

Abstract
KCNT1 gene encodes a sodium-gated potassium channel subunit that plays an important role in regulating excitability in neurons. Quinidine is a partial antagonist of this channel. We report the clinical characteristics of two south Indian children with KCNT1-related epileptic encephalopathy. Both of them had very high seizure burden which were resistant to antiepileptic and dietary therapy. Pharmacological response to quinidine in these children is described. Case 1 had 30% reduction in seizure burden at 20 mg/kg/day and 80% reduction at 36 mg/kg/day; case 2 had 30% reduction at 20 mg/kg/day. Serial electrocardiography was used to monitor the cardiotoxicity. Serum quinidine levels were not measured due to nonavailability. A critical review on the current status of targeted treatment of KCNT1-related epileptic encephalopathies with quinidine is attempted.
Keywords: Epileptic encephalopathy, KCNT1, quinidine
INTRODUCTION
Mutations in KCNT1 gene have been implicated in malignant migrating focal seizures of infancy (MMFSI), autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) and other types of early-onset epileptic encephalopathies. KCNT1 related early infantile epileptic encephalopathies may usually present with multifocal migrating seizures.[1,2,3,4] The seizure burden is usually very high with multiple seizures occurring every hour, which remain resistant to almost all available antiepileptic drugs (AEDs) and also to dietary therapy. The seizure frequency may come down as the affected children become older. However, almost all of them develop significant developmental delay.
Quinidine is a stereoisomer of quinine, which is in clinical use for the last several decades as an antimalarial drug and also as a Class I antiarrhythmic drug. Quinidine has shown to reversibly block rodent KCNT1 channels[5] and is considered to be a potential treatment option for this resistant epileptic encephalopathy. There are some recent case reports describing the efficacy of quinidine in children with KCNT1 gain of function mutation-related resistant epileptic encephalopathies.[2,3,4] The usual dosing range of quinidine is 15–60 mg/kg/day. However, the exact dosing in early infancy, for use as a KCNT1 inhibitor is yet to be established. In view of its potential cardiac effects, children need careful monitoring with repeated ECG evaluation for prolongation of corrected QT interval (QTc), and if possible repeated estimation of serum levels. Other side effects included gastrointestinal intolerance, hepatic dysfunction, leukopenia, cinchonism, and hemolytic anemia.
We report the clinical characteristics of two south Indian children with KCNT1-related epileptic encephalopathy. Both of them had very frequent focal seizures, which were resistant to antiepileptic and dietary therapy. Therapeutic response to quinidine in these children is described in detail. A detailed literature search was done on PubMed to identify all reported cases of KCNT1-related epileptic encephalopathies after 2000. The available literature on the off-label use of quinidine in KCNT1-related epileptic encephalopathies is critically analyzed. This case series has been approved by the Institutional Ethics Committee.
CASE REPORTS
Case 1
4.5 months old boy, with normal perinatal period had his 1st episode of seizure at 15 days of life, which was characterized by clonic jerking of the whole body with blinking of eyes. He was evaluated at a local hospital after 1 month with increased seizure frequency, his magnetic resonance imaging (MRI) and electroencephalography (EEG) were reported to be normal, and he was started on oral phenytoin, with which he had transient improvement. The seizure soon recurred and oral levetiracetam was added and hiked up to 27 mg/kg/day. Later seizure semiology changed to staring look followed by versive head deviation to left with tonic posturing of left upper and lower limbs, which used to last for 1–2 min. He was having 15–20 seizures every day. At 4.5 months of age, the baby was brought to our institute for further evaluation and management. On examination, he was active and alert. He had attained only partial head control with occasional social smile. Video EEG (VEEG) recorded multifocal interictal epileptiform abnormalities and several electroclinical events suggestive of focal seizures of right temporal origin. In view of the persistent onset of seizures from the right temporal region, a resistant structural focal epilepsy due to a cryptogenic lesion was also considered at this stage. 