

Abstract
Background and Aims
Differences in local abundance and ploidy level are predicted to impact the direction of introgression between species. Here, we tested these hypotheses on populations of Betula albosinensis (red birch) and Betula platyphylla (white birch) which were thought to differ in ploidy level, the former being tetraploid and the latter diploid.
Methods
We sampled 391 birch individuals from nine localities in China, and classified them into species based on leaf morphology. Twelve nuclear microsatellite markers were genotyped in each sample, and analysed using principal coordinates analysis and STRUCTURE software. We compared the effects of two different methods of scoring polyploid genotypes on population genetic analyses. We analysed the effect of ploidy, local species abundance and latitude on levels of introgression between the species.
Key Results
Leaf morphology divided our samples into red and white birch, but genetic analyses unexpectedly revealed two groups within red birch, one of which was tetraploid, as expected, but the other of which appeared to have diploid microsatellite genotypes. Five individuals were identified as early-generation hybrids or backcrosses between white birch and red birch and five were identified between red birch and ‘diploid’ red birch. Cline analysis showed that levels of admixture were not significantly correlated with latitude. Estimated genetic differentiation among species was not significantly different between determined tetraploid and undetermined tetraploid genotypes.
Conclusions
Limited hybridization and gene flow have occurred between red birch and white birch. Relative species abundance and ploidy level do not impact the direction of introgression between them, as genetic admixture is roughly symmetrical. We unexpectedly found populations of apparently diploid red birch and this taxon may be a progenitor of allotetraploid red birch populations. Incomplete lineage sorting may explain patterns of genetic admixture between apparently diploid and allotetraploid red birch.
Keywords: Betula, genetic structure, hybridization, microsatellite, morphometric analysis, PCO, symmetric introgression
INTRODUCTION
Introgression, the transfer of genetic materials across species boundaries via hybridization and backcrossing, is a common phenomenon in plants and animals (Arnold, 2006; Rieseberg et al., 2007). It plays an important role in species evolution, introducing diversity and sometimes adaptive alleles into populations (Whitney et al., 2010; Abbott et al., 2013). Understanding the direction of introgression can inform species conservation and management. Several factors are predicted to influence the direction of introgression. Species of high local abundance are likely to produce a lower proportion of hybrid offspring than species of low local abundance, resulting in asymmetrical introgression (Hubbs, 1955; Mayr, 1963; Wirtz, 1999; Burgess et al., 2005; Lepais et al., 2009). Introgression of genes is more likely from diploid to tetraploid populations than vice versa (Stebbins, 1971) because both diploids and hybrid triploids are more likely to produce diploid gametes than tetraploids are to produce haploid gametes. To date, only a few molecular studies testing Stebbins’ hypothesis have been reported: for example, asymmetrical introgression was reported to occur from Betula nana (2x) into B. pubescens (4x) (Wang et al., 2014a; Eidesen et al., 2015; Zohren et al., 2016) and from Miscanthus sinensis (2x) into M. sacchariflorus (4x) (Clark et al., 2015). In addition, asymmetrical hybridization can occur due to demographic history (Buggs, 2007; Currat et al., 2008) and asymmetrical mating barriers (Martin and Willis, 2007). When more than one of these factors is in play for hybridizing species, it may be hard to predict the direction of introgression.
Species of Betula provide an excellent system to investigate the patterns of introgression. This genus includes ~65 species, subspecies and varieties, with distributions spanning from the subtropics to the arctic region (Ashburner and McAllister, 2013). Polyploidy within Betula is common, with nearly 60 % of its species estimated to be polyploids, and the ploidy level ranging from diploidy to dodecaploidy (Ashburner and McAllister, 2013; Wang et al., 2016). Extensive hybridization and introgression within Betula, especially within the subgenus Betula, have been investigated based on morphology, cytogenetics, molecular markers and genome size analysis (Johnsson, 1945; Barnes et al., 1974; Anamthawat-Jónsson and Tómasson, 1990, 1999; Anamthawat-Jónsson and Thórsson, 2003; Anamthawat-Jónsson et al., 2010; Wang et al., 2014a, b; Eidesen et al., 2015; Tsuda et al., 2017). Hybridization and introgression also occur among Betula species of different subgenera (Barnes et al., 1974; Thomson et al., 2015). Triploid individuals have been frequently observed, indicating hybridization between differing ploidy levels (Anamthawat-Jónsson and Tómasson, 1999).
In this study, we selected two species of the subgenus Betula: red birch of section Costatae and white birch of section Betula (Ashburner and McAllister, 2013). In China, both species are widely distributed from the south-west to the north, with white birch having a more northern distribution and often being found at lower altitudes. Their ranges overlap substantially, giving plenty of opportunities for hybridization. In some areas, red birch is limited to sparse mountain-top populations, whereas white birch is widespread and much more abundant on lower slopes. In other areas, the two are intermixed with roughly similar abundance. Birch, like some other plant species, shows shifts in altitudinal range in response to climate change (Liang et al., 2018; Du et al., 2018). As the climate warms, white birch tends to move up to higher altitudes, coming into contact with red birch, giving enhanced potential for hybridization to occur. The two species differ in ploidy level, with red birch reported as tetraploid and white birch as diploid (Ashburner and McAllister, 2013; Wang et al., 2016).
Despite a wide overlapping distribution between red birch and white birch, to our knowledge no studies have been conducted to investigate if hybridization and introgression occur between the two species. If hybridization does occur, we do not know how patterns of introgression might be affected by differences in local abundance and ploidy level. Hence, in the present study, we aimed to address the following questions: (1) Do hybridization and introgression occur between these two widespread species? (2) If introgression occurs, what is its direction? (3) How do species abundance and ploidy level affect the direction of introgression? To this end, we genotyped 391 birch individuals to infer rates of hybridization and direction of introgression between red birch and white birch.
MATERIAL AND METHODS
Sampling
Samples were collected by the research group during the summers of 2016 and 2017. Sampling locations were recorded using a GPS system (UniStrong) (Table 1). For most individuals, we collected a specimen and dried it naturally in a press until DNA extraction. For some individuals where trees were too tall to collect twigs, we just took cambium tissues. We left a minimum of 20–30 m between samples to avoid the chance of repeated sampling of the same clone. In total, we collected 391 individuals: 241 were identified by morphology as red birch and 150 as white birch. We collected from a total of nine populations, six of which contained both red and white birch, and three of which contained only red birch (Fig. 1).
Table 1.
