

Abstract
Motivation
Accurate predictions of protein-binding residues (PBRs) enhances understanding of molecular-level rules governing protein–protein interactions, helps protein–protein docking and facilitates annotation of protein functions. Recent studies show that current sequence-based predictors of PBRs severely cross-predict residues that interact with other types of protein partners (e.g. RNA and DNA) as PBRs. Moreover, these methods are relatively slow, prohibiting genome-scale use.
Results
We propose a novel, accurate and fast sequence-based predictor of PBRs that minimizes the cross-predictions. Our SCRIBER (SeleCtive pRoteIn-Binding rEsidue pRedictor) method takes advantage of three innovations: comprehensive dataset that covers multiple types of binding residues, novel types of inputs that are relevant to the prediction of PBRs, and an architecture that is tailored to reduce the cross-predictions. The dataset includes complete protein chains and offers improved coverage of binding annotations that are transferred from multiple protein–protein complexes. We utilize innovative two-layer architecture where the first layer generates a prediction of protein-binding, RNA-binding, DNA-binding and small ligand-binding residues. The second layer re-predicts PBRs by reducing overlap between PBRs and the other types of binding residues produced in the first layer. Empirical tests on an independent test dataset reveal that SCRIBER significantly outperforms current predictors and that all three innovations contribute to its high predictive performance. SCRIBER reduces cross-predictions by between 41% and 69% and our conservative estimates show that it is at least 3 times faster. We provide putative PBRs produced by SCRIBER for the entire human proteome and use these results to hypothesize that about 14% of currently known human protein domains bind proteins.
Availability and implementation
SCRIBER webserver is available at http://biomine.cs.vcu.edu/servers/SCRIBER/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Numerous protein functions, such as signal transduction, transport, regulation, metabolism, transcription and translation, rely on interactions with proteins, DNA, RNAs and small ligands (Chen and Kurgan, 2009; Cook et al., 2015; Figeys, 2002; Konig et al., 2012; Xie et al., 2011). Elucidation of protein–protein interactions (PPIs) assists development of PPI networks (De Las Rivas and Fontanillo, 2012), facilitates annotation of protein functions (Ahmed et al., 2011; Orii and Ganapathiraju, 2012), provides insights into molecular mechanisms of diseases (Kuzmanov and Emili, 2013; Nibbe et al., 2011), and finds applications in the discovery of novel therapeutics (Petta et al., 2016; Sperandio, 2012). Information about native PPIs is archived in several databases including Mentha (at the protein level) (Calderone et al., 2013), BioLip (at residue level) (Yang et al., 2012) and Protein Data Bank (PDB) (at atomic level) (Berman et al., 2000). However, these resources provide access to only a relatively modest amount of PPIs, e.g. 741 thousand PPIs in Mentha and 21 thousand in BioLip. Significant majority of PPIs remain to be discovered when we factor in that over 133 million proteins were already sequenced (UniProt, 2015) and that PPIs are highly promiscuous (Meng et al., 2016; Patil et al., 2010; Peleg et al., 2014). Computational predictors of PPIs help to bridge this annotation gap. These methods make predictions from either protein structure or protein sequence (Esmaielbeiki et al., 2016; Maheshwari and Brylinski, 2015). A high quality structural model is not available for all proteins in a genome, and methods that incorporate structural features are not always robust to issues with the quality of the structural model (Maheshwari and Brylinski, 2015). Thus, it is important to continue to improve sequence-based PPI predictors that can be applied to all sequenced proteins without the need for computationally costly structurally modeling. The sequence-based methods can be subdivided into protein level approaches, which predict interactions between proteins treated as units, and residue level approaches that predict protein binding residues (PBRs) (Zhang and Kurgan, 2018). We focus on the latter group that provides more detailed information.
Recent survey lists 16 sequence-based predictors of PBRs (Zhang and Kurgan, 2018). They include ISIS (Ofran and Rost, 2007), SPPIDER (Porollo and Meller, 2006), methods by Du et al. (Du et al., 2009) and Chen et al. (Chen and Jeong, 2009), PSIVER (Murakami and Mizuguchi, 2010), predictor by Chen et al. (Chen and Li, 2010), HomPPI (Xue et al., 2011), LORIS (Dhole et al., 2014), SPRINGS (Singh et al., 2014), methods by Wang et al. (Wang et al., 2014) and Geng et al. (Geng et al., 2015), CRF-PPI (Wei et al., 2015), PPIS (Liu et al., 2016), iPPBS-Opt (Jia et al., 2016), SPRINT (Taherzadeh et al., 2016) and SSWRF (Wei et al., 2016). A recent comparative analysis has discovered a substantial flaw shared by these methods (Zhang and Kurgan, 2018). Namely, they are unable to accurately separate residues that bind other molecules, such as DNA, RNA and small ligands, from PBRs. Empirical analysis using a well-annotated dataset shows that the most accurate SSWRF method cross-predicts 28% DNA-binding residues, 32% RNA-binding and 19% small ligand-binding residues as PBRs, and that it predicts the same fraction of PBRs among the native PBRs as among the native nucleic acid-binding residues (Zhang and Kurgan, 2018). Overall, when setting up these methods to predict the correct number of PBRs (equal to the number of native PBRs), they offer sensitivity between 19 and 32% while at the same time cross-predicting a comparable fraction of between 19 and 38% of the other types of binding residues as PBRs (Zhang and Kurgan, 2018). This suggests that these methods essentially predict all binding residues, instead of making predictions for specific interaction partners. This is because they utilize biased training datasets that include only protein-binding proteins, without a sufficient population of residues that bind other protein partners (Zhang and Kurgan, 2018). Inclusion of the latter set of residues is crucial to develop models that accurately differentiate between PBRs and residues that interact with other partners. Moreover, these methods rely on inputs produced with PSI-BLAST that for an average-size protein chain requires over 3 min of runtime, making whole genome-scale use difficult.
We introduce SCRIBER, a novel sequence-based predictor of PBRs. Our aims are to significantly reduce cross-predictions, offer higher predictive quality and reduce runtime when compared with the current methods. We incorporate four innovations to accomplish these aims:
We develop and utilize a new and high-quality dataset that covers interactions with multiple partners including proteins, DNA, RNA and small ligands.
We design an original architecture that employs predictions of binding residues for several partner types (proteins, DNA, RNA and small ligands) to effectively reduce cross-prediction of the output PBRs.
We use novel predictive inputs and effectively combine the novel and previously used input types.
We use much faster and more sensitive HHblits (compared to PSI-BLAST used by the other methods) (Remmert et al., 2012) and several other computationally-efficient tools to ensure that SCRIBER needs low amount of runtime (<1 min for an average size protein).
2 Materials and methods
2.1 Selection of current predictors for comparative analysis
We empirically compare SCRIBER with representative set of current predictors. Similar to the recent comparative review (Zhang and Kurgan, 2018), the criteria used to select these methods are: i) availability of webserver or source code; ii) ability to produce prediction for an average size protein sequence within 30 min; and iii) outputs that include both binary scores (PBR versus non-PBR) and numeric propensity for protein binding. The latter is necessary to compute the commonly used measures of predictive performance. Consequently, we select 7 out of the 16 current predictors that satisfy these criteria: SPPIDER (Porollo and Meller, 2006), PSIVER (Murakami and Mizuguchi, 2010), LORIS (Dhole et al., 2014), SPRINGS (Singh et al., 2014), CRF-PPI (Wei et al., 2015), SPRINT (Taherzadeh et al., 2016) and SSWRF (Wei et al., 2016).