3T MRI brain with contrast did not reveal any structural lesions. The following AEDs were administered: Oxcarbazepine (25.86 mg/kg/day), levetiracetam (43.8 mg/kg/day), clobazam (1.02 mg/kg/day), and phenobarbitone (2.7 mg/kg/day). The frequency of events decreased substantially for initial few days which was followed later by re-emeregence. Repeat VEEG showed very frequent focal seizures arising independently and sequentially from both posterior head regions with frequent migration from one focus to another suggesting MMFSI. Cerebrospinal fluid (CSF) study done was normal (CSF glucose 67.3 mg/dl, CSF protein 45.1 mg/dl, total cells 0), plasma ammonia 56.3 umol/L and plasma lactate 1.2 mmol/L. CSF lactate was 1.3. His seizures continued to be refractory with multiple focal seizures occurring every hour. At 8 months of age, he was initiated on low glycemic index diet according to our institutional protocol and his seizure frequency minimally reduced. Next-generation sequencing identified a heterozygous missense variation in exon 25 of the KCNT1 gene (chr9:138675877; G>G/A; Depth: ×41) that results in the amino acid substitution of glutamine for arginine at codon 950 (p. Arg950GIn; ENST0000037 j757). He continued to have 5–10 seizure events per day and was readmitted at 10 months of age for initiation of quinidine. Oral quinidine (Quinidine sulfate 200 mg tablet, Sandoz) was initiated at 5 mg/kg/day in three divided doses, with serial ECG monitoring for prolonged QTc. Existing AEDs were continued. The dose of oral quinidine was hiked up weekly by around 5 mg/kg, to a maximum dose of 36 mg/kg/day. His seizures came down to >60% of baseline seizure frequency by the time the quinidine dosage reached 20 mg/kg/day. QTc was within acceptable limits even with 30 mg/kg/day, as mentioned in Table 1. With the reduction of seizures, there was an overall improvement in the developmental status. At the last follow-up at 13 months of age, his developmental age was around 6 months; he was able to control his head, was saying monosyllables, had eye contact, and was responding to verbal cues. He was on 36 mg/kg/day of quinidine with around 80% reduction in seizure frequency from baseline. Serum AED levels were not done either before or after starting quinidine.
Table 1.
Corrected QT interval during the titration of quinidine (case 1 and 2)
| Day of quinidine | Dose of quinidine (mg/kg/day) | QTc (ms) | Efficacy and outcome with quinidine |
|---|---|---|---|
| Case 1 | |||
| 1 | 5 | 437 | |
| 4 | 10 | 433 | |
| 13 | 15 | 358 | |
| 20 | 20 | 351 | 30% seizure reduction |
| 57 | 25 | 403 | |
| 75 | 25 | 397 | |
| 89 | 26 | 376 | |
| 103 | 26 | 384 | |
| 123 | 31 | 386 | |
| 135 | 36 | 403 | 80% seizure reduction |
| Case 2 | |||
| 1 | 5 | 390 | |
| 8 | 10 | 405 | |
| 15 | 15 | 380 | |
| 29 | 20 | 378 | 30% seizure reduction |
QTc=Corrected QT interval
Case 2
This case was a 2-month-old baby boy, born to parents of nonconsanguineous marriage. Prenatal history was significant for polyhydramnios at the 7th month of gestational age and administration of injection progesterone for per vaginal bleed. The baby was born at term normally. From day 10 of life, mother noticed focal motor seizures which increased in frequency and severity over the subsequent weeks. He was brought to our hospital at the age of 2 months. He had not attained age-appropriate developmental milestones, and on examination, he was floppy, not fixing and following light and had complete head lag along with truncal hypotonia and brisk reflexes. VEEG showed potentially epileptiform interictal abnormalities over right central region. Three electroclinical and two electrographic events were recorded suggestive of focal seizures of right central onset. 3T MRI brain did not reveal any structural lesions. After optimizing AEDs; levetiracetam (26 mg/kg/day), clobazam (0.94 mg/kg/day), phenobarbitone (4.5 mg/kg/day), sodium valproate (42 mg/kg/day), lacosamide (4.25 mg/kg/day), and topiramate (5.3 mg/kg/day), seizure frequency decreased initially for a few days. Repeat VEEG recorded multifocal interictal epileptiform abnormalities and very frequent focal seizures with migrating character arising from multiple foci [Figures 1–4]. At 4 months of age, he was initiated on ketogenic diet according to the hospital protocol, and his seizure frequency reduced for a brief period with re-emergence later. Next-generation sequencing identified a heterozygous missense variation in exon 24 of the KCNT1 gene (chr9:138671275; G>G/A; Depth: 136x) that results in the amino acid substitution of Threonine for Alanine at codon 934 (p. Ala934Thr; ENST00000371757). Oral quinidine (Quinidine sulfate 200 mg tablet, Sandoz) was initiated at a dose of 5 mg/kg/day, with serial ECG monitoring of QTc interval. Existing AEDs were continued. Dose of oral quinidine was hiked up by around 5 mg/kg every week, with weekly monitoring of ECGs. At the dose of 20 mg/kg/day, his seizures came down to around 70% from baseline. However, during the last follow-up at 7 months of age, he had only 30% reduction in seizure frequency from baseline on the same dose of quinidine. QTc was within accepted limits [Table 1]. Serum quinidine levels were not checked in view of the lack of availability.