Sampling information and genetic diversity of 16 populations of red birch, ‘diploid’ red birch and white birch based on microsatellites. Populations with less than seven samples are not included
| Code | Species* | Location | Latitude (°N) | Longitude (°E) | Gene diversity | Allelic richness |
|---|---|---|---|---|---|---|
| XYB | R (18) | Ningshan County | 33.47 | 108.5 | 0.78 | 5.99 |
| XYB | D (7) | Ningshan County | 33.47 | 108.5 | 0.68 | 4.3 |
| BYS | D (20) | Song County | 33.65 | 111.83 | 0.61 | 3.84 |
| TY | R (38) | Taibai County | 34 | 107.29 | 0.78 | 5.82 |
| TY | W (32) | Taibai County | 34 | 107.29 | 0.59 | 4.06 |
| TTH | R (30) | Feng County | 34.27 | 106.52 | 0.78 | 6.12 |
| TTH | W (23) | Feng County | 34.27 | 106.52 | 0.59 | 4.16 |
| MJS | R (8) | Maiji Mountain | 34.34 | 106.02 | 0.77 | 5.47 |
| SWP | R (24) | Shunwangping | 35.43 | 111.97 | 0.76 | 5.77 |
| SWP | W (15) | Shunwangping | 35.43 | 111.97 | 0.66 | 4.4 |
| PQG | R (32) | Pangquangou | 37.89 | 111.43 | 0.8 | 6.11 |
| PQG | W (21) | Pangquangou | 37.89 | 111.43 | 0.67 | 4.5 |
| BSH | R (20) | Baishi Mountain | 39.21 | 114.69 | 0.8 | 6.11 |
| BSH | W (19) | Baishi Mountain | 39.21 | 114.69 | 0.61 | 4.41 |
| XLS | R (25) | Xiling Mountain | 40.03 | 115.32 | 0.8 | 6.34 |
| XLS | W (30) | Xiling Mountain | 40.03 | 115.32 | 0.65 | 4.65 |
*R, D and W represent red birch, ‘diploid’ red birch and white birch, respectively. The number in parentheses is the sample size. One location (BYS) for red birch and one location (TTH) for ‘diploid’ red birch were not included in this table as sample numbers there are less than seven.
Fig. 1.

Map showing sampling sites. Orange circles and green triangles represent red birch and white birch, respectively. Asterisks indicate populations where ‘diploid’ red birch was found.
Morphometrics
One to five intact and mature leaves from pressed specimens retaining leaves were selected for morphometric analysis. A total of 382 mature leaves from 139 red birch individuals and 218 leaves from 74 white birch individuals were scanned using a Hewlett-Packard printer (LaserJet Pro MFP M128fn) with a resolution of 600 d.p.i. Thirteen landmarks were selected from each scanned leaf as described in previous studies (Jensen et al., 2002; Viscosi et al., 2009; Liu et al., 2018). These were: the distal tip of petiole (1), the junction between the petiole and the leaf blade (2), the first serration on the right-hand side of the leaf (3), the right-hand point of one-fifth of the leaf from the lower side (4), the right-hand point of the widest part of leaf (5), the right-hand point of three-fifths of the leaf from the lower side (6), the right-hand point of four-fifths of each leaf from the lower side (7), the leaf tip (8), the left-hand point of four-fifths of the leaf from the lower side (9), the left-hand point of three-fifths of the leaf from the lower side (10), the left-hand point of the widest part of the leaf (11), the left-hand point of one-fifth of the leaf from the lower side (12) and the first serration on the left-hand side of the leaf (13) (Supplementary Data, Fig. S1). The x and y coordinates of each landmark were recorded on digitized leaves using the program ImageJ (Abràmoff et al., 2004). The 13 landmarks were converted to a configuration of 26 cartesian coordinates in 13 pairs (x, y) for each leaf. Cartesian coordinates of landmarks were recorded on digital images and were stored in a ‘.txt’ file. Principal component analyses (PCAs) on the normalized matrix were conducted on the level of each leaf and each specimen, respectively, using the program MORPHOJ (Klingenberg, 2011).
DNA extraction and microsatellite genotyping
We isolated total genomic DNA from cambial tissue from naturally dried specimens following a modified 2× CTAB (cetyltrimethylammonium bromide) protocol (Wang et al., 2013). The isolated DNA pellets were resuspended in 50–100 µL TE buffer and were assessed with 1.0 % agarose gels. The DNA was diluted to a final concentration of 5–20 ng µL–1 for subsequent use. Twelve microsatellite loci developed for Betula pendula (Kulju et al., 2004), B. pubescens subsp. tortuosa (Truong et al., 2005) and B. maximowicziana (Tsuda et al., 2009) were used for genotyping our samples (Supplementary Data, Table S1). The 5′ terminus of the forward primers was labelled with FAM, HEX or TAM. We amplified each microsatellite marker individually prior to artificially combining them into four multiplexes. In each multiplex, some loci with significant length difference were labelled using the same dye and others with different dyes to avoid potential errors caused by size overlapping. For a subset of samples, polymerase chain reactions (PCRs) were conducted combining between two and four pairs of microsatellites in each multiplex using Qiagen master mix. The final reaction volume was 10 µL, including 5 µL Qiagen PCR Master Mix, 0.2 µL of primers (10 µm each in initial volume), 3.6 µL H2O and 5–20 ng of DNA dissolved in 1.0 µL TE buffer. For red birch, an initial denaturation step at 94 °C for 30 s was followed by 28 cycles of denaturation (94 °C for 30 s), annealing (57 °C for 90 s) and extension (72 °C for 60 s) steps, and a final extension step at 60 °C for 30 min. For white birch, we used the same settings for PCR except a final extension step at 72 °C for 5 min to achieve better results. Fragment lengths were determined by capillary gel electrophoresis using an ABI 3730xl capillary sequencer (Applied Biosystems). We scored alleles using the software GENEMARKER 2.4.0 (Softgenetics) based on relative peak heights and checked these manually. Individuals with more than three missing loci were excluded, resulting in 371 individuals in the final dataset. For tetraploids, where loci were heterozygous we manually estimated the number of copies of each allele using the relative heights of allele peaks. For example, if a tetraploid individual has two alleles A and B, with roughly equal intensity of peak signal; we designated its genotype as AABB. If the signal of allele A is roughly three times as that of allele B, we encoded it as AAAB. For some individuals, the peaks were insufficiently clear to determine allele copy number in this way.
Population structure analyses
We analysed the microsatellite data in STRUCTURE 2.3.4 (Pritchard et al., 2000) to identify the most likely number of genetic clusters (K) with a ploidy level setting of 4. STRUCTURE implements algorithms accounting for genotypic uncertainty when the data include polyploids with partially heterozygous loci. Admixed individuals and putative hybrids could be identified as they have ancestry from different genetic clusters. We created two datasets for STRUCTURE analysis. In dataset 1 (hereafter D1), we did not use our manual estimates of allele copy numbers in partially heterozygous tetraploid individuals. This approach has been adopted in many previous studies. For example, if a tetraploid has three alleles A, B and C, we encoded it as ABC-9; if it has two alleles A and B, we left it as AB-9-9 where -9 represents missing data. In dataset 2 (D2), we used our manual estimates of allele copy number of partially heterozygous tetraploid individuals.
We performed ten replicates of the STRUCTURE analysis (with 1000 000 generations and a burn-in of 100 000 for each run) at each value of K from 2 to 6, under the admixture model, with an assumption of correlated allele frequencies among populations. Individuals were assigned to clusters based on the highest membership coefficient averaged over the ten independent runs. ΔK was calculated based on the rate of change in the log probability of the data between successive K values (Evanno et al., 2005) using the program Structure Harvester (Earl and vonHoldt, 2011). Replicate runs were grouped based on a symmetrical similarity coefficient of >0.9 using the Greedy algorithm in CLUMPP (Jakobsson and Rosenberg, 2007) and visualized in DISTRUCT 1.1 (Rosenberg, 2004). We chose the optimal value of K based on the ΔK analysis of the STRUCTURE outputs.
