from which the readpair could be derived. This inference is made solving an optimization problem that maximizes the coverage of a readpair by a gene or gene pair. As a result, spurious fusion-supporting fragments are significantly reduced. The first stage of AF4 runs independently for each readpair, scaling linearly with available computing resources. In the second stage, AF4 runs a common set of filtering strategies, including constraints on the number of unique supporting fragments, the number of fusion partners, gene pairs located in close proximity in the reference, gene homology and the complexity of the subsequence supporting the fusion event. Because of the improved specificity of the first stage—AF4 typically retains 0.0001–0.1% of the original data—users can also apply more expensive methods to further improve performance.
AF4 supports two modes of operations: target-based and discovery-based (de novo) fusion detection. In targeted mode, it is given an explicit list of possible fusion events in the form of a COSMIC database file (Tate et al., 2019). In de novo mode, it considers all possible pairs of genes listed in the reference transcripts.
Targeted mode is preferred in many practical applications as fusion databases, such as COSMIC include events that have been typically validated and of known clinical relevance. As the fusion database expands, targeted fusion detection is becoming more sensitive and clinically relevant. We thus expect the targeted mode to be used more widely in practice than de novo mode, which is mainly intended for discovery. Although novel fusion discoveries are crucial to expand the knowledge base, such effort may be less relevant to screening or tumor sub-typing purposes for cfNA data; and in practice, the results can contain too many false positives (FPs) to be inspected manually.
Several alignment-free methods have been recently proposed to reduce runtime and improve sensitivity (Bray et al., 2016; Li et al., 2017; Melsted et al., 2017). For example, ChimeRScope (Li et al., 2017) discovers genes a fragment may come from through kmer matching, in a manner similar to AF4. However, its fusion detection mechanism is fairly different: it ranks genes only by the degree at which a gene shares bases with the fragment, ignoring the locations of kmer matches within the fragment. In contrast, AF4 explicitly checks whether a gene pair actually forms a junction and ranks gene pair candidates by their fragment coverage. This mechanism leads to more accurate fusion detection.
We demonstrate AF4 to be a valuable addition to the existing fusion detection tools by applying it to benchmark simulated and real RNA-seq data with known fusions (Liu et al., 2016), clinical cfRNA and cell-line titration cfDNA data. AF4 achieved better F-score when compared with ChimeRScope (Li et al., 2017) on the simulated RNA-seq data. On cfRNA data, AF4 achieved better sensitivity in both targeted and de novo modes than STAR-Fusion (Haas et al., 2017) in retaining fusion fragments. However, a direct comparison of specificity cannot be fairly made as both programs can be adjusted for specificity with different thresholds during filtering (Section 3.2). By changing the reference sequences (and with no algorithmic changes), the targeted mode of AF4 was able to identify real fusions in a series cell-line titration cfDNA datasets comparable to Manta (Liu et al., 2016), a DNA-seq structural variation detection method. In the tested data, AF4 runs 300× faster than ChimeRscope, 12× faster than STAR-Fusion and 20× faster than Manta.
2 Materials and methods
The workflow of AF4 is outlined in Figure 1. There are three components in the workflow: the input component that handles I/O and preprocessing, independent readpair handling in stage one and fusion candidate filtering in stage two.
Fig. 1.
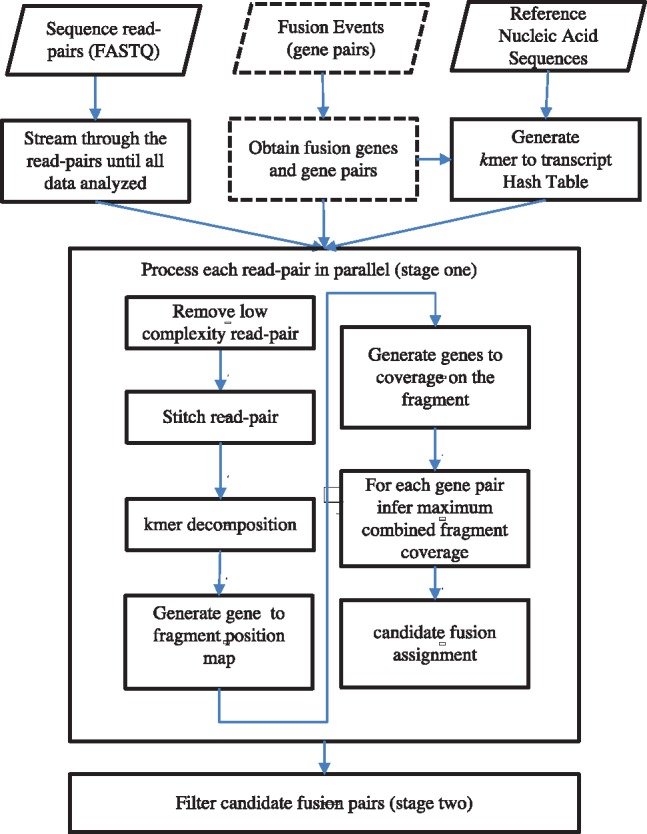
An overview of AF4 workflow. Detailed descriptions are given in the main text
2.1 Input
AF4 takes three inputs:
A list of paired FASTQ files, typically Illumina paired-end sequencing data, in compressed format.
