

Abstract
A combination of bioinformatics, steady-state kinetics, and NMR spectroscopy has revealed the catalytic functions of YcjQ, YcjS and YcjR from the ycj gene cluster in Escherichia coli K-12. YcjS was determined to be a 3-keto-d-glucoside dehydrogenase with a kcat = 22 s−1, and kcat/Km = 2.3 × 104 M−1 s−1 for the reduction of methyl α-3-keto-d-glucopyranoside at pH 7.0 with NADH. YcjS also exhibited catalytic activity for the NAD+-dependent oxidation of d-glucose, methyl β-d-glucopyranoside, and 1,5-anhydro-d-glucitol. YcjQ was determined to be a 3-keto-d-guloside dehydrogenase with kcat = 18 s−1, and kcat/Km = 2.0 × 103 M−1 s−1 for the reduction of methyl α-3-keto-gulopyranoside. This is first reported dehydrogenase for the oxidation of d-gulose. YcjQ also exhibited catalytic activity with d-gulose and methyl β-d-gulopyranoside. The 3-keto products from both dehydrogenases were found to be extremely labile under alkaline conditions. The function of YcjR was demonstrated to be a C-4 epimerase that interconverts 3-keto-d-gulopyranosides to 3-keto-d-glucopyranosides. These three enzymes, YcjQ, YcjR, and YcjS, thus constitute a previously unrecognized metabolic pathway for the transformation of d-gulosides to d-glucosides via the intermediate formation of 3-keto-d-guloside and 3-keto-d-glucoside.
Graphical Abstract
Introduction
We are colonized by billions of microorganisms since birth (1). Apart from the protective effect the gut microbes exert by preventing colonization by pathogens, microbiota serve many important functions in nutrient and drug metabolism, as well as modulation of the gut immune system (2–5). Imbalances, or dysbiosis, in the bacterial composition of the gut can be linked to a number of diseases such as obesity, diabetes, and cancer (6–8). Recent studies have also linked Alzheimer’s disease, Parkinson’s disorder, and psychiatric illnesses to microbial dysbiosis (9–11).
There has been a marked increase in the interest of the gut microbiota due, in part, to the technological advancements in DNA sequencing and gnotobiotics (12–14). A study by Qin et al. found that there are an estimated 3.3 million genes present in our gut metagenome, compared to the 23,000 protein-coding genes in humans (15). Broadly classified, the genes in the metagenome serve either bacterial housekeeping functions or have roles specific to the survival of the bacteria in the gut (15). One of the most obvious functions required for their survival is aiding in the absorption and metabolism of macromolecules. The microbial ‘glycobiome’ contains a large number of genes that facilitate the degradation of complex carbohydrates, which are derived either from the host diet or from the lining of the intestine (16–18). However, functional annotation of these genes has not kept pace with the genome sequencing efforts.
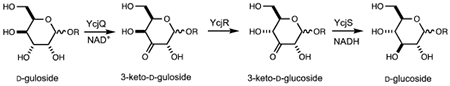
A cluster of 12 genes (ycjM-W and ompG) was identified in Escherichia coli K-12 (Figure 1) and is likely to function as a potential catabolic pathway for unknown carbohydrate(s). This putative carbohydrate metabolic pathway is conserved in a number of other Gram-negative bacteria that are found in the human gut including Salmonella enterica, Shigella dysenteriae, Enterobacter tasmaniensis, and Citrobacter rodentium, among others (www.microbesonline.org). Our previous investigation of this gene cluster indicated that YcjT is a kojibiose phosphorylase, which catalyzes the phosphorolysis of kojibiose (α-(1,2)-d-glucose-d-glucose) to d-glucose and β-d-glucose-1-phosphate (19). YcjT will also accept other substrates in place of d-glucose in the reverse reaction to form disaccharides containing l-sorbose, d-sorbitol, l-iditol, or 1,5-anhydro-d-glucitol coupled with d-glucose. YcjU is a β-phosphoglucomutase, which converts β-d-glucose-1-phosphate to d-glucose-6-phosphate. YcjM was shown to be a α-d-glucosyl-2-glycerate phosphorylase that catalyzes the formation of d-glycerate and α-d-glucose-1-phosphate from α-(1,2)-d-glucose-d-glycerate and phosphate (19).
Figure 1:

The ycj gene cluster from Escherichia coli K-12. The proteins encoded by this cluster include two polysaccharide hydrolases/phosphorylases (YcjM and YcjT), five sugar transporters (YcjN, YcjO, YcjP, YcjV, and OmpG, a porin), two NAD+-dependent dehydrogenases (YcjQ and YcjS), a putative isomerase/epimerase (YcjR), a β-phosphoglucomutase (YcjU), and a LacI-type repressor (YcjW).
The ycj operon also encodes putative sugar transport components (YcjN, YcjO, YcjP, YcjV, and OmpG) and a LacI-type repressor, YcjW. The focus of this report is the catalytic function of the enzymes, YcjQ, YcjR, and YcjS. YcjQ and YcjS are NAD+-dependent sugar dehydrogenases found in different enzyme families, and YcjR is a sugar isomerase/epimerase. Here we show these enzymes function together to convert d-gulosides into d-glucosides through a series of oxidation, epimerization, and reduction chemistry.
Materials and Methods
Materials.
The restriction endonucleases, pfu turbo polymerase, and T4 DNA ligase, used in the cloning of ycjQ, ycjR, and ycjS, were purchased from New England BioLabs. The PCR cleanup and gel extraction kits were bought from Qiagen. The plasmid miniprep kit, buffers, phenylmethylsulfonyl fluoride (PMSF), DNase I, and Chelex resin were obtained from Sigma-Aldrich. Isopropyl-β-d-thiogalactopyranoside (IPTG), NAD+, NADH, NADP+, and NADPH were purchased from Research Products International Corporation. All isotopically-labeled derivatives of d-glucose (1) and d-gulose (8) were purchased from Omicron Biochemicals. Other carbohydrates were obtained from Carbosynth, with the exception of methyl α-d-glucoside (2) and methyl β-d-glucoside (3), which were purchased from Sigma Aldrich.
Synthesis of Substrates for YcjS and YcjQ.
Methyl α/β-d-[3-13C]-glucoside and methyl β-d-guloside (9) were synthesized from d-[3-13C] glucose and d-gulose (8), respectively, by modification of a previously published procedure (20). 3-Keto-d-glucose (5) was synthesized as previously described (21–23). Methyl α-3-keto-d-glucoside (6) and methyl β−3-keto-d-glucoside (7) were synthesized following a previously published protocol (24).
Construction of Sequence Similarity Networks.
Sequence similarity networks (SSN) were generated from a list of the Uniprot IDs assigned to an individual Cluster of Orthologous Genes (COG) using the EFI-EST webtool (http://efi.igb.illinois.edu/efi-est/). Uniprot IDs were downloaded for enzymes annotated to be in cog0673 (YcjS), cog1063 (YcjQ) and cog1082 (YcjR). All network layouts were created and visualized using Cytoscape 3.4 or higher using the organic layout (25).
Cloning of ycjS from E. coli K-12.
The DNA corresponding to ycjS (gi|16129276; UniProt id: P77503) was amplified from Escherichia coli K-12 MG1655 genomic DNA using the following pair of primers:
5′-ACCGTGAATTCATGAAAATCGGCACACAGAATCAGGCG-3′
5′-AATCCAAGCTTTTAGCAGGTACGCAACCAGGC-3′
The gene was cloned into a pET-30a(+) vector by digestion with EcoRI and HindIII restriction enzymes followed by ligation with T4 DNA ligase to create the recombinant plasmid, which was verified by DNA sequencing. Cloning was carried out to facilitate a His6-tag at the amino-terminus of the recombinant protein. BL-21 (DE3) cells were transformed with the plasmid containing ycjS and plated on LB agar supplemented with 50 μg/mL kanamycin.
Expression and Purification of YcjS.
A 15-mL culture from a single colony of BL-21 (DE3) was used to inoculate three 1-L cultures of LB medium supplemented with 50 μg/mL kanamycin in 2.8-L Fernbach flasks. The flasks were incubated at 37 °C with shaking at 150 rpm until the OD600 reached ~0.6. IPTG was added to a final concentration of 0.5 mM to induce protein expression. The cell cultures were allowed to grow overnight at 25 °C. The cells were harvested by centrifugation at 11,000 g for 15 min, and the pellet was stored at −80 °C until needed.
The frozen cell pellet was suspended in 50 mM HEPES/K+, 100 mM KCl, 10 mM imidazole, pH 8.0. Prior to sonication, 0.4 mg/mL of DNase I and 0.1 mg/mL of PMSF were added to the cell lysate. Following sonication, the cell debris was removed by centrifugation at 12,000 g for 15 min and the supernatant fluid containing the crude protein was passed through a 0.45 μm filter before being loaded onto a 5-mL HisTrap column. The protein was eluted from the column by applying a linear gradient of (0 – 0.5 M) imidazole in 50 mM HEPES/K+, 100 mM KCl, pH 8.0. Individual fractions were analyzed by SDS-PAGE, combined, and dialyzed against 50 mM HEPES/K+, 100 mM KCl, pH 8.0. Aliquots were flash-frozen and stored at −80 °C. Protein concentration was determined spectrophotometrically at 280 nm using an extinction coefficient of 38,900 M−1 cm−1 and a molecular weight of 43 kDa that includeed the 5-kDa linker between the His-tag and the protein (https://web.expasy.org/protparam/). Typical yields of purified YcjS were ~10 mg of protein from 1.0 L of cell culture.
Cloning of ycjQ from E. coli K-12.
The following set of primers was used to amplify ycjQ (gi|16129274; UniProt id: P76043) from E. coli K-12 genomic DNA:
5′-ACCGTGAATTCATGAAAAAGTTAGTAGCCACAGCACCGC-3′
5′-AATCCAAGCTTTTAAAACGTAACGCCCATTTTGATGCTCTGTTCCG-3′
The gene was cloned with a His6-tag at the N-terminus. The amplified gene and the pET-30a(+) vector were digested with EcoRI and HindIII and then ligated together with T4 DNA ligase to create the recombinant plasmid. The DNA sequence of the inserted gene was verified by DNA sequencing. E. coli BL-21 (DE3) cells were transformed with the plasmid by electroporation and plated on LB agar that contained 50 μg/mL kanamycin.