We performed principal coordinates (PCO) analysis of D1 and D2 using POLYSAT (Clark and Jasieniuk, 2011) implemented in R 2.15.3 (R Core Team, 2012), based on pairwise genetic distances calculated according to Bruvo et al. (2004). Wright’s fixation index, Fst was also calculated among species for both D1 and D2. Note that POLYSAT assumes that the allele copy number is always ambiguous in microsatellite data for heterozygotic polyploid loci.
The software FSTAT (Goudet, 2001) was used to calculate gene diversity (Nei, 1987) and allelic richness (EI Mousadik and Petit, 1996) using D2. As our dataset includes diploid and tetraploid individuals, we treated tetraploid genotypes as two diploid individuals as described by Tsuda et al. (2017). For a few individuals with any ambiguous locus, if a locus had three alleles we duplicated the last allele while if had two alleles we left it unchanged.
Comparison of level of admixture
We divided our sampling locations into three groups based on the relative abundance of red and white birch estimated in field observations. The first group (Group 1) comprises locations XYB, BYS and MJS where no white birch was observed; the second (Group 2) comprises locations TY, TTH, SWP, PQG and BSH where red and white birch had equal abundance, or red birch was at slightly higher abundance; and the third group (Group 3) is the XLS location where red birch was sparsely distributed at the top of a mountain and white birch was extremely abundant below. As STRUCTURE results of D1 and D2 were different for K = 3 and above, we compared the level of admixture between STRUCTURE results of D1 and D2 when K = 2 using a paired t-test. In addition, we compared if admixture was symmetrical between white birch (2x) and red birch (4x) based on the STRUCTURE result of D1 for K = 3.
All statistical analyses were conducted in R 2.15.3 (R Core Team, 2012). We first conducted the Shapiro–Wilk normality test of genetic admixture values for each location from white birch into red birch and vice versa using the ‘shapiro.test’ function. For values which are not normally distributed, we conducted a Mann–Whitney U-test to test the difference between genetic admixture from white birch into red birch and from red birch into white birch in each location, using the function ‘wilcox.test’. The difference in genetic admixture from red birch into white birch between groups 2 and 3 was also tested using a Mann–Whitney U-test. The effects of groups on ‘genetic admixture from red birch into white birch’ were tested by the Kruskal–Wallis test, using the function ‘kruskal.test’.
To determine if genetic admixture from white birch into red birch is correlated with latitude, we performed a mixed effects model using the lme function in R 2.15.3 (Pinheiro and Bates, 2000) as described in previous studies (Wang et al., 2014a; Zohren et al., 2016).
ITS sequencing
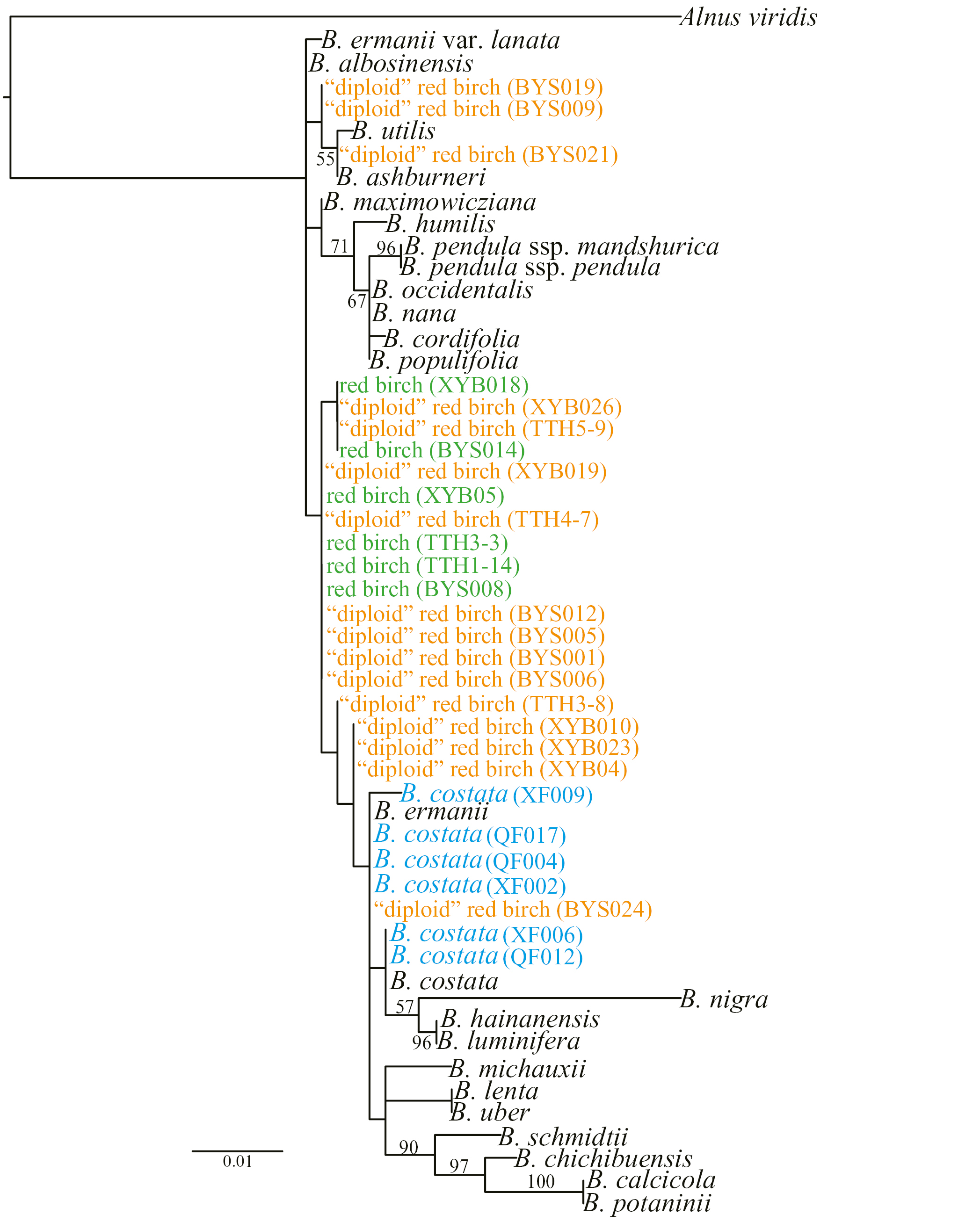
The nuclear ribosomal internal transcribed spacer (nrITS) region (ITS1, 5.8S and ITS2) was amplified using primers ITS4 (White et al., 1990) and ITSLeu (Baum et al., 1998). The volume of the reaction mix was 20 µL, containing: 10 µL of mix (Tiangen), 0.5 µL of each primer (10 mm), 10 µL of ddH2O and 1 µL of diluted DNA (10–20 ng). The PCR was carried out using the same programme as described by Wang et al. (2016). The PCR products were sent to Tsingke Company (Qingdao city) for sequencing. A total of 28 individuals were selected for Sanger sequencing, representing 22 morphologically identified red birch and six B. costata (2x) (GenBank accession numbers MK453256–MK453283). Betula costata is a close relative of red birch and has a more northern distribution (Ashburner and McAllister, 2013). This species was included to check if it formed a cluster with red birch. We incorporated ITS sequences of another 24 ‘verified’ accessions for subsequent analysis. Alnus viridis was chosen as the outgroup. In total, 53 sequences were aligned using BioEdit v7.0.9.0 (Hall, 1999) with default parameters and the alignment edited manually where necessary. A maximum-likelihood (ML) analysis was conducted in RAxML-8.2.11 (Stamatakis, 2014) with a rapid bootstrap analysis with 100 bootstraps and ten searches under a GTR+Γ nucleotide substitution model. The phylogenetic tree was visualized in FigTree v.1.3.1 and edited in Adobe Illustrator CS6 (Adobe Systems).