Figure 1.
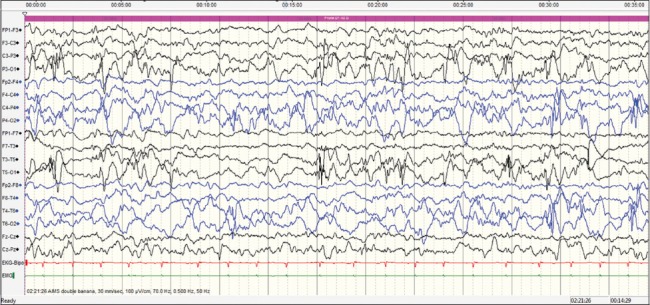
Case 2 interictal electroencephalography showing multifocal, predominantly posterior spike and waves
Figure 4.
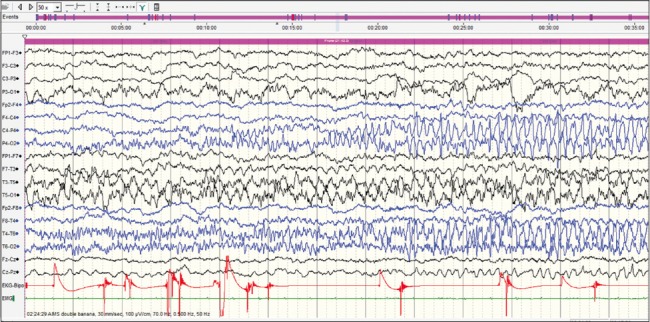
Three minute after the onset. Ictal rhythm is seen, migrating to the right posterior head region
Figure 2.
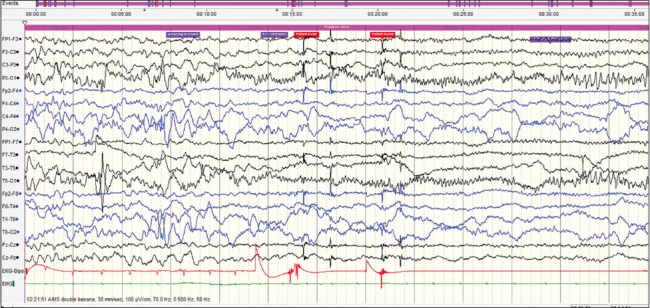
Ictal onset over the left occipital region
Figure 3.