Expression and Purification of YcjQ.
A 10-mL culture of LB medium, started from a single BL-21 (DE3) colony, was grown overnight at 37 °C. The sample was used to inoculate two 1-L cultures of LB medium supplemented with 50 μg/mL kanamycin and 1.0 mM ZnCl2 in 2.8-L Fernbach flasks. The cultures were allowed to grow at 37 °C until the OD600 reached ~0.6. The expression of protein was induced with 0.5 mM IPTG and the cell cultures were allowed to grow overnight at 25 °C. The following day the cells were harvested by centrifugation at 11,000 g for 12 min. The cell pellet was stored at −80 °C until needed.
The frozen cell pellet was thawed and suspended in 50 mM HEPES/K+, 200 mM KCl, 15 mM imidazole, pH 8.0 (binding buffer). DNase (0.4 mg/mL) and PMSF (0.1 mg/mL) were added to the mixture and left to stir for 15 min at 4 °C. The cells were lysed using a Branson Sonifier 450 for three 4-min intervals at 50% output. After sonication, the cell debris was separated from the supernatant fluid by centrifugation at 12,000 g for 20 min. The supernatant solution was passed through a 0.45 µm filter and loaded onto a HisTrap column, which had been equilibrated with binding buffer. The bound protein was then eluted from the column by applying a linear gradient of elution buffer (50 mM HEPES/K+, 200 mM KCl, 0.5 M imidazole, pH 8.0). The fractions were analyzed by SDS-PAGE, pooled, and the imidazole was removed by dialyzing against 50 mM HEPES/K+, 100 mM KCl, pH 8.0. Following dialysis, the protein was concentrated using Vivaspin protein concentrators, dispensed into smaller volumes, flash frozen, and stored at −80 °C. The protein concentration was determined spectrophotometrically at 280 nm using an extinction coefficient of 40,400 M−1 cm−1 and molecular weight of 38.2 kDa, which includes the 5-kDa linker between the His-tag and the protein.
Characterization of YcjS and YcjQ.
All kinetic assays were conducted at 30 °C. A small library of monosaccharides (Schemes S1 and S2) was tested as potential substrates for oxidation by YcjS and YcjQ. The general screening assays contained 2.0 mM substrate, 1.0 mM NAD+, 0.5 µM YcjS or YcjQ in 50 mM CHES/K+, 100 mM KCl, pH 9.5. The increase in absorbance was monitored at 340 nm (Δε = 6,220 M−1 cm−1). The kinetic constants for the oxidation of d-glucose (1), methyl α-d-glucoside (2), methyl β-d-glucoside (3), and 1,5-anhdyro-d-glucitol (4) by YcjS (1.0 µM) were measured with varying concentrations of substrate (0–35 mM), 2.0 mM NAD+ in 50 mM ammonium bicarbonate/K+, 100 mM KCl, pH 8.0 and 50 mM CHES/K+, 100 mM KCl, pH 9.0. Kinetic parameters for the reduction of 3-keto-d-glucose (5) were conducted in 50 mM cacodylate/K+, pH 6.5 in assays containing varying concentrations (0–10 mM) of 3-keto-d-glucose, 250 µM NADH, and 0.1 µM YcjS. Steady-state kinetic parameters for the reduction of methyl α-3-keto-d-glucoside (6) and methyl β−3-keto-d-glucoside (7) (0 – 5 mM) by YcjS were determined with 0.3 µM NADH and 0.1 µM YcjS in 50 mM cacodylate/K+, 100 mM KCl, pH 7.0 and 50 mM ammonium bicarbonate/K+, 100 mM KCl, pH 8.0
Kinetic constants for the oxidation of d-gulose (8) and methyl β-d-guloside (9) catalyzed by YcjQ were measured using varying amounts of substrate (0–20 mM), 2.0 mM NAD+, and 0.5 µM YcjQ in 50 mM HEPES/K+, 100 mM KCl, pH 8.0 or 50 mM CHES/K+, 100 mM KCl, pH 9.0.
The values of kcat and kcat/Km were determined by fitting the initial velocity data to eqn. 1 using GraFit 5 or SigmaPlot 11.0, where v is the initial velocity of the reaction, Et is the enzyme concentration, kcat is the turnover number, and Km is the Michaelis constant. Errors were calculated from the standard deviations of the fit.
| (1) |
Assays for Formate.
The NAD+-dependent formate dehydrogenase (FDH) catalyzes the oxidation of formate to carbon dioxide. The combined assays with FDH and YcjS contained 130 μM d-glucose (1), 4.0 mM NAD+, and 2.0 μM YcjS in 50 mM CHES/K+, pH 9.5, at 30 °C. The formation of NADH was monitored at 340 nm. The enzymatic reaction was allowed to proceed until there was no further increase in the absorbance at 340 nm. A total of 2 U of FDH was added and the reaction monitored at 340 nm for 90 min. In a complementary assay, 250 μM glucose, 4.0 mM NAD+, 1.0 μM YcjS, and 3 U FDH were incubated in 50 mM CHES/K+, pH 9.5, in a volume of 1.0 mL and the change in absorbance was monitored at 340 nm. Formate dehydrogenase was also used to quantify the amount of formate released from the YcjQ-catalyzed reaction with d-gulose (8). The reactions contained 150 µM d-gulose, 4.0 mM NAD+, and 2.0 μM YcjQ in 50 mM CHES/K+, pH 9.5, at 30 °C. The enzymatic reaction was allowed to proceed until there was no further increase in the absorbance at 340 nm. A total of 3 U of FDH was added and the reaction was monitored at 340 nm for an additional 90 min.
NMR Spectroscopy and Mass Spectrometry.
LC-MS experiments, carried out to determine the specific carbon that is oxidized by YcjS, contained the following: 250 µM of deuterium labeled (H1, H2, H3, H4 or H6) d-glucose, 2.0 mM NAD+, 1.7 µM YcjS in 50 mM CHES/K+, pH 9.5. The reactions were incubated for 6 h at 30 °C, and then the enzyme removed using Vivaspin protein filters. The NADH was isolated using reverse phase chromatography and electrospray ionization in the positive mode combined with tandem mass spectrometry was used to identify the mass of the NADH (or NADD) produced during the oxidation of labeled d-glucose. LC-MS was performed using an Agilent 1260 HPLC system connected to a MicroToF-QII mass spectrometer (Bruker Daltonics). Separation was conducted using a C-18 column (3.0 cm × 100 mm, 2.7 µm particles). Buffer A (5 mM ammonium acetate, pH 6.6) and B (75% methanol and 25% H2O) were used for the separation by liquid chromatography.
13C-NMR experiments were utilized to determine the product of the YcjS-catalyzed oxidation of d-glucose. The reaction contained 1.0 mM d-[UL-13C6]-glucose, 3.0 mM NAD+, 5.0 μM YcjS in 50 mM pyrophosphate buffer, pH 9.5. Similar reactions were conducted with d-glucose containing a single 13C-label at positions C1, C2, C3, C4, C5 or C6. The formation of the oxidized products from 13C-labeled d-glucose was also conducted in reactions that contained 1.0 mM d-[1-13C] glucose, 1.0 mM d-[3-13C]-glucose, 300 µM NAD+, 20 mM sodium pyruvate, 40 U lactate dehydrogenase, and 10 µM YcjS in 50 mM cacodylate/K+, pH 6.5. 13C-NMR experiments with enzymatic reactions contained 2.0 mM α-methyl-d-[3-13C]-glucoside, 300 µM NAD+, 5.0 µM YcjS, 20 mM sodium pyruvate, and 40 U lactate dehydrogenase in 50 mM ammonium bicarbonate/K+, pH 8.0.
13C-NMR experiments with YcjQ and d-[UL-13C6]-gulose contained 1.0 mM labeled substrate, 3.0 mM NAD+, 5.0 μM YcjQ in 50 mM pyrophosphate buffer, pH 9.5. In an attempt to determine the product of the YcjQ reaction, 13C-NMR experiments were conducted with methyl β-d-guloside (9). These reactions contained 10 mM methyl β-d-guloside (9), 300 μM NAD+, 20 μM YcjQ, 20 mM sodium pyruvate, and 40 U lactate dehydrogenase in 50 mM ammonium bicarbonate/K+, pH 8.0.
Cloning of ycjR from E. coli K12.
The gene corresponding to ycjR (gi|90111248; UniProt id: P76044) was amplified from E. coli K-12 genomic DNA with the following primer pair:
5′-ACCGTGAATTCATGAAAATCGGCACACAGAATCAGGCG-3′
5′-AATCCAAGCTTTTAGCAGGTACGCAACCAGGC-3′
The vector pET-30a(+) and the amplified gene were digested with EcoRI and HindIII restriction enzymes and ligated together with T4 DNA ligase to create the recombinant plasmid. The ycjR gene was cloned with an N-terminal His6 tag. Following verification by DNA sequencing, BL-21 (DE3) cells were transformed with the plasmid and plated on LB agar with 50 μg/mL kanamycin.
Expression and Purification of YcjR.
A single colony was used to start a 10-mL culture of LB supplemented with 50 μg/mL kanamycin and allowed to grow overnight at 37 °C. The 10-mL LB culture was used to inoculate two 1-L cultures of LB in 2.8-L Fernbach flasks. To each 1-L LB culture, 50 μg/mL kanamycin and 1.0 mM MnCl2 were added. The cultures were allowed to grow at 37 °C until the OD600 reached ~0.6, 1.0 mM IPTG was added to induce protein expression, and then growth was resumed at 25 °C, overnight. The cells were harvested by centrifugation at 11,000 g for 12 min. The cell pellet was stored at −80 °C until needed.
The frozen cell pellet was thawed and re-suspended in binding buffer that contained 20 mM HEPES/K+, 200 mM KCl, 20 mM imidazole, pH 8.0. Before sonication, 0.1 mg/mL PMSF and 0.4 mg/mL DNase I were added to the cell suspension and stirred for 10 min at 4 °C. The cells were lysed using a Branson Sonifier 450 for four 5 min intervals at 50% output. The cell suspension was clarified by centrifugation at 12,000 g for 20 min. The supernatant fluid was passed through a 0.45 μm filter and loaded on a 5-mL HisTrap column, which was previously equilibrated with binding buffer. The protein was eluted from the column using a linear gradient of the elution buffer, which contained 20 mM HEPES/K+, 200 mM KCl, 0.5 M imidazole, pH 8.0. Based on SDS-PAGE, the isolated protein was >95% pure. The appropriate fractions were pooled and concentrated. To remove imidazole, the concentrated protein was passed through a PD-10 column, which had been previously equilibrated with 20 mM HEPES/K+, pH 8.0. Protein concentration was determined spectrophotometrically at 280 nm using an extinction coefficient of 33,000 M−1 cm−1 and a molecular weight of 29.8 kDa, which includes the 5-kDa linker between the protein and the His-tag (https://web.expasy.org/protparam/). The collected fractions were pooled, flash frozen, and stored at −80 °C. Typical yields for this protein were ~60 mg of protein per liter of cell culture.