RESULTS
Morphometric analysis
PCA on leaf morphology identified two distinct clusters. PC1 separated white birch from red birch, summarizing 50.4 % of the variation among leaves (Fig. 2A) and 61.5 % of the variation among individuals (Fig. 2B), whereas PC2 explained 10.3 % of the variation among leaves (Fig. 2A) and 9.4 % of the variation among individuals (Fig. 2B).
Fig. 2.

Principal components analysis performed among leaves (A) and among individuals (B). Each dot in A and B represents each leaf and each individual, respectively.
Microsatellite analysis
Bruvo’s genetic distances among all 371 individuals were calculated and scaled for D1 and D2. PCO analysis based on Bruvo’s genetic distances were conducted for D1 and D2 and gave similar patterns (Fig. 3). The first axis separated white birch from red birch and the second axis unexpectedly separated red birch into two distinct clusters (Fig. 3). On closer examination, it became clear that all individuals in the smaller genetic cluster within red birch possessed no more than two alleles for each of the 12 microsatellite loci: thus they were diploid-like. For the rest of the paper we term these ‘diploid’ red birch (Supplementary Data, Fig. S2).
Fig. 3.

Principal coordinates analysis for microsatellite datasets D1 (A) and D2 (B) based on Bruvo’s distances.
STRUCTURE analysis of D1 and D2 with K = 2 assigned populations that corresponded well to the white birch and red birch morphological groups (Fig. 4). With K = 3, D1 assigned a third population (Fig. 4A), which corresponded well to the ‘diploid’ red birch group whereas D2 showed a more complex pattern within red birch that did not assign the ‘diploid’ red birches to their own population, but seemed to differentiate the parental sub-genomes of the tetraploid red birches, suggesting that one of the sub-genomes was the same as the genome of ‘diploid’ red birch (Fig. 4B). With K = 4, both D1 and D2 assigned the ‘diploid’ red birches to their own population, and both had a pattern of assignments within the remaining red birches, suggesting an allopolyploid origin (Fig. 4). Hence, hereafter we refer to these as ‘allotetraploid’ red birch. Based on the ΔK criterion (Supplementary Data Fig. S3), K = 3 was the most suitable value for D1 and K = 4 for D2.
Fig. 4.

STRUCTURE output for microsatellite datasets D1 (A) and D2 (B) at K = 2, 3 and 4. The optimal cluster number for D1 and D2 is 3 and 4, respectively, based on the ΔK value. Sampling locations are separated by thin black lines and labelled at the bottom of the figure. At K = 2, individuals predominantly coloured green are white birch and individuals predominately coloured yellow are red birch. At K = 4, individuals predominately coloured orange are ‘diploid’ red birch.
STRUCTURE analysis of D1 for K = 3 identified five ‘allotetraploid’ red birch individuals with high levels (>0.1) of admixture from white birch, and one white birch individual with admixture from both ‘diploid’ and ‘allotetraploid’ red birch. All of the admixed red birch individuals were ‘allotetraploid’. This analysis also identified six individuals with high levels of admixture between ‘allotetraploid’ red birch and ‘diploid’ red birch. (Fig. 4A). All five of the individuals with high admixture between ‘allotetraploid’ red birch and white birch were from localities containing both of these species (three from the XLS population and two from TY and BSH) (Fig. 4A). Five of the individuals with high admixture between ‘allotetraploid’ red birch and ‘diploid’ red birch were collected from two sympatric populations (XYB and TTH) and one was from a population (SWP) where pure ‘diploid’ red birch was not detected. There were many cases of low (<0.1) admixture between ‘allotetraploid’ and ‘diploid’ red birch, but fewer between red and white birch.
Levels of admixture of ‘diploid’ red birch alleles into ‘allotetraploid’ red birch decreased from south to north, but this was not significant (P = 0.055) (Fig. 5A). Similarly, there was no significant latitudinal trend of introgression of white birch alleles into ‘allotetraploid’ red birch (P = 0.076) (Fig. 5B).
Fig. 5.

Clines of ‘diploid’ red birch and white birch admixture in ‘allotetraploid’ red birch populations. The horizontal axis and circles show the latitude of each sample populations and logit-transformed STRUCTURE admixture proportions for each ‘allotetraploid’ red birch individual, respectively. Red diamonds indicate the value for each ‘allotetraploid’ red birch population fitted by the mixed effects model. (A) The cline of ‘diploid’ red birch admixture into ‘allotetraploid’ red birch populations, which showed a non-significant but negative correlation with latitude (P = 0.0551). (B) The cline of white admixture into ‘allotetraploid’ red birch populations, which showed a non-significant but negative correlation with latitude (P = 0.0755).
Gene diversity for each locality was 0.76–0.8 for ‘allotetraploid’ red birch, 0.61–0.68 for ‘diploid’ red birch and 0.59–0.67 for white birch. Allelic richness was 5.47–6.34 for ‘allotetraploid’ red birch, 3.84–4.3 for ‘diploid’ red birch and 4.06–4.65 for white birch (Table 1). Tukey post hoc tests showed that gene diversity and allelic richness were higher in ‘allotetraploid’ red birch than in white birch (P = 0) and were similar between ‘diploid’ red birch and white birch (P = 0.79 and 0.39 for gene diversity and allelic richness, respectively).
Comparison of genetic admixture
For the six populations of white birch and red birch with sympatric or parapatric distributions, genetic admixture from white birch into ‘allotetraploid’ red birch shows no significant difference with that from ‘allotetraploid’ red birch into white birch except population BHS (P = 0.009, Mann–Whitney U test). In addition, no significant difference was detected between Groups 1 and 2 but a significant difference was revealed between Groups 1 and 3 (P = 0.009, Dunn post hoc test) and between Groups 2 and 3 (P = 0.01, Dunn post hoc test) as regard with genetic admixture from white birch into ‘allotetraploid’ red birch (Fig. 6).
Fig. 6.

Genetic admixtures between ‘allotetraploid’ red birch and white birch in Groups (Gr) 1, 2 and 3. Admixture values are based on STRUCTURE results for dataset D1 at K = 3. Black and grey represent genetic admixture from ‘allotetraploid’ red birch into white birch and from white birch into ‘allotetraploid’ red birch, respectively.
Paired t-tests showed no significant difference in genetic admixture from white birch into red birch between STRUCTURE results for D1 and D2 for K = 2 (P = 0.062, paired t-test). A marginally significant difference was detected for Group 3 between STRUCTURE results for D1 and D2 for K = 2 (P = 0.024, paired t-test).