2.2 Benchmark dataset
We generate a high-quality dataset to train and test SCRIBER by following procedure introduced in the recent comparative survey of the sequence-based predictors of PBRs (Zhang and Kurgan, 2018). This dataset includes proteins that interact with proteins, RNA, DNA and small ligands, and provides high coverage of native binding residues by combining annotations across multiple complexes that share the same protein. The data was sourced from the BioLip database (Yang et al., 2012) that was extended by the authors to include protein–protein interactions. The BioLip data is compiled using protein complexes from PDB that were solved with resolution ≤2.5 Å. Residues in these complexes are defined as binding if the distance between an atom of these residues and an atom of a given protein partner <0.5 Å plus the sum of the Van der Waal’s radii of the two atoms (Yang et al., 2012). We process the BioLip data to improve quality and uniformly sample proteins. First, we remove protein fragments. Second, we map BioLip sequences into UniProt records to collect binding residues across different complexes where the UniProt protein is shared, i.e. we transfer annotations of binding residues onto the same UniProt sequence. Third, we cluster the UniProt chains with a threshold of 25% similarity using Blastclust (Altschul et al., 1997). We select one protein from each cluster, the one that was the most recently released in UniProt, to ensure uniform sampling of proteins. Finally, we divide the resulting 1291 proteins into the TRAINING and TEST datasets. We ensure that proteins in the TEST dataset have <25% similarity with the proteins in our TRAINING dataset and in the training datasets of the 7 predictors that are included in the comparative analysis (see Section 2.1). We use Blastclust to cluster the 1291 proteins together with the proteins from the training datasets of the 7 methods at 25% similarity. 1120 proteins that share <25% similarity with the training proteins used by the considered 7 predictors (they are in clusters that do not include any proteins from the 7 training datasets) are used to derive the TEST dataset. Since the selected 7 predictors are computationally expensive we limit the size of the TEST dataset to a randomly selected subset of 40% of the 1120 proteins (448 proteins). The remaining part of the set of 1291 proteins (which includes proteins similar to the training datasets of the 7 methods and that share <25% similarity to the TEST proteins) makes up the TRAINING dataset. Table 1 summarizes the TRAINING and TEST datasets and includes information about protein-, RNA-, DNA- and small ligand-binding residues. Supplementary Table S1 compares sizes of datasets used to train and test this and the other seven predictors. It reveals that our datasets are 2.3 and 2.2 times larger compared to the largest previously used training and test datasets, respectively.
Table 1.
Summary of the datasets
| Dataset | Number of proteins | Number and fraction of different types of residues |
Total number of residues | ||||
|---|---|---|---|---|---|---|---|
| Protein-binding | DNA-binding | RNA-binding | Small ligand-binding | Non-binding | |||
| TRAINING | 843 | 32 253 (14.3%) | 1399 (0.6%) | 1508 (0.7%) | 13 643 (6.1%) | 179 615 (79.2%) | 225 299 |
| TEST | 448 | 15 810 (13.6%) | 557 (0.5%) | 696 (0.6%) | 7175 (6.2%) | 93 857 (80.6%) | 116 500 |
2.3 Evaluation setup
SCRIBER and the other 7 predictors output both binary (PBR versus non-PBR) and real-valued predictions (propensity for protein binding). We use the evaluation criteria from (Zhang and Kurgan, 2018). We assess the binary predictions using sensitivity (SN), specificity (SP), precision (PRE), accuracy (ACC), F1-measure (F1), Matthews correlation coefficient (MCC) and cross-prediction rate (CPR):
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
where true positives (TP) and true negatives (TN) are the correctly predicted PBRs and non-PBRs, respectively, false positives (FP) are non-PBRs incorrectly predicted as PBRs, and false negatives (FN) are PBRs incorrectly predicted as non-PBRs. CPR is the fraction of the other types of binding residues (DNA-binding, RNA-binding and small ligand-binding residues) that are cross-predicted as PBRs.
We calibrate the binary predictions to allow for a reliable side-by-side comparison between different predictors on the test dataset. The calibration ensures that the number of predicted PBRs generated by each predictor equals to the number of native PBRs, allowing for side-by-side comparison of binary predictions across different methods. We generate the binary predictions from the propensities using a threshold (residues with putative propensities > threshold are labelled as PBRs and the remaining residues as non-PBRs), and we ensure that the selected threshold provides the desired number of putative PBRs. We note that the entire training process of the SCRIBER model relies on the native annotations, and above calibration is applied only to binarize predictions on the test dataset.
The putative propensities are assessed with the area under receiver operating characteristic curve (AUC), area under precision-recall curve (AUPRC), and area under cross-prediction curve (AUCPC). The receiver operating characteristic curve plots TPR (true positive rate) = TP/(TP+FN) against FPR (false positive rate) = FP/(FP+TN) that are computed by binarizing the propensities using thresholds equal to all unique values of the propensities. The precision-recall curve plots precision against TPR, while cross-prediction curve is a relation of CPR against TPR, both computed using the same thresholds as the receiver operating characteristic curve. Given the imbalanced nature of our datasets (only about 14% of residues are protein binding, see Table 1), we also quantify the AULC (Area Under the Low false positive rate ROC Curve) value. AULC is the area under the receiver operating characteristic where the number of predicted PBRs ≤ number of native PBRs, i.e. where FPR is relatively low. Since AULC values are relatively small, we normalize them by dividing the measured value by the AULC of a random predictor. AULCratio = 1 means that a given method is equivalent to a random predictor while AULCratio > 1 quantifies the rate of improvement over the random predictor.
The design and parametrization of SCRIBER are carried exclusively on the TRAINING dataset using 5-fold cross-validation with the aim to maximize AUC. The final, already parametrized version of the model is trained on the TRAINING dataset is than applied on the TEST dataset, and the corresponding results are compared with the 7 other predictors.
We evaluate significance of the differences in predictive quality measured on the TEST dataset between SCRIBER and each of the other 7 predictors. This quantifies robustness of the improvements offered by SCRIBER by sampling a range of test sets drawn from the TEST dataset. More specifically, we compare results over ten tests, each based on randomly selected 50% of TEST proteins. We use the Anderson-Darling test at 0.05 significance level to check if a given set of measurement is normal. We apply the t-test to quantify significance of differences for normal measurements, otherwise we use the Wilcoxon rank sum test. Differences with P-value < 0.05 are assumed statistically significant.
2.4 Architecture of the SCRIBER predictor
SCRIBER predicts protein-binding resides using a two-layer design (Fig. 1). The first layer converts the input protein sequence into a comprehensive profile that represents structural, evolutionary and physiochemical properties which are relevant to binding. This profile is processed by five machine learning (ML) models to produce putative propensities for protein-, RNA-, DNA-, small ligand- and ‘other’-binding. The ‘other’-binding label combines nucleic acids and small ligand binding to represent all binding residues except for the PBRs. The second layer utilizes these propensities and a ML model to predict propensities for protein-binding, with the goal to reduce the cross-predictions when compared to the propensities produced in the first layer. The predictions are generated for each residue in the input protein chain using information extracted from a sliding window centered on the predicted residue. We do not pad the window at the termini of the sequence, but we rather narrow the window size on the side of the terminus. While multi-layer designs were used in the past, SCRIBER’s architecture is novel in the sense of using predictions of binding to multiple types of partners as inputs to accurately and specifically predict PBRs in the second layer. The other innovative aspects include use of novel predictive inputs, design of predictive inputs that combine structural, physicochemical and evolutionary characteristics of the input sequence, and application of computationally efficient methods to produce the profile. Details are provided in Section 2.5.
Fig. 1.

Flowchart of the SCRIBER predictor. The first layer predicts putative propensities for DNA-binding (red font), RNA-binding (violet), small ligand-binding (orange), other-binding (blue; includes DNA, RNA and small ligands) and protein-binding (green) residues. These propensities are combined in the second layer to re-predict PBRs, with emphasis on reducing cross-predictions between protein binding and other-binding residues
2.5 Design of the first layer
The design of the first layer includes rational delineation of the scope of the profile, selection of methods that are used to compute the profile, construction of a feature vector that is generated from the profile, feature selection and training/optimization of the ML models:
Sequence profile. A recent survey shows that key properties relevant to characterization and prediction of PBRs and nucleic acid-binding residues include amino acid-level propensity for binding, solvent accessibility, evolutionary conservation, hydrophobicity, polarity and charge (Zhang et al., 2017). These are motivated by the observations that the binding residues tend to locate on the protein surface and are typically evolutionarily conserved, and because some amino acid types are more likely to interact with certain protein partners. Moreover, charged residues are important for interactions with DNA and RNA (Ellis et al., 2006; Lejeune et al., 2005) while polar residues are relevant to protein–protein and protein–DNA interactions (Zhang et al., 2017). Correspondingly, the profile includes putative relative solvent accessibility (RSA), evolutionary conservation (ECO), relative amino acid propensity (RAAP) for binding, and the selected relevant physiochemical properties (charge, hydrophobicity and polarity). Importantly, we add a comprehensive set of novel inputs (in the context of this prediction) generated from the protein sequence. This novel part of the profile includes putative protein-binding intrinsically disordered regions, putative secondary structure (SS) and selected physicochemical properties of amino acids (aliphaticity, aromaticity, acidity and size). Inclusion of the intrinsic disorder is motivated by its enrichment in protein–protein, protein–DNA and protein–DNA binding (Dyson, 2012; Peng and Kurgan, 2015; Peng et al., 2014b; Peng et al., 2017; Varadi et al., 2015; Wang et al., 2016; Wu et al., 2015).