Metal Content Analysis.
The metal content of YcjQ and YcjR was determined using a Perkin-Elmer DRCII inductively coupled plasma mass spectrometer (ICP-MS). Samples for ICP-MS were digested with 70% (v/v) nitric acid to prevent precipitation of proteins during measurement, and then refluxed for 30 min. The samples were diluted with deionized water to ensure a final concentration of ~1.0 µM protein and 1% (v/v) nitric acid.
Determination of Catalytic Activity of YcjR.
YcjR was tested for catalytic activity with d-psicose, d-fructose, d-tagatose, d-sorbose, l-xylulose, and l-ribulose using 1H-NMR spectroscopy. The reactions contained 5.0 mM substrate, 0.5 mM MnCl2, and 5.0 µM YcjR in 50 mM ammonium bicarbonate/K+, pH 8.0. After incubation with the enzyme for 2.0 h at 30 °C, the enzyme was removed using a Vivaspin protein filter (molecular weight cut-off of 10 kDa) and the reactions passed over Chelex resin equilibrated with 50 mM ammonium bicarbonate/K+, pH 8.0 to remove Mn2+. The substrate solutions and buffer contained >90% D2O.
Owing to the instability of 3-keto-d-glucose (5), attempts were made to determine the epimerase activity of YcjR with methyl α-3-keto-d-glucoside (6). The reactions contained 2.0 mM unlabeled methyl α-d-glucoside (2), 2.0 mM NAD+, and 5.0 μM YcjS in ammonium bicarbonate/K+, pH 8.0, and incubated for 3 h at 30 °C. To this assay, 5.0 μM YcjR and 0.5 mM MnCl2 were added and the reaction was allowed to proceed for 2 h at 30 °C. The enzymes were removed using the Vivaspin protein filters (molecular cut-off of 10 kDa) and the reactions passed over Chelex resin equilibrated with 50 mM ammonium bicarbonate/K+, pH 8.0. The reactions were subjected to 1H-NMR spectroscopy. All of the buffers, substrate and co-enzyme solutions were made in >90% D2O.
Deuterium-solvent exchange of YcjR with methyl α-3-keto-d-glucoside (6) and methyl β−3-keto-d-glucoside (7) was monitored by 1H-NMR spectroscopy. For this experiment, 20 mM substrate was mixed with YcjR (20 μM) and 0.5 mM MnCl2 in 50 mM phosphate buffer, pH 7.5 in D2O. Aliquots were removed, diluted 10-fold into D2O, and the resulting mixture passed through a small Chelex column that had been pre-washed with D2O.
The steady-state kinetic parameters for YcjQ and YcjR with methyl α-3-keto-d-glucoside (6) and methyl β−3-keto-d-glucoside (7) were determined by UV spectroscopy by monitoring the decrease in absorbance at 340 nm. For YcjQ, 0.5 µM enzyme was mixed with substrate (0–20 mM) in the presence of excess YcjR (20 µM), 0.5 mM MnCl2, and 0.3 mM NADH in 50 mM HEPES/K+, 100 mM KCl, pH 7.0 and 50 mM HEPES/K+, 100 mM KCl, pH 8.0. For YcjR, 0.5 µM enzyme was mixed with various substrate concentrations (0–20 mM) in the presence of excess YcjQ (10 µM), 0.5 mM MnCl2, and 0.3 mM NADH in in 50 mM HEPES/K+, 100 mM KCl, pH 7.0 and 50 mM HEPES/K+, 100 mM KCl, pH 8.0.
Product Identification for Reactions Catalyzed by YcjQ and YcjR.
A solution of 20 mM methyl α-3-keto-d-glucoside (6) or methyl β−3-keto-d-glucoside (7) was mixed with 10 µM YcjQ, 10 μM YcjR, 20 mM NADH, and 250 µM MnCl2 in 50 mM potassium phosphate buffer, pH 7.5 in a final reaction volume of 600 µL D2O. Enzyme was removed using Vivaspin protein filters in aliquots of 150 μL. The flow-through was diluted into 450 µL D2O. The resulting solution was passed through a small column of Chelex resin (0.5 mL) to remove metal ions, and then through a DEAE column (0.2 mL) that had been equilibrated with 500 mM ammonium bicarbonate, pH 8.5, to remove NADH and NAD+. The effluent from the DEAE column was analyzed by 1H-NMR spectroscopy.
Equilibrium Constants for Oxidation and Isomerization Reactions.
The equilibrium constants for the reactions catalyzed by YcjQ, YcjR, and YcjS were obtained in 50 mM CHES/K+, 100 mM KCl, pH 9.0 at a fixed initial concentration of 2.0 mM NAD+. The equilibrium concentration of NADH was determined from the net absorbance at 340 nm using 15 different initial concentrations of methyl β-d-guloside (9) and methyl β-d-glucoside (3) (1–20 mM). The oxidation reactions catalyzed by YcjQ and YcjS were also coupled to the isomerization catalyzed by YcjR (0.5 µM) in the presence of 0.2 mM MnCl2. The equilibrium concentration of NADH for YcjQ/R with methyl β-d-guloside (9) was determined by fitting time courses obtained at 340 nm to equation 2.
| (2) |
The apparent equilibrium constant for each concentration was averaged and the error reported as the standard deviation of the values for the reactions catalyzed by YcjQ, YcjR, and YcjS as shown in Schemes 1a, b, and c.
Scheme 1:
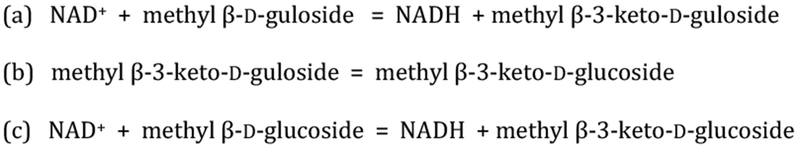
Reactions catalyzed by YcjQ, YcjR, and YcjS, respectively
Results
Determination of Substrate Profile for YcjS.
YcjS belongs to cog0673 and a sequence similarity network (SSN) was constructed at a BLAST E-value of 1 × 10−45 (Figure S1). Based on this SSN, the closest functionally verified homologues to YcjS are UDP-N-acetyl-d-glucosamine-3-dehydrogenase from Methanococcus maripaludis and UDP-N-acetyl-d-glucosaminuronic acid 3-dehydrogenase from Pseudomonas aeruginosa and Thermus thermophilus (26, 27). These three homologues of YcjS are all NAD+-dependent enzymes that oxidize the hydroxyl group at C-3 of their respective substrates.
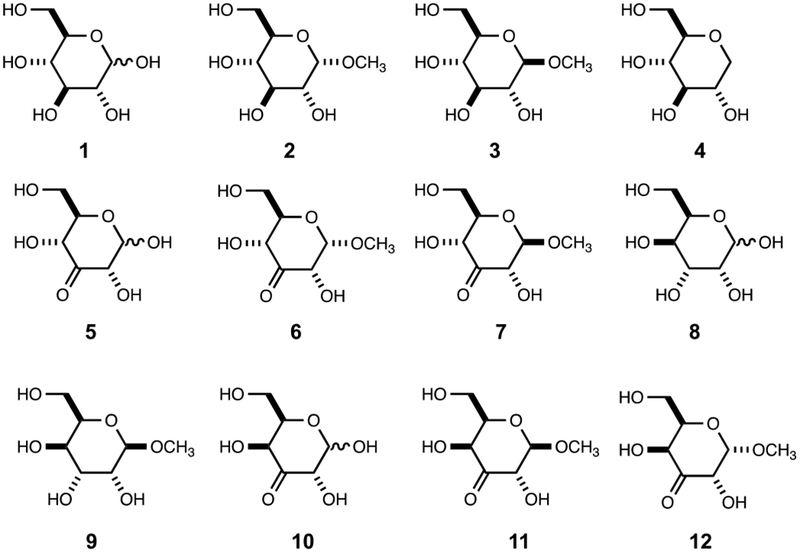
YcjS was purified to homogeneity and tested for catalytic activity against a small and focused library of d- and l-hexoses (Scheme S1) using NAD+ as the oxidant. At pH 9.0, YcjS catalyzed the oxidation of d-glucose (1) with a kcat of 1.14 s−1. YcjS was subsequently screened against a larger library of potential substrates (Schemes S1 and S2). No appreciable activity (<2% relative to d-glucose) was observed with UDP-glucose, UDP-N-acetyl-d-glucosamine, d-glucosamine, N-acetyl-d-glucosamine or UDP-d-galactose. Catalytic activity was observed for methyl α-d-glucoside (2), methyl β-d-glucoside (3), and 1,5-anhydro-d-glucitol (4) (Scheme 2). The kinetic parameters at various values of pH for the oxidation of these substrates by YcjS are collected in Table 1.
Scheme 2:
Substrates and products for YcjS and YcjQ.