Phylogeny based on ITS sequences
A phylogeny based on ITS sequences showed that most of the ‘diploid’ red birch clustered with ‘allotetraploid’ red birch but with low support values (Supplementary Data Fig. S4).
DISCUSSION
Scoring heterozygotic genotypes in polyploids
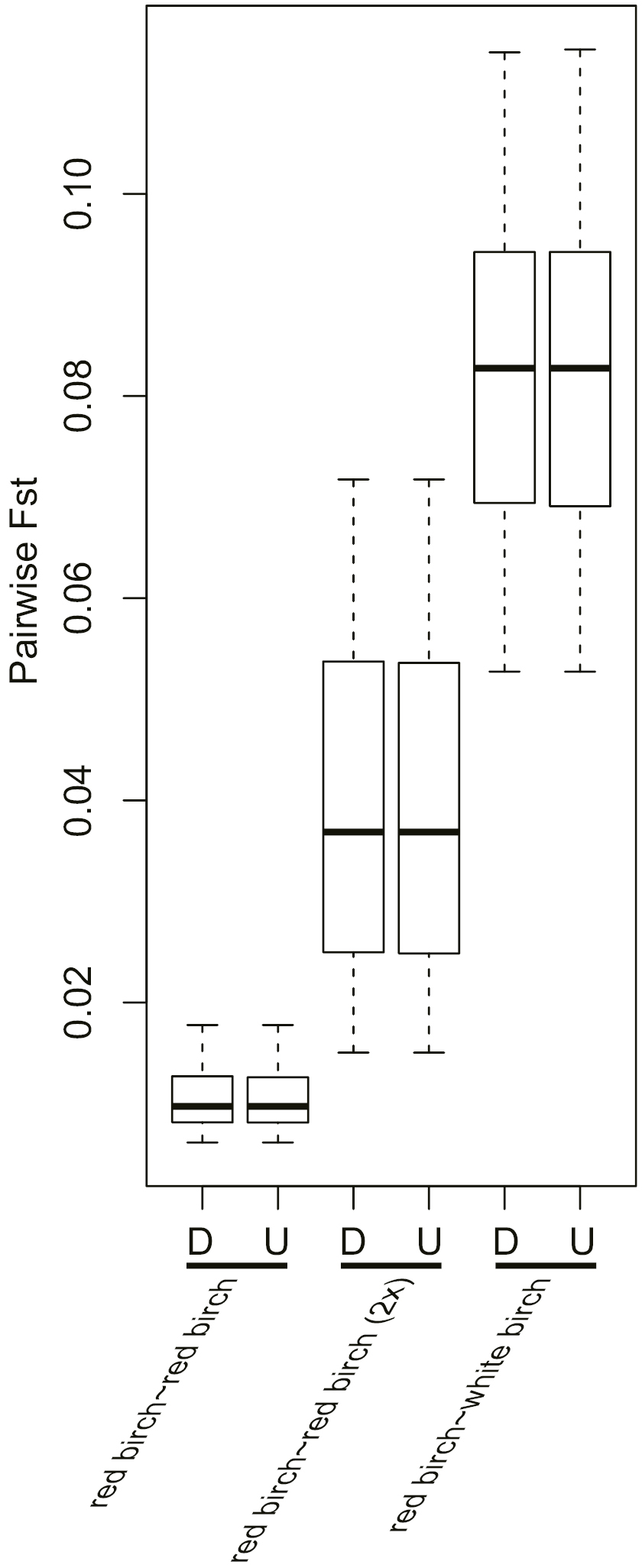
Many previous studies of admixture between diploids and polyploids have scored the alleles present in polyploid individuals without determining the full heterozygote genotypes, as in our dataset D1 (García‐Verdugo et al., 2013; Münzbergová et al., 2013). This may affect the conclusions drawn because the frequency of rare alleles may be over-estimated and that of common alleles under-estimated if the occurrence of multiple alleles in one individual is ignored. For example, when Tsuda et al. (2017) determined full tetraploid genotypes (as in our dataset D2) at seven microsatellite loci, they found that genetic differentiation among B. pendula (2x), B. pubescens (4x) and B. nana (2x) was less pronounced than found by Wang et al. (2014a) who did not fully determine genotypes. It is not clear whether this difference is due to genotyping methods, as the Tsuda et al. (2017) and Wang et al. (2014a) studies are on different samples and different loci.
In our study, using both genotyping methods on the same set of data, we found highly similar genetic differentiation among populations within white, ‘allotetraploid’ red and ‘diploid’ red birch (Supplementary Data Fig. S5). In our STRUCTURE analyses, D1 (undetermined genotypes) and D2 (determined genotypes) both showed distinct clusters for the three species groups at their optimal values of K (K = 3 for D1 and K = 4 for D2) (Fig. 4). They both showed clusters for white birch and red birch at K = 2, with no significant differences in estimated genetic admixture from white birch into red birch in each. A marginally significant difference was only detected for Group 3 where genetic admixture appeared higher for the D2 dataset.
A distinct ‘diploid’ red birch
As mentioned above, STRUCTURE analyses revealed a distinct cluster within red birch at optimal K values for datasets D1 and D2 (Fig. 4). This was further corroborated by PCO analysis of microsatellite data (Fig. 3). In contrast, morphometric analysis revealed only two distinct clusters, corresponding to white birch and red birch, respectively (Fig. 2). Together, these data pointed toward a cryptic lineage of red birch which was previously unrecognized based on morphological characters. Examination of the microsatellite calls showed that at most two alleles were observed for each locus from all these cryptic individuals (Supplementary Data Fig. S2). Hence, we concluded that they may be a diploid cytotype of red birch. STRUCTURE analysis using dataset D2 at K = 3 suggests that this ‘diploid’ red birch may be a progenitor of ‘allotetetraploid’ red birch. If this is the case, the allopolyploidization event was long enough ago for the sub-genome within the allotetraploid to have differentiated from the diploid genome, as it forms a separate group in the STRCUTURE analysis of D2 at K = 4. A phylogeny based on ITS showed that most of the ‘diploid’ red birch individuals clustered with ‘allotetraploid’ red birch (Supplementary Data Fig. S4), indicating a close relationship.
It is possible that this ‘diploid’ red birch is one of two previously described close relatives of red birch: B. ashburneri or B. costata. Betula ashburneri is a multi-stemmed shrub or shrubby tree, reported from south-east Tibet (Ashburner and McAllister, 2013). However, the ‘diploid’ red birch we detected is not a multi-stemmed shrub, suggesting it is not B. ashburneri. Betula costata has its distribution in North China but STRUCTURE analysis including this species showed it to form a distinct genetic cluster to ‘diploid’ red birch (our unpublished data). Based on this evidence, we infer that ‘diploid’ red birch is likely to be a cytotype of B. albosinensis. Multiple cytotypes are common and have been observed for some Betula species such as the hexaploid and the octoploid cytotype of B. chinensis (Ashburner and McAllister, 2013) and the diploid and the tetraploid cytotype of B. pendula (Salojarvi et al., 2017).