A transcriptome FASTA file that lists known transcript sequences. For RNA-seq or cfRNA, we could use the default Gencode transcriptome reference. For analyses in this article, however, we padded 250 intron bases on both 5’ and 3’ regions of every exon in Gencode 26. This reduces FPs that arise from reads spanning intron–exon junctions in unspliced nascent mRNA molecules. For cfDNA, we use the entire genic space including introns.
An optional fusion-gene pair file, used only in targeted mode. We use the COSMIC fusion database (Tate et al., 2019, 297 gene pairs to date), plus structural multiplex cfDNA reference standard (www.horizondiscovery.com) and additional targets, resulting in a total of 655 fusion gene pairs.
2.2 Intuition
When a readpair (r1, r2) supports a fusion event involving genes g1 and g2, one of the three conditions must hold:
r1 and r2 map to different genes but neither r1 nor r2 spans the fusion junction (Fig. 2a).
One of r1 or r2 spans the fusion junction, and the other is covered by one of the genes (Fig. 2b).
Both r1 and r2 span the fusion junction, which happens when the fragment is short and r1 and r2 overlap (Fig. 2c).
Fig. 2.
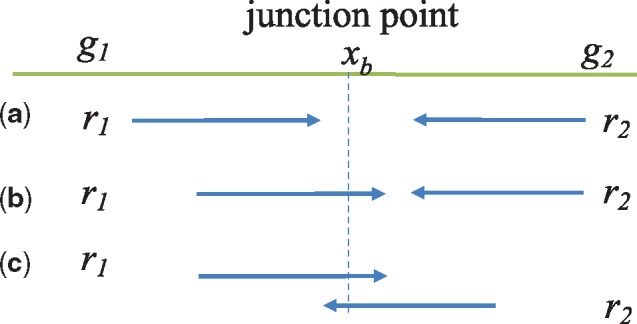
Fusion event involving gene pairs (g1, g2) and readpairs (r1, r2). The green line denotes the fusion transcript derived from genes g1 and g2 with fusion junction point denoted by xb. (a) Neither r1 nor r2 spans xb, (b) r1 but not r1 spans xb and (c) both r1 and r2 span xb
With these observations, we define readpair (r1, r2) supporting a fusion transcript involving gene pair (g1, g2) using parameters α and β.
α fraction of r1 and r2 combined map to g1 and g2. In Figure 2, as the entire r1 and r2 map to the fusion transcript. We set by default to account for sequencing errors.
r1 or r2 share a minimum of β bases on both sides of the fusion junction point. In Figure 2, this means on either side of xb, at least β bases of r1 (r2) map to g1 (g2). We set β = 25 by default.
Although we used paired-end reads as an illustration, it is easily generalizable to single-end inputs.
2.3 Stage 1
AF4 first reads the transcriptome and the optional gene pair files. For every gene implicated in a candidate fusion event, it produces kmers (k = 19 by default) for all possible positions and registers them in a central hash table that maps a kmer to the list of genes that contain the kmer. Then, it drops kmers that map to more than a certain number of genes (by default >2, discussed in Section 4). This policy drops of kmers in both targeted and de novo modes. This stage takes 10 s and produces a hash table of size 4GiB in targeted mode, or 1 min and 35GiB in de novo mode (hardware specified in Section 3).
AF4 then reads the FASTQ file pairs to identify a gene or a gene pair that best supports each readpair (r1, r2). This step works independently and in parallel for every readpair. Algorithm 1 describes our method.
Algorithm 1.
Assign (r1, r2) to a candidate fusion event
1: Drop low-complexity reads.
2: Find a kmer shared between (r1, r2), stitch and trim overhangs to generate a fragment f.
3: Let L be the length of the fragment f and be the set of genes sharing some common kmer with f.
4: for Every gene do
5: set of positions in f covered by g1.
6: for Every gene do
7: set of positions in f covered by g2.
8: Ignore pair (g1, g2) if their coverage do not satisfy concurrently the fusion support criteria (Section 2.2).
9: for Every possible junction position do
10: , representing the combined coverage of g1 and g2 wrt junction q.
11: end for
12: s.t. q is some junction position that maximizes .
13: end for
14: end for
15: s.t. g is the gene that maximizes
16: s.t. (g1, g2) is a gene pair that maximizes .
17: if, then assign the fragment to (g1, g2).