Table 1:
Kinetic constants with YcjS and YcjQ and their respective substrates.
| Enzyme | Substrate | pH | Cofactor | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|---|---|
| YcjS | 1 | 8.0a | NAD+ | 0.65 ± 0.02 | 7.2 ± 0.8 | 90 ± 10 |
| 9.0b | 1.14 ± 0.06 | 8.9 ± 0.1 | 128 ± 7 | |||
| 2 | 8.0a | NAD+ | 0.58 ± 0.01 | 5.6 ± 0.6 | 103 ± 11 | |
| 9.0b | 1.2 ± 0.05 | 5.9 ± 0.7 | 200 ± 25 | |||
| 3 | 8.0a | NAD+ | 0.4 ± 0.01 | 2.0 ± 0.1 | 191 ± 9 | |
| 9.0b | 0.3 ± 0.01 | 1.6 ± 0.2 | 210 ± 20 | |||
| 4 | 8.0a | NAD+ | 0.17 ± 0.01 | 2.1 ± 0.1 | 81 ± 4 | |
| 9.0b | 0.4 ± 0.01 | 3.9 ± 0.4 | 100 ± 10 | |||
| 5 | 6.5c | NADH | 4.7 ± 0.3 | 0.5 ± 0.1 | 8500 ± 200 | |
| 6 | 7.0d | NADH | 22 ± 0.8 | 0.95 ± 0.1 | 23100 ± 2800 | |
| 8.0e | 18 ± 1.0 | 1.2 ± 0.3 | 15000 ± 3800 | |||
| 7 | 7.0d | NADH | 19 ± 1 | 0.80 ± 0.06 | 23400 ± 2200 | |
| 8.0e | 12.5 ± 0.7 | 0.92 ± 0.08 | 13600 ± 1400 | |||
| YcjQ | 8 | 8.0f | NAD+ | 0.18 ± 0.01 | 5.1 ± 0.40 | 31 ± 2 |
| 9.0b | 0.23 ± 0.02 | 5.6 ± 0.8 | 41 ± 6 | |||
| 9 | 8.0f | NAD+ | 0.35 ± 0.03 | 12.5 ± 1.5 | 28 ± 3 | |
| 9.0b | 1.2 ± 0.05 | 12.0 ± 0.8 | 97 ± 6 | |||
| 12g | 7.0h | NADH | 18.5 ± 1.6 | 9.4 ± 1.1 | 2000 ± 290 | |
| 8.0i | 3.8 ± 0.1 | 1.5 ± 0.1 | 2600 ± 160 | |||
| 11g | 7.0h | NADH | 7.0 ± 0.2 | 2.3 ± 0.1 | 3100 ± 200 | |
| 8.0i | 9.7 ± 0.6 | 7.0 ± 0.6 | 1400 ± 160 | |||
| YcjR | 6 | 7.0j | - | - | 240 ± 8k | |
| 8.0l | 19.2 ± 1.7 | 37 ± 4.4 | 520 ± 77 | |||
| 7 | 7.0j | 2.2 ± 0.3 | 34.5 ± 6.7 | 58 ± 14 | ||
| 8.0l | 4.0 ± 0.6 | 31 ± 6.5 | 130 ± 30 | |||
Reactions were carried out at 30 °C in 50 mM NH4+HCO3− buffer, pH 8.0 with 2.0 mM NAD+.
Reactions were carried out at 30 °C in 50 mM Ches/K+ buffer, pH 9.0 with 2.0 mM NAD+.
Reaction was carried out in 50 mM cacodylate/K+, pH 6.5 at 30 °C with 250 µM NADH.
Reactions were carried out at 30 °C in 50 mM cacodylate/K+ , pH 7.0 with 0.3 mM NAD+.
Reactions were carried out at 30 °C in 50 mM NH4+HCO3− buffer, pH 8.0 with 0.3 mM NADH.
Reactions were carried out at 30 °C in 50 mM HEPES/K+ buffer, pH 8.0 with 2.0 mM NAD+.
11 or 12 were generated in situ by the addition of excess YcjR (20 µM), 0.5 mM MnCl2, and 7 or 6, respectively. Concentrations of 11 and 12 were corrected based on the measured equilibrium constant (Keq = 4) between 7 and 11.
Reactions were carried out at 30 °C in 50 mM HEPES/K+ buffer, pH 7.0 with 0.3 mM NADH.
Reactions were carried out at 30 °C in 50 mM HEPES/K+ buffer, pH 8.0 with 0.3 mM NADH.
Reactions were carried out at 30 °C in 50 mM HEPES/K+ buffer, pH 7.0 with 0.3 mM NADH, 0.5 mM MnCl2, and excess YcjQ (10 µM).
Values based on fit of kinetic data to a linear equation with the slope giving the value of kcat/Km.
Reactions were carried out at 30 °C in 50 mM HEPES/K+ buffer, pH 8.0 with 0.3 mM NADH, 0.5 mM MnCl2, and excess YcjQ (10 µM).
Product Identification for YcjS Oxidation of d-Glucose.
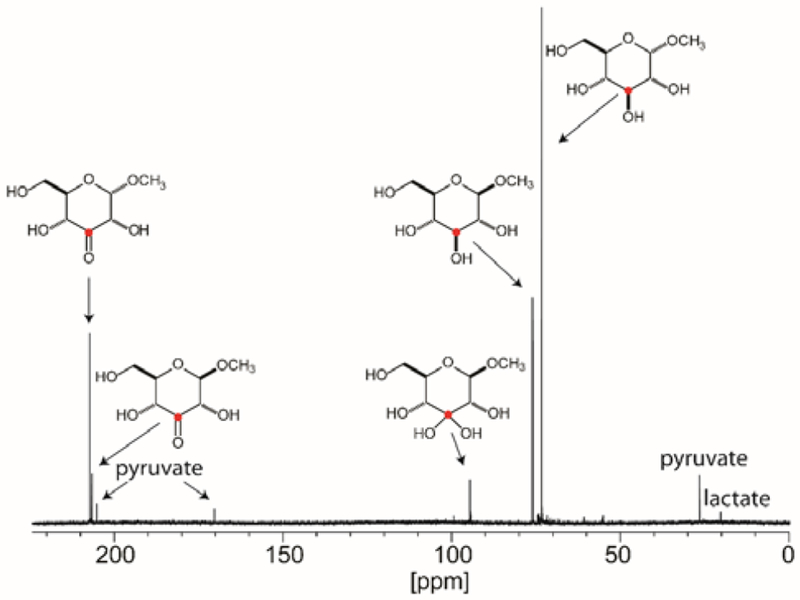
To determine the product of the YcjS-catalyzed reaction with d-glucose, [UL-13C6]-d-glucose was used as the substrate and the reaction mixture examined by 13C-NMR spectroscopy. The NMR spectrum of the reaction products showed a prominent new resonance at 171.03 ppm and numerous other resonances in the range of 60-100 ppm (Figure 2). A similar reaction, using [1-13C]-d-glucose, demonstrated that essentially all of the 13C-label from this substrate was found in the product peak with the chemical shift of 171.03 ppm (Figure S2a) and identified as formate. Formate dehydrogenase (FDH) was used to quantify the amount of formate produced in the YcjS-catalyzed oxidation of d-glucose. The addition of FDH to a reaction mixture containing YcjS and 130 µM d-glucose and an excess of NAD+ produced 257 µM NADH. This result is consistent with the nearly quantitative conversion of C1 from d-glucose to formate after oxidation by YcjS at pH 9.5. Experiments conducted with YcjS, NAD+, and d-glucose containing separate 13C-labels at C-2, C-3, C-4, C-5 or C-6 showed multiple new 13C-NMR resonances for each labeled carbon, indicating that the oxidized d-glucose was highly unstable at pH 9.5 (Figures S2b to S2f).
Figure 2.
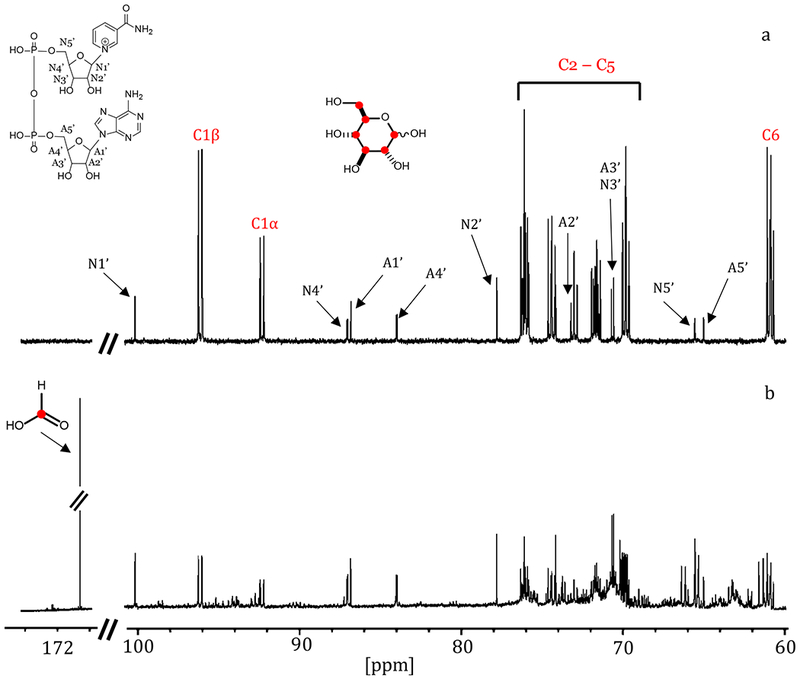
13C-NMR spectrum for the reaction catalyzed by 5.0 µM YcjS with 1.0 mM [UL-13C6]-d-glucose and 2.0 mM NAD+. (a) [UL-13C6]-d-glucose and NAD+ before the addition of YcjS; (b) after incubation with the enzyme. The resonance for formate is observed at 171.03 ppm.
To determine which one of the hydroxyl groups of d-glucose is oxidized by YcjS, the enzyme-catalyzed reaction was conducted using d-glucose that was deuterated at C1, C2, C3, or C4. The reduced nucleotide product was isolated by ion exchange chromatography and the molecular weight determined by mass spectrometry. Only in the presence of [3-2H]-d-glucose was deuterium transferred from the substrate to NAD+ with formation of NAD(2H) and an observed m/z of 667.144 (Figure 3). This result clearly demonstrates that the hydroxyl group at C3 of d-glucose is oxidized in the presence of YcjS and the initial product of the enzymatic reaction is 3-keto-d-glucose (5).