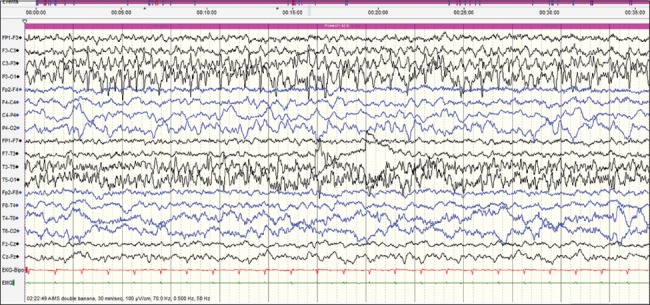
Sixty seconds after onset, well-developed ictal rhythm over the left posterior head regions
DISCUSSION
The initial experience of quinidine in two south Indian children with KCNT1-related early infantile epileptic encephalopathy is reported here. Both the children had the onset of focal seizures in the neonatal period with a crescendo increase in seizure frequency in the initial few months. Initially, the seizures had a more stable focal network origin, raising the clinical suspicion of an undetected focal cortical lesion. However, later during the course, multifocal origin and migrating nature of the focal seizures became more apparent. The seizures were refractory to all available AED and dietary therapy. Targeted treatment with quinidine was tried based on earlier clinical reports.[3,4]
KCNT1 gene encodes a sodium-gated potassium channel subunit, which plays an important role in regulating excitability in neurons.[6] KCNT1 channels are highly expressed in many regions of the mammalian brain, including the frontal and piriform cortices.[7] Missense mutations in KCNT1 gene result in malfunction of sodium-activated potassium channel, resulting in epileptogenesis. Heterozygous KCNT1 mutations had initially described in specific epileptic syndromes such as ADNFLE,[8] MMFSI,[9] and leukoencephalopathy and severe epilepsy.[10] However, a spectrum of clinical phenotypes encompassing resistant focal epilepsies, psychiatric disorders, and early onset epileptic encephalopathies was subsequently being reported. Overall, the developmental and seizure outcomes were less favorable in most of the reported cases with earlier onset in infancy.
Quinidine, a stereoisomer of quinine, is in clinical use for the past several decades as an antimalarial drug and also as a Class I antiarrhythmic drug. On experimental studies, quinidine has shown to be a partial antagonist of KCNT1 channel in addition to alleviating the effects of KCNT1 activating mutation, namely R428Q.[3] Quinidine, in clinical use for the treatment of cardiac arrhythmias for decades, reversibly blocks rodent KCNT1 channels.[5,11] However, in two recent cases of MMFSI due to KCNT1 mutations reported from USA, quinidine resulted in decreased seizure frequency or freedom from seizures along with improved psychomotor development.[3,4] In another case report from Japan, quinidine tried for West syndrome with KCNTI mutation, was found to be effective.[2] However, several subsequent reports failed to demonstrate a major efficacy. Chong et al.[12] reported seizure frequency of 103 + 27.7 per month after quinidine administration, compared to 106 + 13.3 for the same period before. Madaan et al.[13] reported no appreciable reduction in seizure frequency after quinidine in a 6-month-old infant with KCNT1 mutation. The initiation of quinidine at a later age and administration in electroclinical syndromes other than MMFSI are considered to be some of the reasons for the failure of quinidine.[12]
Quinidine is not commercially available in India now. Both the children reported here got the quinidine preparations from overseas. There was an appreciable benefit with improvement in seizure frequency after the introduction of quinidine in both the cases. However, the first child had a better response compared to the 2nd one and neither became completely seizure free. Both of them tolerated the drug well and were continuing the drug at the time of the last contact. Serum drug level monitoring was not done due to the lack of availability.
The recent case reports available in the literature on the usage of quinidine in KCNTI encephalopathy is listed in Table 2.
Table 2.
Use of quinidine in KCNT1 mutation
| Case report | Year of publication | Patient characteristics | Presentation | Number of patient | Mutation | Therapeutic response |
|---|---|---|---|---|---|---|
| Bearden et al.[3] | 2014 | 3 years | Seizure onset at 10 weeks of age | 1 | KCNT1 (c.1283G>A; p.Arg428Gln) | Effective |
| Mikati et al.[4] | 2015 | 11 years/female, 3 years/male | 11-year female: Seizure onset at 18 months of age 3-year male: Seizure onset since neonatal age |
2 | 11-year female: de novo KCNT1 mutation (NM_020822.1:c.2386T>C; p.[Tyr796His], Y796H) 3-year male: De novo KCNT1 mutation (NM_020822.2:c.1887G>C; p.[Lys629Asn], K629N) |
Ineffective for 11-year female Effective for 3-year male, showed 80% seizure reduction |
| Chong et al.[12] | 2016 | 6 years | Seizure onset 6 weeks of age | 1 | Heterozygous de novo KCNT1 R428Q mutation | Ineffective |
| Fukuoka et al.[2] | 2017 | 2.5 years | Seizure onset at 5 month of age | 1 | KCNT1 Mutation (NM_020822.2:c.1955G>T, NP_065873.2:p.G652V) |
Effective |
| Madaan et al.[13] | 2017 | 2 months/boy | Seizure onset day 3 | 1 | Heterozygous missense mutation in exon 10 of KCNT1 gene (chr9:138650308; c. 808C>C/G (p.Q270E)) | Ineffective |
CONCLUSION
KCNT1 related early infantile epileptic encephalopathy is a really difficult to treat epileptic syndrome with a guarded neurodevelopmental outcome. There is a definite need to develop better therapeutic options for this syndrome. This case series adds to the growing literature on the therapeutic outcome of quinidine in KCNT1 related early infantile epileptic encephalopathies. The response is variable and may be related to the type of mutation. Other compounds such as bryostatin-1, which inhibit the increased potassium currents are also currently under investigation in these disorders.[14] Large multicenter registries are needed to prospectively assess the impact of these novel therapies.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
What is known?