Computation of profile. The RSA values are predicted with accurate and fast ASAquick method (Faraggi et al., 2014). The ECO values are computed from the outputs generated with fast and sensitive HHblits (Remmert et al., 2012). The RAAP scores are calculated using an approach described in (Zhang et al., 2017). The putative protein-binding disorder is produced with computationally efficient ANCHOR (Dosztanyi et al., 2009) while secondary structure is predicted with fast version of PSI-PRED that does not utilize multiple alignments (Buchan et al., 2013). The physiochemical properties are quantified using the AAindex resource (Kawashima et al., 2007). We emphasize that the entire profile is generated utilizing computationally efficient methods, resulting in a runtime-efficient implementation of SCRIBER.
Construction of feature vector from the profile. The profile is converted into a fixed-size vector of numeric features. This is necessary to use the ML models. Prediction for a given residue in the input protein chain is based on features that quantify profile-based properties for 1) individual amino acids in the near vicinity of the predicted residue (up to two residues away); 2) averaged properties in 11-residues long sliding window centered on the predicted residue; and 3) relative position of nearest structurally/physicochemically/evolutionarily-defined residue. The first two categories of features are consistent with the design of features for related methods (Peng and Kurgan, 2015; Peng et al., 2017; Zhang and Kurgan, 2018; Zhang et al., 2017). The selected window size corresponds to an average window size used by the current predictors (Zhang and Kurgan, 2018). The innovative aspects of our feature encoding include: i) use of features that quantify the novel parts of the profile; ii) development of features that combine multiple structural, physicochemical and evolutionary properties, e.g. presence of conserved residues on putative protein surface or presence of conserved disordered residues; and iii) design of the relative position-defined features. The latter novel feature set quantifies linear distance (in sequence positions) to the nearest structurally/physicochemically/evolutionarily-defined residue, e.g. distance to the nearest conserved residues or nearest solvent exposed residue. We use the profile to generate total of 1090 features, including 232 features based on the previously used inputs and 858 that rely on the novel part of the profile. Supplementary Table S2 details calculation of these features.
Feature selection. Some of the considered 1090 features may not be relevant to the prediction of PBRs and some could be redundant. We empirically compare three feature selection methods: ML model-specific approach, filter-based selection and wrapper-based selection. Each feature selection approach is parametrized to maximize predictive quality (measured with AUC) based on 5-fold cross validation on the TRAINING dataset. This is done for each of the five predictors used in the first layer (Fig. 1); i.e. predictors of DNA-, RNA-, protein-, small ligand- and other-binding residues. The first approach applies the LASSO method that embeds feature selection into optimization of the regression model, which is used for the prediction of PBRs (use of regression is motivated in the next paragraph). We use LASSO implementation in MATLAB and we parametrize the number of regularization coefficients. The second approach is a simple filter that does not rely on the use of the ML model. First, we quantify predictive value of each feature based on the AUC computed when this feature is used individually to make predictions of PBRs. Next, we select a subset of features for which AUCs are greater that a parametrized threshold value. The third approach is the wrapper-based feature selection that was used in several related studies (Hu et al., 2018; Meng and Kurgan, 2018; Mizianty et al., 2010; Yan et al., 2016; Yan and Kurgan, 2017) and which chooses feature sets that secure highest predictive performance of the ML model. First, like in the filter approach, we rank features by their AUCs when they are used separately to make predictions. Next, we incrementally add to the set of selected features using this ranked list. More specifically, starting with the top-ranked (the most predictive) feature, we add the next-ranked feature to the current feature set only if this results in a higher AUC than the AUC obtained before this feature was added (i.e. when this inclusion improves predictive quality); otherwise the next-ranked feature is removed. We scan the sorted feature set once. The same three feature selection methods are used to design the second layer, and thus the corresponding empirical results are compared in Section 2.6.
Selection and training of ML models. We pick logistic regression as the prediction model because: i) it has been recently used to make accurate predictions of various types of functional residues including PBRs (Zhang et al., 2017), DNA- and RNA-binding residues (Yan and Kurgan, 2017), intrinsically disordered residues (Meng and Kurgan, 2016; Obradovic et al., 2005; Peng et al., 2014a; Peng et al., 2017), protease cleavage sites (Song et al., 2018) and post-translational modification sites (Li et al., 2018); ii) it outputs real numbers in the 0 to 1 range that intuitively quantify propensity for protein-binding; iii) of simplicity of this linear model which decreases chances of overfitting the TRAINING dataset; and iv) it is computationally efficient to train from the TRAINING dataset and to generate predictions, when compared to other popular models such as support vector machines and neutral networks. The runtime efficient training is critical because SCRIBER requires completion of 18 feature selection experiments (three feature selection methods for each of the six models: five in the first layer + one in the second layer) that rely on calculation of thousands of regression models in the cross-validation setting. Moreover, the fast predictions facilitate applications on a genomic scale and allow us to apply SCRIBER to analyze PBRs in the human proteome.
2.6 Design of the second layer
The five predictions from the first layer are used as inputs to the second layer that applies a separate regression model to (re-)predict PBRs with the goal to reduce cross-predictions with the other types of binding residues. We exploit an empirical observation that correctly predicted interacting amino acids typically cluster in the protein sequence (Zhang et al., 2017). For instance, residues predicted as DNA-binding the first-layer regression that are nearby many other AAs that are also predicted to interact with DNA and fewer residues predicted as PBRs are more likely to in fact interact only with DNA; this way we can eliminate the cross-predicted PBRs. Correspondingly, we encode the five predictions using features that quantify the five sets of propensities generated in the first layer for both individual residues located nearby the currently predicted amino acid and an aggregated propensity (using average and standard deviation) in a sequence window centered on that residue. Supplementary Table S3 details the corresponding set of 175 features. Like in the first layer, we perform three feature selections to optimize the regression model in the second layer, i.e. to maximize its AUC in the cross-validation on the TRAINING dataset. We empirically compare predictive performance offered by the corresponding models to select the best option. After parametrization on the TRAINING dataset was completed, we apply the three models to make predictions on the TEST dataset. The model that relies on the filter-based feature selection secures the lowest predictive performance, with AUC = 0.665 and AUPRC = 0.266. The second best LASSO selection-based predictor secures AUC = 0.691 and AUPRC = 0.266. The wrapper selection-derived model secures the best performance with AUC = 0.715 and AUPRC = 0.287. Consequently, SCRIBER is implemented using the latter approach that selected 81, 88, 225, 223 and 249 features to make predictions of DNA-, RNA-, protein-, small ligand- and other-binding residues in the first layer, respectively, and 46 features for the model in the second layer. Figure 2, which directly compares the three sets of results, also reveals that the wrapper-based selection produces the model with the lowest amount of cross-predictions; i.e. CPR = 0.116 for SCRIBER versus 0.127 for the LASSO-based predictor and 0.129 for the filter-based predictor.
Fig. 2.