In Line 1, we drop low-complexity readpairs, for which the frequency of any two nucleotides reaches 90% of the sequence length.
As the library is expected to contain many short molecules, r1 and r2 are likely to overlap. In addition, if the molecule is smaller than a read length, r1 or r2 may further contain bases that are not part of the molecule. Therefore, we stitch r1 and r2 into a fragment (Line 2 and Fig. 3). We produce kmers at all positions for the two reads, then find some common kmer that anchors the suffix–prefix alignment of r1 and r2 (Fig. 3a). If such a kmer is found and the section shared between (r1, r2) has sufficient similarity (e.g. Hamming distance of the section length), we stitch the two sequences into one. If the end of r1/r2 extends beyond the end of r2/r1 (overhang), f becomes the overlapping region (Fig. 3b). If r1 and r2 cannot be stitched, either because they are not overlapping or they have too many sequencing errors, we concatenate the readpair in form r1N where N is an artificial base distinct from real bases, and is a reverse complement of r2 (Fig. 3c). The N character prevents generation of kmers.
Fig. 3.
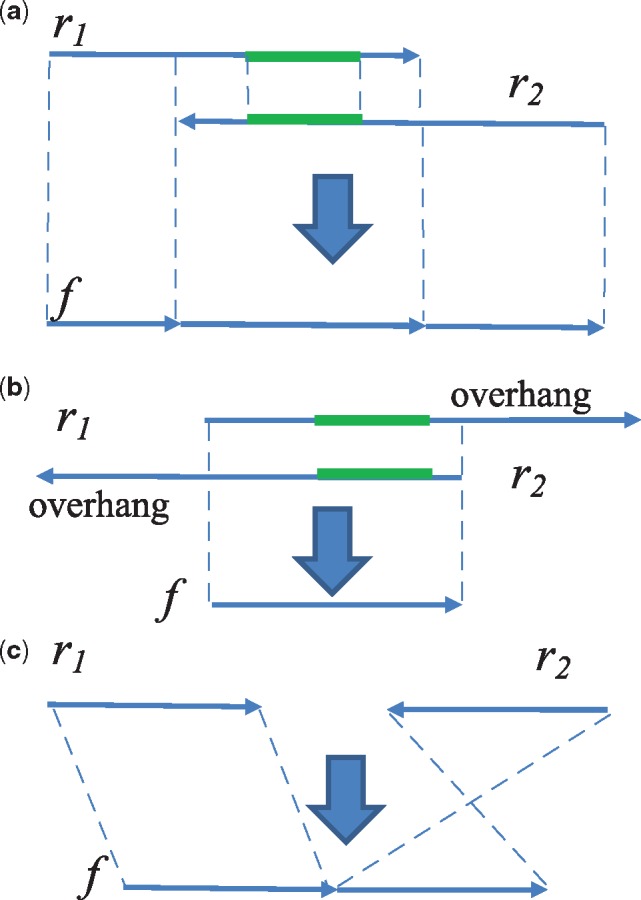
Fragment generation by stitching and overhang trimming of readpair (r1, r2). (a) r1 and r2 are represented as arrows facing each other denoting the forward and reverse complement strands. The green bars denote one of the shared kmers between them, which is an anchor for suffix—prefix alignment. The stitched fragment is a concatenation of prefix of r1, overlap and suffix of r2. (b) When r1 and/or r2 extends beyond the region of the other read, the overhang is trimmed, and f is the overlap. (c) When r1 and r2 cannot be merged, f is a concatenation of r1 and reverse complement of r2
Lines 4–16 detect fusions as described in Section 2.2. The key idea here is to approximate the coverage of fragment f by gene g without aligning them to a reference. Instead, if the kmer of f at position x matches any kmer of g, we assume that g covers positions ( means a half-open range, ) of f. If multiple kmers of f match those of g, we blindly merge the matched ranges without verifying if these kmers are sequentially aligned on the gene. Next, we pick the pair of genes that provide the most coverage of the fragment, among those that satisfy the fusion criteria defined in Section 2.2.
Figure 4 illustrates this process. , . A junction in range will maximize the combined coverage, thus .
Algorithm 1 may look expensive since it seems to examine all pairs of genes that share any segment with the fragment. However, in practice it can be implemented efficiently, for the following reasons:
given a pair of genes, the best junction point can be computed in time linear to the fragment length—to calculate , we first sort the regions of each gene in an increasing order of positions from 5’ to 3’ w.r.t. f. Then, we only need to consider range boundaries between g1 and g2, assuming there’s at most one fusion junction spanned by f.
The average number of genes that needs to be examined per fragment is small due to the conditions imposed in Line 8. In targeted mode, fragments match less than one gene on average, so the overhead of this process is negligible. In de novo mode, fragments match more genes depending on samples, but we found it still takes less than 5% of the CPU time overall, since this codepath exhibited high memory-access locality. We examine the performance of AF4 in more detail in Section 3.4.