Gain-of-function mutations in KCNT1 gene cause early onset infantile epileptic encephalopathies with resistant focal seizures, which might have a migrating character. Targeted treatment with quinidine is reported to benefit these children.
What is new?
We report the first clinical series of targeted treatment of KCNT1-related resistant epileptic encephalopathy with quinidine from India. The available literature on the off-label use of quinidine in KCNT1-related epileptic encephalopathies is critically analyzed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Ohba C, Kato M, Takahashi N, Osaka H, Shiihara T, Tohyama J, et al. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia. 2015;56:e121–8. doi: 10.1111/epi.13072. [DOI] [PubMed] [Google Scholar]
- 2.Fukuoka M, Kuki I, Kawawaki H, Okazaki S, Kim K, Hattori Y, et al. Quinidine therapy for west syndrome with KCNTI mutation: A case report. Brain Dev. 2017;39:80–3. doi: 10.1016/j.braindev.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM, et al. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol. 2014;76:457–61. doi: 10.1002/ana.24229. [DOI] [PubMed] [Google Scholar]
- 4.Mikati MA, Jiang YH, Carboni M, Shashi V, Petrovski S, Spillmann R, et al. Quinidine in the treatment of KCNT1-positive epilepsies. Ann Neurol. 2015;78:995–9. doi: 10.1002/ana.24520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang B, Gribkoff VK, Pan J, Damagnez V, Dworetzky SI, Boissard CG, et al. Pharmacological activation and inhibition of slack (Slo2.2) channels. Neuropharmacology. 2006;51:896–906. doi: 10.1016/j.neuropharm.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharjee A, Kaczmarek LK. For K+channels, Na+is the new Ca2+ Trends Neurosci. 2005;28:422–8. doi: 10.1016/j.tins.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the slack potassium channel in the rat central nervous system. J Comp Neurol. 2002;454:241–54. doi: 10.1002/cne.10439. [DOI] [PubMed] [Google Scholar]
- 8.Sarah EH, Katherine RS, Melanie B, Lino N. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44:1188–90. doi: 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- 9.Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44:1255–9. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanderver A, Simons C, Schmidt JL, Pearl PL, Bloom M, Lavenstein B, et al. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr Neurol. 2014;50:112–4. doi: 10.1016/j.pediatrneurol.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK, et al. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci. 2003;23:11681–91. doi: 10.1523/JNEUROSCI.23-37-11681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong PF, Nakamura R, Saitsu H, Matsumoto N, Kira R. Ineffective quinidine therapy in early onset epileptic encephalopathy with KCNT1 mutation. Ann Neurol. 2016;79:502–3. doi: 10.1002/ana.24598. [DOI] [PubMed] [Google Scholar]
- 13.Madaan P, Jauhari P, Gupta A, Chakrabarty B, Gulati S. A quinidine non responsive novel KCNT1 mutation in an Indian infant with epilepsy of infancy with migrating focal seizures. Brain Dev. 2018;40:229–32. doi: 10.1016/j.braindev.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Delanty N, Cavallleri G. Genomics-guided precise anti-epileptic drug development. Neurochem Res. 2017;42:2084–8. doi: 10.1007/s11064-017-2312-y. [DOI] [PubMed] [Google Scholar]