Comparison of predictive performance on the TEST dataset between different designs of the SCRIBER model and different feature selections. Bars for the AUC and AUPRC are quantified with the left y-axis, while lines and markers for the AUCPC and CPR with the right y-axis
3 Results
3.1 Improvement in predictive performance due to the use of novel design strategies
SCRIBER relies on three novel design ideas: i) ‘novel features’: use of features that quantify new inputs that we included in the sequence profile; ii) ‘combined features’: use of innovative features that combine information across different protein properties included in the profile; and iii) ‘second layer’: use of the second layer that combines putative propensities for protein, DNA, RNA and small ligand binding generated in the first layer to improve prediction of PBRs. We perform an ablation study that quantifies improvements brought by these innovative ideas by comparing the complete SCRIBER model with its several versions that exclude these ideas. More specifically we compare SCRIBER with MODEL1 that excludes all three ideas (no novel features, no combined features and no second layer), i.e. we use the prediction of PBRs generated in the first layer using the remaining features; MODEL2 that does not use the combined features and the second layer but uses the novel features; and MODEL3 that does not use the second layer but applies both novel and combined features. Supplementary Table S4 provides a complete set of results for these four models on the TEST dataset, while Figure 2 summarizes the four arguably key measures: AUC, AUPRC (area under precision-recall curve), CPR (cross-prediction rate) and AUCPC (area under cross-prediction curve). We observe a substantial improvement in predictive performance when moving from the simplest MODEL1 that excludes all novel design ideas to MODEL2 that includes just the novel features. AUC and AUPRC improve from 0.647 to 0.691 and from 0.219 to 0.256, respectively, and cross predictions are reduced with AUCPC decreasing from 0.374 to 0.340. MODEL3 that includes both novel and combined features provides improvements compared to MODEL2, with AUC and AUPRC going up to 0.712 and 0.281, respectively, and AUCPC going further down to 0.313. Finally, SCRIBER that differs from MODEL3 by inclusion of the second layer provides slightly higher overall predictive quality (AUC = 0.715 for SCRIBER versus 0.712 for MODEL3; AUPRC = 0.287 versus 0.281) while significantly reducing the cross predictions (AUCPC = 0.282 for SCRIBER versus 0.313 for MODEL3, P-value = 0.00026; CPR = 0.116 versus 0.125, P-value = 0.04). This is because the main objective behind the design of the second layer is to decrease the cross-predictions rather than to improve prediction of PBRs. Moreover, Supplementary Table S4 shows that SCRIBER significantly outperforms MODEL1 and MODEL2 on all measures of predictive quality (P-values ≤ 0.0026). Overall, the consistent improvements over the consecutive versions (Fig. 2) clearly demonstrate that each of the three novel designs strongly contributes to the SCRIBER’s predictive performance.
3.2 Comparative assessment of predictive performance
Comparison with the seven representative predictors of PBRs (Table 2) reveals that SCRIBER produces the most accurate predictions. These improvements are statistically significant when compared with each of the seven predictors and for all 12 evaluation measures (P-values < 0.007). Compared to the second best SSWRF, SCRIBER provides AUC = 0.72 versus 0.69 (P-value = 0.00002), AUPRC = 0.29 versus 0.26 (P-value = 0.00005), AULCratio = 3.7 versus 3.1 (P-value = 0.000003), MCC = 0.23 versus 0.18 (P-value = 0.00000009) and F1 = 0.33 versus 0.29 (P-value = 0.0000001). Moreover, when setup to predict the correct number of PBRs (equal to the number of native PBRs), SCRIBER improves sensitivity by 5% (0.334 versus 0.288) while also providing s slightly higher specificity (0.896 versus 0.891). The corresponding ROC curves and precision-recall curves are shown in Supplementary Figure S1A and B, respectively. They demonstrate that SCRIBER provides the highest TPRs for FPRs < 0.73 and the highest precision over the entire rage of recall, both with a wide margin ahead of the second best SSWRF method. Overall, these results suggest that SCRIBER significantly outperforms the current sequence-based predictors of PBRs.
Table 2.
Comparison of the predictive performance of SCRIBER with the seven representative predictors of PBRs on the TEST dataset
| Predictor | Sensitivity | Specificity | Precision | Accuracy | F1 | MCC | AUC | AUPRC | AULC | AULCratio | AUCPC | CPR | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SPPIDER | Average | 0.202 | 0.870 | 0.194 | 0.781 | 0.198 | 0.071 | 0.517 | 0.159 | 0.015 | 1.791 | 0.596 | 0.332 |
| Stdev | ±0.011 | ±0.003 | ±0.010 | ±0.004 | ±0.010 | ±0.009 | ±0.005 | ±0.007 | ±0.001 | ±0.099 | ±0.007 | ±0.013 | |
| P-value | 5.40E-15 | 2.45E-11 | 1.81E-15 | 1.19E-11 | 2.84E-15 | 6.81E-16 | 1.24E-19 | 3.82E-16 | 2.55E-08 | 4.26E-14 | 4.86E-22 | 1.85E-19 | |
| SPRINT | Average | 0.183 | 0.873 | 0.183 | 0.781 | 0.183 | 0.057 | 0.570 | 0.167 | 0.012 | 1.523 | 0.663 | 0.380 |
| Stdev | ±0.010 | ±0.004 | ±0.009 | ±0.005 | ±0.010 | ±0.008 | ±0.008 | ±0.008 | ±0.001 | ±0.096 | 0.006 | ±0.012 | |
| P-value | 2.90E-16 | 3.02E-10 | 3.13E-16 | 2.61E-11 | 2.93E-16 | 1.19E-16 | 1.42E-16 | 1.71E-15 | 2.29E-12 | 4.15E-15 | 9.27E-24 | 2.41E-21 | |
| PSIVER | Sverage | 0.191 | 0.874 | 0.191 | 0.783 | 0.191 | 0.066 | 0.581 | 0.170 | 0.013 | 1.607 | 0.537 | 0.250 |
| Stdev | ±0.012 | ±0.003 | ±0.012 | ±0.005 | ±0.012 | ±0.012 | ±0.010 | ±0.009 | ±0.001 | ±0.118 | ±0.011 | ±0.010 | |
| P-value | 3.70E-15 | 5.02E-10 | 4.51E-15 | 6.38E-11 | 3.96E-15 | 1.82E-15 | 2.78E-15 | 6.96E-15 | 9.74E-12 | 1.38E-14 | 1.62E-19 | 5.44E-17 | |
| SPRINGS | Average | 0.229 | 0.882 | 0.228 | 0.796 | 0.229 | 0.111 | 0.625 | 0.201 | 0.015 | 2.164 | 0.500 | 0.236 |
| Stdev | ±0.013 | ±0.003 | ±0.012 | ±0.004 | ±0.012 | ±0.011 | ±0.007 | ±0.011 | ±0.001 | ±0.118 | ±0.007 | ±0.008 | |
| P-value | 1.14E-12 | 1.75E-07 | 1.01E-12 | 1.69E-08 | 1.05E-12 | 4.41E-13 | 3.99E-13 | 4.02E-12 | 1.38E-08 | 2.62E-12 | 2.54E-19 | 4.44E-17 | |
| LORIS | Average | 0.264 | 0.887 | 0.263 | 0.805 | 0.263 | 0.151 | 0.656 | 0.228 | 0.017 | 2.726 | 0.439 | 0.195 |
| Stdev | ±0.010 | ±0.003 | ±0.010 | ±0.004 | ±0.010 | ±0.009 | ±0.006 | ±0.010 | ±0.001 | ±0.101 | ±0.004 | ±0.003 | |
| P-value | 1.90E-10 | 5.77E-05 | 2.19E-10 | 9.53E-06 | 2.00E-10 | 1.35E-10 | 2.25E-10 | 9.14E-10 | 1.66E-05 | 2.73E-09 | 3.48E-17 | 1.27E-15 | |
| CRFPPI | Average | 0.268 | 0.887 | 0.264 | 0.805 | 0.266 | 0.154 | 0.681 | 0.238 | 0.017 | 2.671 | 0.448 | 0.208 |
| Stdev | ±0.011 | ±0.003 | ±0.010 | ±0.004 | ±0.010 | ±0.009 | ±0.006 | ±0.012 | ±0.001 | ±0.099 | ±0.007 | ±0.009 | |
| P-value | 8.36E-10 | 5.41E-05 | 2.54E-10 | 1.16E-05 | 4.28E-10 | 2.17E-10 | 1.64E-06 | 6.22E-08 | 4.41E-05 | 1.11E-09 | 3.55E-17 | 1.98E-14 | |
| SSWRF | Average | 0.288 | 0.891 | 0.286 | 0.811 | 0.287 | 0.178 | 0.687 | 0.256 | 0.018 | 3.093 | 0.423 | 0.209 |
| Stdev | ±0.010 | ±0.002 | ±0.010 | ±0.003 | ±0.010 | ±0.009 | ±0.006 | ±0.013 | ±0.001 | ±0.121 | ±0.006 | ±0.005 | |
| P-value | 1.30E-07 | 3.83E-03 | 1.25E-07 | 1.15E-03 | 1.22E-07 | 8.98E-08 | 1.52E-05 | 4.89E-05 | 7.27E-03 | 3.14E-06 | 4.81E-16 | 3.70E-16 | |
| SCRIBER | Average | 0.334 | 0.896 | 0.332 | 0.821 | 0.333 | 0.230 | 0.715 | 0.287 | 0.020 | 3.725 | 0.282 | 0.116 |
| Stdev | ±0.013 | ±0.004 | ±0.013 | ±0.007 | ±0.013 | ±0.016 | ±0.013 | ±0.012 | ±0.001 | ±0.258 | ±0.014 | ±0.009 |
Note: The methods are sorted by their AUC value in the ascending order. The binary predictions for all methods are calibrated to allow for direct side-by-side comparison, such that the number of putative PBRs each method generates equals to the number of the native PBRs. The results are computed over 10 subsets of randomly selected 50% of TEST proteins to assess robustness of the predictions and evaluate statistical significance of differences between SCRIBER and the other methods (Section 2.3 gives details). We report the corresponding averages, standard deviations (stdev) and P-values; differences with P-value <0.05 are assumed statistically significant and are given in bold italics font. The best value for each measure of predictive quality are shown with bold font.