2.4 Stage 2
Stage one obtained potential candidate fragments and their supporting gene fusion pairs. In stage two, like other fusion detection methods (Haas et al., 2017; Nicorici et al., 2014), AF4 runs a set of empirically derived methods to remove spurious gene pairs. Given a candidate fragment f and the corresponding candidate fusion gene pair (g1, g2), we run the following filters:
Low-complexity sequences: If the subsequences of f covered by g1 or g2 has low complexity, f is dropped.
Nearby genes: If g1 and g2 are on the same chromosome and are less than 100 000 bps apart, f is dropped. This policy prevents the inference of read-through events or the overlapping genes.
PCR duplicates: Fragments with the same UMIs are collapsed into one. To allow for sequencing errors, UMIs that are within two-Hamming distance apart are considered the ‘same’. Unlike other systems that collapse UMIs early in the pipeline, AF4 performs it here, since the first stage processes each readpair independently. After removal of PCR duplicates, any fusion gene pairs supported by <2 distinct fragments are dropped. When the data has no UMIs, we merge highly similar fragments, where by default 95% similarity threshold is used.
Genes with too many partners: If a gene g appears in too many gene fusion pairs, all fragments that contain g are dropped. The default number of maximum gene fusion pairs g involved in is set to be 5.
These criteria are empirically determined and are not always appropriate. AF4 supports reading candidate fragments and gene pairs from a checkpoint file and running dataset specific filtering algorithms quickly.
3 Results
AF4 is written in Go (www.golang.org). The experiments were run on an Ubuntu 16.04 machine with Linux 4.4.0, Xeon E5-2690@2.6GHz * 28 (two hyperthreads/core, 56 total CPUs), 256GiB of memory and XFS (en.wikipedia.org/wiki/XFS) on NVMe for storage. In this section, the same default parameter values were used in all cases to avoid overfitting to an individual dataset. The scripts used to generate the results in this section are available at: https://github.com/grailbio/bio/tree/master/fusion/benchmark.
3.1 Simulated and real RNA-seq data
Although AF4 was designed to handle cfNA data, it is applicable directly to RNA-seq data. To demonstrate this, we ran AF4 on widely used datasets in methods comparison (Liu et al., 2016). They consist of 15 datasets—three read lengths (50 bp, 75 bp and 100 bp) and five different coverage levels (5×, 20×, 50×, 100× and 200×). Each dataset contains fragments that match 150 possible fusion events.
Table 1 shows the results of AF4, STAR-Fusion (Haas et al., 2017) and ChimeRScope (Li et al., 2017), a recently developed alignment-free method that was shown to have an overall better performance than other methods. The most recent version of STAR-Fusion (v.1.5.0) was obtained from github (github.com/STAR-Fusion/STAR-Fusion/) along with the required reference folder (GRCh38_v27_CTAT_lib_Feb092018). We used the provided docker image to run STAR-Fusion. Note that ChimeRScope only reported 135 out of 150 possible fusion events due to the constraint that ChimeRScope is built on RefSeq annotation but the 15 excluded fusion events were simulated from Ensembl and involved non-coding regions by RefSeq annotation. AF4 reported on all 150 and achieved a higher F-score than ChimeRscope and STAR-Fusion for both targeted (over 0.98) and de novo modes (over 0.96). As expected, the de novo mode produced higher number of FPs.
Table 1.
AF4 for simulated datasets (Liu et al., 2016)
| Samples | AF4 |
ChimeRScope | STAR-Fusion | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Targeted |
De novo
|
||||||||||
| Length (bp) | Coverage | TP | FP | FN | F-score | TP | FP | FN | F-score | F-score | F-score |
| 50 | 5× | 146 | 0 | 4 | 0.986 | 143 | 1 | 7 | 0.973 | 0.948 | 0.416 |
| 50 | 20× | 147 | 0 | 3 | 0.990 | 144 | 2 | 6 | 0.973 | 0.954 | 0.855 |
| 50 | 50× | 147 | 0 | 3 | 0.990 | 144 | 2 | 6 | 0.973 | 0.947 | 0.867 |
| 50 | 100× | 147 | 0 | 3 | 0.990 | 144 | 3 | 6 | 0.970 | 0.908 | 0.868 |
| 50 | 200× | 147 | 0 | 3 | 0.990 | 144 | 3 | 6 | 0.970 | 0.905 | 0.875 |
| 75 | 5× | 144 | 0 | 6 | 0.980 | 141 | 1 | 9 | 0.966 | 0.948 | 0.700 |
| 75 | 20× | 147 | 0 | 3 | 0.990 | 143 | 1 | 7 | 0.973 | 0.949 | 0.865 |
| 75 | 50× | 147 | 0 | 3 | 0.990 | 143 | 1 | 7 | 0.973 | 0.957 | 0.872 |
| 75 | 100× | 147 | 0 | 3 | 0.990 | 144 | 2 | 6 | 0.973 | 0.954 | 0.875 |
| 75 | 200× | 147 | 0 | 3 | 0.990 | 144 | 3 | 6 | 0.970 | 0.947 | 0.875 |
| 100 | 5× | 144 | 0 | 6 | 0.980 | 140 | 0 | 10 | 0.966 | 0.940 | 0.692 |
| 100 | 20× | 147 | 0 | 3 | 0.990 | 143 | 1 | 7 | 0.973 | 0.957 | 0.878 |
| 100 | 50× | 147 | 0 | 3 | 0.990 | 143 | 1 | 7 | 0.973 | 0.957 | 0.875 |
| 100 | 100× | 147 | 0 | 3 | 0.990 | 143 | 1 | 7 | 0.973 | 0.957 | 0.872 |
| 100 | 200× | 147 | 0 | 3 | 0.990 | 143 | 1 | 7 | 0.973 | 0.957 | 0.877 |
Note: Each dataset contains fused reads of 150 gene pairs. In the targeted mode, the names of the 655 target gene pairs (including all 150 target ones) are given in the command line, along with the transcriptome. In the de novo mode, only the transcriptome is given.