Figure 3:
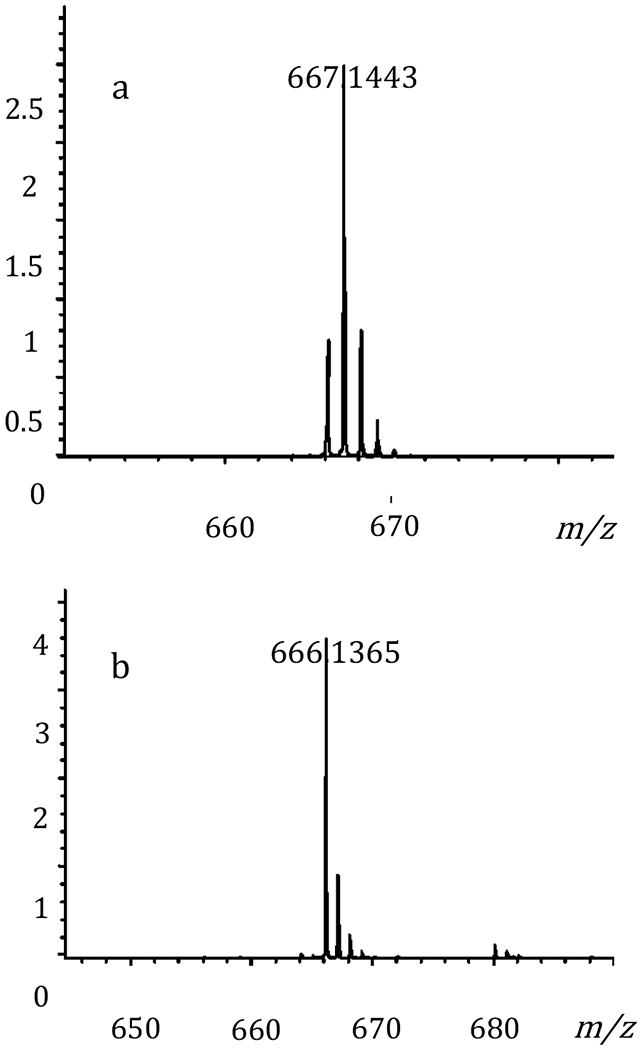
Mass spectra of the reduced pyridine nucleotide after reaction with deuterated d-glucose, NAD+, and YcjS. (a) [3-2H]-d-glucose and (b) [1-2H]-d-glucose.
Due to the inherent instability of the 3-keto-d-glucose (5) product (28), 13C-NMR experiments for product identification were subsequently conducted using methyl α/β-[3-13C]-d-glucoside as the substrate. The NMR spectrum of the product formed from the oxidation of this substrate, exhibits new resonances at 207.26 and 206.58 ppm, which correspond to the expected chemical shifts of the methyl α-3-keto-d-glucose and methyl β−3-keto-d-glucoside, respectively. A new resonance at 94.6 ppm corresponds to the hydrated form of methyl β−3-keto-d-glucoside (Figure 4). Methyl α-3-keto-d-glucoside (6) and methyl β−3-keto-d-glucoside (7) were chemically synthesized and used as substrates for reduction by YcjS at pH 7.0 and 8.0. YcjS readily reduces both compounds at essentially the same rate. The kinetic constants for the reduction of compounds 5-7 are summarized Table 1.
Figure 4:
13C-NMR spectrum of the YcjS-catalyzed reaction (20 μM) starting with 5.0 mM methyl α/β-d-[3-13C]-glucoside and 300 μM NAD+ in the presence of 20 mM pyruvate, 40 U lactate dehydrogenase and 50 mM phosphate/K+ buffer, pH 8.0.
Catalytic Activity of YcjQ.
YcjQ is annotated as a zinc-dependent dehydrogenase and a member of cog1063 by NCBI. YcjQ was purified to homogeneity and found to have a metal content of 0.7 equivalents of zinc/enzyme subunit. The sequence similarity network (Figure S3) for YcjQ contains several proteins whose catalytic properties have been determined previously: scyllo-inosose-3-dehydrogenase from Thermotoga maritima, l-galactonate-5-dehydrogenase from Bacteroides vulgatus, galactitol-1-phosphate-5-dehydrogenase from E. coli K-12 MG1655, and sorbitol dehydrogenase from Bacillus subtilis (29–32). However, there are no experimentally verified homologues close to the cluster containing YcjQ. YcjQ was screened for catalytic activity against the library of hexoses (Scheme S1) and activity was observed only with d-gulose (8). Among the other compounds tested as potential substrates (Scheme S2), YcjQ exhibited catalytic activity with methyl β-d-guloside (9). The full set of kinetic parameters for these two substrates are presented in Table 1. The product from the oxidation of d-gulose (8) by YcjQ was examined by 13C-NMR spectroscopy. When [UL-13C6]-d-gulose, NAD+, and YcjQ were incubated at pH 9.5 for 5.0 h, the most prominent new resonance is that for formate, along with multiple other resonances formed from the instability of the enzyme-catalyzed product (Figure S4). A triplet with a coupling constant of 42 Hz for the oxidized carbon is present at 213.07 ppm. In the presence of formate dehydrogenase, YcjQ, 150 µM d-gulose and an excess of NAD+, a total of 304 µM NADH was formed. The quantitative formation of formate from the oxidation of d-gulose (8) by YcjQ at pH 9.5 clearly indicates that this substrate is oxidized at C3 to form 3-keto-d-gulose (10).
Functional Characterization of YcjR.
The SSN for YcjR (cog1082), constructed at an E-value of 1 × 10−25 (Figure S5) reveals that the closest functionally verified homologues to this enzyme are d-tagatose-3-epimerase from Rhodobacter sphaeroides , d-psicose-3-epimerase from Agrobacterium tumefaciens, and l-ribulose-3-epimerase from Rhizobium loti (33–35). YcjR was purified to homogeneity and ICP-MS of the isolated enzyme indicated that the metal content was 0.8 equivalents of Mn2+ per protein monomer. In an attempt to characterize the putative epimerase activity of YcjR, the enzyme was initially incubated with some of the known substrates for the closest homologues to this protein. Using 1H-NMR spectroscopy to detect the formation of new products, no new resonances were observed when 5.0 μM YcjR was incubated with d-psicose, d-fructose, d-tagatose, d-sorbose, l-xylulose, or l-ribulose for up to 2 h (data not shown). Since YcjS and YcjQ both catalyze the oxidation of their respective sugar substrates at C3, it is thus highly likely that YcjR will use one or both of these oxidized products as a substrate and catalyze an epimerization of stereochemistry at either C2 or C4. When YcjR was incubated with chemically synthesized methyl α-3-keto-d-glucoside (6) in D2O, the proton attached to C4 was quantitatively exchanged with solvent (Figure 5).
Figure 5:
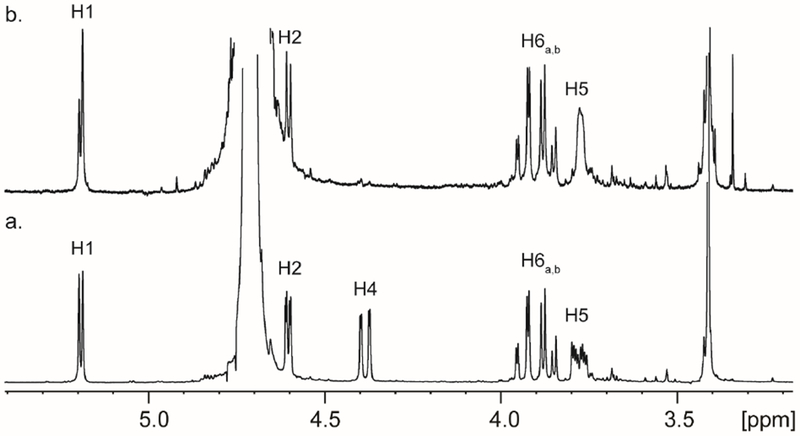
(a) 1H-NMR spectrum of methyl α-3-keto-d-glucoside (6) in D2O. (b) 1H-NMR spectrum of methyl α-3-keto-d-glucoside (6) in D2O after incubation with 20 μM YcjR for 120 min.
Epimerization of Methyl β−3-Keto-d-Glucoside by YcjR.
YcjR catalyzed the exchange of the H4 proton with solvent in methyl α-3-keto-d-glucoside (6), but no new resonances were distinguishable in the 1H-NMR spectrum. We therefore added YcjR, YcjQ and NADH to a solution of methyl β−3-keto-d-glucoside (7) at pH 7.5 in an attempt to shift the equilibrium to the reduced methyl β-d-guloside (9) product. The 1H-NMR spectrum of the methyl β−3-keto-d-glucoside (7) is presented in Figure 6a. Shown in Figure 6b is the 1H-NMR spectrum of the reaction mixture after this substrate was incubated with YcjR, YcjQ, and NADH for 3 h. The 1H-NMR spectrum of the product shown is clearly that of β-methyl-d-guloside (9) where the proton at H4 has exchanged with deuterium from the solvent. This is further confirmed by the loss of proton coupling to H3 from a triplet to a doublet and the coupling of H5 changing from a doublet of triplets to a doublet of doublets. The 1H-NMR spectrum of chemically synthesized methyl β-d-guloside (9) is presented for comparison in Figure 6c. The transformation of methyl β−3-keto-d-glucoside (7) to methyl β-d-guloside (9) requires the presence of YcjR (Figure 6d). Similar results were obtained starting from NADH, methyl α-3-keto-d-glucoside (6), YcjR, and YcjQ but a chemical synthesis of methyl α-d-guloside was not achieved for a direct comparison with the enzyme-catalyzed product (Figure S6).
Figure 6.
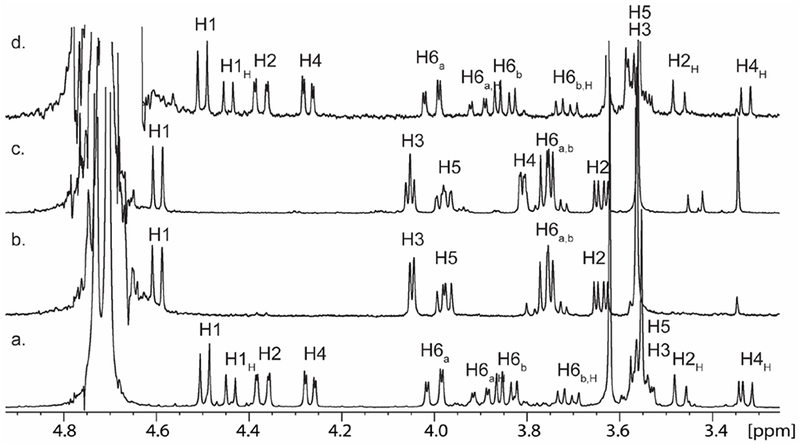
(a) 1H-NMR spectrum of chemically synthesized methyl β-3-keto-glucoside (7); (b) 1H-NMR spectrum of the reaction mixture after YcjR, YcjQ, and NADH were added to methyl β-3-keto-glucoside (7); (c) 1H-NMR spectrum of chemically synthesized methyl β-guloside (9); and (d) 1H-NMR spectrum of methyl β-3-keto-glucoside (7) after the addition of YcjQ and NADH. In all experiments, NADH and NAD+ were removed by ion exchange chromatography.