3.3 Assessment of cross-predictions
The low predictive performance of current methods stems from the observation that they incorrectly recognize residues that bind other types of protein partners as PBRs, i.e. they produce a large number of cross-predictions. One of the main strengths of SCRIBER is that it offers by far the lowest levels of cross-predictions, with AUCPC = 0.28 and CPR = 0.12 (Table 2), i.e. it incorrectly predicts only 12% of residues that bind other types of protein partners as PBRs. This rate is comparable to the SCRIBER’s overall false positive rate = 1 – specificity = 10.4% (Table 2). To compare, the second lowest CPR = 0.20 is by LORIS and the highest CPR = 0.38 by SPRINT, compared to their false positive rates at 11.3% and 12.7%, respectively. Correspondingly, SCRIBER reduces the cross-predictions rates by between 41% and 69% compared to the current predictors. The corresponding CPR curves (see Supplementary Fig. S1C), clearly shows that SCRIBER generates much lower CPRs across the entire range of sensitivity, with a wide margin to the second best SSWRF.
CPR values should be substantially lower than the corresponding sensitivity since only than the rates of correct predictions for the native PBRs are higher than the rates of cross-predictions for the other types of binding residues. This is true for SCRIBER for which the ratio of sensitivity to CPR = 0.334/0.116 = 2.9 (Table 2). Only three other methods maintain ratio greater than 1: CRFPPI with ratio = 1.3 and SWRF and LORIS with ratios = 1.4. The other four methods have the ratios = 0.97 (SPRINGS), 0.76 (PSIVER), 0.61 (SPPIDER) and 0.48 (SPRINT). This means that they predict more PBRs among the other types of binding residues than among the native PBRs. This observation confirms results in (Zhang and Kurgan, 2018) and reveals that these tools in fact indiscriminately predict all types of binding residues as PBRs.
We investigate the cross-predictions for specific types of residues including native DNA-binding, RNA-binding, small ligand-binding, non-binding and all residues (see Fig. 3). The rates across all types of binding residues are by far the lowest for SCRIBER, with largest improvements for the DNA-binding, small ligand-binding and RNA-binding residues. On average, the RNA-binding residues are the most difficult to differentiate from PBRs, with SCRIBER’s CPR = 0.178, which still is much lower than CPR = 0.239 for the second best SPRINT. Overall, we conclude that only SCRIBER is truly capable of specifically predicting PBRs and differentiating them from the other types of binding residues.
Fig. 3.
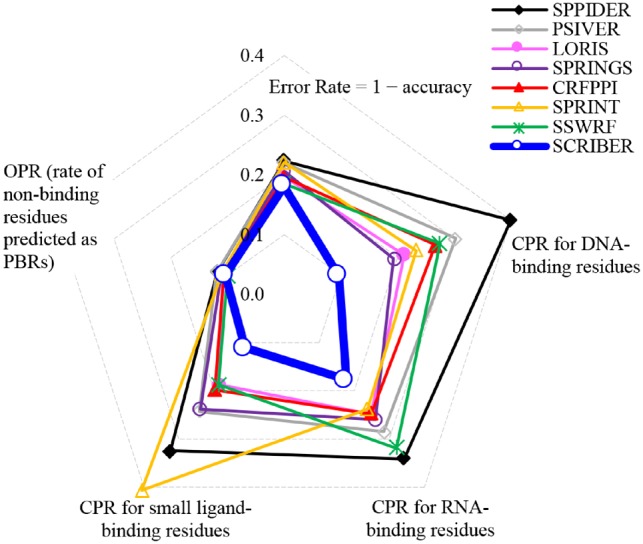
Cross-prediction rates (CPRs) for the native DNA, RNA and small ligand binding residues, over-prediction rates (OPRs) for the native non-binding residues, and Error Rates = 1 – accuracy on the TEST dataset
3.4 Comparative assessment of false positive predictions
PBRs are annotated using a threshold that is applied to the distance between atoms from the two proteins, resulting in a somehow arbitrary inclusion/exclusion of residues that are close to that cut-off. Moreover, PBRs tend to cluster in the sequence since this proximity often translates into proximity in structure. This suggests that some of the false positives that are nearby native PBRs in the sequence could be in fact involved in binding. The corresponding conjecture is that the residues in close proximity in the sequence to the native PBRs are more likely to in fact bind proteins when compared to the residues that are farther away.
Figure 4 shows distance from the nearest native PBRs in the TEST dataset for the false positives generated by all evaluated methods (each method is calibrated to predict the same number of PBRs = number of native PBRs). Inset in the figure reveals that SCRIBER predicts arguably higher quality false positives. 50% (60%) of its predicted PBRs are no farther than 1 residue away (3 residues away) from a native PBR, compared to 43% (51%) for the second best SSWRF and an average of 33% (41%) for the seven current predictors. The entire SCRIBER's curve has a much higher slope compared to the curves of the other methods, showing that its false positives are closer to the native PBRs. The curves do not reach the fraction of 1 because the remaining residues are predicted in proteins that do not have native PBRs (the distance is undefined). The corresponding fraction of PBRs residues that are predicted for proteins that do not interact with proteins (while they may interact with the other types of protein partners) is the best and equals 9.5% for SCRIBER, while is equals 12% for the second best SSWFR and 21.7% for the third best SPPIDER. Altogether, these results strongly suggest that SCRIBER predicts higher quality false positives.
Fig. 4.
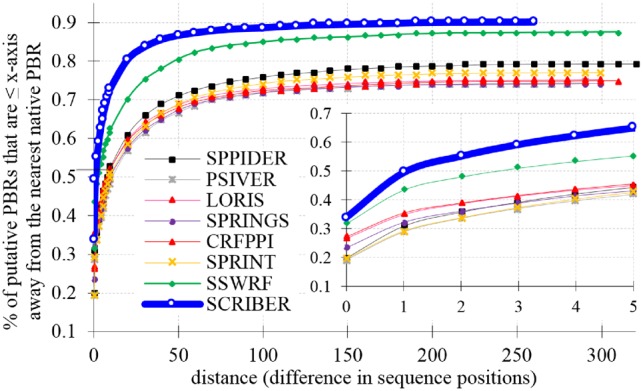
Distance between the predicted PBRs and the nearest native PBRs measured as the number of positions in the sequence for the proteins in the TEST dataset. The y-axis shows fractions of putative PBRs that are ≤ distance shown on the x-axis away from the nearest PBR. Inset in the lower right corner shows the results for the low values of the distance
3.5 Case study
Supplementary Figure S2 illustrates and compares predictions from SCRIBER and the second best SSWRF for the NADP reductase taken from the TEST dataset (Uniprot ID: O29370). SCRIBER’s and SSWRF’s AUC for this protein is similar to the AUCs on the TEST dataset. Moreover, this protein includes PBRs and residues that bind NADP (a small ligand), which allows us to study the cross-predictions. SSWRF (green markers) successfully predicts six native PBRs (correctly focusing on the cluster of native PBRs at the C-terminus) and some of its predictions are close to the native PBRs, but at the expense of also predicting four NADP-binding residues as PBRs. SCRIBER (dark blue markers) correctly predicts 12 native PBRs at the cost of 2 incorrect predictions located at the native NADP site. Moreover, these two false positives are in the cluster of PBRs at the C-terminus and are only 1 or 2 residues away from a native PBR. When comparing SCRIBER with the prediction from its first layer (light blue markers), this case study shows that addition of the second layer has substantially reduced the cross-predictions (from 7 to 2) at the expense of a modest reduction of the correct predictions of PBRs (from 17 to 12). Overall, the SCRIBER’s outputs offer a reasonably accurate approximation of the location of the native PBRs while excluding the other types of binding events.