We further demonstrate the utility of AF4 using four breast cancer RNA-seq data (Liu et al., 2016) in targeted mode, where the real fusion events can be found in Edgren et al., 2011, Table 1. Because of the updated annotations of gene names, the genes TMEM49, WDR67 and KIAA0406 have been replaced with VMP1, TBC1D31 and TTI1, respectively. ENSG00000236127 was eliminated from evaluation since no proper annotation replacement was found. Using default parameters, 23 out of 26 total validated fusions have been identified and one Fp gene pair HMGA2/LP was reported. Upon inspection, we found that the two false negative gene pairs DHX35/ITCH and CCDC85C/SETD3 were excluded because of they failed by the ‘Nearby genes’ criterion imposed by AF4 (Section 2.4). The results, using default parameters, have one more true positive prediction than ChimeRScope, which was shown to have better predictions than others alignment-based methods that typically reported 5–20 true positive predictions in these data (Li et al., 2017, Table 4).
Table 4.
Efficacy of filtering policies implemented in AF4
| Samples | Stage one | Low cmpl seq | Nearby genes | PCR dups | Abund partners | Final |
|---|---|---|---|---|---|---|
| T1 | 345 | 1 | 0 | 243 | 66 | 35 |
| T2 | 287 | 5 | 0 | 202 | 41 | 39 |
| T4 | 709 | 5 | 43 | 519 | 65 | 77 |
| T7 | 397 | 4 | 8 | 302 | 0 | 83 |
| T10 | 704 | 3 | 3 | 571 | 0 | 127 |
| T13 | 866 | 3 | 0 | 717 | 0 | 146 |
| T14 | 679 | 3 | 3 | 559 | 0 | 114 |
| Prc101 (T) | 29 754 | 0 | 0 | 25 592 | 0 | 4162 |
| Prc101 (D) | 296 113 | 1172 | 6038 | 257 979 | 29 838 | 1481 |
| Prc108 (T) | 284 | 0 | 28 | 203 | 0 | 53 |
| Prc108 (D) | 325 417 | 794 | 6047 | 261 012 | 56 199 | 1634 |
| HC332 (T) | 64 | 0 | 0 | 59 | 0 | 5 |
| HC332 (D) | 140 189 | 407 | 3127 | 121 074 | 14 156 | 1681 |
| HC118 (T) | 116 | 0 | 0 | 111 | 0 | 5 |
| HC118 (D) | 449 265 | 723 | 9030 | 420 528 | 17 305 | 1880 |
| HC160 (T) | 2 | 1 | 0 | 1 | 0 | 0 |
| HC160 (D) | 22 173 | 174 | 539 | 18 658 | 1693 | 1154 |
Note: ‘Stage one’ column shows the number of fusion candidate fragments found in the first stage. Columns ‘Low cmpl seq’ (low-complexity sequences), ‘Nearby genes’, ‘PCR dups’ (PCR duplicates), and ‘Abund partners’ (genes with too many partners) show the number of candidates dropped due to the named policies defined in Section 2.4. ‘Final’ column shows the final number of fusion fragments reported.
3.2 cfRNA data
We have obtained 40 ml of whole blood collected in Streck tubes, separated into plasma and extracted cfRNA samples from two Stage IV Prostate cancer patients and three healthy controls from Conversant Bio and generated cfRNA sequencing data with UMIs added. For the two cancer patients, it has been determined that TMPRSS2-ETV4 and TMPRSS2-ERG fusion were the most likely real fusion events, because TMPRSS2 fusion are detected in 50% of prostate cancer (Tomlins et al., 2008). The three healthy individuals had a primary diagnosis of normal, and therefore, should contain no real fusion events and serve as negative controls.