In addition to identifying the products of the reaction catalyzed by YcjR, the kinetic constants for the reaction were determined using UV spectroscopy by following the oxidation of NADH to NAD+ at 340 nm when YcjR was added to a reaction mixture containing methyl β−3-keto-glucoside (7) or methyl α-3-keto-glucoside (6) and an excess of YcjQ. The results of these experiments are summarized in Table 1.
Equilibrium Constants for Interconversion of Substrates and Products Catalyzed by YcjQ, YcjR and YcjS.
The reactions catalyzed by YcjQ, YcjR, and YcjS strongly suggest that the equilibrium constant for the collective conversion of d-gulosides to d-glucosides favors the formation of the d-glucoside. To measure the equilibrium constants for these transformations, YcjQ was mixed with a fixed concentration of NAD+ and variable amounts of methyl β-d-guloside (9), and the reaction allowed to reach equilibrium. The net concentration of NADH and methyl β−3-keto-d-guloside (11) formed was obtained from the change in absorbance at 340 nm and the average equilibrium constant of 3.6 (± 0.2) × 10−4 for the transformation shown in Scheme 1a was calculated. To obtain the equilibrium constant for the isomerization reaction catalyzed by YcjR (Scheme 1b), the dehydrogenase YcjQ was mixed with NAD+, variable amounts of methyl β-d-guloside (9) and YcjR was added to pull the reaction toward the formation of methyl β−3-keto-d-glucoside (11). The net concentration of NADH and the equilibrium constant for the reaction catalyzed by YcjQ was used to calculate the equilibrium concentrations of methyl β−3-keto-d-guloside (11) and methyl β−3-keto-d-glucoside (7) as 3.9 ± 0.2. The equilibrium constant for the oxidation of methyl β-d-glucoside (3) was determined (Scheme 1c) using the same protocol for the oxidation of methyl β-d-guloside (9) by YcjQ. The equilibrium constants for YcjQ, YcjR, and YcjS are presented in Table 2.
Table 2.
: Equilibrium constants for the conversion of 9 to 3a
| Reaction | Enzyme | Keq |
|---|---|---|
| 9 to 11 | YcjQ | 3.6 ± 0.2 × 10−4 |
| 11 to 7b | YcjR | 3.9 ± 0.2 |
| 7 to 3c | YcjS | 6.4 ± 0.3 × 103 |
| 9 to 3 | YcjQ, YcjR, YcjS | 9.0 ± 0.9d |
Reactions were carried out at 30 °C in 50 mM CHES/K+ buffer, pH 9.0 with 2.0 mM NAD+.
Reaction was followed by monitoring the oxidation 9 to 11 and conversion of 11 to 7 with excess YcjR present. The equilibrium constant of 11 to 7 was calculated as described in the text.
Reaction was followed by monitoring the oxidation of 3 to 7.
Calculated based on the individual equilibrium constants measured for each step of the conversion of 9 to 3.
Based on these equilibrium constants, it was likely that methyl β-d-guloside (9) could be converted to methyl β-d-glucoside (3) by the combined actions of YcjQ, YcjR, and YcjS. In this experiment, methyl β-d-guloside (9) was mixed with YcjQ, YcjR, and YcjS at pH 7.5 in D2O. The 1H-NMR spectrum of the reaction mixture (Figure 7b) after the addition of the three enzymes shows nearly complete conversion of methyl β-d-guloside (Figure 7a) to methyl β-d-glucoside (Figure 7c). In Figure 7b, it is clear that the resonance for H4 has been lost because of the enzyme-catalyzed exchange with deuterium from solvent and the loss of coupling to H5 and H3.
Figure 7:
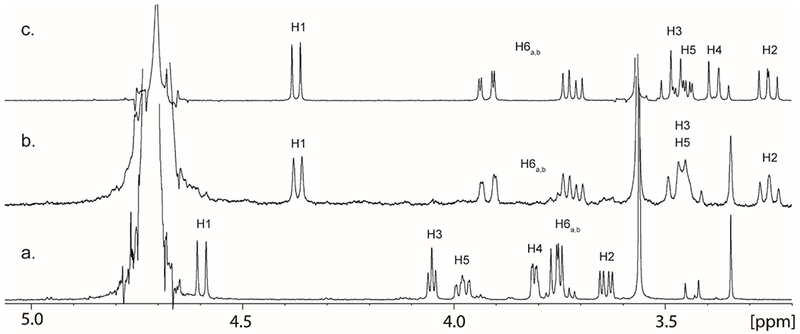
(a) 1H-NMR spectrum of chemically synthesized methyl β-d-guloside (9) ; (b) 1H-NMR spectrum of the reaction mixture after YcjR, YcjQ, YcjS, and NAD+ were incubated with methyl β-d-guloside (9) to yield methyl β-d-glucoside (3) ; (c) 1H-NMR spectrum of commercially purchased methyl β-d-glucoside (3).
Discussion
Functional Characterization of YcjS.
YcjS was shown to oxidize d-glucose (1) to 3-keto-d-glucose and reduce 3-keto-d-glucose back to d-glucose. Formation of NAD(2H) from d-[3-H2]-glucose confirmed the C3 hydroxyl group is oxidized yielding 3-keto-d-glucose (5) as the reaction product. This conclusion was further supported by 13C-NMR experiments with methyl α/β-d-[3-13C]-glucoside, which shows new resonances at 207.25 and 206.58 ppm, corresponding to the methyl α- and β−3-keto products, respectively. At elevated pH, 3-keto-d-glucose is highly unstable (28). This was demonstrated by the formation of formate from [UL-13C6]-d-glucose as well as d-glucose containing 13C-labels at C-1, C-2, C-3, C-4, C-5, or C-6 positions (Figure S2). The proposed mechanism for the formation of formate from d-glucose is presented in Scheme 3.
Scheme 3.
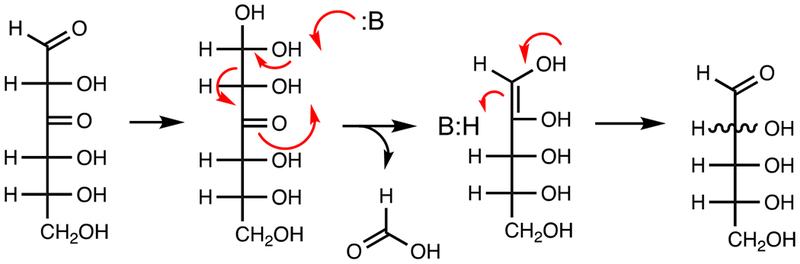
Proposed mechanism for the formation of formic acid from 3-keto-d-glucose (5).
YcjS belongs to cog0673 and shares 28% sequence identity with UDP N-acetyl-d-glucosamine-3-dehydrogenase and 30% identity with UDP-N-acetyl-d-glucosaminuronic acid dehydrogenase (Uniprot: G3XD23)(26, 27). These dehydrogenases belong to the glucose-fructose oxidoreductase family (GFOR) (36). An InterPro search of the YcjS sequence indicates a predicted Rossman fold domain in the N-terminal (5 – 158 amino acids) region of the protein, which is responsible for binding NAD. A sequence alignment of UDP-N-acetyl-d-glucosaminuronic acid dehydrogenase (WlbA) from P. aeruginosa with YcjS shows that the amino acid residues in WlbA that bind to UDP (Tyr-12, Lys-169, Arg-160, Thr-243, and Arg-245), the N-acetyl group (Tyr-156 and Arg-245), or the C-6’ carboxylate group of the hexose (Arg-160, Tyr-164, Lys-169, and Asn-181) are not conserved in YcjS (Figure S7)(27). In WlbA the hydroxyl group at C3’ that becomes oxidized is 3.0 Å away from C4 of the nicotinamide ring of NAD and hydrogen bonds with Lys-101 and His-185 (27). Both of these residues are conserved in YcjS (Lys-103 and His-189). The lysine residue is part of the characteristic consensus sequence (Glu-Lys-Pro) found in all members of the GFOR family, including YcjS. This motif interacts with the nicotinamide ring of NAD+ (37). YcjS contains the second predicted GFOR family consensus sequence, Gly-Gly-X3-Asp-X3-(Tyr/His), found in the active site as 180Gly-Gly-Pro-Leu-Ile-Asp-Ile-Gly-Ile-His189. These residues have been proposed to bind and orient the substrate for catalysis (38). In the structure of another YcjS homologue, KijD10 (PDB id: 3RC1) the aspartate residue from the conserved motif interacts with the C2’ hydroxyl group of the nicotinamide ribose and was suggested to have a role in binding with C4” of the bound hexose substrate based on modeling of dTDP-3-keto-6-deoxy-d-galactose in the active site (39).
The NAD-dependent YcjS was shown to oxidize the hydroxyl group at C3 of d-glucose. With UDP N-acetyl-d-glucosamine dehydrogenase, the proposed enzymatic product (3-keto-UDP-GlcNAc) could not be isolated by HPLC (40). UDP was detected, which was proposed to form because of the inherent instability of the 3-keto product (40). This observation agrees with our results where the labile 3-keto product leads to the formation of formate under alkaline conditions, followed by further degradation to uncharacterized products. In a study reporting the role of NtdC in the biosynthesis of kanosamine, the authors report the instability of the enzymatic product, 3-keto-d-glucose-6-phosphate (28).
Functional Characterization of YcjQ.
YcjQ belongs to cog1063 and the SSN constructed for this cog did not show any functionally verified homologs close to YcjQ. Testing YcjQ against a library of different sugars showed catalytic activity only with d-gulose (8). To the best of our knowledge, this is the first reported enzyme that is capable of oxidizing d-gulose, a very rare hexose sugar. From the degradation pattern of the enzymatic product and the formation of formate it was demonstrated that the product of the YcjQ and d-gulose (8) reaction is 3-keto-d-gulose (10). Attempts to chemically synthesize the 3-keto product were unsuccessful.
Functional Characterization of YcjR.