3.6 Comparative evaluation of runtime
Runtime is an important consideration, particularly when considering large-scale applications that target big protein families or genomes. The main bottleneck of the existing tools is that they require PSSM generated with PSI-BLAST (Altschul et al., 1997). Furthermore, four of these methods (SPRINGS, LORIS, CRF-PPI and SSWRF) also use solvent accessibility produced by SANN (Joo et al., 2012). We use the time to compute the results with PSI-BLAST alone and with PSI-BLAST and SANN to approximate the lower bound of the runtime for these methods. We note that SCRIBER applies much faster HHblits and ASAquick to compute the same information. We consider complete prediction process (including computation of the entire profile, calculation of features and generation of propensities with the six regressions) to quantify runtime for SCRIBER. All computations were done on the same hardware (PC with i5 CPU and 8GB RAM) allowing us to directly compare the results. We focus on relative differences in the runtime rather than absolute values since the latter depend on the hardware used.
We measure runtime in the function of chain length for 200 proteins from the TEST dataset that uniformly sample the sequence length. Figure 5 summarizes the measured time for SCRIBER, PSI-BLAST (that estimates lower bound of runtime for SPPIDER, SPRINT and PSIVER) and PSI-BLAST+SANN (that approximates lower bound of runtime for SPRINGS, LORIS, CRF-PPI and SSWRF). The three runtime measurements scale linearly with the protein length, with SCRIBER having the smallest/best slope of the linear fit. SCRIBER also offers by far the fastest predictions, with runtime between 4 and 7.1 times faster (depending on the chain length) than PSI-BLAST+SANN and 3 to 5.5 times faster than PSI-BLAST. For an average length chain (approx. 300 residues) SCRIBER takes about 45 s per proteins, compared to PSI-BLAST that needs 194 s and PSI-BLAST+SANN that requires 246 s. Since the latter two are just lower bounds of the actual runtimes, these results indisputably show that SCRIBER offers substantially faster predictions.
Fig. 5.
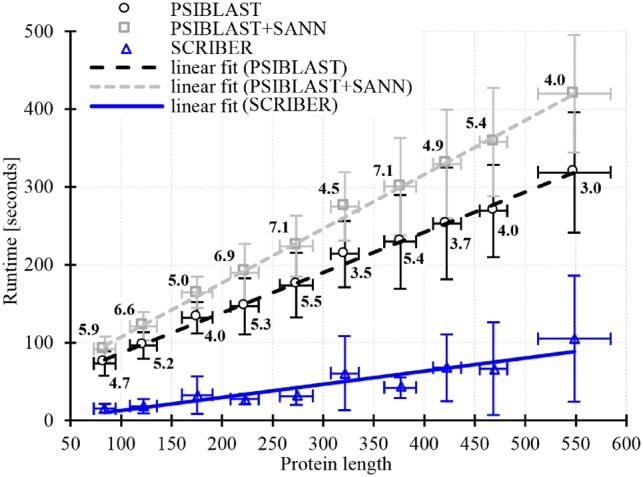
Runtime of SCRIBER and the lower bound of runtime for the other seven methods. SPPIDER, SPRINT and PSIVER runtime is estimated by the calculation of the PSSM with PSI-BLAST that they utilize to make predictions. Runtime of SPRINGS, LORIS, CRF-PPI and SSWRF is estimated by the combined time to calculate PSSM with PSI-BLAST and to predict RSA with SANN that these methods apply to predict PBRs. We use the uniprot90 database with 3 iterations of PSI-BLAST to calculate PSSMs, and default parameters and nndb database to run SANN. The y-axis shows runtime in seconds. The x-axis quantifies protein length. Each point reports median measurement of runtime and chain length, measured on 20 proteins drawn from the TEST dataset in the corresponding length range. Standard deviations of the chain length and runtime are denoted by whiskers. The lines show linear fit into the measured median values. Values next to the PSI-BLAST and PSI-BLAST+SANN lines reflect the ratio of these measurements to the corresponding value for SCRIBER, e.g. 4.7 in the lower left corner means that the PSI-BLAST’s runtime for short chains is 4.7 times higher than the SCRIBER’s runtime
3.7 Protein binding domains in the human proteome
First, we investigate whether SCRIBER accurately predicts protein-binding domains in the TEST dataset. We use Pfam (El-Gebali et al., 2019) to annotate total of 600 domains in the TEST proteins, and we map the native and putative PBRs into these domains. Figure 6 compares the corresponding fractions of domains with a given minimal number of native (black line) and putative (dark blue line) PBRs. We performed the Kolmogorov-Smirnov test to investigate whether these cumulative distributions for the native and putative PBRs are statistically different. The test rejected the hypothesis that they are different (P-value = 0.08), suggesting that SCRIBER relatively accurately estimates number of PBRs per domain. Given this result, we use the computationally efficient SCRIBER to predict PBRs in the complete human proteome (18 568 human proteins) collected from UniProt (UniProt, 2015), which we also annotated with 44 886 domains collected from Pfam (see the light blue line in Fig. 6). Past analyses of structures of protein–protein complexes show that these binding interfaces cover more than 37 amino acids (only 3% were shown to be smaller), while large interfaces cover at least 112 amino acids and are primarily found among homodimers (Bahadur et al., 2004; Bahadur and Zacharias, 2008). We use these two cut-offs to annotate the corresponding protein-binding domains and protein-binding domains that have large interfaces (see two vertical dashed lines in Fig. 6). The fractions of the corresponding protein-binding domains in the TEST datasets that are estimated using the native and putative PBRs are very similar: 21% versus 23% for all protein-binding domains, and 0% versus 2% for the protein-binding domains that have large interfaces. Using this analysis, we estimate that about 14% of the currently known domains in human proteins are protein-binding, and about 4% have large interfaces and thus are likely to be formed by homodimers. The pre-computed putative PBRs and domain annotations for the human proteome are available on the SCRIBER’s website at http://biomine.cs.vcu.edu/servers/SCRIBER/.
Fig. 6.
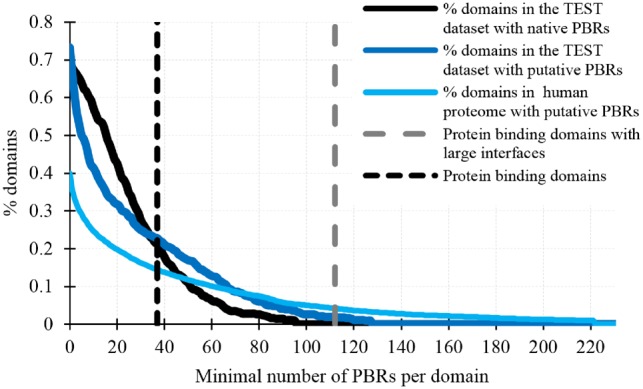
Fraction of domains that have a given minimal number of native and putative PBRs domains in the TEST dataset and the human proteome
3.8 SCRIBER webserver
A webserver that implements SCRIBER is freely available at http://biomine.cs.vcu.edu/servers/SCRIBER/. SCRIBER requires only the FASTA-formatted protein sequences as input. Users should provide email address where we send link to the results, once the prediction is completed. The same link is also provided in the browser window, but the window has to be open for the duration of the prediction. A single request can service batch predictions of up to ten protein chains. The server takes about 30 s to predict a query sequence about 200 residues. The server outputs the putative propensities for protein-, DNA-, RNA- and small ligand-binding generated by its first layer, and the putative protein-binding propensities and annotated PBRs produced by the second layer. The results are available via an HTML page and a parsable comma-separated text file. We archive the results, which can be accessed via the link, for at least one month.