Chromosome counting or genome size analysis is needed to confirm the ploidy level of ‘diploid’ red birch. At this stage, we cannot rule out the possibility of it being an autotetraploid as microsatellite alleles may seem similar between the diploid cytotype and the autotetraploid cytotype. A similar case has been proposed by Zohren et al. (2016) where a UK birch individual appeared to be diploid based on restriction-site associated DNA (RAD) data but had a tetraploid genome size. If there is a ploidy level difference between ‘allotetraploid’ red and ‘diploid’ red birch, this would help explain their differentiation. The ‘diploid’ red birches were detected in three locations, with three individuals, seven individuals and 20 individuals sampled from TTH, XYB and BYS, respectively. In contrast, ‘allotetraploid’ red birch was present in all locations we sampled. This indicated that the ‘diploid’ red birch is possibly rare compared with ‘allotetraploid’ red birch and may need conservation.
Limited hybridization and genetic admixture
Five individuals were likely to be early-generation hybrids or backcrosses between white birch and ‘allotetraploid’ red birch, as they had an admixture proportion in STRUCTURE (dataset D1, K = 3) of >0.1. One of these had an admixture proportion of >0.2. These are a much lower proportion of early-generation hybrids or backcrosses than found in a study of hybridization between B. lenta and B. alleghanensis and between B. papyrifera and B. alleghanensis (Thomson et al., 2015). The ploidy level difference between white birch and ‘allotetraploid’ red birch may be responsible for such a low incidence of hybridization, as triploid individuals are generally less fertile (Levin, 1975). Interestingly, we found less evidence for admixture between ‘diploid’ red birch and white birch than we did between ‘allotetraploid’ red birch and white birch. This could suggest that the ploidy level difference is not an important factor in differentiation, or that the ‘diploid’ red birch is autopolyploid.
A possible alternative explanation for the signal of admixture that we found in our STRUCTURE analyses is incomplete lineage sorting (ILS) rather than introgressive hybridization. ILS is likely among species with long generation times and a large effective population size (Bouillé and Bousquet, 2005; Chen et al., 2010). Unlike hybridization, ILS is not expected to give a strong geographical signal (Barton, 2001). In our STRUCTURE analyses we found that ‘allotetraploid’ red birches tended to have higher levels of admixture from white birch in locations where white birch was also present, and lower levels in locations XYB and BYS where white birch was absent. This suggests introgressive hybridization, not ILS. In contrast, ‘allotetraploid’ red birch individuals had a low level of apparent admixture from ‘diploid’ red birch in all populations, even when ‘diploid’ red birch was absent; this suggests ILS.
Further research with a more comprehensive sampling is needed to understand the relative importance of ILS versus hybridization in this system. For example, we detected one ‘allotetraploid’ red birch in population SWP with a high level of apparent admixture from ‘diploid’ red birch, despite the lack of ‘diploid’ red birch individuals in the local area. This could be due to ILS as stated above or the presence of hybrid or early backcrosses in the absence of one parental species, which has been demonstrated in some plant species, such as oaks (Dodd and Afzal-Rafii, 2004) and pines (Lanner and Phillips, 1992). Three hypotheses can explain the presence of hybrids in the absence of one parental species: long-distance dispersal of pollen, local extinction of one parental species and sampling bias. Long-distance dispersal of pollen is likely for birches as they are wind-pollinated species and their pollen can remain viable for a long period (Hjelmroos, 1991; Skjøth et al., 2007; Ashburner and McAllister, 2013). Range expansion of one species into the range of another has the potential to cause local extinction of the declining species in birch (Wang et al., 2014a; Eidesen et al., 2015; Zohren et al., 2016). This type of history would be expected to cause gradients of genetic admixture into the zone of expansion (Buggs, 2007). However, based on the currently available evidence, the demographic history of ‘diploid’ red birch and ‘allotetraploid’ red birch remains unresolved. Our third hypothesis is that ‘diploid’ red birch was not sampled from SWP due to its rarity and the difficulty of distinguishing its morphological characteristics from ‘allotetraploid’ red birch among the widespread stands of birch on the mountain. Hence, a more comprehensive sampling is needed to determine the presence or absence of ‘diploid’ red birch at the SWP site, and more generally to assess the roles of ILS and hybridization in the genetic structure of the system.
Patterns of introgression
The relative abundance of hybridizing species can impact hybridization dynamics (Hubbs, 1955; Nason et al., 1992; Burgess et al., 2005; Lepais et al., 2009). A species with lower abundance is expected to receive more pollen from a species of higher abundance and F1 hybrids will also tend to receive pollen from the more abundant species. Hence, backcrossing and introgression tends to occur toward the more abundant species (Lepais et al., 2009). In this study, we did not observe such a pattern. For example, in the XLS population where white birch is much more abundant than ‘allotetraploid’ red birch, the level of introgression from ‘allotetraploid’ red birch into white birch is not higher than vice versa. In addition, we also failed to observe higher levels of introgression from white birch into ‘allotetraploid’ red birch in populations where ‘allotetraploid’ red birch has a higher or roughly equal abundance than white birch. One possible explanation is that birch trees can vary a great deal in the amounts of pollen they produce (Bradshaw, 1981). Individuals in a species of a lower abundance may produce more pollen per tree than a species of a higher abundance (Healy, 1997). In the XLS population, ‘allotetraploid’ red birch is much rarer than white birch, but ‘allotetraploid’ red birch occurs higher up the mountain and at lower population density. The lack of directional admixture may be due to higher pollen production by the ‘allotetraploid’ red birch trees which have less light competition. Plants in shadier conditions tend to invest more resources in vegetative growth and decrease their allocation to sexual production (Charnov, 1982; Solomon, 1985; Jacquemyn et al., 2010). We also observed that many birch individuals do not bear flowers or fruits in shady conditions (our field observations). Another possible reason is that F1 hybrids were selected against and failed to reach the adult age. Genetic admixture seems symmetrical between ‘allotetraploid’ red birch and white birch in all except the BSH population where genetic admixture from white birch into ‘allotetraploid’ red birch is slightly higher than vice versa (P = 0.009). This result is in contrary to many studies showing asymmetrical or unidirectional introgression from diploid species into polyploid species (Field et al., 2011; Zohren et al., 2016).
CONCLUSIONS
In the present study, we detected limited hybridization and gene flow between red birch and white birch, but species abundance and ploidy level did not appear to impact the direction of introgression in the locations studied. We unexpectedly discovered the occurrence of red birch trees that are genetically differentiated from other red birch trees, and have genotypes that suggest that they are diploid rather than tetraploid. Tetraploid red birch may have a hybrid origin, with ‘diploid’ red birch as one progenitor. Patterns of apparent admixture between ‘diploid’ red birch and ‘allotetraploid’ red birch may be due to ILS, but further research is needed on the differences between the two, and on their evolutionary history.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Figure S1: The 13 landmarks used for PCA of leaf from red birch and white birch, respectively. Figure S2: An example of allele profiles for each of the 12 microsatellite loci for red birch (A), ‘diploid’ red birch (B) and white birch (C). Figure S3: ΔK values of STRUCTURE output of datasets D1 (A) and D2 (B), respectively. Figure S4: Phylogenetic tree based on ITS sequences using the maximum likelihood (ML) approach. Only bootstrap values above 50 % are shown. Figure S5: Genetic differentiation within populations of ‘allotetraploid’ red birch, between ‘allotetraploid’ red birch and ‘diploid’ red birch, and between ‘allotetraploid’ red birch and white birch. Fst values were calculated based on datasets with tetraploid genotypes determined (D) and undetermined (U). Table S1: Summary of the 12 polymorphic microsatellite loci used for all samples.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We are grateful to Prof. Richard Nichols from the School of Biological and Chemical Sciences, Queen Mary University of London, and Dr Yongpeng Ma at Kunming Institute of Botany Chinese Academy of Sciences for valuable comments on the manuscript. This work was funded by the National Natural Science Foundation of China (31600295 and 31770230), the Natural Science Foundation of Shandong province (ZR2016CQ09), and Funds of Shandong ‘Double Tops’ Program (SYL2017XTTD13).