The SSN for cog1082 reveals that the three closest homologues to YcjR are annotated as d-tagatose-3-epimerase, d-psicose-3-epimerase, and l-ribulose-3-epimerase (33–35). However, YcjR shares a sequence identity of ≤30% with these three enzymes. All three homologues epimerize the stereochemistry of the hydroxyl group attached to C3 of their respective substrates, which contain a keto-group at C2. Like these homologues, YcjR is a manganese-dependent enzyme. We have demonstrated that YcjR epimerizes the stereochemistry of the hydroxyl group attached to C4 position of the 3-keto products of YcjS and YcjQ. This set of reactions provides a novel metabolic pathway for the transformation of d-gulosides to d-glucosides (Scheme 4).
Scheme 4.
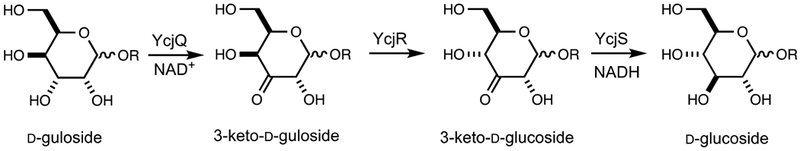
Enzymatic Formation of 3-Keto-d-glucosides.
3-Keto-d-glucosides have important applications as food additives, antioxidants, and aminoglycoside antibiotics (41, 42). A FAD-dependent glucoside-3-dehydrogenase that oxidizes the C3 hydroxyl group of hexoses and its derivatives was first reported in Agrobacterium tumefaciens and has since been identified in Halomonas (Deleya sp. α-15), Agaricus bisporus, Flavobacterium saccharophilum, Cytophaga marinoflava, Stenotrophomonas maltophilia, and Sphingobacterium faecium (41–50). Studies conducted on Agrobacterium tumefaciens showed that the FAD-dependent glucoside-3-dehydrogenase, most likely present in the periplasmic space of the bacterium, converts disaccharides, such as sucrose to 3-keto-sucrose, which is then transported inside the cell (42, 51–53). The cytoplasmic enzyme, 3-ketoglucosidase, catalyzes the hydrolysis of 3-keto sucrose to form fructose and 3-keto-d-glucose (5), which in turn gets reduced to glucose by the NADPH-dependent 3-ketoglucose reductase (54, 55). Ampomah et. al, reported that the thuEFGKAB operon found in A. tumefaciens was responsible for the transport of various disaccharides in their 3-keto forms (56). Disruption of thuAB genes affected the bacterium’s ability to utilize sucrose as a carbon source, but the mutant was able to grow on other disaccharides such as trehalose, sucrose, cellobiose, and lactose, indicating that the bacterium has multiple pathways dedicated to the transport and utilization of disaccharides. However, the function of ThuAB, from the putative trehalose-utilization pathway, has not been experimentally verified (56). While ThuB belongs to cog0673, the other FAD-dependent enzymes from Halomonas (Deleya sp. α-15) and Agaricus bisporus belong to cog2303.
There are a number of other NAD-dependent glucoside-3-dehydrogenase enzymes that catalyze the conversion of hexoses to their 3-keto derivative. Some of the enzymes that convert sugar-nucleotides to 3-hexulose sugar nucleotides include UDP N-acetyl-d-glucosamine dehydrogenase, and UDP-N-acetyl-d-glucosaminuronic acid dehydrogenases, from Pseudomonas aeruginosa PAO1 and Thermus thermophilus (27, 40). Levoglucosan (1,6-anhydro-β-d-glucopyranose) dehydrogenase from Arthrobacter sp. I-552 can oxidize levoglucosan, a product of burnt biomass, to 3-keto levoglucosan (57). In the kanosamine biosynthetic pathway, NtdC, a sugar dehydrogenase, from Bacillus subtilis catalyzes the oxidation of the hydroxyl group at C-3 of d-glucose-6-phosphate (28). KijD10, a C-3” ketoreductase from Actinomadura kijaniata, is an NADPH-dependent enzyme that catalyzes the reduction of dTDP-3,4-diketo-2,6-dideoxy-d-glucose to dTDP-4-keto-2,6-dideoxy-d-glucose (39).
Conclusion.
In this paper we have characterized the catalytic properties of YcjQ, YcjR, and YcjS from the ycj gene cluster in E. coli. It was shown that YcjS and YcjQ are able to catalyze the oxidation of the hydroxyl group at C3 of d-glucose and d-gulose, respectively, in addition to the corresponding methyl glycosides. YcjR was shown to catalyze the epimerization at C-4 of methyl 3-keto-d-glucoside and 3-keto-d-guloside. However, very little is currently known about the occurrence of d-gulose or d-gulosides in nature, and thus the specific physiological substrates for this metabolic pathway are unknown. 6-Deoxy-d-gulose has been found in the lipopolysaccharide O-antigen of Yersinia enterocolitica (58). d-Gulose has been identified in the extracellular matrix of Volvox carteri and in the exoglycolipid of an antigen derived from Chlamydia trachomatis (59, 60). d-Gulose has also been reported to be an integral component of the extracellular polysaccharide (EPS) of Caulobacter crecentus CB2A (61) where it is attached to the carbohydrate backbone via a 1,4-linkage to a d-glucosyl moiety. It is thus possible that disaccharide metabolites similar to (1,4)-d-gulose-d-glucose may be the actual physiological substrates for the transformations catalyzed by YcjQ, YcjR, and YcjS.
Supplementary Material
Acknowledgments
Funding
This work was supported by grants from the Robert A. Welch Foundation (A-840) and the National Institutes of Health (GM122825).
Footnotes
Supporting Information
Sequence similarity networks for YcjS, YcjQ, and YcjR; library of compounds used for activity screening; 13C-NMR spectrum of glucose with YcjS; 13C-NMR spectrum of d-gulose with YcjQ; 1H-NMR spectrum of methyl α-d-guloside (12) formed from methyl α-d-3-keto-glucoside (6) and YcjQ and YcjR activity; multiple sequence alignment with YcjS and UDP-N-acetyl-d-glucosaminuronic acid dehydrogenase.
The authors declare no competing financial interest.
REFERENCES
- (1).Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, and Nelson KE (2006) Metagenomic analysis of the human distal gut microbiome, Science 312, 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kelly D, Conway S, and Aminov R (2005) Commensal gut bacteria: mechanisms of immune modulation, Trends Immunology 26, 326–333. [DOI] [PubMed] [Google Scholar]
- (3).Ley RE, Peterson DA, and Gordon JI (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine, Cell 124, 837–848. [DOI] [PubMed] [Google Scholar]
- (4).Ley RE, Turnbaugh PJ, Klein S, and Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity, Nature 444, 1022–1023. [DOI] [PubMed] [Google Scholar]
- (5).Sousa T, Paterson R, Moore V, Carlsson A, Abrahamsson B, and Basit AW (2008) The gastrointestinal microbiota as a site for the biotransformation of drugs, International Journal of Pharmceuticals 363, 1–25. [DOI] [PubMed] [Google Scholar]
- (6).Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, Baselga J, Liu C, Shivdasani RA, Ogino S, Birren BW, Huttenhower C, Garrett WS, and Meyerson M (2012) Genomic analysis identifies association of Fusobacterium with colorectal carcinoma, Genome Research 22, 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Walters WA, Xu Z, and Knight R (2014) Meta-analyses of human gut microbes associated with obesity and IBD, FEBS Letters 588, 4223–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, and Chervonsky AV (2008) Innate immunity and intestinal microbiota in the development of Type 1 diabetes, Nature 455, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hill-Burns EM, Debelius JW, Morton JT, Wissemann WT, Lewis MR, Wallen ZD, Peddada SD, Factor SA, Molho E, Zabetian CP, Knight R, and Payami H (2017) Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome, Movement Disorders 32, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Malan-Muller S, Valles-Colomer M, Raes J, Lowry CA, Seedat S, and Hemmings SMJ (2018) The gut microbiome and mental health: implications for anxiety- and trauma-related disorders, OMICS 22, 90–107. [DOI] [PubMed] [Google Scholar]
- (11).Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S, Zetterberg H, Blennow K, Bendlin BB, and Rey FE (2017) Gut microbiome alterations in Alzheimer’s disease, Scientific Reports 7, 13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Land M, Hauser L, Jun SR, Nookaew I, Leuze MR, Ahn TH, Karpinets T, Lund O, Kora G, Wassenaar T, Poudel S, and Ussery DW (2015) Insights from 20 years of bacterial genome sequencing, Functional Integrative Genomics 15, 141–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Martin R, Bermudez-Humaran LG, and Langella P (2016) Gnotobiotic rodents: an in vivo model for the study of microbe-microbe interactions, Frontiers in Microbiology 7, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Williams SC (2014) Gnotobiotics, Proceedings of the National Academy of Sciences U S A 111, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC, Bork P, Ehrlich SD, and Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing, Nature 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Flint HJ, Bayer EA, Rincon MT, Lamed R, and White BA (2008) Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis, Nature Reviews Microbiology 6, 121–131. [DOI] [PubMed] [Google Scholar]
- (17).Hooper LV, Midtvedt T, and Gordon JI (2002) How host-microbial interactions shape the nutrient environment of the mammalian intestine, Annual Review of Nutrition 22, 283–307. [DOI] [PubMed] [Google Scholar]
- (18).Xu J (2004) Message from a human gut symbiont: sensitivity is a prerequisite for sharing, Trends in Microbiology 12, 21–28. [DOI] [PubMed] [Google Scholar]
- (19).Mukherjee K, Narindoshvili T, and Raushel FM (2018) Discovery of a kojibiose phosphorylase in Escherichia coli K-12, Biochemistry 57, 2857–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li CW, Dong HJ, and Cui CB (2015) The synthesis and antitumor activity of twelve galloyl glucosides, Molecules 20, 2034–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kartha KPR (1986) Iodine, a novel catalyst in carbohydrate reactions I. -isopropylidination of carbohydrates, Tetrahedron Letters 27, 3415–3416. [Google Scholar]