4 Summary and conclusions
Recent years have witnessed the development of over a dozen sequence-based predictors of PBRs. However, these methods are only modestly accurate and produce many cross-predictions. SCRIBER outperforms the current methods by providing both statistically significantly better predictive performance and significantly reduced cross-predictions. Empirical analysis demonstrates that SCRIBER is the only method that can accurately differentiate PBRs from the other types of binding residues. The empirical tests also reveal that the novel design ideas implemented in SCRIBER strongly contribute to its high predictive performance. These innovations includes use of novel and combined input features and application of putative propensities for protein-, DNA-, RNA- and small ligand-binding to reduce cross-predictions of PBRs. We show that SCRIBER predicts higher quality false positives (located closer to the native PBR) than the current predictors; i.e. about 60% of PBRs predicted by SCRIBER are no farther than 3 residues away from a native PBR. Furthermore, our conservative estimates demonstrate that SCRIBER’s predictions are generated at least three times faster than the results of the current tools. Altogether, we conclude that SCRIBER delivers accurate, partner type-specific and runtime-efficient sequence-based predictions of PBRs.
Recent literature shows that the current sequence-based predictors of PBRs find various practical applications including estimation of protein–protein binding affinity (Lu et al., 2018) and functional characterization of a wide array of proteins (Banadyga et al., 2017; Burgos et al., 2015; Mahboobi et al., 2015; Mahita and Sowdhamini, 2017; Ntostis et al., 2015; Wiech et al., 2015; Yang et al., 2017; Yoshimaru et al., 2017). They are also applied in the context of personalized medicine as part of a platform for prediction of functional effects for single point variants or mutations (Hecht et al., 2015). Availability of more accurate and faster tools, such as SCRIBER, is likely to attract additional users and open other areas of applications.
One of the SCRIBER’s limitations is that it does not provide information about the binding partner(s) for the predicted PBRs. The corresponding partner-specific methods predict residues involved in a particular PPI; i.e. inter-protein residue-residue contacts. A few such predictors that rely on machine learning-derived models were published in recent years (Ahmad and Mizuguchi, 2011; Fout et al., 2017; Minhas et al., 2014; Sanchez-Garcia et al., 2019). They make accurate predictions when using protein structures as the input while their sequence-based versions are substantially less accurate (Sanchez-Garcia et al., 2019). These versions use rather simple profiles that include the sequence itself, evolutionary information and putative solvent accessibility. SCRIBER-inspired architecture that uses more comprehensive profile should provide improvements when used for the partner-specific predictions. We are also planning to extend SCRIBER to predict interactions with other biomolecules, such as DNA, RNA and small ligands. This extension should be addressed with multi-labels models to accommodate for the fact that some amino acids interact with multiple types of partners. So far only a few methods that predict binding residues for multiple partner types are available (Carson et al., 2010; Peng and Kurgan, 2015; Peng et al., 2017; Su et al., 2018; Wang et al., 2010; Yan and Kurgan, 2017; Zhang et al., 2017), and none of them covers such a wide range of partners or applies multi-label models.
Supplementary Material
Acknowledgements
We are grateful to Dr Chen Wang for his contribution to setup the webserver.
Funding
This work was supported in part by the National Science Foundation (grant 1617369), National Natural Science Foundation of China (grant 61802329), the Robert J. Mattauch Endowment funds, the Innovation Team Support Plan of University Science and Technology of Henan Province (grant 19IRTSTHN014), the Science and Technology Department of Henan Province (grant 192102310478), and by the Nanhu Scholars Program for Young Scholars of the Xinyang Normal University.
Conflict of Interest: none declared.
References
- Ahmad S., Mizuguchi K. (2011) Partner-aware prediction of interacting residues in protein–protein complexes from sequence data. PLoS One, 6, e29104.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed K.S. et al. (2011) Improving the prediction of yeast protein function using weighted protein–protein interactions. Theor. Biol. Med. Model, 8, 11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F. et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadur R.P. et al. (2004) A dissection of specific and non-specific protein–protein interfaces. J. Mol. Biol., 336, 943–955. [DOI] [PubMed] [Google Scholar]
- Bahadur R.P., Zacharias M. (2008) The interface of protein–protein complexes: analysis of contacts and prediction of interactions. Cell. Mol. Life Sci., 65, 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banadyga L. et al. (2017) Ebola virus VP24 interacts with NP to facilitate nucleocapsid assembly and genome packaging. Sci. Rep., 7, 7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M. et al. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan D.W.A. et al. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res., 41, W349–W357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos E.S. et al. (2015) Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J. Biol. Chem., 290, 9674–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone A. et al. (2013) Mentha: a resource for browsing integrated protein-interaction networks. Nat. Methods, 10, 690–691. [DOI] [PubMed] [Google Scholar]
- Carson M.B. et al. (2010) NAPS: a residue-level nucleic acid-binding prediction server. Nucleic Acids Res., 38, W431–W435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Kurgan L. (2009) Investigation of atomic level patterns in protein–small ligand interactions. PLoS One, 4, e4473.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Li J. (2010) Sequence-based identification of interface residues by an integrative profile combining hydrophobic and evolutionary information. BMC Bioinformatics, 11, 402.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X-w., Jeong J.C. (2009) Sequence-based prediction of protein interaction sites with an integrative method. Bioinformatics, 25, 585–591. [DOI] [PubMed] [Google Scholar]
- Cook K.B. et al. (2015) High-throughput characterization of protein-RNA interactions. Brief. Funct. Genomics, 14, 74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Las Rivas J., Fontanillo C. (2012) Protein–protein interaction networks: unraveling the wiring of molecular machines within the cell. Brief. Funct. Genomics, 11, 489–496. [DOI] [PubMed] [Google Scholar]
- Dhole K. et al. (2014) Sequence-based prediction of protein–protein interaction sites with L1-logreg classifier. J. Theor. Biol., 348, 47–54. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z. et al. (2009) ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics, 25, 2745–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X. et al. (2009) Improved prediction of protein binding sites from sequences using genetic algorithm. Protein J., 28, 273–280. [DOI] [PubMed] [Google Scholar]
- Dyson H.J. (2012) Roles of intrinsic disorder in protein–nucleic acid interactions. Mol. Biosyst., 8, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S. et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res., 47, D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J.J. et al. (2006) Protein–RNA interactions: structural analysis and functional classes. Proteins, 66, 903–911. [DOI] [PubMed] [Google Scholar]
- Esmaielbeiki R. et al. (2016) Progress and challenges in predicting protein interfaces. Brief. Bioinf., 17, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraggi E. et al. (2014) Accurate single-sequence prediction of solvent accessible surface area using local and global features. Proteins, 82, 3170–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figeys D. (2002) Functional proteomics: mapping protein–protein interactions and pathways. Curr. Opin. Mol. Ther., 4, 210–215. [PubMed] [Google Scholar]
- Fout A. et al. (2017) Protein interface prediction using graph convolutional networks. Advances in Neural Information Processing Systems, pp. 6530–6539. [Google Scholar]
- Geng H. et al. (2015) Prediction of protein–protein interaction sites based on naive Bayes classifier. Biochem. Res. Int., 2015, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht M. et al. (2015) Better prediction of functional effects for sequence variants. BMC Genomics, 16, S1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G. et al. (2018) Quality assessment for the putative intrinsic disorder in proteins. Bioinformatics, 35, 1692–1700. [DOI] [PubMed] [Google Scholar]
- Jia J. et al. (2016) iPPBS-Opt: a sequence-based ensemble classifier for identifying protein–protein binding sites by optimizing imbalanced training datasets. Molecules, 21, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo K. et al. (2012) solvent accessibility prediction of proteins by nearest neighbor method. Proteins Struct. Funct. Bioinf., 80, 1791–1797. [DOI] [PubMed] [Google Scholar]
- Kawashima S. et al. (2007) AAindex: amino acid index database, progress report 2008. Nucleic Acids Res., 36, D202–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J. et al. (2012) Protein–RNA interactions: new genomic technologies and perspectives. Nat. Rev. Genet., 13, 77–83. [DOI] [PubMed] [Google Scholar]