We applied AF4 to these data using both the targeted and de novo mode using default parameters. We also tested the STAR-Fusion program (Haas et al., 2017), which is one of few programs that can work with data of this size in a reasonable runtime.
Table 2 shows the results on the inputs of 350 to 450 million readpairs. It contains the runtime, the number of supporting fragments for the fusions (TMPRSS2-ETV4 and TMPRSS2-ERG) in the two prostate cancer patient samples as an indicator of sensitivity and the number of reported fusion events as an indicator of FPs.
Table 2.
Results of AF4 and STAR-Fusion on cfRNA data
| Samples | #readpairs (millions) | Coverage | Number of TMPRSS2-ETV4/ERG supporting fragments |
Number of reported fusion events |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AF4 |
STAR-Fusion |
AF4 |
STAR-Fusion |
|||||||||
| Targeted |
De novo
|
Targeted | De novo | |||||||||
| Final | Prelim | Final | Prelim | Final | Prelim | Final | Prelim | |||||
| Prc101 | 373.8 | 29 132 | 19 | 345 | 19 | 345 | 0 | 31 | 9 | 239 | 19 | 979 |
| Prc108 | 464.5 | 36 200 | 9 | 114 | 0 | 116 | 0 | 13 | 12 | 140 | 13 | 787 |
| HC118 | 399.8 | 31 158 | — | — | — | — | — | — | 1 | 289 | 5 | 930 |
| HC160 | 348.9 | 27 191 | — | — | — | — | — | — | 0 | 303 | 21 | 254 |
| HC332 | 443.2 | 34 540 | — | — | — | — | — | — | 1 | 350 | 18 | 840 |
Note: The coverage is calculated by the formula #readpairs × 166/2.13, where 2.13 (million bp) is the panel size and 166 (bp) is used as the average fragment length. The middle section of the table shows the number of readpairs that support either the TMPRSS2-ETV4 or the TMPRSS2-ERG fusion event. As these two fusions are not expected in the healthy controls, they are marked by ‘—’. The right most part of the table shows the number of unique fusion events reported.
For the two real fusions (middle section of Table 2), we report both the preliminary and final results for both programs. Preliminary results of AF4 are the output from the first stage of the program, whereas for STAR-Fusion, there were two preliminary filtered results, one based on FFPM filtering (fusion fragments per million total reads) that relies on the expression value (file with suffix.preliminary.filtered.FFPM) and the other is based on splice information (file with suffix.wSpliceInfo.wAnnot.pass). We used the result reported in the FFPM file in the table. As shown in the table, AF4 identified more fusion-supporting fragments compared to STAR-Fusion in the preliminary output and in most cases much less spurious fusions in both targeted and de novo mode. The preliminary results indicate the method’s capability to identify read-pairs derived from real fusion events. Also, the chimeric read counts considered by STAR-Fusion were around 20 and 33 million, respectively, 100 times more than AF4, indicating a higher specificity of AF4. In the final results (after filtering in stage two), AF4 reported both real fusions in targeted mode but missed one in de novo mode. Upon inspection, TMPRSS2-ERG was filtered out in the second stage because ERG has more than five partners. On the other hand, STAR-Fusion filtered out both real fusions, likely due to aggressive filtering as indicated by very low number of total reported fusions.
Note that the parameters of both programs can be tuned to reduce the number of FP fusions, while retaining the real fusions. For example, by raising the minimum number of unique fusions supporting fragments from 2 to 5, AF4 will still retain the real fusions but reduce the number of total reported fusions to be under 10. Nonetheless, we report the results by using the default of both programs to reflect the needs to empirically adjust the parameters of the program to different data type to achieve acceptable performance.
3.3 cfDNA data
Fusion detection in DNA-seq data is more challenging. Because fusion junctions may occur within introns, typically over half of existing gene panels are devoted to capture fusions. Computationally, this translates to a larger search space and higher number of FPs. Typically, fusion detection programs intended for RNA-seq data are not directly applicable to DNA-seq data. However, we show the targeted mode of AF4 may be applicable to cfDNA data.
We have created a proxy for circulating tumor DNA derived from plasma from human subjects by titrating genomic DNA carrying known genetic variations into a well characterized genome in a bottle (GIB) sample (NA12878) at six concentrations and then sheared them. Horizon control HD753 (https://www.horizondiscovery.com/structural-multiplex-reference-standard-hd753) was used as the spike in sample, which contains two fusion variants, CCDC6/RET and SLC34A2/ROS1. Titration percentages varied between.001 and.01.