LITERATURE CITED
- Abbott R, Albach D, Ansell S, et al. 2013. Hybridization and speciation. Journal of Evolutionary Biology 26: 229–246. [DOI] [PubMed] [Google Scholar]
- Abràmoff MD, Magalhães PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics International 11: 36–42. [Google Scholar]
- Anamthawat-Jónsson K, Tómasson T. 1990. Cytogenetics of hybrid introgression in Icelandic birch. Hereditas 112: 65–70. [Google Scholar]
- Anamthawat-Jónsson K, Tómasson T. 1999. High frequency of triploid birch hybrid by Betula nana seed parent. Hereditas 130: 191–193. [Google Scholar]
- Anamthawat-Jónsson K, Thórsson AT. 2003. Natural hybridisation in birch: triploid hybrids between Betula nana and B. pubescens. Plant Cell Tissue and Organ Culture 75: 99–107. [Google Scholar]
- Anamthawat-Jónsson K, Thórsson AT, Temsch EM, Greilhuber J. 2010. Icelandic birch polyploids – the case of perfect fit in genome size. Journal of Botany 347254. [Google Scholar]
- Arnold M. 2006. Evolution through genetic exchange. Oxford: Oxford University Press. [Google Scholar]
- Ashburner K, McAllister HA. 2013. The genus Betula: a taxonomic revision of birches. London: Kew Publishing. [Google Scholar]
- Barnes BV, Bruce PD, Sharik TL. 1974. Natural hybridization of yellow birch and white birch. Forest Science 20: 215–221. [Google Scholar]
- Barton NH. 2001. The role of hybridisation in evolution. Molecular Ecology 10: 551–568. [DOI] [PubMed] [Google Scholar]
- Baum DA, Small RL, Wendel JF. 1998. Biogeography and floral evolution of Baobabs (Adansonia, Bombacaceae) as inferred from multiple data sets. Systematic Biology 47: 181–207 [DOI] [PubMed] [Google Scholar]
- Bouillé M, Bousquet J. 2005. Trans-species shared polymorphisms at orthologous nuclear gene loci among distant species in the conifer Picea (Pinaceae): implications for the long-term maintenance of genetic diversity in trees. American Journal of Botany 92: 63–73. [DOI] [PubMed] [Google Scholar]
- Bradshaw RHW. 1981. Modern pollen-representation factors for woods in south-east England. Journal of Ecology 69: 45–70. [Google Scholar]
- Bruvo R, Michiels NK, D’Souza TG, Schulenburg H. 2004. A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Molecular Ecology 13: 2101–2106. [DOI] [PubMed] [Google Scholar]
- Buggs RJA. 2007. Empirical study of hybrid zone movement. Heredity 99: 301–312. [DOI] [PubMed] [Google Scholar]
- Burgess KS, Morgan M, Deverno L, Husband BC. 2005. Asymmetrical introgression between two Morus species (M. alba, M. rubra) that differ in abundance. Molecular Ecology 14: 3471–3483. [DOI] [PubMed] [Google Scholar]
- Charnov EL. 1982. The theory of sex allocation. Princeton: Princeton University Press. [PubMed] [Google Scholar]
- Chen J, Kallman T, Gyllenstrand N, Lascoux M. 2010. New insights on the speciation history and nucleotide diversity of three boreal spruce species and a Tertiary relict. Heredity 104: 3–14. [DOI] [PubMed] [Google Scholar]
- Clark LV, Jasieniuk M. 2011. POLYSAT: an R package for polyploid microsatellite analysis. Molecular Ecology Resources 11: 562–566. [DOI] [PubMed] [Google Scholar]
- Clark LV, Stewart JR, Nishiwaki A, et al. 2015. Genetic structure of Miscanthus sinensis and Miscanthus sacchariflorus in Japan indicates a gradient of bidirectional but asymmetric introgression. Journal of Experimental Botany 66: 4213–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currat M, Ruedi M, Petit RJ, Excoffier L. 2008. The hidden side of invasions: massive introgression by local genes. Evolution 62: 1908–1920. [DOI] [PubMed] [Google Scholar]
- Dodd RS, Afzal-Rafii Z. 2004. Selection and dispersal in a multispecies oak hybrid zone. Evolution 58: 261–269. [PubMed] [Google Scholar]
- Du HB, Liu J, Li MH, et al. 2018. Warming-induced upward migration of the alpine treeline in the Changbai Mountains, northeast China. Global Change Biology 24: 1256–1266. [DOI] [PubMed] [Google Scholar]
- EI Mousadik A, Petit RJ. 1996. High level of of genetic differentiation for alleloc richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic of Morocco. Theoretical and Applied Genetics 92: 832–839. [DOI] [PubMed] [Google Scholar]
- Eidesen PB, Alsos IG, Brochmann C. 2015. Comparative analyses of plastid and AFLP data suggest different colonization history and asymmetric hybridization between Betula pubescens and B. nana. Molecular Ecology 24: 3993–4009. [DOI] [PubMed] [Google Scholar]
- Earl DA, vonHoldt BM. 2011. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources 4: 359–361. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Field D, Ayre D, Whelan R, Young A. 2011. Patterns of hybridization and asymmetrical gene flow in hybrid zones of the rare Eucalyptus aggregata and common E. rubida. Heredity 106: 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Verdugo C, Calleja JA, Vargas P, Silva L, Moreira O, Pulido F. 2013. Polyploidy and microsatellite variation in the relict tree Prunus lusitanica L.: how effective are refugia in preserving genotypic diversity of clonal taxa?. Molecular Ecology 22: 1546–1557. [DOI] [PubMed] [Google Scholar]
- Goudet J. 2001. FSTAT (version 2.9.3): a program to estimate and test gene diversities and fixation indices Avaliable from www.unil.ch/izea/softwares/fasta.html.