- Kuzmanov U., Emili A. (2013) Protein–protein interaction networks: probing disease mechanisms using model systems. Genome Med., 5, 37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune D. et al. (2005) Protein-nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins, 61, 258–271. [DOI] [PubMed] [Google Scholar]
- Li F. et al. (2018) Quokka: a comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics, 34, 4223–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G.-H. et al. (2016) Prediction of protein–protein interaction sites with machine-learning-based data-cleaning and post-filtering procedures. J. Membr. Biol., 249, 141–153. [DOI] [PubMed] [Google Scholar]
- Lu B. et al. (2018) ProBAPred: inferring protein–protein binding affinity by incorporating protein sequence and structural features. J. Bioinform. Comput. Biol., 16, 1850011. [DOI] [PubMed] [Google Scholar]
- Mahboobi S.H. et al. (2015) The interaction of RNA helicase DDX3 with HIV-1 Rev-CRM1-RanGTP complex during the HIV replication cycle. PLoS One, 10, e0112969.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari S., Brylinski M. (2015) Predicting protein interface residues using easily accessible on-line resources. Brief. Bioinform., 16, 1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahita J., Sowdhamini R. (2017) Integrative modelling of TIR domain-containing adaptor molecule inducing interferon-beta (TRIF) provides insights into its autoinhibited state. Biol. Direct., 12, 9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F., Kurgan L. (2016) DFLpred: high-throughput prediction of disordered flexible linker regions in protein sequences. Bioinformatics, 32, i341–i350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F., Kurgan L. (2018) High-throughput prediction of disordered moonlighting regions in protein sequences. Proteins, 86, 1097–1110. [DOI] [PubMed] [Google Scholar]
- Meng F. et al. (2016) Compartmentalization and functionality of nuclear disorder: intrinsic disorder and protein–protein interactions in intra-nuclear compartments. Int. J. Mol. Sci., 17, E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhas F. et al. (2014) PAIRpred: partner-specific prediction of interacting residues from sequence and structure. Proteins, 82, 1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizianty M.J. et al. (2010) Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics, 26, i489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y., Mizuguchi K. (2010) Applying the Naïve Bayes classifier with kernel density estimation to the prediction of protein–protein interaction sites. Bioinformatics, 26, 1841–1848. [DOI] [PubMed] [Google Scholar]
- Nibbe R.K. et al. (2011) Protein–protein interaction networks and subnetworks in the biology of disease. Wiley Interdiscip. Rev. Syst. Biol. Med., 3, 357–367. [DOI] [PubMed] [Google Scholar]
- Ntostis P. et al. (2015) Evidence for association of the rs605059 polymorphism of HSD17B1 gene with recurrent spontaneous abortions. J. Matern Fetal Neonatal Med., 28, 2250–2253. [DOI] [PubMed] [Google Scholar]
- Obradovic Z. et al. (2005) Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins, 61, 176–182. [DOI] [PubMed] [Google Scholar]
- Ofran Y., Rost B. (2007) ISIS: interaction sites identified from sequence. Bioinformatics, 23, e13–e16. [DOI] [PubMed] [Google Scholar]
- Orii N., Ganapathiraju M.K. (2012) Wiki-pi: a web-server of annotated human protein–protein interactions to aid in discovery of protein function. PLoS One, 7, e49029.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil A. et al. (2010) Hub promiscuity in protein–protein interaction networks. Int. J. Mol. Sci., 11, 1930–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg O. et al. (2014) Evolution of specificity in protein–protein interactions. Biophys. J., 107, 1686–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z., Kurgan L. (2015) High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res., 43, e121.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z. et al. (2014a) Genome-scale prediction of proteins with long intrinsically disordered regions. Proteins, 82, 145–158. [DOI] [PubMed] [Google Scholar]
- Peng Z. et al. (2014b) A creature with a hundred waggly tails: intrinsically disordered proteins in the ribosome. Cell Mol. Life Sci., 71, 1477–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z. et al. (2017) Prediction of disordered RNA, DNA, and protein binding regions using DisoRDPbind. Methods Mol. Biol., 1484, 187–203. [DOI] [PubMed] [Google Scholar]
- Petta I. et al. (2016) Modulation of protein–protein interactions for the development of novel therapeutics. Mol. Ther., 24, 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porollo A., Meller J. (2006) Prediction-based fingerprints of protein–protein interactions. Proteins Struct. Funct. Bioinf., 66, 630–645. [DOI] [PubMed] [Google Scholar]
- Remmert M. et al. (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods, 9, 173–175. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia R. et al. (2019) BIPSPI: a method for the prediction of partner-specific protein–protein interfaces. Bioinformatics, 35, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G. et al. (2014) SPRINGS: prediction of protein–protein interaction sites using artificial neural networks. PeerJ PrePrints 2:e266v2, https://doi.org/10.7287/peerj.preprints.266v2. [Google Scholar]
- Song J. et al. (2018) PROSPERous: high-throughput prediction of substrate cleavage sites for 90 proteases with improved accuracy. Bioinformatics, 34, 684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio O. (2012) Editorial: toward the design of drugs on protein–protein interactions. Curr. Pharm. Des., 18, 4585.. [DOI] [PubMed] [Google Scholar]
- Su H. et al. (2018) Improving the prediction of protein-nucleic acids binding residues via multiple sequence profiles and the consensus of complementary methods. Bioinformatics, 35, 930–936. [DOI] [PubMed] [Google Scholar]
- Taherzadeh G. et al. (2016) Sequence-based prediction of protein–peptide binding sites using support vector machine. J. Comput. Chem., 37, 1223–1229. [DOI] [PubMed] [Google Scholar]
- UniProt C.U. (2015) a hub for protein information. Nucleic Acids Res., 43, D204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M. et al. (2015) Functional advantages of conserved intrinsic disorder in RNA-binding proteins. PLoS One, 10, e0139731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. et al. (2016) Disordered nucleiome: abundance of intrinsic disorder in the DNA- and RNA-binding proteins in 1121 species from Eukaryota, Bacteria and Archaea. Proteomics, 16, 1486–1498. [DOI] [PubMed] [Google Scholar]
- Wang D.D. et al. (2014) Fast prediction of protein–protein interaction sites based on extreme learning machines. Neurocomputing, 128, 258–266. [Google Scholar]
- Wang L. et al. (2010) BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst. Biol., 4, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z.-S. et al. (2016) Protein–protein interaction sites prediction by ensembling SVM and sample-weighted random forests. Neurocomputing, 193, 201–212. [Google Scholar]
- Wei Z.-S. et al. (2015) A cascade random forests algorithm for predicting protein–protein interaction sites. IEEE Trans. Nanobiosci., 14, 746–760. [DOI] [PubMed] [Google Scholar]
- Wiech E.M. et al. (2015) Molecular modeling and computational analyses suggests that the Sinorhizobium meliloti periplasmic regulator protein ExoR adopts a superhelical fold and is controlled by a unique mechanism of proteolysis. Protein Sci., 24, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. et al. (2015) In various protein complexes, disordered protomers have large per-residue surface areas and area of protein-, DNA- and RNA-binding interfaces. FEBS Lett., 589, 2561–2569. [DOI] [PubMed] [Google Scholar]
- Xie Z. et al. (2011) Systematic characterization of protein–DNA interactions. Cell. Mol. Life Sci., 68, 1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L.C. et al. (2011) HomPPI: a class of sequence homology based protein–protein interface prediction methods. BMC Bioinformatics, 12, 244.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J. et al. (2016) Molecular recognition features (MoRFs) in three domains of life. Mol. Biosyst., 12, 697–710. [DOI] [PubMed] [Google Scholar]
- Yan J., Kurgan L. (2017) DRNApred fast sequence-based method that accurately predicts and discriminates DNA- and RNA-binding residues. Nucleic Acids Res., 45, e84.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. (2012) BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res., 41, D1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.M. et al. (2017) Co-chaperone BAG2 determines the pro-oncogenic role of Cathepsin B in triple-negative breast cancer cells. Cell Rep., 21, 2952–2964. [DOI] [PubMed] [Google Scholar]
- Yoshimaru T. et al. (2017) A-kinase anchoring protein BIG3 coordinates oestrogen signalling in breast cancer cells. Nat. Commun., 8, 15427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Kurgan L. (2018) Review and comparative assessment of sequence-based predictors of protein-binding residues. Brief. Bioinform., 19, 821–837. [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. (2017) Comprehensive review and empirical analysis of hallmarks of DNA-, RNA- and protein-binding residues in protein chains. Brief. Bioinform, doi: 10.1093/bib/bbx168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.