Table 3 shows the results for AF4 and Manta (Liu et al., 2016), with AF4 running in targeted mode using default parameters. For Manta, raw cfDNA sequencing data were trimmed to remove adapter sequences and aligned using BWA. PCR duplicates were identified using an internal tool that uses the fragment start and end position along the genome, along with the pair of UMIs that are annealed to each end of the read to disambiguate collision events. Aligned and deduplicated reads were then processed by Manta using the command line: ‘python configManta.py –tumorBam –runDir –referenceFasta –generateEvidenceBam –exome’, where hg19 was used as the reference. AF4 achieved comparable or better sensitivity compared to Manta. For Manta outputs, we removed non-fusion related outputs and merged all fusions involving the same two genes. However, the FPs in Manta are too high to be used to confidently call genuine fusion events. By further filtering using the target gene pairs as used by AF4, nearly all FPs were eliminated. This demonstrated that the strategy of targeting clinical relevant fusions is highly effective.
Table 3.
Results of AF4-targeted mode and Manta on cfDNA titration data for identified fusion events
| Samples (titration) | readpairs (millions) | Coverage | AF4 |
Manta |
||||
|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TP | FP | FN | |||
| T1 (0.001) | 1000.9 | 78 004 | 1 | 13 | 1 | 1 | 3774 | 1 |
| T2 (0.002) | 938.8 | 73 165 | 1 | 11 | 1 | 0 | 2884 | 2 |
| T4 (0.004) | 1141.2 | 88 939 | 2 | 16 | 0 | 2 | 2723 | 0 |
| T7 (0.006) | 998.1 | 77 786 | 2 | 16 | 0 | 1 | 2353 | 1 |
| T10 (0.008) | 951.4 | 74 147 | 2 | 15 | 0 | 2 | 2455 | 0 |
| T13 (0.01) | 1033.2 | 80 522 | 2 | 22 | 0 | 2 | 2832 | 0 |
| T14 (0.01) | 1014.2 | 79 041 | 2 | 17 | 0 | 2 | 3108 | 0 |
Note: The coverage is calculated by the formula #readpairs (millions) × 166/2.13, where 2.13 (million bp) is the panel size and 166 (bp) is used as the average fragment length. Some titrations have two to three replicates as shown in the first column.
Note that due to the large search space and the fact that screening or tumor sub-typing is the main-intended use for cfDNA data, only targeted mode of AF4 is intended to be used for this data type. Although Manta can be used to discover novel fusion events, the excessive number of FPs makes it impractical without additional post-filtering.
3.4 Performance
Table 4 shows the number of fragments retained or dropped by various stages in the AF4 pipeline, using the samples from the previous sections. AF4 yields very few candidate fragments after the first stage—<0.0001% in targeted mode and 0.1% in de novo mode. Moreover, the vast majority of the candidates from the first stage are PCR duplicates that can be removed. That leaves just a few candidates, even for very large datasets.
Table 5 summarizes the performance of the systems studied in the previous sections. We omitted simulated datasets (Section 3.1) as AF4 takes about 10 s in targeted mode to 60 s in de novo mode. We could not run ChimeRScope ourselves due to licensing reasons, but Li et al., (2017) reported that its read throughput is of >5000 reads per minute/CPU. Extrapolating, AF4 looks to be 300 times faster than ChimeRScope. AF4 is significantly faster than STAR-Fusion, by 12× to 23×. Its performance is on par with Manta; however, this does not include the time spent generating the alignments which took over 6 h for the samples in our datasets.
Table 5.
Runtimes of AF4, STAR-Fusion and Manta in seconds (wall-clock time), where the same number of cores were used
| Samples | #readpairs (millions) | AF4 | AF4 | STAR- Fusion | Manta |
|---|---|---|---|---|---|
| Targeted | De novo | ||||
| Prc101 | 373.8 | 362 | 481 | 8475 | — |
| Prc108 | 464.5 | 340 | 462 | 16 075 | — |
| HC118 | 399.8 | 346 | 470 | 8801 | — |
| HC160 | 348.9 | 373 | 381 | 7903 | — |
| HC332 | 443.2 | 382 | 391 | 12 666 | — |
| T1 (0.001) | 1000.9 | 1000 | — | — | 1280 |
| T2 (0.002) | 938.8 | 938 | — | — | 1160 |
| T4 (0.004) | 1141.2 | 1268 | — | — | 2016 |
| T7 (0.006) | 998.1 | 1124 | — | — | 1160 |
| T10 (0.008) | 951.4 | 1104 | — | — | 1040 |
| T13 (0.01) | 1033.2 | 1217 | — | — | 1190 |
| T14 (0.01) | 1014.2 | 1117 | — | — | 1490 |
Note: Samples are from Sections 3.2 and 3.3. Times for Manta exclude the alignment and de-duplication steps.