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98. [Google Scholar]
- Healy WH. 1997. Thinning New England oak stands to enhance acorn production. Northern Journal of Applied Forestry 14:152–156. [Google Scholar]
- Hjelmroos M. 1991. Evidence of long-distance transport of Betula pollen. Grana 30: 215–228. [Google Scholar]
- Hubbs CL. 1955. Hybridization between fish in nature. Systematic Zoology 4: 1–20. [Google Scholar]
- Jacquemyn H, Brys R, Jongejans E. 2010. Size-dependent flowering and costs of reproduction affect population dynamics in a tuberous perennial woodland orchid. Journal of Ecology 98: 1204–1215. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA. 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806. [DOI] [PubMed] [Google Scholar]
- Jensen RJ, Ciofani KM, Miramontes LC. 2002. Lines, outlines, and landmarks: morphometric analyses of leaves of Acer rubrum, Acer saccharinum (Aceraceae) and their hybrid. Taxon 51: 475–492. [Google Scholar]
- Johnsson H. 1945. Interspecific hybridization within the genus Betula. Hereditas 31: 163–176. [DOI] [PubMed] [Google Scholar]
- Klingenberg CP. 2011. MORPHOJ: an integrated software package for geometric morphometrics. Molecular Ecology Resources 11: 353–357. [DOI] [PubMed] [Google Scholar]
- Kulju KKM, Pekkinen M, Varvio S. 2004. Twenty-three microsatellite primer pairs for Betula pendula (Betulaceae). MolecularEcology Notes 4: 471–473. [Google Scholar]
- Lanner RM, Phillips AMI. 1992. Natural hybridization and introgression of pinyon pines in northwestern Arizona. International Journal of Plant Sciences 153: 250–257. [Google Scholar]
- Lepais O, Petit RJ, Guichoux E, Lavabre JE, Alberto F, Kremer A, Gerber S. 2009. Species relative abundance and direction of introgression in oaks. Molecular Ecology 18: 2228–2242. [DOI] [PubMed] [Google Scholar]
- Levin D. 1975. Minority cytotype exclusion in local plant populations. Taxon 24: 35–43. [Google Scholar]
- Liang QL, Xu XT, Mao KS, Wang MC, Wang K, Xi ZX, Liu JQ. 2018. Shifts in plant distributions in response to climate warming in a biodiversity hotspot, the Hengduan Mountains. Journal of Biogeography 45: 1334–1344. [Google Scholar]
- Liu Y, Li YJ, Song JL, Zhang RP, Yan Y, Wang YY, Du FK. 2018. Geometric morphometric analyses of leaf shapes in two sympatric Chinese oaks: Quercus dentata Thunberg and Quercus aliena Blume (Fagaceae). Annals of Forest Science 75: 90. [Google Scholar]
- Münzbergová Z, Šurinová M, Castro S. 2013. Absence of gene flow between diploids and hexaploids of Aster amellus at multiple spatial scales. Heredity 110: 123–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NH, Willis JH. 2007. Ecological divergence associated with mating system causes nearly complete reproductive isolation between sympatric Mimulus species. Evolution 61: 68–82. [DOI] [PubMed] [Google Scholar]
- Mayr E. 1963. Animal species and evolution. Cambridge: Harvard University Press. [Google Scholar]
- Nason JD, Ellstrand NC, Arnold ML. 1992. Patterns of hybridization and introgression in populations of oaks, manzanitas and irises. American Journal of Botany 79: 101–111. [Google Scholar]
- Nei M. 1987. Molecular evolutionary genetics. New York: Columbia University Press. [Google Scholar]
- Pinheiro J, Bates D. 2000. Mixed-effects models in S and S-PLUS. New York: Springer. [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team 2012. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Rieseberg LH, Kim SC, Randell RA, Whitney KD, Gross BL, Lexer C, Clay K. 2007. Hybridization and the colonization of novel habitats by annual sunflowers. Genetica 129: 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA. 2004. DISTRUCT: a program for the graphical display of population structure. Molecular Ecology Notes 4: 137–138. [Google Scholar]
- Salojarvi J, Smolander OP, Nieminen K, et al. 2017. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nature Genetics 49: 904–912. [DOI] [PubMed] [Google Scholar]
- Skjøth CA, Sommer J, Stach A, Smith M, Brandt J. 2007. The long-range transport of birch (Betula) pollen from Poland and Germany causes significant pre‐season concentrations in Denmark. Clinical and Experimental Allergy 37: 1204–1212. [DOI] [PubMed] [Google Scholar]
- Solomon BP. 1985. Environmentally influenced changes in sex expression in an andromonoecious plant. Ecology 66: 1321–1332. [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins GL. 1971. Chromosomal evolution in higher plants. London: Edward Arnold. [Google Scholar]
- Thomson AM, Dick CW, Pascoini AL Dayanandan S. 2015. Despite introgressive hybridization, North American birches (Betula spp.) maintain strong differentiation at nuclear microsatellite loci. Tree Genetics and Genomes 11: 1–12. [Google Scholar]
- Truong C, Palme AE, Felber F, Naciri-Graven Y. 2005. Isolation and characterization of microsatellite markers in the tetraploid birch, Betula pubescens ssp. tortuosa. Molecular Ecology Notes 5: 96–98. [Google Scholar]
- Tsuda Y, Lascoux M, Tsuda Y, Sebastiani F, Vendramin GG, Semerikov V. 2017. Multispecies genetic structure and hybridization in the Betula genus across Eurasia. Molecular Ecology 26: 589–605 [DOI] [PubMed] [Google Scholar]
- Tsuda Y, Ueno S, Ide Y, Tsumura Y. 2009. Development of 14 EST-SSRs for Betula maximowicziana and their applicability to related species. Conservation Genetics 10: 661–664. [Google Scholar]
- Viscosi V, Fortini P, Slice DE, Loy A, Blasi C. 2009. Geometric morphometric analyses of leaf variation in four oak species of the subgenus Quercus (Fagaceae). Plant Biosystems 143: 575–587. [Google Scholar]
- Wang N, Thomson M, Bodles WJA, et al. 2013. Genome sequence of dwarf birch (Betula nana) and cross-species RAD markers. Molecular Ecology 22: 3098–3111. [DOI] [PubMed] [Google Scholar]
- Wang N, Borrell JS, Bodles WJA, Kuttapitiya A, Nichols RA, Buggs RJA. 2014a . Molecular footprints of the Holocene retreat of dwarf birch in Britain. Molecular Ecology 23: 2771–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Borrell JS, Buggs RJA. 2014b . Is the Atkinson discriminant function a reliable method for distinguishing between Betula pendula and B. pubescens? New Journal of Botany 4: 90–94. [Google Scholar]
- Wang N, McAllister HA, Bartlett P, Buggs RJA. 2016. Molecular phylogeny and genome size evolution of the genus Betula (Betulaceae). Annals of Botany 117: 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TJ, Bruns T, Lee S, Taylor T. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfland DH, Sninsky JJ, White TJ, eds. PCR protocols: a guide to methods and applications. New York: Academic press, 315–322. [Google Scholar]
- Whitney KD, Randell RA, Rieseberg LH. 2010. Adaptive introgression of abiotic tolerance traits in the sunflower Helianthus annuus. New Phytologist 187: 230–239. [DOI] [PubMed] [Google Scholar]
- Wirtz P. 1999. Mother species–father species: unidirectional hybridization in animals with female choice. Animal Behaviour 58: 1–12. [DOI] [PubMed] [Google Scholar]
- Zohren J, Wang N, Kardailsky I, et al. 2016. Unidirectional diploid-tetraploid introgression among British birch trees with shifting ranges shown by restriction site-associated markers. Molecular Ecology 25: 2413–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.