- (22).Lourenco EC, Maycock CD, and Rita Ventura M (2009) Synthesis of potassium (2R)-2-O-alpha-d-glucopyranosyl-(1-->6)-alpha-d-glucopyranosyl-2,3-dihydroxypropa noate a natural compatible solute, Carbohydrate Research 344, 2073–2078. [DOI] [PubMed] [Google Scholar]
- (23).Morris PE, Hope KD, and Kiely DE (1989) The Isomeric Composition of d-Ribo-Hexos-3-Ulose (3-Keto-D-Glucose) in Aqueous-Solution, Journal of Carbohydrate Chemistry 8, 515–530. [Google Scholar]
- (24).Jager M, Hartmann M, de Vries JG, and Minnaard AJ (2013) Catalytic regioselective oxidation of glycosides, Angewandte Chemie International Edition Engl 52, 7809–7812. [DOI] [PubMed] [Google Scholar]
- (25).Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Research 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Larkin A, and Imperiali B (2009) Biosynthesis of UDP-GlcNAc(3NAc)A by WbpB, WbpE, and WbpD: enzymes in the Wbp pathway responsible for O-antigen assembly in Pseudomonas aeruginosa PAO1, Biochemistry 48, 5446–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Thoden JB, and Holden HM (2010) Structural and functional studies of WlbA: A dehydrogenase involved in the biosynthesis of 2,3-diacetamido-2,3-dideoxy-D-mannuronic acid, Biochemistry 49, 7939–7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Vetter ND, Langill DM, Anjum S, Boisvert-Martel J, Jagdhane RC, Omene E, Zheng H, van Straaten KE, Asiamah I, Krol ES, Sanders DA, and Palmer DR (2013) A previously unrecognized kanosamine biosynthesis pathway in Bacillus subtilis, Journal of America Chemical Society 135, 5970–5973. [DOI] [PubMed] [Google Scholar]
- (29).Benavente R, Esteban-Torres M, Kohring GW, Cortes-Cabrera A, Sanchez-Murcia PA, Gago F, Acebron I, de las Rivas B, Munoz R, and Mancheno JM (2015) Enantioselective oxidation of galactitol 1-phosphate by galactitol-1-phosphate 5-dehydrogenase from Escherichia coli, Acta Crystallographica Section D Biological Crystallography 71, 1540–1554. [DOI] [PubMed] [Google Scholar]
- (30).Hobbs ME, Williams HJ, Hillerich B, Almo SC, and Raushel FM (2014) l-Galactose metabolism in Bacteroides vulgatus from the human gut microbiota, Biochemistry 53, 4661–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ng K, Ye R, Wu XC, and Wong SL (1992) Sorbitol dehydrogenase from Bacillus subtilis. Purification, characterization, and gene cloning, Journal of Biological Chemistry 267, 24989–24994. [PubMed] [Google Scholar]
- (32).Rodionova IA, Leyn SA, Burkart MD, Boucher N, Noll KM, Osterman AL, and Rodionov DA (2013) Novel inositol catabolic pathway in Thermotoga maritima, Environmental Microbiology 15, 2254–2266. [DOI] [PubMed] [Google Scholar]
- (33).Kim K, Kim HJ, Oh DK, Cha SS, and Rhee S (2006) Crystal structure of D-psicose 3-epimerase from Agrobacterium tumefaciens and its complex with true substrate d-fructose: a pivotal role of metal in catalysis, an active site for the non-phosphorylated substrate, and its conformational changes, Journal of Molecular Biology 361, 920–931. [DOI] [PubMed] [Google Scholar]
- (34).Uechi K, Sakuraba H, Yoshihara A, Morimoto K, and Takata G (2013) Structural insight into l-ribulose 3-epimerase from Mesorhizobium loti, Acta Crystallographica Section D Biological Crystallography 69, 2330–2339. [DOI] [PubMed] [Google Scholar]
- (35).Zhang L, Mu W, Jiang B, and Zhang T (2009) Characterization of d-tagatose-3-epimerase from Rhodobacter sphaeroides that converts d-fructose into d-psicose, Biotechnology Letters 31, 857–862. [DOI] [PubMed] [Google Scholar]
- (36).Taberman H, Parkkinen T, and Rouvinen J (2016) Structural and functional features of the NAD(P) dependent Gfo/Idh/MocA protein family oxidoreductases, Protein Science 25, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kingston RL, Scopes RK, and Baker EN (1996) The structure of glucose-fructose oxidoreductase from Zymomonas mobilis: an osmoprotective periplasmic enzyme containing non-dissociable NADP, Structure 4, 1413–1428. [DOI] [PubMed] [Google Scholar]
- (38).Carbone V, Hara A, and El-Kabbani O (2008) Structural and functional features of dimeric dihydrodiol dehydrogenase, Cell and Molecular Life Sciences 65, 1464–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kubiak RL, and Holden HM (2011) Combined structural and functional investigation of a C-3”-ketoreductase involved in the biosynthesis of dTDP-L-digitoxose, Biochemistry 50, 5905–5917. [DOI] [PubMed] [Google Scholar]
- (40).Namboori SC, and Graham DE (2008) Enzymatic analysis of uridine diphosphate N-acetyl-D-glucosamine, Analytical Biochemistry 381, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Maeda A, Adachi S, and Matsuno R (2001) Improvement of selectivity in 3-ketocellobiose production from cellobiose by Agrobacterium tumefaciens, Biochemical Engineering Journal 8, 217–221. [Google Scholar]
- (42).Stoppok E, Matalla K, and Buchholz K (1992) Microbial modification of sugars as building blocks for chemicals., Applied Microbiology and Biotechnology 36, 604–610. [DOI] [PubMed] [Google Scholar]
- (43).Bernaerts MJ, and De Ley J (1958) 3-Ketoglycosides, new intermediates in the bacterial catabolism of disaccharides, Biochimica et Biophysica Acta 30, 661–662. [DOI] [PubMed] [Google Scholar]
- (44).Bernaerts MJ, and De Ley J (1960) The structure of 3-ketoglycosides formed from disaccharides by certain bacteria, Journal of General Microbiology 22, 137–146. [DOI] [PubMed] [Google Scholar]
- (45).Kojima K, Tsugawa W, and Sode K (2001) Cloning and expression of glucose 3-dehydrogenase from Halomonas sp. alpha-15 in Escherichia coli, Biochemical and Biophysical Research Communications 282, 21–27. [DOI] [PubMed] [Google Scholar]
- (46).Morrison SC, Wood DA, and Wood PM (1999) Characterization of a glucose 3-dehydrogenase from the cultivated mushroom ( Agaricus bisporus ), Applied Microbiology and Biotechnology 51, 58–64. [Google Scholar]
- (47).Takeuchi M, Ninomiya K, Kawabata K, Asano N, Kameda Y, and Matsui K (1986) Purification and properties of glucoside 3-dehydrogenase from Flavobacterium saccharophilum, The Journal of Biochemistry 100, 1049–1055. [DOI] [PubMed] [Google Scholar]
- (48).Tsugawa W, Horiuchi S, Tanaka M, Wake H, and Sode K (1996) Purification of a marine bacterial glucose dehydrogenase from Cytophaga marinoflava and its application for measurement of 1,5-anhydro-d-glucitol, Applied Biochemistry and Biotechnology 56, 301–310. [DOI] [PubMed] [Google Scholar]
- (49).Zhang J, Chen W, Ke W, and Chen H (2014) Screening of a glucoside 3-dehydrogenase-producing strain, Sphingobacterium faecium, based on a high-throughput screening method and optimization of the culture conditions for enzyme production, Applied Biochemistry and Biotechnology 172, 3448–3460. [DOI] [PubMed] [Google Scholar]
- (50).Zhang JF, Zheng YG, Xue YP, and Shen YC (2006) Purification and characterization of the glucoside 3-dehydrogenase produced by a newly isolated Stenotrophomonas maltrophilia CCTCC M 204024, Applied Microbiology and Biotechnology 71, 638–645. [DOI] [PubMed] [Google Scholar]
- (51).Fensom AH, Kurowski WM, and Pirt SJ (2007) The use of ferricyanide for the production of 3-keto sugars by non-growing suspensions of Agrobacterium tumefaciens, Journal of Applied Chemistry and Biotechnology 24, 457–467. [Google Scholar]
- (52).Hayano K, and Fukui S (1967) Purification and properties of a 3-ketosucrose forming enzymes from the cells of A. tumefaciens, Journal of Biological Chemistry. 242, 3655–3672. [PubMed] [Google Scholar]
- (53).Schuerman PL, Liu JS, Mou H, and Dandekar AM (1997) 3-Ketoglycoside-mediated metabolism of sucrose in E. coli as conferred by genes from Agrobacterium tumefaciens, Applied Microbiology and Biotechnology 47, 560–565. [DOI] [PubMed] [Google Scholar]
- (54).Hayano K, and Fukui S (1970) Alpha-3-ketoglucosidase of Agrobacterium tumefaciens, Journal of Bacteriology 101, 692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Hayano K, Tsubouchi Y, and Fukui S (1973) 3-Ketoglucose reductase of Agrobacterium tumefaciens, Journal of Bacteriology 113, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ampomah OY, Avetisyan A, Hansen E, Svenson J, Huser T, Jensen JB, and Bhuvaneswari TV (2013) The thuEFGKAB operon of Rhizobia and Agrobacterium tumefaciens codes for transport of trehalose, maltitol, and isomers of sucrose and their assimilation through the formation of their 3-keto derivatives, Journal of Bacteriology 195, 3797–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Nakahara K, Kitamura Y, Yamagishi Y, Shoun H, and Yasui T (1994) Levoglucosan dehydrogenase involved in the assimilation of levoglucosan in Arthrobacter sp. I-552, Bioscience, Biotechnology, and Biochemistry 58, 2193–2196. [DOI] [PubMed] [Google Scholar]
- (58).Zhang L, Radziejewska-Lebrecht J, Krajewska-Pietrasik D, Toivanen P, and Skurnik M (1997) Molecular and chemical characterization of the lipopolysaccharide O-antigen and its role in the virulence of Yersinia enterocolitica serotype O:8, Molecular Microbiology 23, 63–76. [DOI] [PubMed] [Google Scholar]
- (59).Mengele R, and Sumper M (1992) Gulose as a constituent of a glycoprotein, FEBS Letters 298, 14–16. [DOI] [PubMed] [Google Scholar]
- (60).Stuart ES, Tirrell SM, and MacDonald AB (1987) Characterization of an antigen secreted by Chlamydia-infected cell culture, Immunology 61, 527–533. [PMC free article] [PubMed] [Google Scholar]
- (61).Ravenscroft N, Walker SG, Dutton GG, and Smit J (1991) Identification, isolation, and structural studies of extracellular polysaccharides produced by Caulobacter crescentus, Journal of Bacteriology 173, 5677–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.