The vast majority of AF4’s runtime is spent in the first stage; the second stage runs in a few seconds for all the datasets. For the first stage, AF4 spends 40–50% of its time in kmer-to-gene-list map lookups. This map is so large that it always causes TLB- and CPU-cache misses, and this operation happens at every position of every fragment.
Interestingly, the core AF4 algorithms—computing the coverage by genes, examining pairwise combinations of genes, and picking the best fusion—takes an insignificant fraction of time: 0% in targeted mode and 3% in de novo mode. In targeted (or de novo) mode, one fragment finds on average 0.5 (or 2, respectively) gene that has any kmer in common. Thus, many fragments are dropped without running our main fusion detection code.
4 Discussion
We have originally developed AF4 to efficiently handle cfRNA data but we observed that AF4 could be directly applied to tissue RNA-seq data. Moreover, it is encouraging to see that the targeted mode of AF4 can be used for cfDNA fusion detection: previously, alignment-based algorithms were typically needed to handle DNA-seq data due to the much larger search space.
Fusion detection methods use varied filtering criteria for different data types. These programs, including AF4, are typically rich in parameters that require tuning. Therefore, AF4 is not intended to fully replace other fusion detection methods, but it would be a valuable addition due to its linear scalability, high sensitivity in retaining relevant fusion fragments, and effective optimization strategy to remove the majority of spurious fragments. Users have an option to use the output of the first stage of AF4 in combination with more sophisticated filtering methods, which should be able to handle a much-reduced data size. It is also worth noting that different data types may have different properties, therefore requiring different parameter settings. Our results showing superiority on these benchmark datasets does not imply that the superior performance will generalize to all other datasets.
In order to select default parameters for AF4, we identified kmer size and the number of occurrences of the kmer shared across genes in the transcriptome data and identified the reasonable choices of the former to be 17–19 and the latter to be 2–3. With k = 19, the default value, this approach allows efficient coverage computation with negligibly low false-match rate. Both of these parameters can be adjusted: when k decreases, the method would be more sensitive in picking up candidate fusion fragments, however, the average number of genes a kmer occurs in would increase. Correspondingly, the number of occurrences of a kmer shared across genes should be increased to avoid of kmers being dropped. The filtering thresholds in the second stage of AF4 would be more relevant to the particular application and users have the option to learn empirically the optimal settings to have a balance between sensitivity and specificity.
One drawback of AF4 is its difficulty in detection fusion events where single nucleotide polymorphisms (SNPs) exist near a junction, which limits its ability to identify fusion spanning fragments, whereas alignment-based methods would likely be able to identify these fragments by tolerating mismatches. However, based on the design of the method, this can be addressed by generating additional reference sequences by incorporating common SNPs on the exon boundaries without needing to change the program. Some improvements can be built-in further in AF4 such as to include more sophisticated filtering criteria and to incorporate alignment-based strategies in stage two to improve specificity.
Acknowledgments
We thank Darya Filippova, Alex Fields, Marius Eriksen, Arash Jamshidi, Alex Aravanis, Nathan Hunkapiller, Ognjen Nikolic and Megan Hall for their valuable suggestions and support of this work.
Funding
Grail Inc funded this work. No external grants or NIH grants were used.
Conflict of interest: the authors are employed by Grail, Inc.
References
- Bray N.L. et al. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525.. [DOI] [PubMed] [Google Scholar]
- Chen K. et al. (2012) BreakFusion: targeted assembly-based identification of gene fusions in whole transcriptome paired-end sequencing data. Bioinformatics, 28, 1923–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson J., Park B.H. (2018) Circulating tumor DNA: measurement and clinical utility. Ann. Rev. Med., 69, 223–234. [DOI] [PubMed] [Google Scholar]
- Edgren H. et al. (2011) Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol., 12, R6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B. et al. (2017). STAR-Fusion: fast and accurate fusion transcript detection from RNA-seq. https://www.biorxiv.org/content/10.1101/120295v1. [Google Scholar]
- Jia W. et al. (2013) SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-seq data. Genome Biol., 14, R12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Salzberg S.L. (2011) TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol., 12, R72.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. et al. (2016) Comparative assessment of methods for the fusion transcripts detection from RNA-seq data. Sci. Rep., 6, 21597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2017) ChimeRScope: a novel alignment-free algorithm for fusion transcript prediction using paired-end RNA-seq data. Nucleic Acids Res., 45, e120.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. et al. (2016) Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res., 44, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melsted P. et al. (2017) Fusion detection and quantification by pseudoalignment. https://www.biorxiv.org/content/10.1101/166322v1. [Google Scholar]
- Nicorici D. et al. (2014) Fusioncatcher—a tool for finding somatic fusion genes in paired-end RNA-sequencing data. https://www.biorxiv.org/content/10.1101/011650v1. [Google Scholar]
- Tate J.G. et al. (2019) COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res., 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins S.A. et al. (2008) Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia, 10, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]